
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral spirosiladiphosphines: ligand development and applications in Rh-catalyzed asymmetric hydrosilylation/cyclization of 1,6-enynes with enhanced reactivity†
Fei
Hou
ab,
Minjie
Liu
ab,
Tong
Ru
ab,
Zequn
Tan
ab,
Yingtang
Ning
 *ab and
Fen-Er
Chen
*ab
*ab and
Fen-Er
Chen
*ab
aEngineering Center of Catalysis and Synthesis for Chiral Molecules, Fudan University, 200433 Shanghai, China. E-mail: ningyt@fudan.edu.cn; rfchen@fudan.edu.cn
bShanghai Engineering Center of Industrial Catalysis for Chiral Drugs, 200433 Shanghai, China
First published on 28th March 2023
Abstract
Spirodiphosphines have been successfully applied in various asymmetric catalytic transformations. However, controlling the coordinating conformations by the direct displacement of the spiro atom remains elusive. Herein, we report the application of Si-centered spirodiphosphine (Si-SDP) ligands in the enantioselective hydrosilylation/cyclization of 1,6-enynes. The Si-SDPs showed superior reactivity to existing C2-symmetric diphosphines, allowing the generation of a range of chiral pyrrolidines with high yields and enantioselectivity (up to 96% yield and 92% ee) at room temperature with low catalyst loading. The mechanistic observations were consistent with the modified Chalk–Harrod mechanism, and the high reactivity of Si-SDPs was further leveraged for the room-temperature Rh-catalyzed hydrosilylation of alkynes.
Introduction
Chiral diphosphines with C2 symmetry represent a privileged class of ligands in asymmetric catalysis.1 These atropos ligands (Fig. 1), exemplified by BINAP (L1)2 and spirodiphosphines (SDPs, L2),3 help to improve catalyst activity and induce enantioselectivity. In particular, SDPs derived from the rigid 1,1′-spirobiindane backbone are extraordinarily successful.3 The larger bite angles, accompanied by the conformational rigidity and chemical robustness of SDPs,4 have contributed to tremendous advances in enantioselective transformations, including asymmetric hydrogenation,5 allylic alkylation,4b hydrosilylation/cyclization,6 and others.7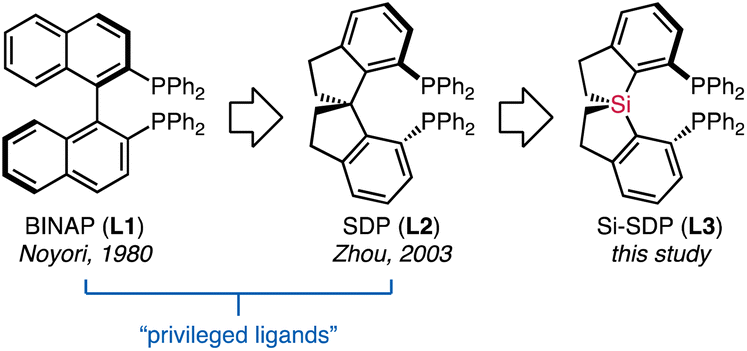 | ||
| Fig. 1 Examples of C2-symmetric diphosphines. | ||
Effective control over the ligand conformation in chelating complexes is considered a key concept for ligand design.8 Steric modulation of SDPs (L2) has primarily relied on the installation of additional substituents, resulting in ligands with various steric properties.9 On the other hand, as the rotation of the spirocyclic framework is restricted by the tetrahedral structure of the spiro carbon atom, replacing this conjunct atom would provide straightforward access to regulating the conformations. In this context, Wang reported the asymmetric synthesis of spirosilabiindanes by hydrosilylation.10 This protocol allows for the preparation of monodentate phosphoramidites, which promote asymmetric hydrogenation reactions of olefins and asymmetric intramolecular carboamination reactions.10a,11 However, diphosphine ligands based on the spirosilabiindanes backbone (Si-SDPs, L3) remain unexplored.21 The elongated C–Si bonds in these ligands should create a robust coordination space distinct from the carbon-centered L2. Our preliminary computations suggested that a significantly larger P–M–P bite angle than SDP (L2) can be achieved with Si-SDP L3 (Fig. S1†). We are interested in the effect of such regulated geometry, which is indulgent to the required orbital overlap during various common steps in organometallic chemistry.12
In the current study, we chose the Rh-catalyzed asymmetric silylcyclization of 1,6-enynes13–15 as an optimal reaction to challenge the effect of Si-SDPs (Scheme 1A). This reaction, originally developed by Ojima,13 is a synthetically useful transformation with the one-step formation of a carbon–carbon bond and a carbon–silicon bond. The asymmetric versions were independently reported by Widenhoefer17 and Zhou6 (Scheme 1A). The implementation of C2-symmetric diphosphines, especially Zhou's SDP ligand L2, delivered high levels of stereocontrol at 70 °C. However, mild yields and relatively high catalyst loadings (5 mol%) of the reaction limit its further applications. The reaction pathway was assumed to follow a modified Chalk–Harrod mechanism18 interrupted by β-migratory insertion into the Rh–C bond.13b We envisioned that the coordination of L3 with a large bite angle could accelerate the reductive elimination and migratory insertion steps12 in the catalytic cycle, as well as favoring the formation of the sterically crowded intermediate required for silylcyclization. Our results demonstrate the superior reactivity of Si-SDPs compared to the corresponding SDPs, affording chiral pyrrolidines16 that are ubiquitous in natural products and pharmaceuticals at room temperature with low catalyst loadings (Scheme 1B).
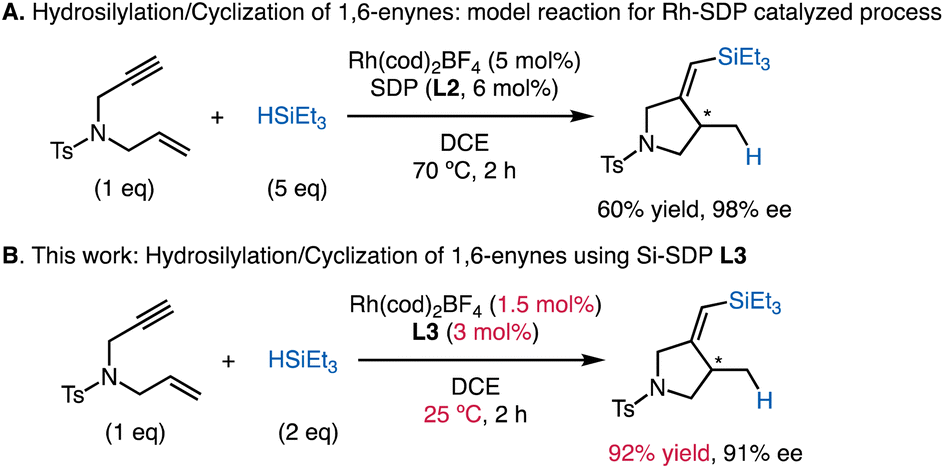 | ||
| Scheme 1 Rh-catalyzed hydrosilylation/cyclization. | ||
Results and discussion
Effect of ligands
To verify the catalyst activity generated from Si-centered spiro diphosphines, we prepared Si-SDPs (L3–L6, Table 1) that are relatively air-stable and easy to handle, based on reported procedures5a for other diphosphines.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reaction at 50 °C | Reaction at 25 °C | ||||||||
| Entry | Ligand | Yieldb (%) | ee%c (3a) | Entry | Ligand | Yieldb (%) | ee%c (3a) | ||
| 3a | 4a | 3a | 4a | ||||||
| a Reactions were performed with 1a (0.2 mmol), 2a (0.4 mmol), Rh(cod)2BF4 (1.5 mol%), and L1–L9 (3 mol%) in DCE (2.0 mL). b NMR yield using dibromomethane as an internal standard. c ee values were determined by chiral HPLC of the desilylated compounds. | |||||||||
| 1 | L1 | 30 | 67 | n.d. | 10 | L1 | — | — | — |
| 2 | L2 | 42 | — | 95 | 11 | L2 | 27 | — | 94 |
| 3 | L3 | 95 | — | 91 | 12 | L3 | 92 | — | 91 |
| 4 | L4 | 82 | — | 90 | 13 | L4 | 78 | — | 90 |
| 5 | L5 | 89 | — | 89 | 14 | L5 | 78 | — | 90 |
| 6 | L6 | 77 | — | 91 | 15 | L6 | 92 | — | 89 |
| 7 | L7 | — | 66 | — | 16 | L7 | — | — | — |
| 8 | L8 | — | 84 | — | 17 | L8 | — | — | — |
| 9 | L9 | — | 20 | — | 18 | L9 | — | — | — |
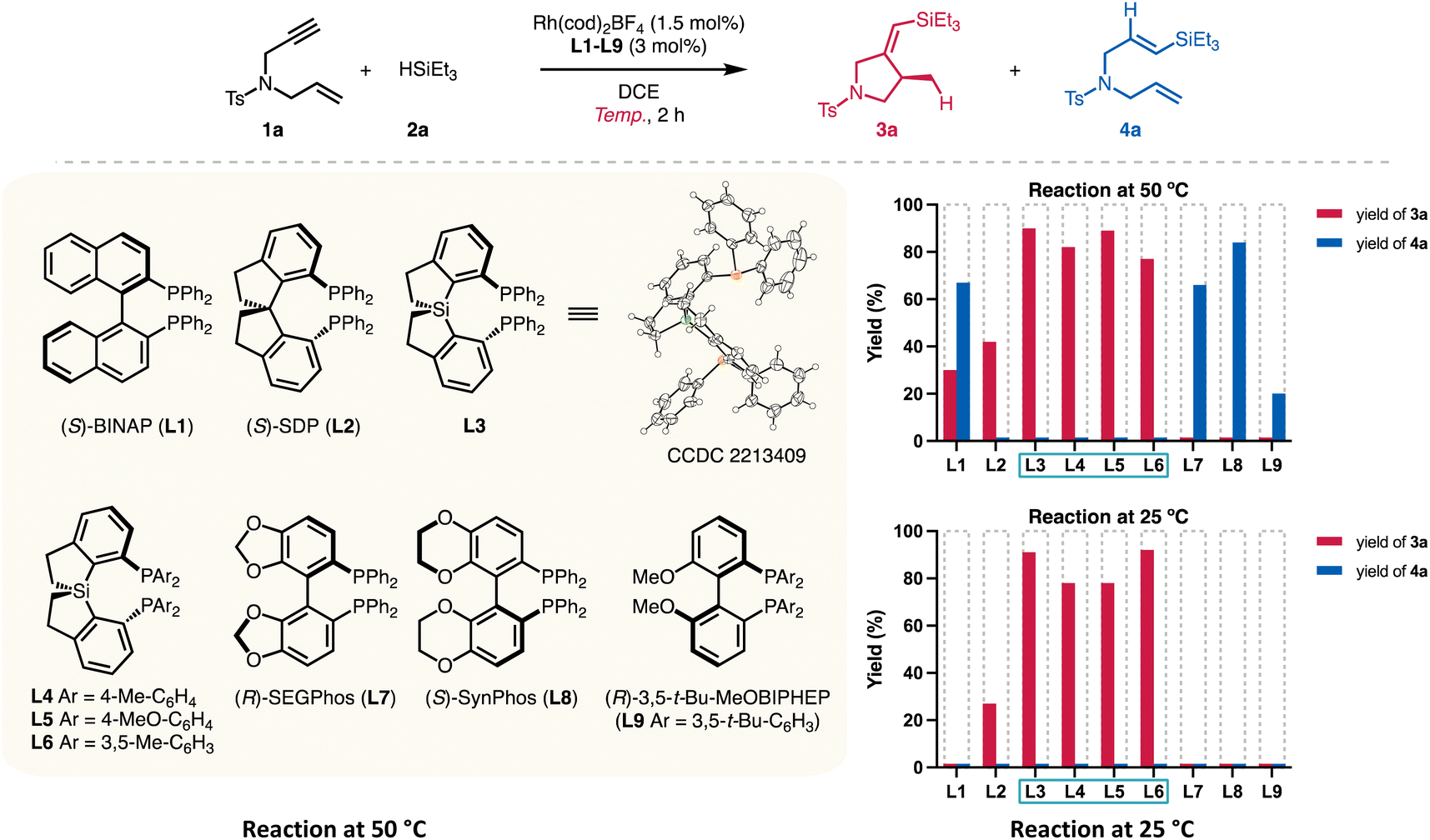
Different diphosphines L1–L9 were evaluated using the reaction of enyne 1a and triethylsilane (2a) (Table 1). As Zhou reported that SDP L2 displayed the highest yield (62%) at a reaction temperature of 50 °C,6 we first examined the performance of L1–L9 at the same temperature using a reduced amount of Rh(cod)2BF4 (1.5 mol%). The reaction using BINAP (L1) gave a mixture of the cyclized product 3a (30% yield) and the hydrosilylation product 4a (67% yield, entry 1). SDP (L2) showed good selectivity to silylcyclization, and 3a was obtained in 42% yield (entry 2). All the tested Si-centered spirodiphosphine ligands gave the desired 3a in high yields (77–90%) with high ee values (entries 3–6). Ligand L3, bearing phenyl groups on the phosphorous atoms, showed the highest yield (95%) and enantioselectivity (91%). Other C2-symmetric ligands L7–L9 failed to provide the cyclized products. Instead, the hydrosilylation product 4a was obtained in 20–84% yields (entries 7–9), indicating distinct chemoselectivity and high reactivity enabled by the spirosilabiindane structures.
A stark contrast in catalyst activity arose by lowering the reaction temperature (entries 10–18). Only the ligands L2–L6 possessing spirocyclic scaffolds promoted the conversions of 1a at 25 °C. SDP ligand L2 gave the cyclized product 3a in 27% yield at room temperature for 2 h (entry 11). On the other hand, full conversion was observed for L3 at 25 °C, giving 3a in 92% yield and 91% ee (entry 12). Other spirodiphosphines with a Si center atom (entries 13–15) also allowed the room-temperature silylcyclization reaction to occur with good yields (78–92%) and ee values (89–90% ee). Ligands L7–L9, which afforded hydrosilylation product 4a at 50 °C, exhibited marginal reactivity (entries 16–18).
Reaction scope
The substrate scope of the reaction enabled by the L3-modified Rh catalyst is outlined in Scheme 2. Chiral 1-sulfonyl pyrrolidines with different substituents on the sulfonyl groups reacted with HSiEt3 to afford the cyclized products in satisfactory yields. The enantioselectivity of the reaction was determined based on the desilylated products for improved handling. Alkyl (3b), chlorine (3c), and phenyl (3d) groups were well tolerated, and the products were isolated in 90–96% yields. We tested the electron-donating groups (3e and 3f) and electron-withdrawing groups (3g–3i) on the phenyl group, and there were no significant electronic effects of these substituents on the reaction yields and enantioselectivity. Including a methyl group (3j) or a bromide atom (3k) in the meta-position of the phenyl ring also results in the corresponding products with good yields. The reductively labile bromide survived under the current conditions. We explored two examples with multiple substituents (3l and 3m), and both reactions proceeded smoothly. Naphthyl-substituted enyne underwent smooth silylcyclization (3n, 75% yield, and 87% ee), and comparable yield and selectivity were observed by introducing an additional NMe2 substituent (3o). We also examined several heterocyclic substituents, and both the thiophenyl (3p) and pyridyl (3q) rings were compatible. The L3-modified catalyst was also successfully applied to substrates having aliphatic sulfonyl groups, forming 3r and 3s in good yields. We found that the substrate with internal alkynes reacted with triethoxysilane smoothly, affording 3t in 60% yield and 92% ee. Notably, no conversion was observed between the same enyne (1a) with triethylsilane 2a.The catalyst employing L3 is also suitable for the asymmetric synthesis of substituted cyclopentanes. The silylcyclization afforded 3u containing a quaternary carbon atom in 80% ee under the standard reaction conditions.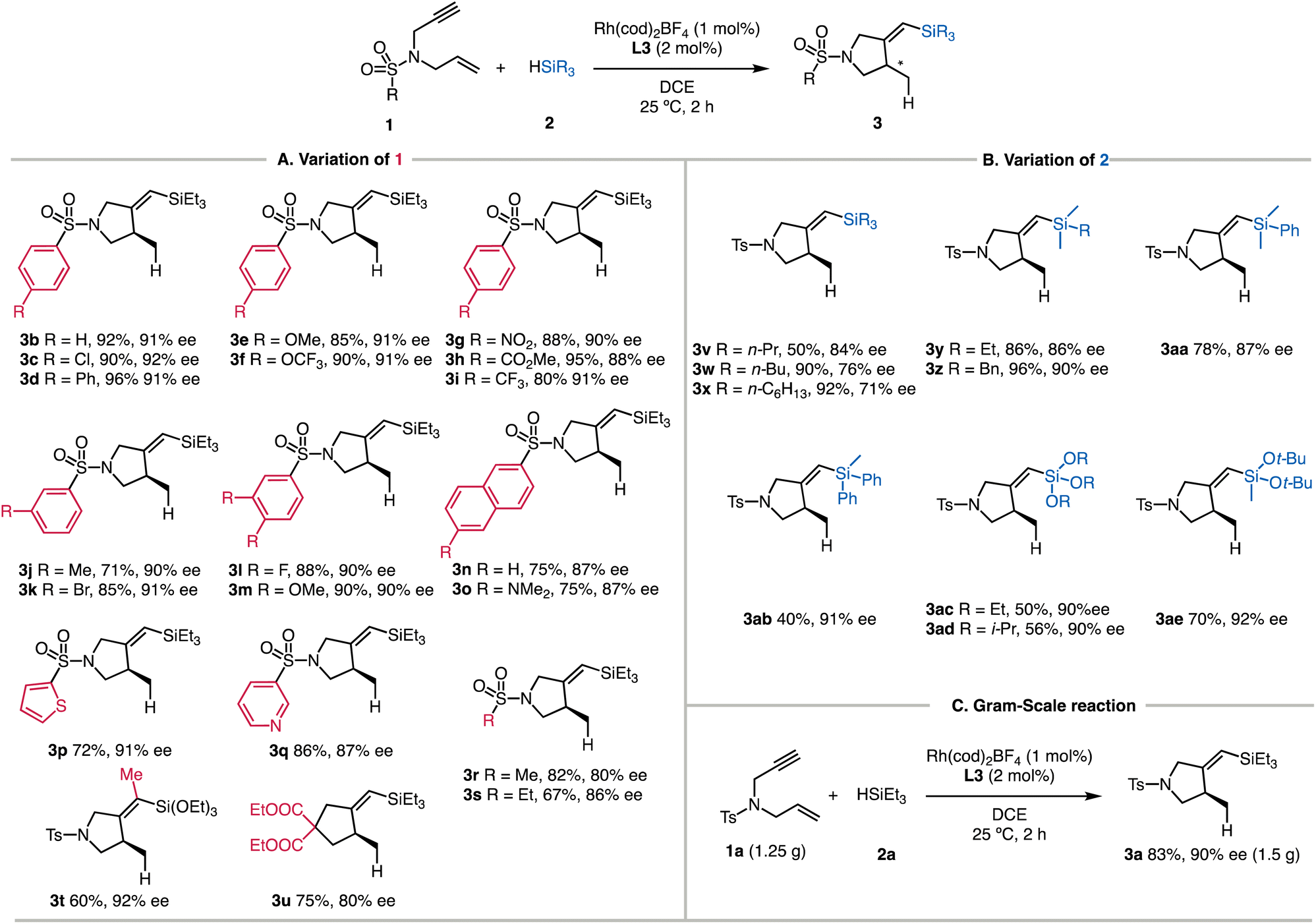 | ||
| Scheme 2 Scope of Rh-L3 catalyzed silylcyclization a–c.a Reactions were performed with 1a (0.2 mmol), 2a (0.4 mmol), Rh(cod)2BF4 (1.5 mol%), and L3 (3 mol%) in DCE (2.0 mL).b Yields of isolated 3.c ee values were determined by chiral HPLC of the desilylated compounds. | ||
Using enyne 1a, we next evaluated the influence of different silanes (Scheme 2B). Different alkyl substitution patterns on the silicon atom (3v–3z) posed no difficulty for the reactions. The benzyl group remained intact to deliver 3z in 96% yield and 90% ee. Attaching a phenyl group to the silicon atom also gave the corresponding 3aa in 78% yield and 87% ee, but the silane with multiple phenyl groups led to lower reactivity (3ab, 40% yield, and 91% ee). Trialkoxysilanes were also applicable, giving the desired 3ac and 3ad in moderate yields, which are facile counterparts for coupling reactions (ESI†).19 Sterically hindered silane also reacted well to generate 3ae. In order to capitalize on the superior reactivity, the scalability of the developed reaction is illustrated in Scheme 2C. When 1.25 g of 1a was employed, the silylcyclization with 2a produced 3a in 83% yield with 90% ee.
Mechanistic insights
To further probe the high activity of the Si-centered ligand L3, we performed several experiments concerning the mechanistic details. The hydrosilylation of alkyne 5 was feasible, but the addition of the H–Si bond to alkene 7 could not operate even at 50 °C (Scheme 3A). Thus, the hydrosilylation/cyclization process is likely triggered by the migratory insertion of the alkyne into the Rh–Si bond (modified Chalk–Harrod mechanism). Among the different Rh species tested, only the cationic Rh(cod)2BF4 and Rh(cod)2OTf showed conversions of the substrates (Scheme 3B). HRMS analysis also confirmed the formation of Rh-L3 and the subsequent coordination of 1a (Fig. S8†). In addition, we obtained the cryogenic structure of L3 coordinating with PdCl2 (Scheme 3D). The tailored P–Pd–P bite angle for L3 (101.0°) is larger than those of BINAP (L1, 92.7°) and SDP (L2, 96.0°) (Fig. S1†). We hypothesize that this large bite angle of L3 could be replicated in analogous Rh complexes, which would underlie the high reactivity of L3.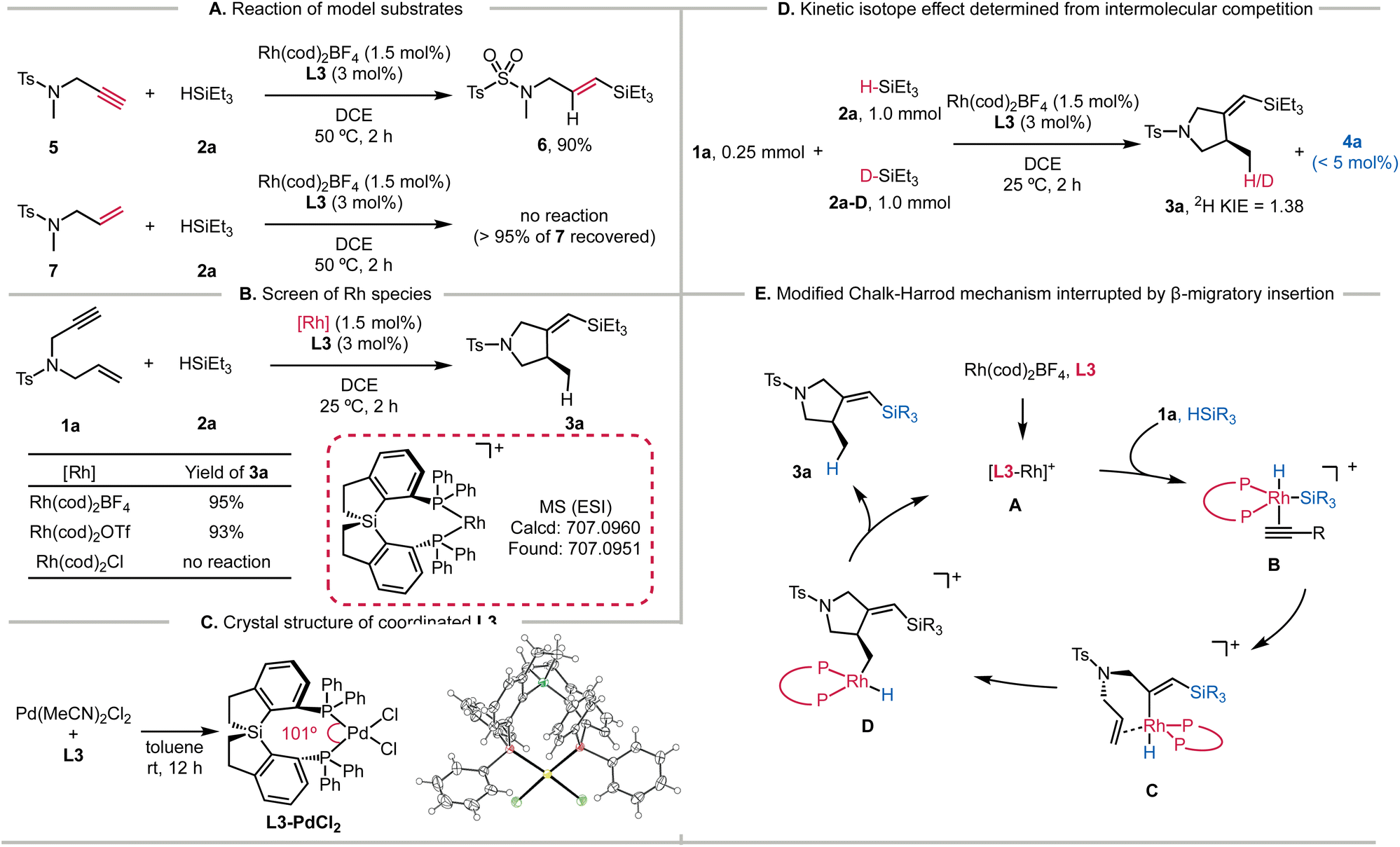 | ||
| Scheme 3 Mechanistic insights into Rh-L3 catalyzed silylcyclization. | ||
The deuterium labeling experiment suggested that the alkyne C–H bond remained intact during the enyne cyclization (Scheme S2†). Moreover, we observed a secondary deuterium kinetic isotope effect (2H KIE) of 1.38 from intermolecular competition experiments (Scheme 3D). Additionally, we measured the initial rates of the reaction by varying the concentrations of 1a and 2a, revealing that the reaction was first order to the concentration of 1a and zero order to that of 2a (Fig. S9†). As such, the oxidative addition of the H–Si bond across the cationic Rh species is not involved in the rate-determining step. Interestingly, a marginal amount of hydrosilylation product 4a was detected with excess HSiEt3 (2a) (Scheme 3D). These results are consistent with the catalytic cycle shown in Scheme 3E. Intermediate A formed by mixing L3 and Rh(cod)2BF4 reacted with silanes in the presence of 1a to generate intermediate B. The migratory insertion of the alkyne into the Rh–Si bond forms intermediate C, which is followed by cyclization achieved by the insertion of the alkene into the new Rh–C bond. The resulting Rh-alkyl intermediate D having the pyrrolidine skeleton is ready to produce 3a and regenerate the active Rh species Avia reductive elimination.
Application of Si-SDP to room-temperature hydrosilylation
Finally, we sought to examine the reactivity of Rh/L3-catalyzed hydrosilylation of alkynes, given the broad application of this transformation.20 Room-temperature hydrosilylation of 5 using L3 produced 6 in a 92% yield within 2 h (Scheme 4A). Using L2 gave a limited yield of 22% under the same conditions, which supports the superior reactivity of Si-centered L3 in Rh-catalyzed hydrosilylation. The higher reactivity of Si-SDP (L3) in this hydrosilylation process is qualitatively supported by our preliminary DFT calculations, using silane and acetylene as simplified substrates. While such simplifications would underestimate the energy barriers, the computational results suggested that the modified Chalk–Harrold mechanism is viable (Scheme 4B). For the process using SDP (L2), oxidative addition of the silane is followed by migratory insertion of the alkyne into the Rh–Si bond. The transition state of this migratory insertion step (TSC-2) has an energy barrier of 8.2 kcal mol−1; whereas, in the reaction employing Si-centered L3, the migratory insertion viaTSSi-2 is energetically less demanding (5.0 kcal mol−1). The P–M–P angle increased from 100.3° to 101.1° for L2 and from 107.1° to 111.8° for L3. The large bite angle of L3 would thus accelerate the migratory insertion to enable room-temperature hydrosilylation.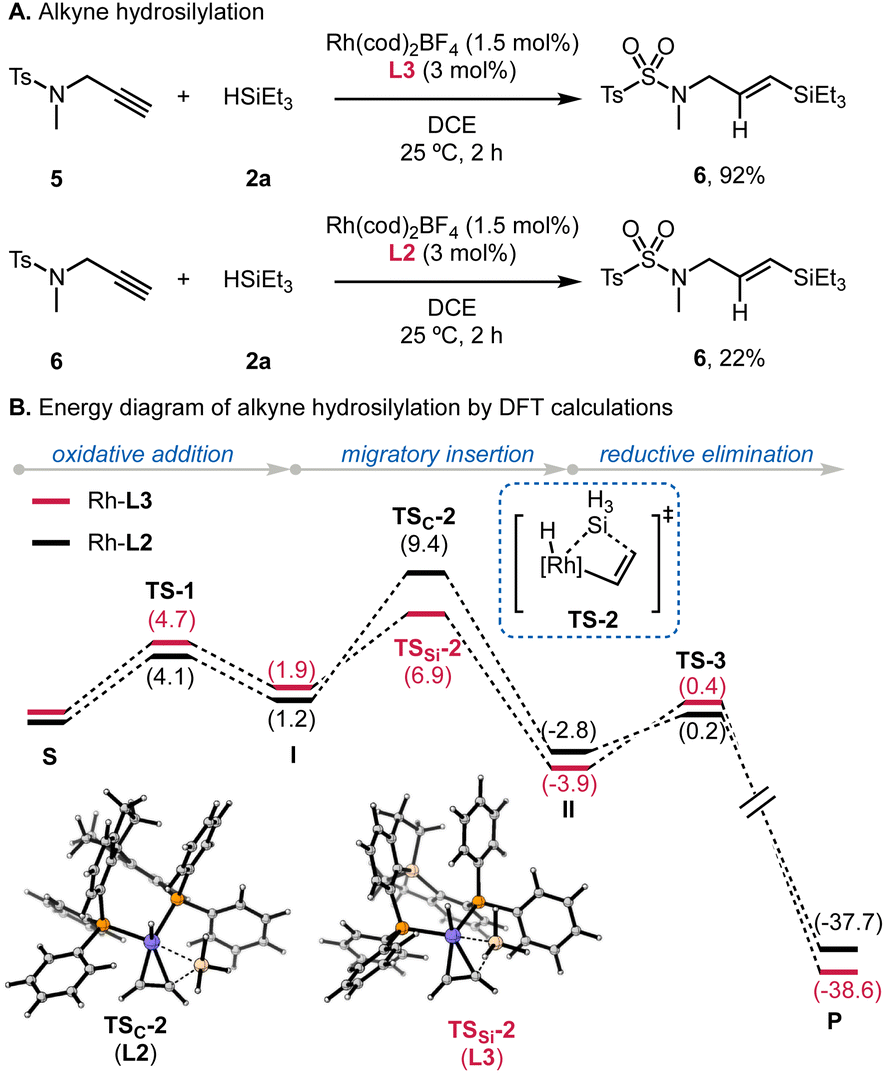 | ||
| Scheme 4 Room-temperature hydrosilylation of alkynes. | ||
Conclusions
We have synthesized a series of Si-centered spirodiphosphines (Si-SDPs), and their efficiency as ligands in Rh-catalyzed silylcyclization of 1,6-enynes was examined. These Si-SDPs suggested higher reactivity than other C2-symmetric ligands. The reaction tolerates a wide range of substrates, affording cyclized pyrrolidines in high yields and satisfactory enantioselectivity at room temperature. Further experiments revealed the crucial role of Si-SDP in accelerating the catalytic pathway composed of a modified Chalk–Harrod mechanism interrupted by β-migratory insertion. With the Si-centered spiro scaffolds of C2-symmetry, we anticipate that Si-SDPs will find broad applications in asymmetric catalysis.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
H. F., Y. N., and F.-E. C. conceived the research. H. F., M. L., T. R., and Z. T. performed the laboratory experiments. H. F. and Z. T. synthesized the ligands. H. F. and Y. N. conducted the DFT calculations. H. F., Y. N. and F. -E. C. prepared the manuscript with feedback from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (22077018) is gratefully acknowledged.Notes and references
- (a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed; (b) Q.-L. Zhou, Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim, 2011 CrossRef.
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- (a) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed; (b) J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581–593 CrossRef CAS PubMed; (c) K. Ding, Z. Han and Z. Wang, Chem.–Asian J., 2009, 4, 32–41 CrossRef CAS PubMed; (d) V. B. Birman, A. L. Rheingold and K.-C. Lam, Tetrahedron: Asymmetry, 1999, 10, 125–131 CrossRef CAS.
- (a) F. Ozawa, A. Kubo, Y. Matsumoto, T. Hayashi, E. Nishioka, K. Yanagi and K. Moriguchi, Organometallics, 1993, 12, 4188–4196 CrossRef CAS; (b) J.-H. Xie, H.-F. Duan, B.-M. Fan, X. Cheng, L.-X. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2004, 346, 625–632 CrossRef CAS.
- (a) J.-H. Xie, L.-X. Wang, Y. Fu, S.-F. Zhu, B.-M. Fan, H.-F. Duan and Q.-L. Zhou, J. Am. Chem. Soc., 2003, 125, 4404–4405 CrossRef CAS PubMed; (b) C. Liu, J.-H. Xie, Y.-L. Li, J.-Q. Chen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 593–596 CrossRef CAS PubMed.
- B.-M. Fan, J.-H. Xie, S. Li, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 1275–1277 CrossRef CAS PubMed.
- (a) X. Wang, Z. Han, Z. Wang and K. Ding, Acc. Chem. Res., 2021, 54, 668–684 CrossRef CAS PubMed; (b) J. Zeng, W. Fang, B. Lin, G.-Q. Chen and X. Zhang, Org. Lett., 2022, 24, 869–874 CrossRef CAS PubMed; (c) J.-W. Park, Z. Chen and V. M. Dong, J. Am. Chem. Soc., 2016, 138, 3310–3313 CrossRef CAS PubMed; (d) S. G. Sethofer, S. T. Staben, O. Y. Hung and F. D. Toste, Org. Lett., 2008, 10, 4315–4318 CrossRef CAS PubMed.
- (a) J. M. Crawford and M. S. Sigman, Synthesis, 2019, 51, 1021–1036 CrossRef CAS PubMed; (b) Q. Peng, F. Duarte and R. S. Paton, Chem. Soc. Rev., 2016, 45, 6093–6107 RSC; (c) J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Chem. Rev., 2014, 114, 2824–2880 CrossRef CAS PubMed.
- (a) X.-H. Huo, J.-H. Xie, Q.-S. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2007, 349, 2477–2484 CrossRef CAS; (b) S. Li, J.-W. Zhang, X.-L. Li, D.-J. Cheng and B. Tan, J. Am. Chem. Soc., 2016, 138, 16561–16566 CrossRef CAS PubMed; (c) G.-Q. Chen, B.-J. Lin, J.-M. Huang, L.-Y. Zhao, Q.-S. Chen, S.-P. Jia, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 8064–8068 CrossRef CAS PubMed; (d) A. J. Argüelles, S. Sun, B. G. Budaitis and P. Nagorny, Angew. Chem., Int. Ed., 2018, 57, 5325–5329 CrossRef PubMed; (e) L. Yin, J. Xing, Y. Wang, Y. Shen, T. Lu, T. Hayashi and X. Dou, Angew. Chem., Int. Ed., 2019, 58, 2474–2478 CrossRef CAS PubMed; (f) Z. Zheng, Y. Cao, Q. Chong, Z. Han, J. Ding, C. Luo, Z. Wang, D. Zhu, Q.-L. Zhou and K. Ding, J. Am. Chem. Soc., 2018, 140, 10374–10381 CrossRef CAS PubMed; (g) R. Zhang, S. Ge and J. Sun, J. Am. Chem. Soc., 2021, 143, 12445–12449 CrossRef CAS PubMed.
- (a) X. Chang, P.-L. Ma, H.-C. Chen, C.-Y. Li and P. Wang, Angew. Chem., Int. Ed., 2020, 59, 8937–8940 CrossRef CAS PubMed; (b) K. Tamao, K. Nakamura, H. Ishii, S. Yamaguchi and M. Shiro, J. Am. Chem. Soc., 1996, 118, 12469–12470 CrossRef CAS.
- L. Yang, W.-Q. Xu, T. Liu, Y. Wu, B. Wang and P. Wang, Chem. Commun., 2021, 57, 13365–13368 RSC.
- (a) J. E. Marcone and K. G. Moloy, J. Am. Chem. Soc., 1998, 120, 8527–8528 CrossRef CAS; (b) K. J. Cavell, Coord. Chem. Rev., 1996, 155, 209–243 CrossRef CAS; (c) P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530 RSC; (d) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed; (e) Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890–1901 RSC.
- (a) I. Ojima, R. J. Donovan and W. R. Shay, J. Am. Chem. Soc., 1992, 114, 6580–6582 CrossRef CAS; (b) I. Ojima, A. T. Vu, S.-Y. Lee, J. V. McCullagh, A. C. Moralee, M. Fujiwara and T. H. Hoang, J. Am. Chem. Soc., 2002, 124, 9164–9174 CrossRef CAS PubMed.
- For selected reviews and examples: (a) P. Cao, B. Wang and X. Zhang, J. Am. Chem. Soc., 2000, 122, 6490–6491 CrossRef CAS; (b) C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813–834 CrossRef CAS PubMed; (c) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884–4908 RSC; (d) I. D. G. Watson and F. D. Toste, Chem. Sci., 2012, 3, 2899–2919 RSC; (e) S. E. Denmark and J. H.-C. Liu, J. Am. Chem. Soc., 2007, 129, 3737–3744 CrossRef CAS PubMed; (f) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed; (g) B. M. Trost, Acc. Chem. Res., 1990, 23, 34–42 CrossRef CAS; (h) H.-Y. Jang and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 7875–7880 CrossRef CAS PubMed; (i) B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1985, 107, 1781–1783 CrossRef CAS; (j) K. H. Park, I. G. Jung, S. Y. Kim and Y. K. Chung, Org. Lett., 2003, 5, 4967–4970 CrossRef CAS PubMed; (k) T. Nishimura, T. Kawamoto, M. Nagaosa, H. Kumamoto and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 1638–1641 CrossRef CAS PubMed.
- For recent examples: (a) M. Dong, L. Qi, J. Qian, S. Yu and X. Tong, J. Am. Chem. Soc., 2023, 145, 1973–1981 CrossRef CAS PubMed; (b) Z. Chen, Y.-F. Li, S.-Z. Tan, Q. Ouyang, Z.-C. Chen, W. Du and Y.-C. Chen, Chem. Sci., 2022, 13, 12433–12439 RSC; (c) E. Hayashi, N. Akiyama, K. Kakiuchi, T. Kawai and T. Morimoto, Chem.–Asian J., 2023, 18, e202201241 CrossRef CAS PubMed; (d) S.-H. Hou, X. Yu, R. Zhang, C. Wagner and G. Dong, J. Am. Chem. Soc., 2022, 144, 22159–22169 CrossRef CAS PubMed; (e) X. Han, Q. Gaignard-Gaillard, P. Retailleau, V. Gandon and A. Voituriez, Chem. Commun., 2022, 58, 3043–3046 RSC.
- (a) T. Shi, G. Yin, X. Wang, Y. Xiong, Y. Peng, S. Li, Y. Zeng and Z. Wang, Green Synth. Catal., 2023, 4, 20–34 CrossRef; (b) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC.
- H. Chakrapani, C. Liu and R. A. Widenhoefer, Org. Lett., 2003, 5, 157–159 CrossRef CAS PubMed.
- (a) S. B. Duckett and R. N. Perutz, Organometallics, 1992, 11, 90–98 CrossRef CAS; (b) S. Sakaki, M. Sumimoto, M. Fukuhara, M. Sugimoto, H. Fujimoto and S. Matsuzaki, Organometallics, 2002, 21, 3788–3802 CrossRef CAS.
- S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982–994 CrossRef CAS PubMed.
- (a) X. Zeng, Chem. Rev., 2013, 113, 6864–6900 CrossRef CAS PubMed; (b) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (c) R. J. Hofmann, M. Vlatković and F. Wiesbrock, Polymers, 2017, 9, 534 CrossRef PubMed.
- During the preparation of this manuscript, the preparation of the diphosphine ligands was reported by Wang et al: P. Wang, Y. Wu, Spiro-dihydrobenzothiole diphosphine compound, and preparation method and application thereof, Chinese Pat., CN114478632, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2213409 and 2220312. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06349b |
| This journal is © The Royal Society of Chemistry 2023 |