
Regulating the electrical double layer to prevent water electrolysis for wet ionic liquids with cheap salts†
Jiedu
Wu
 ab,
Jinkai
Zhang
a,
Ming
Chen
*ac,
Jiawei
Yan
b,
Bingwei
Mao
b and
Guang
Feng
ac
ab,
Jinkai
Zhang
a,
Ming
Chen
*ac,
Jiawei
Yan
b,
Bingwei
Mao
b and
Guang
Feng
ac
aState Key Laboratory of Coal Combustion, School of Energy and Power Engineering, Huazhong University of Science and Technology (HUST), 430074 Wuhan, China. E-mail: mchen@hust.edu.cn
bState Key Laboratory of Physical Chemistry of Solid Surfaces, and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, 361005 Xiamen, China
cInstitute of Interdisciplinary Research for Mathematics and Applied Science, Huazhong University of Science and Technology (HUST), 430074 Wuhan, China
First published on 24th October 2023
Abstract
Hydrophobic ionic liquids (ILs), broadly utilized as electrolytes, face limitations in practical applications due to their hygroscopicity, which narrows their electrochemical windows via water electrolysis. Herein, we scrutinized the impact of incorporating cheap salts on the electrochemical stability of wet hydrophobic ILs. We observed that alkali ions effectively manipulate the solvation structure of water and regulate the electrical double layer (EDL) structure by subtly adjusting the free energy distribution of water in wet ILs. Specifically, alkali ions significantly disrupted the hydrogen bond network, reducing free water, strengthening the O–H bond, and lowering water activity in bulk electrolytes. This effect was particularly pronounced in EDL regions, where most water molecules were repelled from both the cathode and anode with the disappearance of the H-bond network connectivity along the EDL. The residual interfacial water underwent reorientation, inhibiting water electrolysis and thus enhancing the electrochemical window of wet hydrophobic ILs. This theoretical proposition was confirmed by cyclic voltammetry measurements, demonstrating a 45% enhancement in the electrochemical windows for salt-in-wet ILs, approximating the dry one. This work offers feasible strategies for tuning the EDL and managing interfacial water activity, expanding the comprehension of interface engineering for advanced electrochemical systems.
1. Introduction
Room-temperature ionic liquids (RTILs), lauded for their excellent thermal stability, non-volatility, non-flammability, and notably wide electrochemical windows, have been exceptionally used as electrolytes in diverse electrochemical energy storage (EES) devices,1–4 such as supercapacitors,2,3 batteries,5 and solar cells.6 However, there is still a gap between well-controlled laboratory systems and practical systems.3,7 One such challenge stems from their hygroscopic nature, where ILs will become wet when exposed to the atmosphere, and water in wet ILs is difficult to eradicate.8 This moisture absorption generally leads to narrowed electrochemical windows (EWs), owing to the electrolysis of water, which in turn hastens the performance degradation of EES devices.9–13Since the EES system performance is largely controlled by electrochemical interfaces, extensive studies have been dedicated to understanding the water effect on the IL mixtures/electrode interfaces.4,12–15 It has been established that the interfacial behavior of water molecules is governed by a combination of the electrostatic force which drives dipolar solvents to positions with intense electrical fields, the interaction of solvent molecules with their ionic surroundings, and the steric hindrance near the electrodes.9,12,16–18 By finely tuning the hydrophilic and hydrophobic properties of ILs, it has been revealed that water molecules in hydrophilic ILs are repelled from the negatively charged electrode, thereby maintaining the EWs. Conversely, in hydrophobic ILs, which are widely used as electrolytes in EES systems, water molecules are inclined to accumulate on charged electrode surfaces and be decomposed electrochemically, resulting in a narrowed EW and hampering their application in practical systems.19
A recent report suggested that incorporating lithium salts could boost the electrochemical stability of wet hydrophobic ILs by regulating the electrical double layer (EDL).20 Nevertheless, the scarcity of lithium in the Earth's crust (lower than 20 ppm, i.e., 0.002%) limits its viability as a long-term solution.21 As alternatives, abundant elements (e.g., sodium and potassium) hold great promise as additives to regulate the EDL. In this regard, we explored the effects of addition of cheap alkali salts (i.e., sodium and potassium salts) to wet hydrophobic 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)-imide ([Emim][TFSI]) with the aid of molecular dynamics (MD) simulations, density functional theory (DFT) calculations and cyclic voltammetry (CV) measurements. Our simulations and experiments found that the addition of cheap alkali salts has effectively modulated the EDL structure, thereby widening the electrochemical window of wet hydrophobic RTILs. This research elucidates the mechanisms underpinning EDL regulation and the expansion of the EWs, paving the way for the enhanced protection and application of ILs.
2. Methods
2.1. MD simulation
Molecular dynamics simulations were utilized to explore the impact of added salts on the wet ILs. Initially, we conducted MD simulations of mixtures of hydrophobic ILs, water, and salts in their bulk states with the objective of elucidating changes in the solvation structure. Subsequently, the same electrolytes were confined between two atomically flat graphite electrode surfaces to investigate the salt effect on the EDL structure change (ESI Fig. 1†). An all-atom model with unit charge was taken for hydrophobic ILs ([Emim][TFSI]);22,23 whereas water was represented via the SPC/E model.24 The OPLS force field was used for alkali ions;25 carbon atoms comprising electrodes were modeled using the force fields referenced in an earlier study.26 It is noteworthy that the explicit treatment of electrostatic interactions and polarization effects is crucial for comprehending the complex role of long-range strong electrostatics under heterogeneous conditions.27–29 However, implementing such treatment with polarizable force fields poses significant computational challenges. An alternative approach with a scaled-charge force field, which accounts for electronic polarization by scaling the charges on the ions, was also employed to investigate the effect of alkali salts on the solvation structure of water molecules.30,31 The scaling factor was set to 0.8, consistent with previous work.30,31 We ensured that the sizes of all the simulated systems were sufficiently extensive to reproduce the bulk-like behavior of the electrolytes in the region central to the two electrodes (ESI Fig. 1†). Detailed specifications pertaining to the number of species and inter-electrode distances for all the systems are provided in ESI Table 1.†All simulations were performed in the NVT ensemble with the MD package GROMACS.32 Temperature was controlled through the Nosé–Hoover thermostat33,34 at 333 K with a coupling constant of 1.0 ps. The cutoff distance for the van der Waals term was set at 1.2 nm through direct summation. Long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) method.35 We employed an FFT grid spacing of 0.1 nm in conjunction with cubic interpolation for computing the reciprocal space electrostatic interaction. A cutoff length of 1.2 nm was utilized for real-space electrostatic interaction. The leapfrog integration algorithm was used to solve the equations of motion with a time step of 2 fs. Specifically, in order to accurately account for the electrode polarization effects in the presence of electrolytes, the constant potential method (CPM) was implemented to allow the fluctuations of the charges on electrode atoms.36–38
To guarantee accuracy, the charges on the electrode were updated on the fly at every simulation step. Each simulation began with a heating phase at 500 K for 3 ns and then annealed to 333 K over a period of 2 ns, followed by another 10 ns to reach equilibrium. Thereafter, a 20 ns production was performed for analysis. Each case was repeated five times with varying initial configurations to certify the accuracy and robustness of the simulation results.
2.2. PMF calculation
The potential of mean force (PMF), representing the variation of free energy, was evaluated using the well-established umbrella sampling technique.39,40 Specifically, the molecule was pulled by applying a force to the graphene electrode along the z-axis to generate 45 configurations (Δz = 0.05 nm as measured between the centers-of-mass of the molecule and the graphene electrode). Each configuration was sampled over a period of 11 ns, with the first nanosecond utilized for equilibration. Then the PMF was calculated via weighted histogram analysis.39,402.3. Interaction energy calculation
The interaction energies were systematically evaluated using MD-generated trajectories. Specifically, the A–B interaction energy, decomposed into van der Waals and coulombic interactions, was calculated between component A and component B surrounding A within the first solvation shell. Such a methodological approach has been widely employed in previous simulation work.41,422.4. DFT calculations
To ascertain the oxidation stability of water, DFT calculations were performed with the Gaussian 09 program (revision, D.01).43 Initially, geometry optimizations for both alkali ion-free/bound water configurations were performed with the B3LYP hybrid functional and the 6-311G** basis set. Subsequently, single-point energy calculations were carried out for these optimized structures to obtain the highest occupied molecular orbital (HOMO) levels. To account for the implicit solvation effects of the IL environment, the SMD solvation model was employed in all calculations.44 Specifically, the index of refraction and the dielectric constants were set to 1.4225 and 12.25, respectively. The macroscopic surface tension was set to 56.13 cal mol−1 Å−2. Meanwhile, the fraction of non-hydrogen atoms that are aromatic carbon atoms and the fraction of non-hydrogen atoms that are electronegative halogen atoms were set to 0.1304 and 0.2609, respectively.44The coordination structure is important because the solvation structure modifies the energy levels. Thus, the HOMO levels within varying coordination environments have been investigated with the aid of MD + DFT calculations, which were widely used in determining the electrochemical stability of electrolytes.45,46 Specifically, we considered alkali ion-free/bound water molecules coordinated with 1 or 2 anions. We generated 50 distinct configurations randomly sampled from MD simulations for each coordination scenario. Subsequently, single-point energy calculations were conducted at the B3LYP/6-311G** level without further optimization. The average HOMO level over the 50 configurations was then calculated for each coordination structure in the SMD model. Then, to provide a more rigorous estimate of the oxidation stability of alkali ion-free/bound water, the vertical ionization potential (VIP) was calculated by evaluating the free energy change for alkali ion-free/bound water with different coordination structures upon losing one electron.46
2.5. Experimental materials and measurements
The ionic liquid [Emim][TFSI] (99%) utilized in this study was purchased from IoLiTec, and NaTFSI (99.5%) was procured from DoDoChem. Prior to each experimental procedure, ILs were meticulously purified with ultrapure water (Milli-Q, 18.2 MΩ cm) followed by vacuum drying at 80 °C for 24 hours in an argon-filled glovebox (Linde Industrial Gases, 99.999%) to eliminate residual water content to the greatest extent, and the resultant dry ILs were then utilized for further investigations.The capacity of pure IL [Emim][TFSI] to absorb water from the environment was evaluated under constant humidity conditions (48.20 ± 2.48% for [Emim][TFSI], as detailed in ESI Fig. 2†). Wet ILs were produced by the addition of ultrapure water to the ILs. Salt-in-wet ILs were formulated by incorporating ultrapure water and NaTFSI into the ILs in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, subsequently stirring the mixtures until a uniform solution was obtained. The water content in the samples was determined by employing a Karl Fischer coulometer (Metrohm, KF-831).47
:1, subsequently stirring the mixtures until a uniform solution was obtained. The water content in the samples was determined by employing a Karl Fischer coulometer (Metrohm, KF-831).47
Cyclic voltammetry measurements were performed in a glovebox using an Autolab electrochemical workstation (Eco Chemie, The Netherlands). Highly oriented pyrolytic graphite (HOPG) was used as a working electrode, with its surface prepared by a tape-peeling method to ensure cleanliness for electrochemical measurements.20 Silver wire and platinum wire were used as the reference electrode and counter electrode, respectively. Measurements were carried out in a sealed electrochemical cell to isolate it from external influences. CV measurements were typically concluded within a half-hour timespan, thereby rendering any alterations in the water content negligible during the measurement.
3. Results and discussion
3.1 Microscopic understanding of the salt effect on water in RTILs
We first investigated the alkali metal ion effect on the solvation structure of water in wet [Emim][TFSI] in the bulk state. Compared with the radial distribution function (RDF) of the mass-center of water and water in various electrolytes (Fig. 1a), it was evident that the solvation structure of water underwent significant disruption upon the addition of M+ (Na+, K+) ions. Instead, alkali cations form strong interactions with water molecules (Fig. 1b), breaking the interaction between H2O and H2O. Moreover, the solvation structure of water–water is destroyed to different extents by different alkali metal ions, leading to the formation of alkali ion-bound water (water is considered to be bound to M+ within the first solvation shell; otherwise, it is labeled as alkali ion-free water20,48–50). Notably, a sharp peak of RDF between H2O and H2O peaking at 0.25 nm was detected in wet ILs, which was separated into two peaks (0.25 nm and 0.35 nm) in the Na+-in-wet IL system. As for K+-in-wet ILs, though the peak location of RDF between H2O and H2O remained unchanged compared with wet ILs, the magnitude of RDF experienced a substantial decline. Consequently, the coordination number of one molecule of water around another molecule of water decreases significantly with the addition of alkali cations (Fig. 1a). This phenomenon was also supported by a sharp decrease in the proportion of alkali ion-free water (Fig. 1c), and the diminished amount of water clusters (ESI Fig. 3†), accompanied by a solvated state dominated by a solitary water molecule surrounding an alkali metal ion (ESI Fig. 4†). The disruption of the solvation structure of water molecules induced by the introduction of M+ (Na+, K+) ions mirrors a phenomenon analogous to that observed with lithium ions.20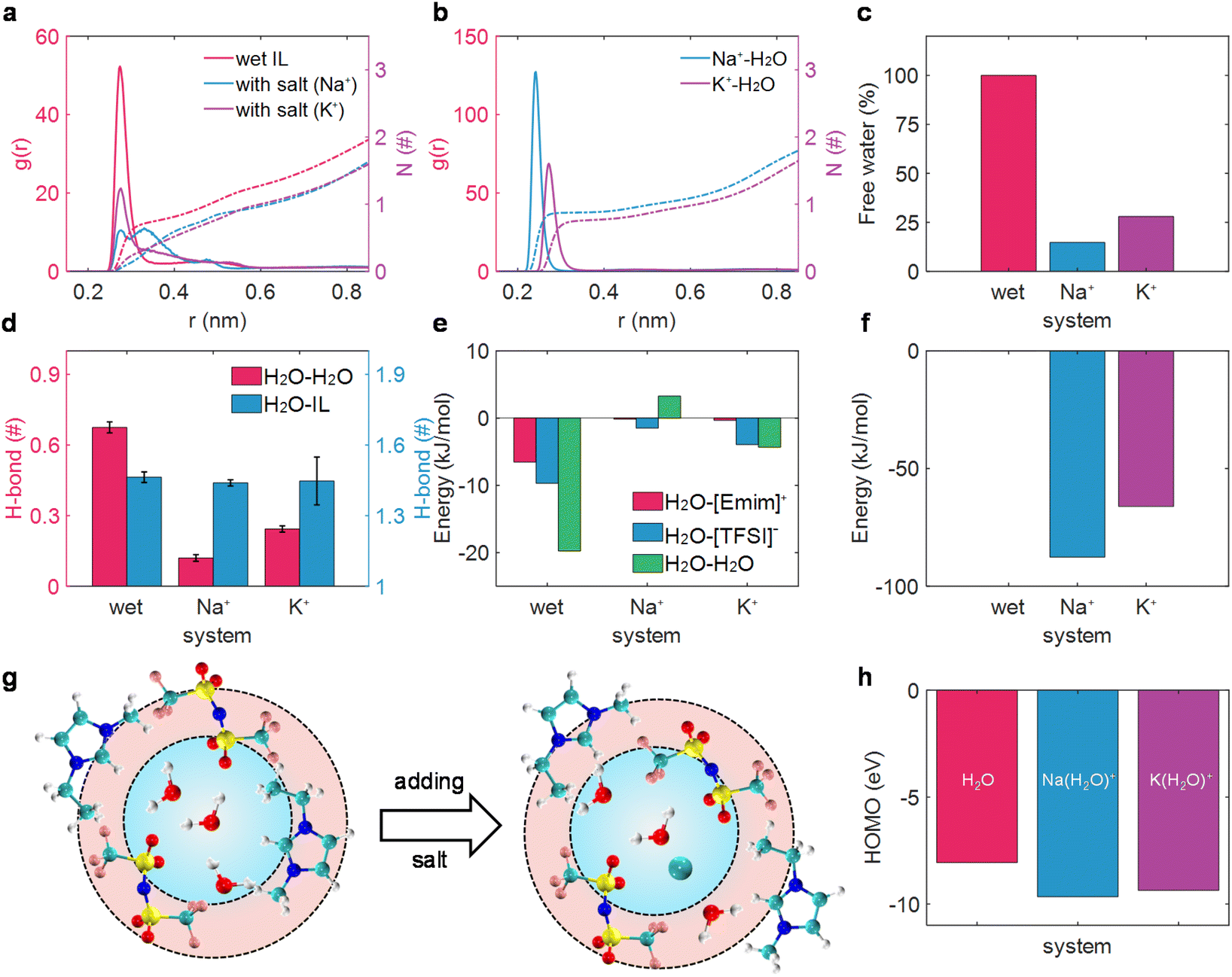 | ||
| Fig. 1 Microscopic understanding of the salt effect on water in RTILs. (a) The RDF (left) and coordination number (right) of the mass-center of water with water in wet [Emim][TFSI] and salt-in-wet [Emim][TFSI]. (b) The RDF (left) and coordination number (right) of the mass-center of water with alkali ion salt-in-wet [Emim][TFSI]. (c) Proportion of free water in wet [Emim][TFSI] and salt-in-wet [Emim][TFSI]. (d) Hydrogen bond between water molecules (left) and water and ILs (right). (e) Interaction energy between water and [Emim]+ (red), [TFSI]− (blue), and water (green). (f) Interaction energy between alkali ions and water molecules. (g) Schematic of the solvation structure of water-in-wet [Emim][TFSI] and salt-in-wet [Emim][TFSI]. (h) HOMO energy levels of H2O and M(H2O)+. | ||
We then delved into the alkali ion effect on the hydrogen bond (H-bond) of water molecules, an aspect intrinsically linked to water activity.20,51,52 Essentially, an increased quantity of H-bonds signifies a weak O–H bond strength in thermodynamic terms,52 and the H-bond in the water cluster could facilitate the efficient separation of water electrolysis products, thus promoting water electrolysis kinetically, following the Grotthuss mechanism.51 The H-bond was defined by the geometrical criterion, as elaborated in previous works.20,53 As illustrated in Fig. 1d, alkali ions had a negligible influence on the H-bond between water and ionic liquids, while exhibiting a profound effect on the H-bond between water molecules. Specifically, each water molecule could form 0.67 H-bonds with another water molecule in wet ILs. Nevertheless, with the addition of M+ (Na+, K+) ions, the hydrogen bonds between water molecules are substantially disrupted, reducing to fewer than 0.25 H-bonds per water molecule, thus strengthening the O–H bond and suppressing the electrolysis product separation.51,52 A similar disruption of the clustering and H-bond of water molecules has also been detected with the scaled-charge force field (ESI Fig. 5†). In this sense, the activity of water might decrease to some extent in salt-in-wet IL electrolytes from both thermodynamic and kinetic perspectives.
The reduced amount of water clusters and the formed alkali ion-bound water could be fundamentally attributed to the interaction energy between water and the other components as well as the water itself. As presented in Fig. 1e and f, for wet ILs, the interaction between water molecules in the first solvation shell is about −20 kJ mol−1. As a salt was added to wet ILs, the interaction between water and the alkali metal ion increased to be the strongest (around −87 kJ mol−1 for water and Na+ and −66 kJ mol−1 for water and K+). Conversely, the interaction between water and IL ions decreased nearly to zero, and even the interaction between water molecules turned a bit repulsive with the addition of Na+ (Fig. 1e). The resultant interaction makes water molecules isolated from each other and associated with Na+ or K+ (Fig. 1b), leading to a sharp decrease in the proportion of free water (Fig. 1c). The motif of the salt effect on the solvation structure of water in wet ILs is depicted in Fig. 1g.
Except for the solvation structure change of water, the addition of alkali cations may significantly alter the oxidation stability of water, which can be qualitatively estimated with the highest occupied molecular orbital (HOMO).45,54,55 The calculated HOMO level of the alkali ion-free water with the implicit solvation model is ca. −8.06 eV. However, the HOMO levels of alkali ion-bound water decrease considerably, reaching around −9.66 eV for Na+-bound water and around −9.37 eV for K+-bound water. Even when coordinated with different solvation structures, a decrease in HOMO levels and an increase in vertical ionization potentials have also been detected for the alkali ion-bound water (ESI Fig. 6†). The lower HOMO levels and higher vertical ionization potentials suggested that a greater amount of energy is necessary for the oxidation reaction when water is bound to alkali cations.56,57
Consequently, our combined application of MD simulations and DFT calculations has demonstrated that the added alkali salt could not only alter the solvation structure of water and reduce the activity of water in wet ILs to a degree but also affect the electrochemical properties of water, lowering the HOMO levels and enhancing oxidation stability. This comprehensive understanding indicates that the addition of an alkali salt may be beneficial for improving the electrochemical stability of wet ILs and optimize the performance of electrolytes.
3.2 Interfacial ion and water distributions
The elucidation of interfacial processes is intrinsically tied to the understanding of the interfacial structure between the electrode and the electrolyte.58 Hence, taking sodium salt as an example, the effect of sodium salt on the electrode/electrolyte interfacial structure, including the ion distribution, the state of interfacial water (alkali ion-free/bound water), and the inheritance of changes in the solvation structure of water molecules, was then dissected via constant potential MD simulations.4,19The EDL potential (ΦEDL) is defined as the potential across the EDL relative to its value at zero charge of the electrode (PZC). As shown in Fig. 2, it was revealed that the added salt exhibits a minimal impact on the distribution of the cation and anion of ILs (Fig. 2a and b), while it has a quite pronounced impact on the water distribution (Fig. 2c and d). Explicitly, water preferentially gathers at the charged electrode surface in wet [Emim][TFSI] (Fig. 2c), in alignment with prior work on the water-in-hydrophobic IL mixture system.9,10,59 In contrast, when a salt is introduced into wet [Emim][TFSI], the majority of water molecules are markedly excluded from the electrode surface (Fig. 2d). This exclusion can be ascribed to the association between Na+ and water, as demonstrated by the similarly positioned peaks in the number density distribution of Na+ and water molecules (Fig. 2d). A more detailed portrayal of the distribution of ionic liquids, water molecules, and Na+ ions, as well as their evolution with the applied voltage, is shown in ESI Fig. 7–10.†
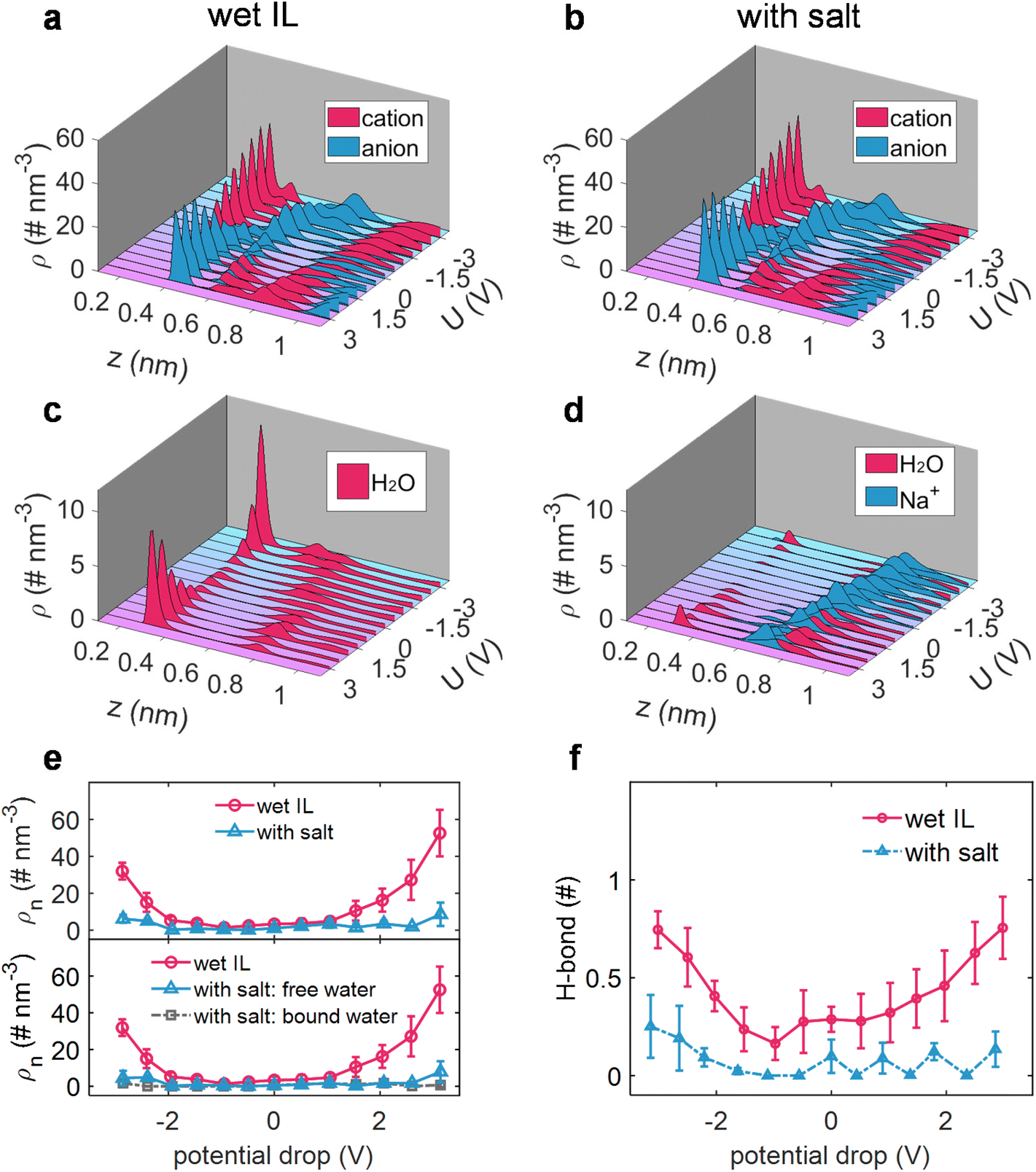 | ||
| Fig. 2 Effect of adding a salt on water adsorption near the electrode surface. The number density of the cation and anion in wet [Emim][TFSI] (a) and the salt-in-wet [Emim][TFSI] electrolyte (b). The number density of water and Na+ in wet [Emim][TFSI] (c) and the salt-in-wet [Emim][TFSI] electrolyte (d). (e) Electrosorption of water from wet [Emim][TFSI] in wet [Emim][TFSI] and salt-in-wet [Emim][TFSI] electrolytes. The top and bottom panels are the total and free/bound adsorbed water, respectively. (f) The H-bond of each interfacial water molecule as a function of applied voltage. | ||
We, subsequently, quantified the water adsorption in an adlayer (0–0.35 nm), where water could be contact-adsorbed on the electrode surface and directly participate in electrolysis.12,19,20 The cumulative number of water molecules (incorporating both free and bound water) in response to varying polarizations within the adlayer, before and after adding a salt, is depicted in Fig. 2e. Interestingly, the electrosorbed water is nearly eliminated across the entire voltage region after adding the salt. Although a certain amount of water remains adsorbed in the adlayer under a specific bias voltage, a fraction of it is bound to Na+ ions, further reducing the quantity of free water and consequently limiting its participation in the potential electrolysis processes.
Moreover, the H-bond of interfacial water has also been significantly altered with the addition of sodium salt. As illustrated in Fig. 2f, analogous to the effect of sodium salt on the H-bond network of water in the bulk electrolyte, the H-bond between interfacial water and water is disrupted under both negative and positive polarizations. This observation suggests that the destructive effect on the water–water H-bond network in the bulk electrolyte, induced by the addition of sodium ions, is similarly reflected in the interfacial region, leading to a decrease in the activity of interfacial water.
Further modeling was carried out to assess the universality of the observed effects with alkali ions (Li+ and K+), where water molecules were found to be repelled from the electrodes in the presence of alkali ions (ESI Fig. 11†). Therefore, it can be inferred that the introduction of a salt into wet ILs could not only repel water from both negative and positive electrode surfaces but also break the H-bond of water in the adlayer region. These features synergistically contribute to potentially safeguarding the wet ILs from electrolysis and preventing the reduction of the electrochemical windows of wet ILs.
3.3 Mechanism of adding a salt on the distribution of interfacial water
To elucidate the underlying mechanism of how sodium salt affects water distribution near the electrode, we then performed PMF analysis through the umbrella sampling method.20,39,51 It is well established that a pronounced minimum with a negative free energy of PMF in the adlayer compared to the bulk state (considered as the region beyond ∼1.5 nm from the electrode surface, where PMF decays to zero) would trigger an accumulation in the adlayer, while a positive free energy profile responds to metastable adsorption, causing a depletion within the adlayer.51For a free water molecule under negative polarization, a distinct minimum of PMF is visible at approximately 0.3 nm from the electrode (Fig. 3a), corresponding to an accumulation of water at the negatively polarized EDL (Fig. 2e). Simultaneously, a similar negative minimum of PMF was observed for a free water molecule at positive electrodes (Fig. 3b). Specifically, the potential well near the positive electrode was much deeper than that at the negative electrode, leading to the asymmetric adsorption of water in the wet ILs (Fig. 2e).
 | ||
| Fig. 3 The tendency for free and bound water electrosorption at electrodes. Potential of mean force (PMF) of water under negative polarization (a) and positive polarization (b) in [Emim][TFSI] as a function of distance from the electrode. PMF of bound water under negative polarization (c) and positive polarization (d) in [Emim][TFSI] as a function of distance from the electrode. The pink-shaded region is considered as the interfacial region (z < 0.35 nm). | ||
Regarding the salt-in-wet IL scenario, water molecules are bound with Na+, with a coordination number of one (see Fig. 1b and ESI Fig. 4†). So, where does Na+-bound water locate itself? A markedly different motif is observed for Na+-bound water (Fig. 3c and d). At negative polarization (−2 V), a positive minimum of PMF is found near the electrode (around 20 kJ mol−1 at 0.3 nm), leading to metastable adsorption. Concurrently, the Na+-bound water induces a pronounced potential well at 0.7 nm, causing Na+-bound water molecules to preferentially remain far away from the electrode. As the EDL potential shifts positively (2 V), the potential well exhibits a distinct valley at approximately 0.75 nm, in accordance with the previous report for lithium salt in wet [Pyr13][TFSI].20 Therefore, the Na+-bound water in [Emim][TFSI] is reluctant to stay near the electrode surface, resulting in a depletion of interfacial water.
This preference for Na+-bound water away from the electrode can be linked to the free energy distribution of Na+ in [Emim][TFSI] (ESI Fig. 12†). A substantially positive potential well for Na+ is identified in the adlayer under both negative and positive electrodes, implying metastable adsorption. It is worth noting that the lowest potential well under negative polarization occurred at ca. 0.75 nm, quite close to the location of the [TFSI]− anion (spanning from 0.6 to 0.74 nm, ESI Fig. 7†). Under positive polarization, the potential well is located between the first and second anion layers (ESI Fig. 8†). The preference for Na+ ions to remain outside the interfacial region is likely due to the strong interaction between Na+ and the TFSI− anion (∼−370 kJ mol−1).
Briefly, the PMF curves demonstrated that the wet ILs exhibit water accumulation at polarized electrodes; nevertheless, the presence of Na+ has regulated the free energy distribution of interfacial water in wet ILs. This modulation results in a significant exclusion of water from the electrode under both negative and positive polarizations.
3.4 Salt effect on the activity of interfacial water
We then directed our attention to the structure of water (including alkali ion-free and bound water) within the interfacial domain in wet ILs and salt-in-wet ILs, aiming to elucidate the effects of sodium salt on the activity of interfacial water. The detailed examinations of the atom density profile, orientation distribution, and the H-bond network along the EDL of water molecules are shown in Fig. 4a–h.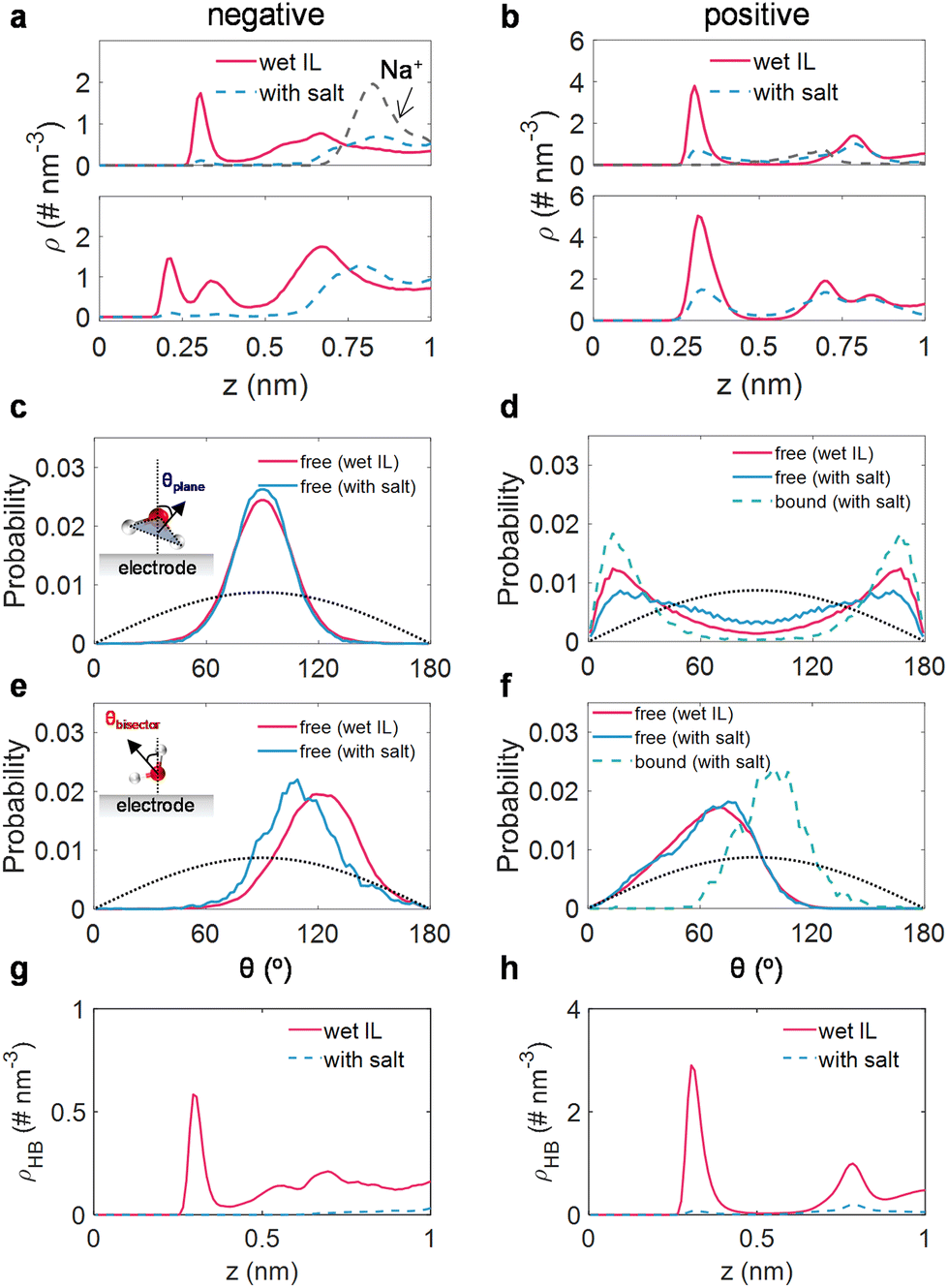 | ||
| Fig. 4 The interfacial water structure at the electrified surface. The oxygen/hydrogen atom number densities (ρ) of water in wet [Emim][TFSI] (red solid line) and in salt-in-wet ILs (blue dashed line) under negative (a) and positive (b) polarizations. The black dotted line is the atom number density of Na+ in salt-in-wet [Emim][TFSI]. Orientations of the interfacial water plane in wet [Emim][TFSI] (red solid lines) and in salt-in-wet [Emim][TFSI] (blue lines for free water; green dashed lines for bound water) under negative (c) and positive (d) polarizations. Orientations of the interfacial water bisector in wet [Emim][TFSI] (red solid lines) and in salt-in-wet [Emim][TFSI] (blue lines for free water; green dashed lines for bound water) under negative (e) and positive (f) polarizations. θplane is the angle formed between the normal of the electrode surface and the normal of the water plane; θbisector is defined as the angle between the normal of the electrode surface and the water bisector. The black dotted line represents the orientation of bulk water. The H-bond network of interfacial water in the EDL region under negative (g) and positive (h) polarizations. | ||
Under negative polarization, a prominent peak for oxygen atoms and two peaks for hydrogen atoms of interfacial water in wet [Emim][TFSI] were identified (Fig. 4a). Meanwhile, the orientation of the water plane (peak at around 90°, Fig. 4c) and the orientation of the water bisector (peak located at ∼120°, Fig. 4e) illustrate that most water molecules adopt a perpendicular orientation to the electrode surface, with the hydrogen atom directed towards the surface. Such orientations are propitious for the hydrogen evolution reaction.60 Nevertheless, upon the introduction of a salt, the Na+ ions are disinclined to stay close to the electrode (Fig. 4a), thus, the peaks for oxygen atoms and hydrogen atoms decrease sharply, resulting in the repulsion of water. Simultaneously, the residual trace water molecules are in the Na+-free state. Though the plane orientation of these Na+-free water molecules remains virtually unaltered (Fig. 4c), the bisector orientation of the residual trace water molecules shifts to 110° (Fig. 4e), signifying that the water molecule reorients itself, with its hydrogen atoms moving away from the electrode. This reconfiguration of interfacial water hinders electron transfer from the electrode to interfacial water, impeding the hydrogen evolution reaction.20 We also detected the H-bond network connectivity of water in the EDL region. Typically, the H-bond network in the EDL region for salt-in-wet [Emim][TFSI] is considerably sparse compared to that in wet ILs, potentially obstructing water electrolysis product transfer via kinetics pathways.53
In terms of the positive polarization, the nearly indistinguishable peak locations for oxygen and hydrogen atom density distributions (around 0.3 nm, Fig. 4b) suggest a configuration parallel to the electrode surface. This configuration is further evidenced by the orientation of the water plane peaking at 13/167° and the orientation of the water bisector peaking at around 80° (Fig. 4d and f). By adding sodium salt, the interfacial water can be classified into free and Na+-bound water. As for free water molecules, they maintain a parallel configuration, exhibiting a minor alteration in plane orientation (Fig. 4d) and an almost identical bisector orientation (Fig. 4f) compared to free water molecules in wet ILs. While the bound water molecules in the adlayer manifest a more ordered organization, characterized by a narrower plane orientation compared to free water in wet ILs (Fig. 4d), the peak of bisector orientation migrates from 80° to 100° (Fig. 4f). This re-arrangement of water tends to lose electrons to the electrode, benefiting from the inhibition of water oxidation.20 Moreover, akin to the negative polarization, the H-bond network connectivity in the EDL region under positive polarization is substantially disrupted.
3.5 Experimental verification of the electrochemical window
Incorporating a salt into wet ILs has the theoretical potential to effectively modulate the solvation structure of water and regulate the EDL structure, ultimately safeguarding the wet ILs from electrolysis and preventing the reduction of electrochemical windows in wet ILs. To substantiate this hypothesis, we conducted CV measurements for pure [Emim][TFSI], wet [Emim][TFSI], and salt-in-wet [Emim][TFSI] with highly oriented pyrolytic graphite (HOPG) electrodes (Fig. 5). Details of the measurements can be found in the Methods section.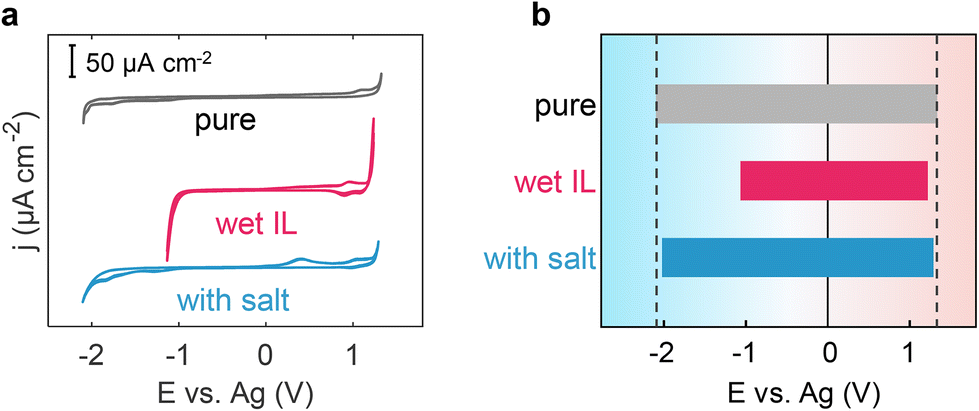 | ||
| Fig. 5 Electrochemical windows for electrolytes. (a) Cyclic voltammograms of HOPG in pure [Emim][TFSI], wet [Emim][TFSI] and wet [Emim][TFSI] upon adding a salt. Scan rate: 100 mV s−1. (b) Electrochemical windows for pure [Emim][TFSI], wet [Emim][TFSI] and wet [Emim][TFSI] upon adding a salt. The cathodic and anodic limits were determined according to a cut-off of ±30 μA cm−2. | ||
As depicted in Fig. 5, the CV curve of pure [Emim][TFSI] exhibits an absence of distinct faradaic current peaks, with its EW spanning an impressive 3.41 V, from −2.09 V to 1.32 V. However, when [Emim][TFSI] becomes wet (containing 8464 ppm water, in close proximity to 8446 ppm set in the MD simulation), the faradaic current starts to appear at ca. −1.0 V, primarily attributed to water reduction. Meanwhile, in the anodic region, the oxidation process occurs at a more negative potential than that in pure ILs. Our focus in this investigation centers on the electrochemical behavior of interfacial water within ionic liquids (ILs), leading to the establishment of specific cut-off potentials (Fig. 5). Notably, this alteration results in a discernible reduction in the electrochemical window, contracting the cathodic and anodic voltage limits to the range of −1.06–1.21 V (Fig. 5b). Upon the addition of sodium salt, the EW discernibly expands to 3.29 V, resulting in a 45% increase in the electrochemical window of the wet ILs and almost fully restoring the range of both positive and negative polarizations to −2.01–1.28 V (Fig. 5b). These findings, derived from both simulation and experimental data, corroborate that introducing a salt into wet hydrophobic [Emim][TFSI] effectively broadens its EW.
4. Conclusions
We have explored the effect of cheap alkali salts on the electrochemical stability of wet hydrophobic ILs from thermodynamic and kinetic perspectives. Our theoretical investigation reveals that alkali ions exhibit a pronounced hydration effect, which profoundly disrupts the water–water interaction, resulting in a decrease of free water and an almost complete disappearance of the H-bond. Consequently, the O–H bond strength in water is enhanced thermodynamically, making the splitting of the O–H bond a more energy-intensive process. The corresponding decrease in the HOMO level of bound water further complements the thermodynamic enhancement of electrolyte oxidation stability.Notably, these effects manifest prominently in the EDL regions. Alkali ions are found to be capable of efficiently redefining the EDL structure, primarily by modulating the free energy distribution of water in the wet ILs. This modification leads to a substantial reduction of water present in the adlayer under both negative and positive polarizations. Concurrently, the introduction of alkali salts leads to a virtual annihilation of the H-bond network connectivity across the entire voltage range within the EDL region. These depletions of water molecules and H-bond network connectivity synergistically increase the energy barrier for water electrolysis in a kinetic sense. In addition, the introduced alkali ions induce modifications in the configuration of adsorbed water, altering its orientation and atomic position to thermodynamically shield it from electrolysis.
The theoretical predictions were corroborated by experimental measurements, revealing a substantial 45% enhancement in the EW of wet ILs, approaching the limits of dry ILs. This research, underpinned by the concept of cheap alkali salts, broadens our understanding of salt-in-wet IL electrolytes. It provides a strategic guideline for the nuanced regulation of the electrical double layer by fine-tuning the free energy distribution and controlling the activity of interfacial water. These findings not only deepen the scientific comprehension of interface engineering but also furnish practical strategies for the improvement of electrochemical systems.
Author contributions
M. C. conceived this research. M. C. and J. K. Z carried out all simulations; J. D. W., J. W. Y. and B. W. M. carried out the experiment. M. C. drafted the manuscript. All authors contributed to the analysis and discussion of the data and revision of the manuscript.Data availability
The data that support the findings of this study are available from the corresponding author upon request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge the funding support from the National Natural Science Foundation of China (52106090, 52161135104, and 22072123) and the Program for HUST Academic Frontier Youth Team. M. C. also thanks the China Postdoctoral Science Foundation (2022T150228). The authors acknowledge the Wuhan Supercomputing Center and the HPC Platform of HUST for providing computational resources that have contributed to the research results reported within this paper.References
- S. Kondrat, G. Feng, F. Bresme, M. Urbakh and A. A. Kornyshev, Chem. Rev., 2023, 123, 6668–6715 CrossRef CAS.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS.
- G. Jeanmairet, B. Rotenberg and M. Salanne, Chem. Rev., 2022, 122, 10860 CrossRef CAS.
- D. M. Piper, T. Evans, K. Leung, T. Watkins, J. Olson, S. C. Kim, S. S. Han, V. Bhat, K. H. Oh, D. A. Buttry and S.-H. Lee, Nat. Commun., 2015, 6, 6230 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- S. Zhang, J. Sun, X. Zhang, J. Xin, Q. Miao and J. Wang, Chem. Soc. Rev., 2014, 43, 7838–7869 RSC.
- V. A. Azov, K. S. Egorova, M. M. Seitkalieva, A. S. Kashin and V. P. Ananikov, Chem. Soc. Rev., 2018, 47, 1250–1284 RSC.
- R. Qiao, Curr. Opin. Electrochem., 2019, 13, 11–17 CrossRef CAS.
- K. Motobayashi and M. Osawa, Electrochem. Commun., 2016, 65, 14–17 CrossRef CAS.
- Y. Zhong, J. Yan, M. Li, L. Chen and B. Mao, ChemElectroChem, 2016, 3, 2221–2226 CrossRef CAS.
- G. Feng, X. Jiang, R. Qiao and A. A. Kornyshev, ACS Nano, 2014, 8, 11685–11694 CrossRef CAS.
- Q. Zheng, Z. A. H. Goodwin, V. Gopalakrishnan, A. G. Hoane, M. Han, R. Zhang, N. Hawthorne, J. D. Batteas, A. A. Gewirth and R. M. Espinosa-Marzal, ACS Nano, 2023, 17(10), 9347 CrossRef CAS PubMed.
- B. Docampo-Alvarez, V. Gomez-Gonzalez, H. Montes-Campos, J. M. Otero-Mato, T. Mendez-Morales, O. Cabeza, L. J. Gallego, R. M. Lynden-Bell, V. B. Ivanistsev, M. V. Fedorov and L. M. Varela, J. Phys.: Condens. Matter, 2016, 28, 464001–464008 CrossRef CAS.
- T. Kobayashi, A. Kemna, M. Fyta, B. Braunschweig and J. Smiatek, J. Phys. Chem. C, 2019, 123, 13795–13803 CrossRef CAS.
- M. Chen, G. Feng and R. Qiao, Curr. Opin. Colloid Interface Sci., 2020, 47, 99–110 CrossRef CAS.
- Y. A. Budkov, A. L. Kolesnikov, Z. A. H. Goodwin, M. G. Kiselev and A. A. Kornyshev, Electrochim. Acta, 2018, 284, 346–354 CrossRef CAS.
- D.-E. Jiang and J. Wu, Nanoscale, 2014, 6, 5545–5550 RSC.
- S. Bi, R. Wang, S. Liu, J. Yan, B. Mao, A. A. Kornyshev and G. Feng, Nat. Commun., 2018, 9, 5222–5230 CrossRef PubMed.
- M. Chen, J. Wu, T. Ye, J. Ye, C. Zhao, S. Bi, J. Yan, B. Mao and G. Feng, Nat. Commun., 2020, 11, 5809 CrossRef CAS PubMed.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- J. N. C. Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 16893–16898 CrossRef CAS.
- J. N. C. Lopes, J. Deschamps and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 11250–11250 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- K.-j. Jeong, S. Jeong, S. Lee and C. Y. Son, Adv. Mater., 2023, 35, 2204272 CrossRef CAS PubMed.
- J. G. McDaniel and C. Y. Son, J. Phys. Chem. B, 2018, 122, 7154–7169 CrossRef CAS PubMed.
- C. Y. Son and Z.-G. Wang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020615118 CrossRef CAS PubMed.
- T. C. Lourenço, Y. Zhang, L. T. Costa and E. J. Maginn, J. Chem. Phys., 2018, 148, 193834 CrossRef PubMed.
- B. Doherty, X. Zhong, S. Gathiaka, B. Li and O. Acevedo, J. Chem. Theory Comput., 2017, 13, 6131–6145 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P.-L. Taberna, C. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- C. Merlet, B. Rotenberg, P. A. Madden, P. L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306–310 CrossRef CAS PubMed.
- J. Vatamanu, M. Vatamanu and D. Bedrov, ACS Nano, 2015, 9, 5999–6017 CrossRef CAS PubMed.
- P. V. Klimovich, M. R. Shirts and D. L. Mobley, J. Comput. Aided Mol. Des., 2015, 29, 397–411 CrossRef CAS PubMed.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed.
- V. V. Chaban, N. A. Andreeva and E. E. Fileti, New J. Chem., 2018, 42, 18409–18417 RSC.
- H. Zhang, W. Feng, C. Li and T. Tan, J. Phys. Chem. B, 2010, 114, 4876–4883 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09 Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- V. S. Bernales, A. V. Marenich, R. Contreras, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2012, 116, 9122–9129 CrossRef CAS PubMed.
- E. R. Fadel, F. Faglioni, G. Samsonidze, N. Molinari, B. V. Merinov, W. A. Goddard III, J. C. Grossman, J. P. Mailoa and B. Kozinsky, Nat. Commun., 2019, 10, 3360 CrossRef PubMed.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- H. R. Westrich, Chem. Geol., 1987, 63, 335–340 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- C.-Y. Li, M. Chen, S. Liu, X. Lu, J. Meng, J. Yan, H. D. Abruña, G. Feng and T. Lian, Nat. Commun., 2022, 13, 5330 CrossRef CAS PubMed.
- N. Dubouis, A. Serva, R. Berthin, G. Jeanmairet, B. Porcheron, E. Salager, M. Salanne and A. Grimaud, Nat. Catal., 2020, 3, 656 CrossRef CAS.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS PubMed.
- P. Li, Y. Jiang, Y. Hu, Y. Men, Y. Liu, W. Cai and S. Chen, Nat. Catal., 2022, 5, 900 CrossRef CAS.
- M. Chen, J. Zhang, X. Ji, J. Fu and G. Feng, Curr. Opin. Electrochem., 2022, 34, 101030 CrossRef CAS.
- S. Hou, X. Ji, K. Gaskell, P.-f. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129–16137 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- Y. Fang, S.-Y. Ding, M. Zhang, S. N. Steinmann, R. Hu, B.-W. Mao, J. M. Feliu and Z.-Q. Tian, J. Am. Chem. Soc., 2020, 142, 9439–9446 CrossRef CAS.
- J. Meng, M. Ye, Y. Wang, Y. Sun, X. Zhang, K. Shi and X. Yan, Sci. China: Chem., 2022, 65, 96–105 CrossRef CAS.
- J. Vatamanu and O. Borodin, J. Phys. Chem. Lett., 2017, 8, 4362–4367 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr04700h |
| This journal is © The Royal Society of Chemistry 2023 |