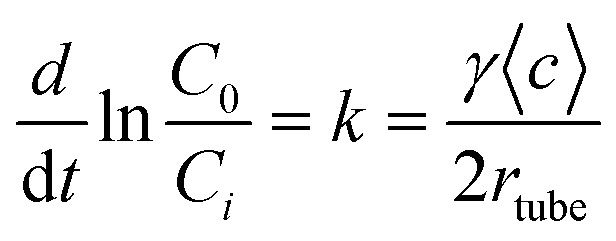Comprehensive study about the heterogeneous conversion of NH3 on nanoscale TiO2 particles: insight into the mechanism and atmospheric implications
Received
22nd October 2022
, Accepted 17th November 2022
First published on 18th November 2022
Abstract
The heterogeneous reactions of NH3 on TiO2 were comprehensively investigated using in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and a flow tube reactor. The effect of SO2 and illumination on the formation of surface and gaseous products was explored. The results reveal that surface hydroxyls and H2O play important roles in the adsorption of NH3 while adsorbed sulfur species like sulfite and sulfate can significantly promote the conversion of NH3 to NH4+ under dark conditions. The reaction mechanism under light conditions is quite different from that under dark conditions. NH3 can be directly converted into gaseous NO and NO2 and particulate nitrate on the surface of TiO2 by photooxidation. Surface sulfate formed from the photooxidation of SO2 can combine with NH3 to form ammonium sulfate, which inhibits the oxidation of NH3 and alters the production ratios of NO/NO2. The uptake coefficients of NH3 on TiO2 are in the range of 1.5–2.5 × 10−5 regardless of illumination while SO2 shows an inhibition effect on the initial uptake of NH3 due to competition in adsorption on surface hydroxyls. These results demonstrate that the heterogeneous reaction of NH3 on TiO2 can be a significant sink of NH3 while its transformation pathway greatly depends on the environmental factors in the atmosphere.
Environmental significance
As the main alkaline constituent in the atmosphere, NH3 has significant impacts on the formation of secondary aerosol as well as nucleation events. However, the heterogeneous reactions of NH3 on particles are not fully understood and still not considered in the air quality model, which greatly restricted the formulation of ammonia emission control strategies. In this study, a systematic laboratory study of the heterogeneous reaction of NH3 on nanoscale TiO2 under air pollution complex conditions is conducted. It was found that the heterogeneous transformation mechanism of ammonia and its effect on secondary pollution formation greatly depends on environmental conditions such as irradiation and coexisting SO2. These results are helpful for understanding NH3 migration and transformation in the atmosphere.
|
1. Introduction
Atmospheric particulate matter (APM) has been a prominent air pollution problem, especially in south Asia and East Asia.1,2 Exposure to fine particulate matter (PM2.5, particulate matter with an aerodynamic diameter less than 2.5 μm) has a serious health risk by causing respiratory and cardiovascular diseases. A large number of field observations have indicated that secondary inorganic aerosol (SIA, e.g., sulfate, nitrate, ammonium) accounts for 25–60% of PM2.5 in China.3–6 Thus, lots of attention have been paid to the transformation of gaseous precursors to secondary inorganic aerosol in laboratory research in recent decades. The formation pathways of sulfate and nitrate have been widely investigated. For example, the well-known conversion pathways of SO2 to sulfate include gas-phase oxidation by the OH radical or stabilized Criegee intermediate to form sulfuric acid,7,8 aqueous-phase oxidation in cloud and fog droplets,9,10 and heterogeneous reactions on surfaces of atmospheric particles.11,12 For the formation pathways of nitrates, it includes the homogeneous oxidation of NO2 by OH radicals to form HNO3 and by O3 to form N2O5 (ref. 13) which then transforms to nitrate on the surface of particles,14 as well as the direct transformation of NO2 to nitrates through heterogeneous reactions.15 Compared with sulfate and nitrate, there is still a lack of understanding of the formation pathway of ammonium aerosol.
NH3 has significant impacts on atmospheric chemistry since it is the main alkaline constituent in the atmosphere. It is reported that global ammonia emission has increased more than 2–5 times since preindustrial times.16 The main emission sources of NH3 include livestock and human excreta, fertilizer application, agricultural activities, nitrogen-fixing plants, the chemical industry, traffic, and so on.17,18 The average urban concentration of NH3 in Beijing was about 36 ppb while the maximum value reached 800 ppb in 2013.19 NH3 has been considered a key species for neutralizing H2SO4 and HNO3 in the atmosphere and forming (NH4)2SO4/NH4HSO4 and NH4NO3, respectively.20,21 Several recent model assessment studies showed that the large atmospheric NH3 emissions in northern China can reduce the effectiveness of existing PM2.5 control strategies through SO2 and NOx emission reductions,22,23 while ammonia emission reduction can be considered as an effective measure to reduce PM2.5.24 Field observation and laboratory simulation indicated that NH3 can promote the formation of secondary pollutants including sulfate, nitrate, and HONO.25–30 In addition, NH3 has been found to react with carbonyl compounds in secondary organic aerosol (SOA) which results in the formation of nitrogen-containing organic compounds (NOC) and significantly affects regional air quality.31–37 These results demonstrate that besides acid–base neutralization reactions in the gas phase, heterogeneous and multiphase reactions of ammonia should also be investigated systematically.
Mineral aerosols, which originate from arid and semi-arid regions with an annual flux of about 1600–2000 Tg, represent one of the largest mass fractions of global aerosols.38 Mineral dust particles can transport over thousands of kilometers and alter the chemical balance of the atmosphere by providing a reactive substrate or medium for gaseous pollutants. These reactions can not only change atmospheric trace gas distributions but also modify the chemical composition and mixing state of particles. TiO2 is an important component of mineral dust particles, with a content of 1–10 wt%, and has strong photocatalytic activity.39 Kebede et al.40 found that heterogeneous photooxidation of NH3 over TiO2 under atmospherically relevant concentrations leads to the formation of NOx. In addition, NH4+ in NH4NO3 coated on TiO2 can be converted to NOx upon irradiation and can be significantly affected under complex air pollution conditions.41 Therefore, the heterogeneous reactions process of NH3 on TiO2 under complex air pollution conditions need further study.
In this study, we comprehensively studied the heterogeneous reaction process of NH3 on TiO2 using in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and a wall-coated flow tube. It mainly focused on the role of irradiation and the interaction between SO2 and NH3 in heterogeneous reactions. The relevant research results are helpful to further understand the atmospheric migration and transformation of NH3 and the environmental impacts of mineral dust.
2. Experimental section
In situ DRIFTS
The heterogeneous reactions of NH3 on TiO2 particles were measured using a DRIFTS instrument (NEXUS 670, Thermo Nicolet Instrument Corporation), equipped with an in situ reaction chamber and a high-sensitivity mercury cadmium telluride (MCT) detector cooled with liquid N2. The TiO2 sample (about 15 mg) was finely ground and placed into a ceramic crucible in the in situ chamber. The total flow rate was 100 mL min−1 in all flow systems. The reference spectrum was measured after the pretreated sample was cooled to 298 K in a synthesized air stream. The infrared spectra were collected and analyzed using a data acquisition computer with OMNIC 6.0 software (Nicolet Corp.). All spectra reported here were recorded at a resolution of 4 cm−1 for 100 scans in the spectral range of 4000 to 600 cm−1. The spectra are presented in the Kubelka–Munk (K–M) scale, which can reduce or eliminate the mirror effect and give a better linear relation with concentration.42 Before DRIFTS measurement, the TiO2 sample was pretreated in an in situ infrared cell by heating in 100 mL min−1 of synthesized air (20% O2 + 80% N2) at 473 K for 2 h. UV light was introduced into the DRIFTS reaction cell via a UV fiber (Φ = 5 mm). The light acquired with a mercury lamp (λ > 200 nm, CHF-XM-500 W, the Trusttech. Co. Ltd., Beijing) was used as the light resource. The spectrum of the light in the DRIFTS experiments was measured with a fiber optic spectrometer (BLUE-Wave-UVNb, Stellar Net Inc., USA).
Coated-wall flow tube reactor
The uptake of NH3 on TiO2 was studied in a horizontal cylindrical coated-wall flow tube reactor, which has been described in detail elsewhere.43–45 The experiments were performed at ambient pressure and maintained at 298 K by a circulating water bath through the outer jacket of the flow tube reactor. The tube with the deposited TiO2 sample was introduced into the main reactor along its axis. Synthetic air was used as the carrier gas, which was introduced into the flow tube reactor in a total flow rate of 1800 mL min−1, ensuring a laminar regime at ambient pressure. NH3 was introduced into the gas flow by a movable injector with a 0.3 cm radius. The reactor was surrounded by 6 UV lamps (365 nm, T5, UVA, 8 W) with a broad UV emission spectrum between 330 and 420 nm. The UV lamps were installed into a stainless light-tight box covering the main reactor tube. The irradiance intensity in the reactor could be regulated by switching off some lamps. When all lamps were switched on, the irradiance intensity in the reactor was 1.27 W m−2 at 370 nm. All reactions were conducted under dry conditions with relative humidity (RH) lower than 5%.
Uptake coefficient
The kinetic behavior can be well described by assuming a pseudo first-order reaction with respect to the gas-phase reactant concentration. The first-order rate constant (k) is related to the geometric uptake coefficient (γgeo) using eqn (1):43| | 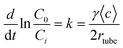 | (1) |
where rtube, t and 〈c〉 are the flow tube radius, the exposure time, and the reactant average molecular velocity, respectively. C0 and Ci are the reactant concentration at t = 0 and t = i, respectively.
If the loss of reactant at the TiO2 surface is too rapid to be recovered with the reactant supply, a radial concentration gradient in the gas phase will be formed, which may cause diffusion limitations. Therefore, a correction for diffusion in the gas phase should be taken into account and the Cooney–Kim–Davis (CKD) method was used to correct uptake coefficients.46,47
Materials
TiO2 particles were purchased from commercial sources (P25: 80% anatase, 20% rutile, Degussa). A total mass of 1.0 g of TiO2 powder was dissolved in 20.0 mL of water. This suspension was dripped into a quartz tube (20.0 cm length, 1.1 cm i.d.) and dried overnight in an oven at 373 K. The resulting homogeneous film covered the entire inner area of the tube. The Brunauer–Emmett–Teller (BET) area of the sample is 54.6 m2 g−1. NH3 (N2 balance, Beijing Huayuan), SO2 (N2 balance, Beijing Huayuan), O2 (99.999%, Beijing Huayuan) and N2 (99.999%, Beijing Huayuan) were used as-received. Gas analyzers (THERMO 42i, 43) were used to monitor the concentrations of NOx and SO2, respectively. NH3 was monitored by a Quantum Cascade Tunable Infrared Laser Differential Absorption Spectrometer (QC-TILDAS, Aerodyne Research).
3. Results and discussion
3.1 DRIFTS study the heterogeneous reaction of NH3 on TiO2
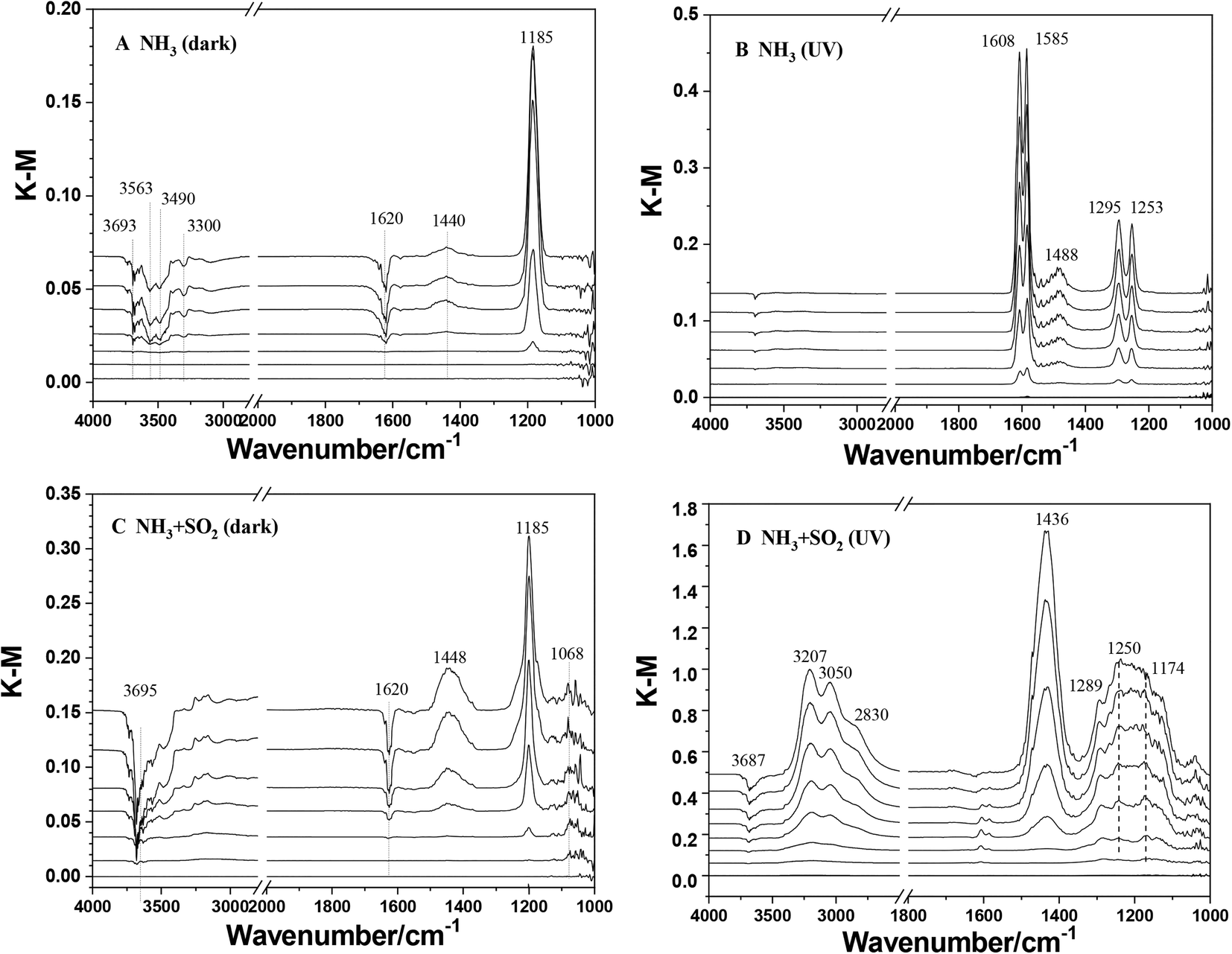
The heterogeneous reactions of NH3 on TiO2 were studied using in situ DRIFTS. Fig. 1 shows the DRIFTS spectra of TiO2 exposed to NH3 with or without SO2 and UV irradiation. In the case of ammonia individual dark reaction (Fig. 1A), a dominant peak at 1185 cm−1 and a weak peak at 1440 cm−1 were observed. These two peaks can be assigned to NH3(ads) adsorbed on Lewis acid sites and NH4+ adsorbed on Brønsted acid sites, respectively.30 These results indicate that the adsorption of NH3 on TiO2 under dark conditions leads to the formation of adsorbed NH3 species as the major products and a small amount of NH4+ species. Meanwhile, several negative peaks decreasing in intensity at 1620, 3300, 3490, 3563, and 3693 cm−1 were observed. The peak at 3693 cm−1 is attributed to surface OH groups,48 suggesting that surface OH groups were involved in the adsorption of NH3 on TiO2. Other peaks at 1620, 3300, 3490, and 3563 cm−1 are attributed to surface water. It has been reported that NH3 would react with water to produce NH4+viareaction (2) or bond to water molecules through a hydrogen bond between the H atom of H2O and the N atom of NH3.40,49,50| | | NH3 + H2O(ads) → NH4+(ads) + OH−(ads) | (2) |
Thus, the adsorption of NH3 on the TiO2 surface not only generates NH4+ but also increases the surface alkalinity by reacting with surface water.
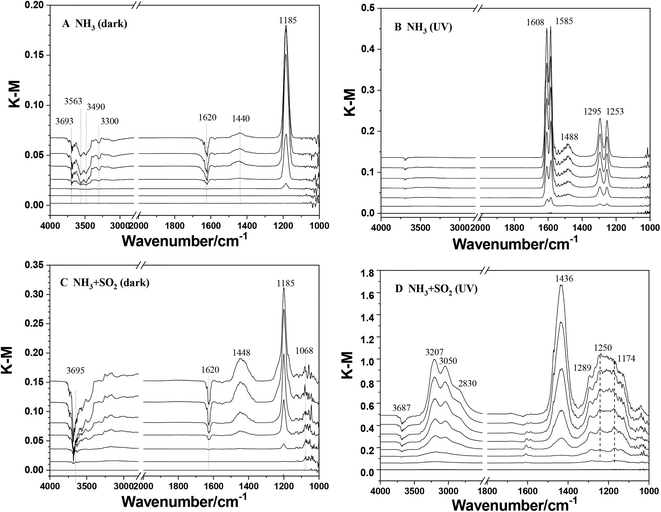 |
| | Fig. 1
In situ DRIFTS spectra of TiO2 exposed to various gases as a function of time (0, 10, 30, 60, 90, 120, and 180 min). (A) 5 ppm NH3 under dark conditions; (B) 5 ppm NH3 under UV irradiation; (C) 5 ppm NH3 + 5 ppm SO2 under dark conditions; (D) 5 ppm NH3 + 5 ppm SO2 under UV irradiation. | |
Compared to the dark reaction, the adsorption of NH3 on TiO2 upon UV irradiation showed different results. As seen in Fig. 1B, several peaks at 1253, 1295, 1488, 1585, and 1608 cm−1 increased in intensity with the reaction proceeding. These peaks are assigned to surface nitrate,51 which indicate the conversion of NH3 to nitrate on TiO2 upon UV irradiation. The decrease in the intensity of the peak at 3693 cm−1 in the photoreaction is much weaker than that in the dark reaction. This suggested that surface OH groups were not the main sites for the photooxidation reaction of NH3. Moreover, the negative peaks attributed to surface water are also not observed. Yamazoe et al.52 proposed that the reaction between NH3 with a photoinduced hole on TiO2 can result in the formation of the amino radical, ˙NH2:
| | | NH3 + h+ → ˙NH2(ads) + H+ | (3) |
The H
+ generated in this reaction can neutralize the hydroxyl ions generated in
reaction (2), which results in no net consumption of water. Kebede
et al.40 suggested another possible reaction where ˙OH radicals generated on the TiO
2 surface can react with NH
3 by abstracting a hydrogen atom to form the amino radical:
| | | NH3 + ˙OH → ˙NH2(ads) + H2O(ads) | (4) |
The formed H
2O in this reaction can also compensate for the consumption of H
2O in
reaction (2). The formed ˙NH
2 on the surface can be oxidized by O
2 to produce gaseous NO and NO
2 and surface nitrate, which will be discussed later.
When SO2 was introduced simultaneously with NH3, the reaction pathway of NH3 was changed. In the SO2 + NH3 dark reaction (Fig. 1C), both peaks at 1185 and 1448 cm−1 were also observed with greater intensity compared to those in NH3 individual dark reaction (Fig. 1A), indicating the promotion effect of SO2 on the adsorption of NH3 on TiO2. A weak peak at 1068 cm−1 attributed to sulfite species and no peak attributed to sulfate was observed. This suggests that NH3 has little effect on the heterogeneous oxidation of SO2 on TiO2.53 Two negative peaks at 3693 and 3628 cm−1 attributed to surface OH groups were also observed. Meanwhile, the decreasing intensity of the peak at 1620 cm−1 assigned to surface water is stronger than that in the NH3 reaction alone. This could be due to the involvement of H2O in the heterogeneous reaction of SO2 on TiO2. Nanayakkara et al.54 proposed that the complex between SO2 and H2O is in equilibrium with bisulfite on TiO2 according to the reaction:
| | | SO2·H2O(ads) ↔ HSO3−H+(ads) | (5) |
The H
+ in HSO
3−H
+ may further interact with NH
3 to generate ammonium ions, which is consistent with the intensity enhancement of the infrared peak at 1448 cm
−1 attributed to NH
4+ adsorbed on Brønsted acid sites (
Fig. 1C).
In the case of the NH3 + SO2 reaction upon UV irradiation (Fig. 1D), several peaks at 1174, 1250, and 1289 cm−1 were observed with increasing intensity with reaction time. These peaks are assigned to surface sulfate species, indicating the promotion effect of UV irradiation on the conversion of SO2 to sulfate or H2SO4 on TiO2:53
| | | SO2 + ˙OH + O2 → HO2˙ + SO3 | (6) |
| | | SO3 + H2O(ads) → H2SO4(ads) | (7) |
Meanwhile, a dominate peak at 1436 cm
−1 and several peaks at 2830, 3050, and 3207 cm
−1 with remarkable intensity were observed. These peaks can be assigned to NH
4+ in (NH
4)
2SO
4 due to the reaction between H
2SO
4 and NH
3:
55| | | H2SO4(ads) + 2NH3 → (NH4)2SO4(ads) | (8) |
The peaks of surface nitrate observed in
Fig. 1B were not observed in the presence of SO
2 which indicates the inhibition effect of coexisting SO
2 on the photooxidation of NH
3 to nitrate.
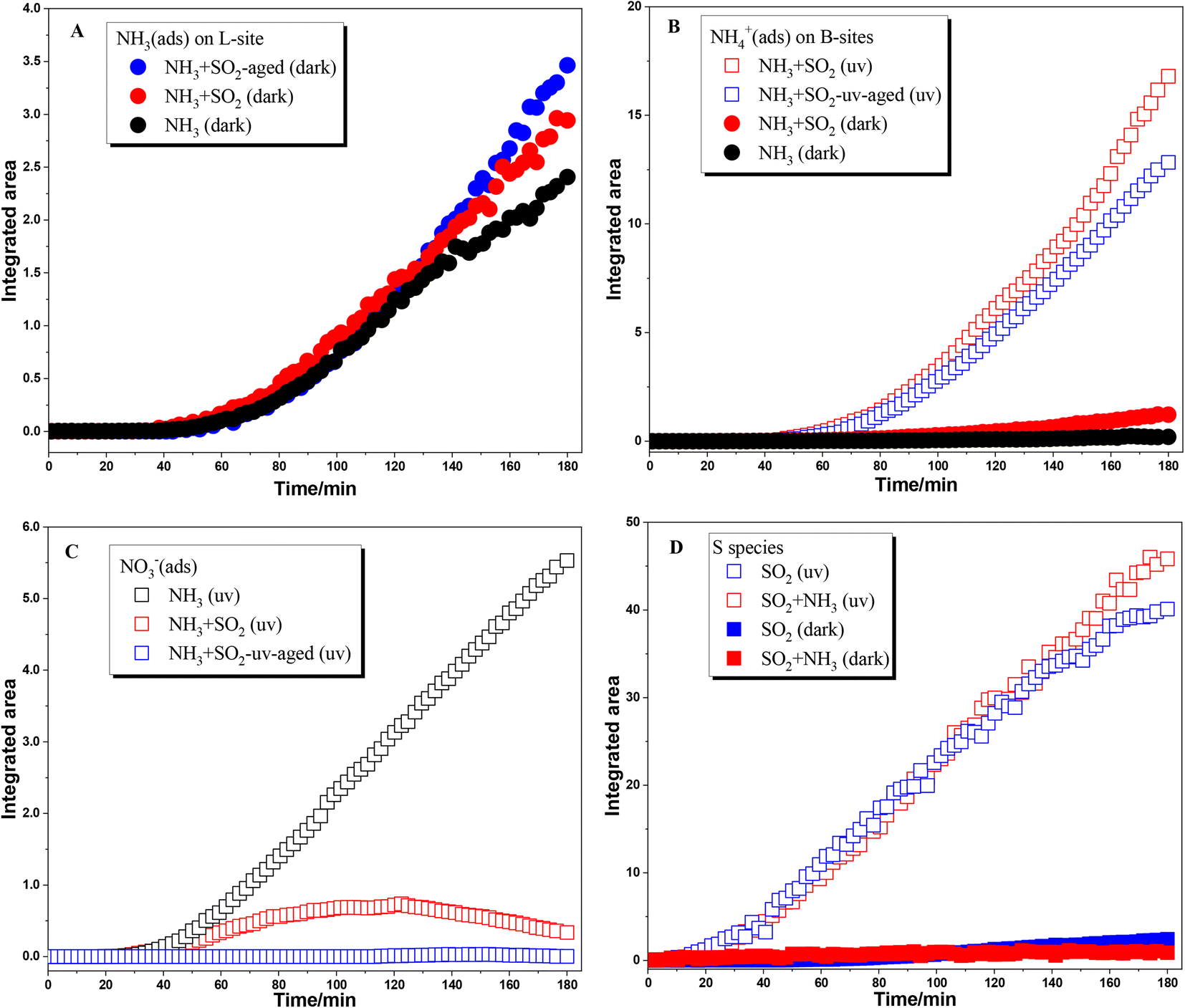
The surface products in different reactions are compared by integrating the IR peak area and are shown in Fig. 2. The infrared peak integral area of the surface product became more obvious after 40 min of the reaction, which may be due to the low infrared sensitivity of ammonia species adsorbed on the TiO2 surface in the beginning. Fig. 2A shows the comparison of the integrated area of peaks due to NH3(ads) on Lewis acid sites. Because these species were not observed in the reaction upon UV irradiation, here we only compared the heterogeneous reactions of NH3 on TiO2 under dark conditions. These reactions were analyzed including individual NH3 adsorption on fresh TiO2 particles and SO2-aged TiO2 particles, and mixing NH3 and SO2 adsorption on TiO2 particles. In the initial 120 min, the amount of NH3(ads) on Lewis acid sites showed little difference among these three reactions. This may be due to the formation of NH3(ads) mainly through the interaction of NH3 with surface OH or H2O. After 120 min, the increase rate of NH3(ads) in both NH3 + SO2 aged TiO2 and NH3 + SO2 systems is greater than that in the individual NH3 system. These results suggest that the coexistence of SO2 or SO2 aging treatment can slightly promote the adsorption of NH3 on TiO2.
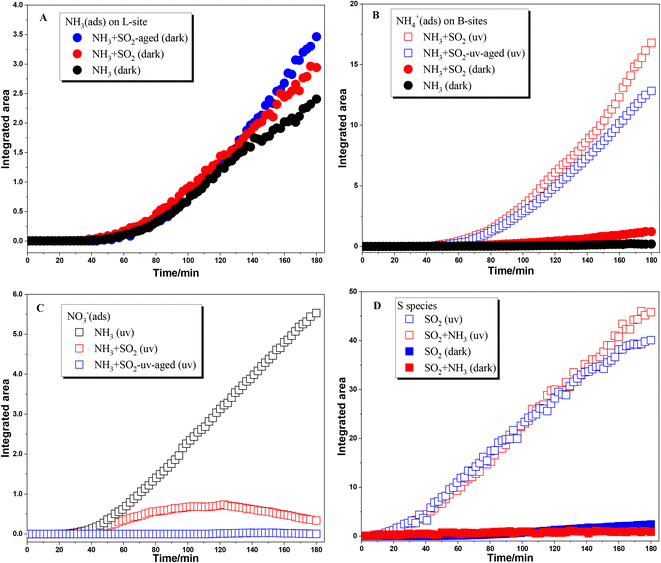 |
| | Fig. 2 Dynamic change of surface products formed in heterogeneous reactions of NH3 on TiO2 with/without SO2 and UV irradiation. (A) NH3(ads) on Lewis acid sites; (B) NH4+ on Brønsted acid sites; (C) surface nitrate species; (D) surface S species. | |
As for NH4+ species adsorbed on Brønsted acid sites, coexisting SO2 also promotes the formation of NH4+ species under dark conditions compared to individual NH3 adsorption (seen in Fig. 2B). This is because the sulfite/bisulfite species formed during SO2 adsorption on the surface of TiO2 could also serve as Brønsted acid sites for NH4+ adsorption. Moreover, the formation of NH4+ species was greatly enhanced when NH3 was adsorbed on SO2 photochemically aged TiO2 or coexisting SO2 on TiO2 in the presence of UV irradiation. The increase in the formation of NH4+ species was attributed to the presence of surface sulfate or H2SO4 which can readily combine with NH3. It should be noted that individual NH3 adsorption on fresh TiO2 does not produce surface NH4+ species and only surface nitrate was formed upon UV irradiation. Thus, in the absence of SO2, the reaction mechanism of NH3 on TiO2 upon UV irradiation is photooxidation, but it goes through an acid–base neutralization reaction in the presence of SO2. Fig. 2C shows the formation of nitrate in different photochemical reactions. The nitrate species shows a continuous increase in the photooxidation of NH3 on fresh TiO2 while no nitrate was observed in the adsorption of NH3 on SO2 photochemically aged TiO2. When SO2 was coexisting with NH3, the formation of nitrate increased in the initial step of reaction and decreased in the later step. This is not unexpected. In the photochemical reaction of coexisting SO2 and NH3 on TiO2, there are enough surface sites for oxidation of both SO2 and NH3. Then nitrate can form in the initial stage. With the continuous increase of surface sulfate, the generated nitrate is gradually replaced or decomposed.40 Therefore, in the photoreaction where SO2 and NH3 coexist, the surface nitrate tends to increase first and then decrease.
NH3 shows little effect on the formation of surface sulfur species in both dark reactions and photoreactions (Fig. 2D). In a previous study, Yang et al.30 found that NH3 can promote the formation of sulfate on the TiO2 surface under dark conditions, in which much higher concentrations of SO2 (200 ppm) and NH3 (100 ppm) were used compared to 5 ppm in the present study. The adsorption of SO2 on the oxide surface is mainly through the interaction with surface OH or H2O. When the concentration of SO2 and NH3 is relatively low, enough OH on the surface makes the competition between them not obvious. As for the photoreaction, the oxidation of SO2 is attributed to surface reactive oxygen species (ROS) like ˙OH while NH3 mainly participates in the subsequent neutralization reaction.
3.2 Flow tube reactor study for the uptake of NH3 on TiO2
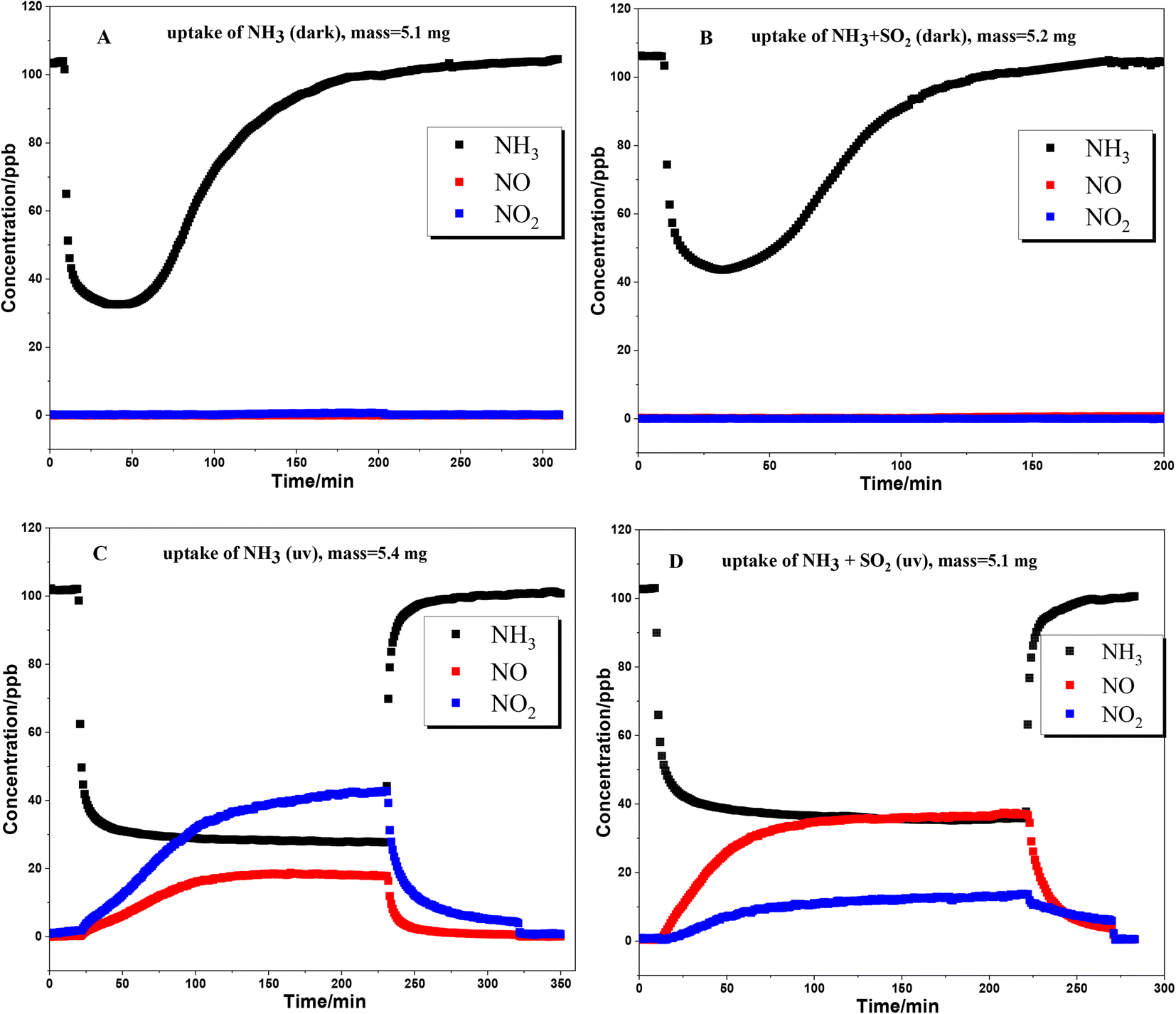
The uptake of NH3 on TiO2 particles was further studied with a flow tube reactor. Fig. 3 shows the results of NH3 uptake on TiO2 particles under various conditions. Under dark conditions (Fig. 3A and B), whether SO2 is present or not, an abrupt decrease in NH3 concentration was observed when TiO2 was exposed to NH3, indicating strong interaction between NH3 and TiO2 particles. It should be noted that the concentration of NH3 reached the minimum point after half an hour, and then gradually increased. The lowest concentration of NH3 in the presence of SO2 is higher than that in the absence of SO2. This could be due to the competition effect between SO2 and NH3 since surface OH groups are the main adsorption sites for both NH3 and SO2. For the products, no NOx formed under dark conditions, suggesting that the oxidation of NH3 on the TiO2 surface hardly occurred under dark conditions.
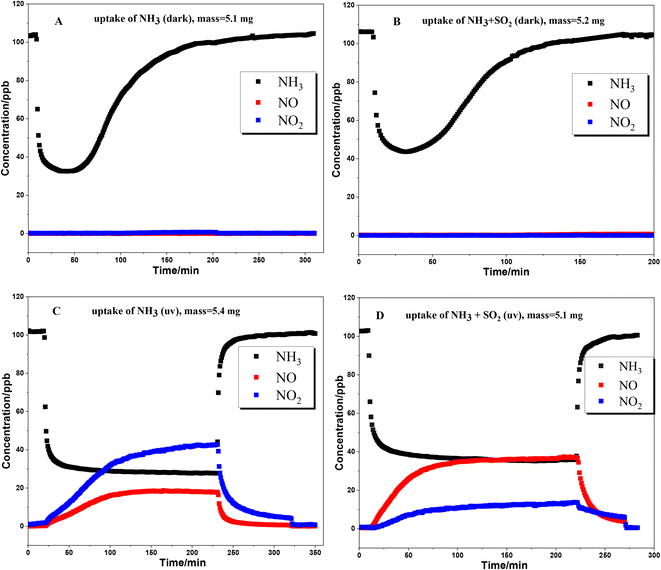 |
| | Fig. 3 Uptake of NH3 on TiO2 under various conditions. (A) dark condition; (B) dark condition with 200 ppb SO2 coexisting; (C) UV irradiation condition; (D) UV irradiation with 200 ppb SO2 coexisting. Sample masses: 5.1–5.4 mg. | |
When the uptake of NH3 took place in the presence of UV irradiation (Fig. 3C and D), an abrupt decrease in NH3 concentration was also observed at the beginning. However, the concentration of NH3 had not achieved the minimum value and gradually reached a plateau after about 90 min. The formation of NO and NO2 was accompanied by the uptake of NH3. The concentrations of NO and NO2 also reached a plateau simultaneously with NH3. These results suggested that NH3 was converted to NO and NO2 through the heterogeneous photocatalytic oxidation reaction on the TiO2 surface. Kebede et al.40 proposed a net photooxidation reaction mechanism of NH3 on TiO2 as follows:
| | | Ti4+–O− + NO → Ti4+–ONO− → Ti3+ + NO2 | (10) |
The intermediate nitrite was not observed in the present study which may be due to a higher concentration (5 ppm) than that (0.74 ppm) used in Kebede
et al. In addition, surface nitrate species were the dominant surface products in the present study, which could be due to the further photooxidation of NOx to nitrate on TiO
2:
| | | 2NO2 + O2 + 2e− → 2NO3−(ads) | (11) |
| | | NO + O2 + e− → NO3−(ads) | (12) |
It is interesting to note that coexisting SO
2 can greatly change the distribution of products. Compared to NH
3 individual uptake, the presence of 100 ppb SO
2 caused the concentration of NO to be higher than that of NO
2 (
Fig. 3C and D). In order to reveal the effect of SO
2 on the photooxidation of NH
3 on the TiO
2 surface, the yields of NO and NO
2 from NH
3 conversion at different coexisting SO
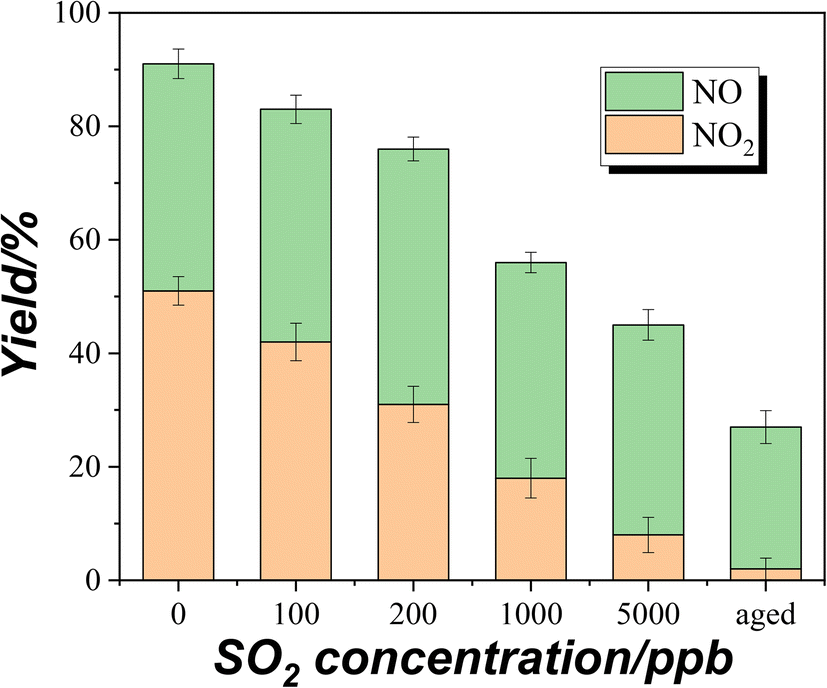
2 concentrations are compared, as shown in
Fig. 4. These values were based on the plateau concentrations in uptake experiments. The aged samples were prepared by exposing the TiO
2 samples to 5 ppm SO
2 for 4 h upon UV irradiation. The total yields of NO and NO
2 were about 90% in the absence of SO
2. Combined with the DRIFTS result (
Fig. 1B), it suggests that partial NOx has been converted to surface nitrate. It should be noted that the nitrate on the surface of TiO
2 can produce NO and NO
2 through photolysis.
40 Hence, there could be an equilibrium between surface nitrate and gaseous NOx (NO + NO
2). When the concentration of coexisting SO
2 increased, the total yields of NO and NO
2 from NH
3 conversion decreased. The photooxidation of SO
2 on TiO
2 leads to the formation of H
2SO
4/sulfate which can combine with NH
3 to form ammonium sulfate, as seen in
Fig. 1D. Therefore, NH
3 is trapped on the surface as ammonium, instead of being converted to NOx as well as nitrate. On the other hand, the photooxidation of SO
2 to H
2SO
4/sulfate will consume ROS on the surface. This could decrease the oxidation of NH
3 to NO and the consequent oxidation of NO to NO
2.
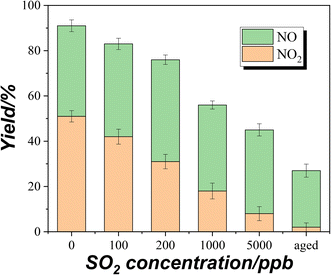 |
| | Fig. 4 The yields of NO and NO2 from NH3 conversion at different SO2 concentrations. | |
3.3 The uptake coefficients of NH3 on TiO2
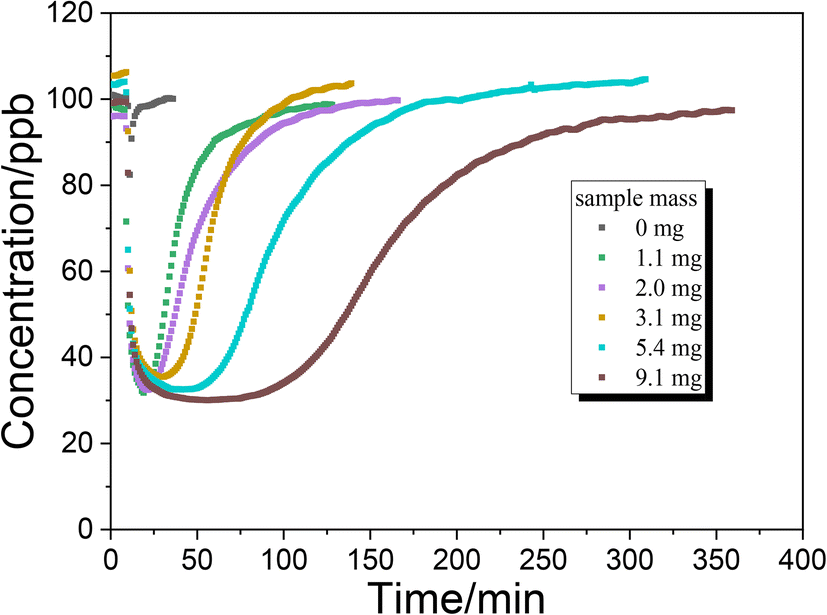
Fig. 5 shows the uptake process of NH3 on TiO2 with various masses under dark conditions. The time for NH3 concentration to reach the minimum value under dark conditions is prolonged with increasing sample mass. When the sample mass exceeds 5 mg, the time to reach the minimum concentration exceeds 30 minutes. It is possible that the diffusion of NH3 to the underlayers is much slower than the NH3 reaction on surface sites. In other words, the diffusion of NH3 may be the rate determining step for uptake on TiO2. To calculate the uptake coefficient in the dark reaction, the minimum concentration of NH3 is also selected as a convention which represents the initial uptake coefficient. In the presence of UV irradiation, the concentration of NH3 had not achieved the minimum value and gradually reached a plateau (Fig. 3C and D). As for the photoreaction, the plateau concentration of NH3 is selected for the calculation of the uptake coefficient which represents the steady-state uptake coefficient. The measured uptake coefficients (γgeo) of NH3 on TiO2 under various conditions are in the range of 2.0–2.5 × 10−5 while the linear mass dependence of γgeo was not observed. Coexisting SO2 exhibits an inhibition effect on the γgeo of NH3 on TiO2 because of the competitive effect between SO2 and NH3 on the surface hydroxyl groups. Upon UV irradiation, the measured steady-state uptake coefficients of NH3 on TiO2 are also in the range of 2.0–2.5 × 10−5 with no linear mass dependence.
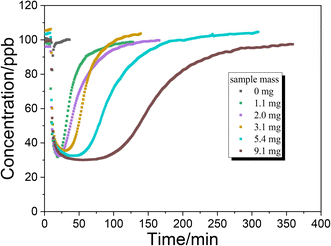 |
| | Fig. 5 Uptake of NH3 on TiO2 with various masses under dark conditions. | |
4. Conclusion and environmental implications
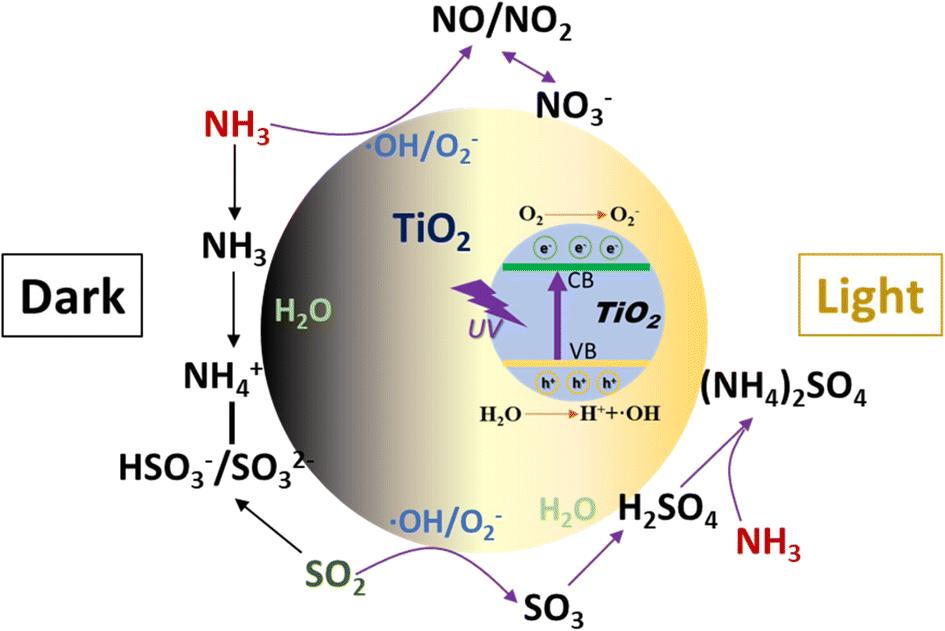
The heterogeneous reactions of NH3 on TiO2 with or without irradiation were investigated using DRIFTS and a flow tube reactor. Under dark conditions, adsorbed NH3 on Lewis acid sites was found to be the dominant surface species regardless of the coexisting SO2. The adsorption of SO2 produces sulfite and bisulfite species which can act as Brønsted acid sites to enhance the formation of NH4+ on the surface. Upon irradiation, NH3 can be converted to NO, NO2, and nitrate on TiO2 without the formation of surface adsorbed NH3 and NH4+ species. However, the presence of SO2 can promote the formation of NH4+ due to the combination of H2SO4/sulfate with NH3 to form (NH4)2SO4, which inhibits the photochemical oxidation of NH3. The main heterogeneous reaction pathways of NH3 on TiO2 are summarized in Scheme 1.
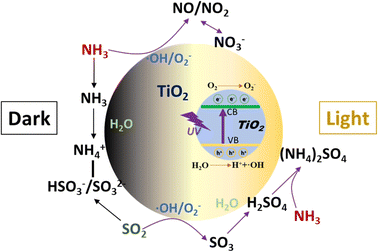 |
| | Scheme 1 The heterogeneous reaction pathways of NH3 on TiO2. | |
The initial uptake coefficients of NH3 on TiO2 are in the range of 2.0–2.5 × 10−5 and independent of the sample mass in the range of 1–10 mg. SO2 showed an inhibition effect on the uptake coefficients of NH3 under both dark and photoreaction conditions, because of the competition effect between SO2 and NH3 adsorption. These results show that the conversion of NH3 on the surface of TiO2 is an important sink. It also demonstrates that the importance of the synergistic effect of coexisting pollutants in the heterogeneous reaction is worthy of further study.
As an important alkaline gas in the atmosphere, NH3 is the main precursor of ammonium aerosol, which has an important impact on the formation of secondary aerosol.3,26 However, at present, there is little understanding of the atmospheric chemical process of NH3, and model research always applies the thermodynamic equilibrium parameter of NH3 with main acidic substances such as H2SO4 and HNO3, resulting in inaccurate evaluation of the contribution of NH3. China has been facing a serious problem of atmospheric fine particle and NH3 emissions from various sources are huge, resulting in very high NH3 concentration in ambient atmosphere.17,19,21 The results of this study show that the heterogeneous process participated by NH3 can contribute to not only ammonium aerosol but also nitrate and NOx, which is also closely related to the formation process of sulfate. Therefore, much attention should be paid to the atmospheric chemical processes involved in ammonia in future atmospheric chemistry and models. At the same time, it also reminds us that in the control strategy of atmospheric fine particles, we should not only control SO2 and NOx, but also control NH3 emission.22 In addition, TiO2 is widely used as a photocatalyst material in the environment for the removal of ammonia.52,56,57 These catalysts showed good catalytic removal activity to NH3 in experimental tests. However, the synergistic effect of coexisting pollutants should be fully considered when they are further applied to the actual atmospheric environment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (21922610, 22188102, and 22276205). The authors appreciate the Ozone Formation Mechanism and Control Strategies Project of the Research Center for Eco-Environmental Sciences, CAS (RCEES-CYZX-2020), and the Young Talent Project of the Center for Excellence in Regional Atmospheric Environment, CAS (CERAE201801).
References
-
WHO, Ambient air pollution: a global assessment of exposure and burden of disease, World Health Organization, Geneva, 2016 Search PubMed.
- C. L. Reddington, L. Conibear, C. Knote, B. J. Silver, Y. J. Li, C. K. Chan, S. R. Arnold and D. V. Spracklen, Exploring the impacts of anthropogenic emission sectors on PM2.5 and human health in South and East Asia, Atmos. Chem. Phys., 2019, 19, 11887–11910 CrossRef CAS.
- R. K. Pathak, W. S. Wu and T. Wang, Summertime PM2.5 ionic species in four major cities of China: nitrate formation in an ammonia-deficient atmosphere, Atmos. Chem. Phys., 2009, 9, 1711–1722 CrossRef CAS.
- F. Yang, J. Tan, Q. Zhao, Z. Du, K. He, Y. Ma, F. Duan, G. Chen and Q. Zhao, Characteristics of PM2.5 speciation in representative megacities and across China, Atmos. Chem. Phys., 2011, 11, 5207–5219 CrossRef CAS.
- X. Zhang, Y. Wang, T. Niu, X. Zhang, S. Gong, Y. Zhang and J. Sun, Atmospheric aerosol compositions in China: spatial/temporal variability, chemical signature, regional haze distribution and comparisons with global aerosols, Atmos. Chem. Phys., 2012, 12, 779–799 CrossRef CAS.
- R.-J. Huang, Y. Zhang, C. Bozzetti, K.-F. Ho, J.-J. Cao, Y. Han, K. R. Daellenbach, J. G. Slowik, S. M. Platt, F. Canonaco, P. Zotter, R. Wolf, S. M. Pieber, E. A. Bruns, M. Crippa, G. Ciarelli, A. Piazzalunga, M. Schwikowski, G. Abbaszade, J. Schnelle-Kreis, R. Zimmermann, Z. An, S. Szidat, U. Baltensperger, I. E. Haddad and A. S. H. Prévôt, High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 2014, 514, 218–222 CrossRef CAS PubMed.
- M. Kulmala, L. Pirjola and J. Mäkelä, Stable sulphate clusters as a source of new atmospheric particles, Nature, 2000, 404, 66–69 CrossRef CAS PubMed.
- M. Kulmala, How particles nucleate and grow, Science, 2003, 302, 1000 CrossRef CAS PubMed.
- E. Harris, B. Sinha, D. van Pinxteren, A. Tilgner, K. W. Fomba, J. Schneider, A. Roth, T. Gnauk, B. Fahlbusch and S. Mertes, Enhanced role of transition metal ion catalysis during in-cloud oxidation of SO2, Science, 2013, 340, 727–730 CrossRef CAS.
- J. Xue, Z. Yuan, S. M. Griffith, X. Yu, A. K. Lau and J. Z. Yu, Sulfate formation enhanced by a cocktail of high NOx, SO2, particulate matter and droplet pH during haze-fog events in megacities in China: An Observation-Based Modeling investigation, Environ. Sci. Technol., 2016, 50, 7325–7334 CrossRef CAS.
- C. R. Usher, A. E. Michel and V. H. Grassian, Reactions on mineral dust, Chem. Rev., 2003, 103, 4883–4939 CrossRef CAS PubMed.
- J. N. Crowley, M. Ammann, R. A. Cox, R. G. Hynes, M. E. Jenkin, A. Mellouki, M. J. Rossi, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V–heterogeneous reactions on solid substrates, Atmos. Chem. Phys., 2010, 10, 9059–9223 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 2004, 4, 1461–1738 CrossRef CAS.
- W. L. Chang, P. V. Bhave, S. S. Brown, N. Riemer, J. Stutz and D. Dabdub, Heterogeneous Atmospheric Chemistry, Ambient Measurements, and Model Calculations of N2O5: A Review, Aerosol Sci. Technol., 2011, 45, 665–695 CrossRef.
- V. H. Grassian, Chemical reactions of nitrogen oxides on the surface of oxide, carbonate, soot, and mineral dust particles: Implications for the chemical balance of the troposphere, J. Phys. Chem. A, 2002, 106, 860–877 CrossRef CAS.
- S. Reis, R. W. Pinder, M. Zhang, G. Lijie and M. A. Sutton, Reactive nitrogen in atmospheric emission inventories, Atmos. Chem. Phys., 2009, 9, 7657–7677 CrossRef CAS.
- X. Huang, Y. Song, M. Li, J. Li, Q. Huo, X. Cai, T. Zhu, M. Hu and H. Zhang, A high-resolution ammonia emission inventory in China, Global Biogeochem. Cycles, 2012, 26, GB1030 Search PubMed.
- S. N. Behera, M. Sharma, V. P. Aneja and R. Balasubramanian, Ammonia in the atmosphere: a review on emission sources, atmospheric chemistry and deposition on terrestrial bodies, Environ. Sci. Pollut. Res., 2013, 20, 8092–8131 CrossRef CAS.
- Z. Meng, X. Xu, W. Lin, B. Ge, Y. Xie, B. Song, S. Jia, R. Zhang, W. Peng, Y. Wang, H. Cheng, W. Yang and H. Zhao, Role of ambient ammonia in particulate ammonium formation at a rural site in the North China Plain, Atmos. Chem. Phys., 2018, 18, 167–184 CrossRef CAS.
- F. J. Dentener and P. J. Crutzen, A three-dimensional model of the global ammonia cycle, J. Atmos. Chem., 1994, 19, 331–369 CrossRef CAS.
- Z. Y. Meng, W. L. Lin, X. M. Jiang, P. Yan, Y. Wang, Y. M. Zhang, X. F. Jia and X. L. Yu, Characteristics of atmospheric ammonia over Beijing, China, Atmos. Chem. Phys., 2011, 11, 6139–6151 CrossRef CAS.
- X. Fu, S. X. Wang, J. Xing, X. Y. Zhang, T. Wang and J. M. Hao, Increasing Ammonia Concentrations Reduce the Effectiveness of Particle Pollution Control Achieved via SO2 and NOx Emissions Reduction in East China, Environ. Sci. Technol. Lett., 2017, 4, 221–227 CrossRef CAS.
- Y. Wang, Q. Q. Zhang, K. He, Q. Zhang and L. Chai, Sulfate-nitrate-ammonium aerosols over China: response to 2000-2015 emission changes of sulfur dioxide, nitrogen oxides, and ammonia, Atmos. Chem. Phys., 2013, 13, 2635–2652 CrossRef.
- M. Liu, X. Huang, Y. Song, J. Tang, J. Cao, X. Zhang, Q. Zhang, S. Wang, T. Xu, L. Kang, X. Cai, H. Zhang, F. Yang, H. Wang, J. Z. Yu, A. K. H. Lau, L. He, X. Huang, L. Duan, A. Ding, L. Xue, J. Gao, B. Liu and T. Zhu, Ammonia emission control in China would mitigate haze pollution and nitrogen deposition, but worsen acid rain, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7760–7765 CrossRef CAS.
- W. Yang, Q. Ma, Y. Liu, J. Ma, B. Chu, L. Wang and H. He, Role of NH3 in the Heterogeneous Formation of Secondary Inorganic Aerosols on Mineral Oxides, J. Phys. Chem. A, 2018, 122, 6311–6320 CrossRef CAS PubMed.
- R.-J. Huang, J. Duan, Y. Li, Q. Chen, Y. Chen, M. Tang, L. Yang, H. Ni, C. Lin, W. Xu, Y. Liu, C. Chen, Z. Yan, J. Ovadnevaite, D. Ceburnis, U. Dusek, J. Cao, T. Hoffmann and C. D. O'Dowd, Effects of NH3 and alkaline metals on the formation of particulate sulfate and nitrate in wintertime Beijing, Sci. Total Environ., 2020, 717, 137190 CrossRef CAS.
- W. Xu, Y. Kuang, C. Zhao, J. Tao, G. Zhao, Y. Bian, W. Yang, Y. Yu, C. Shen, L. Liang, G. Zhang, W. Lin and X. Xu, NH3-promoted hydrolysis of NO2 induces explosive growth in HONO, Atmos. Chem. Phys., 2019, 19, 10557–10570 CrossRef CAS.
- S. Ge, G. Wang, S. Zhang, D. Li, Y. Xie, C. Wu, Q. Yuan, J. Chen and H. Zhang, Abundant NH3 in China Enhances Atmospheric HONO Production by Promoting the Heterogeneous Reaction of SO2 with NO2, Environ. Sci. Technol., 2019, 53, 14339–14347 CrossRef CAS.
- L. Li, Z. Duan, H. Li, C. Zhu, G. Henkelman, J. S. Francisco and X. C. Zeng, Formation of HONO from the NH3-promoted hydrolysis of NO2 dimers in the atmosphere, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7236–7241 CrossRef CAS PubMed.
- W. Yang, H. He, Q. Ma, J. Ma, Y. Liu, P. Liu and Y. Mu, Synergistic formation of sulfate and ammonium resulting from reaction between SO2 and NH3 on typical mineral dust, Phys. Chem. Chem. Phys., 2016, 18, 956–964 RSC.
- Y. Liu, J. Liggio, R. Staebler and S. M. Li, Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 2015, 15, 13569–13584 CrossRef CAS.
- C. M. Stangl and M. V. Johnston, Aqueous Reaction of Dicarbonyls with Ammonia as a Potential Source of Organic Nitrogen in Airborne Nanoparticles, J. Phys. Chem. A, 2017, 121, 3720–3727 CrossRef CAS PubMed.
- G. Zhang, X. Lian, Y. Fu, Q. Lin, L. Li, W. Song, Z. Wang, M. Tang, D. Chen, X. Bi, X. Wang and G. Sheng, High secondary formation of nitrogen-containing organics (NOCs) and its possible link to oxidized organics and ammonium, Atmos. Chem. Phys., 2020, 20, 1469–1481 CrossRef CAS.
- J. R. Horne, S. Zhu, J. Montoya-Aguilera, M. L. Hinks, L. M. Wingen, S. A. Nizkorodov and D. Dabdub, Reactive uptake of ammonia by secondary organic aerosols: Implications for air quality, Atmos. Environ., 2018, 189, 1–8 CrossRef CAS.
- K. Wu, S. Zhu, Y. Liu, H. Wang, X. Yang, L. Liu, D. Dabdub and C. D. Cappa, Modeling Ammonia and Its Uptake by Secondary Organic Aerosol Over China, J. Geophys. Res., 2021, 126, e2020JD034109 CAS.
- Z. Feng, F. Zheng, C. Yan, P. Fu, Y. Zhang, Z. Lin, J. Cai, W. Du, Y. Wang, J. Kangasluoma, F. Bianchi, T. Petäjä, Y. Wang, M. Kulmala and Y. Liu, The impact of ammonium on the distillation of organic carbon in PM2.5, Sci. Total Environ., 2022, 803, 150012 CrossRef CAS PubMed.
- T. Chen, Y. Liu, Q. Ma, B. Chu, P. Zhang, C. Liu, J. Liu and H. He, Significant source of secondary aerosol: formation from gasoline evaporative emissions in the presence of SO2 and NH3, Atmos. Chem. Phys., 2019, 19, 8063–8081 CrossRef CAS.
- R. Gieré and X. Querol, Solid Particulate Matter in the Atmosphere, Elements, 2010, 6, 215–222 CrossRef.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Titanium dioxide photocatalysis in atmospheric chemistry, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS.
- M. A. Kebede, M. E. Varner, N. K. Scharko, R. B. Gerber and J. D. Raff, Photooxidation of Ammonia on TiO2 as a Source of NO and NO2 under Atmospheric Conditions, J. Am. Chem. Soc., 2013, 135, 8606–8615 CrossRef CAS PubMed.
- Q. Ma, C. Zhong, J. Ma, C. Ye, Y. Zhao, Y. Liu, P. Zhang, T. Chen, C. Liu, B. Chu and H. He, Comprehensive Study about the Photolysis of Nitrates on Mineral Oxides, Environ. Sci. Technol., 2021, 55, 8604–8612 CrossRef CAS.
- T. Armaroli, T. Becue and S. Gautier, Diffuse reflection infrared spectroscopy (DRIFTS): Application to the in situ analysis of catalysts, Oil Gas Sci. Technol., 2004, 59, 215–237 CrossRef CAS.
- Y. Liu, C. Han, J. Ma, X. Bao and H. He, Influence of relative humidity on heterogeneous kinetics of NO2 on kaolin and hematite, Phys. Chem. Chem. Phys., 2015, 17, 19424–19431 RSC.
- C. Han, Y. Liu and H. He, Role of organic carbon in heterogeneous reaction of NO2 with soot, Environ. Sci. Technol., 2013, 47, 3174–3181 CrossRef CAS PubMed.
- Q. Ma, T. Wang, C. Liu, H. He, Z. Wang, W. Wang and Y. Liang, SO2 initiates the efficient conversion of NO2 to HONO on MgO surface, Environ. Sci. Technol., 2017, 51, 3767–3775 CrossRef CAS.
- D. M. Murphy and D. W. Fahey, Mathematical treatment of the wall loss of a trace species in denuder and catalytic converter tubes, Anal. Chem., 1987, 59, 2753–2759 CrossRef CAS.
- D. O. Cooney, S.-S. Kim and E. J. Davis, Analyses of mass transfer in hemodialyzers for laminar blood flow and homogeneous dialysate, Chem. Eng. Sci., 1974, 29, 1731–1738 CrossRef CAS.
- C. Liu, Q. Ma, H. He, G. He, J. Ma, Y. Liu and Y. Wu, Structure–activity relationship of surface hydroxyl groups during NO2 adsorption and transformation on TiO2 nanoparticles, Environ. Sci.: Nano, 2017, 2388–2394 RSC.
- C. Lee, G. Fitzgerald, M. Planas and J. J. Novoa, Ionization of Bases in Water: Structure and Stability of the NH4+···OH- Ionic Forms in Ammonia−Water Clusters, J. Phys. Chem., 1996, 100, 7398–7404 CrossRef CAS.
- D. J. Donaldson, Adsorption of Atmospheric Gases at the Air−Water Interface. I. NH3, J. Phys. Chem. A, 1999, 103, 62–70 CrossRef CAS.
- G. M. Underwood, T. M. Miller and V. H. Grassian, Transmission FT-IR and Knudsen cell study of the heterogeneous reactivity of gaseous nitrogen dioxide on mineral oxide particles, J. Phys. Chem. A, 1999, 103, 6184–6190 CrossRef CAS.
- S. Yamazoe, T. Okumura, Y. Hitomi, T. Shishido and T. Tanaka, Mechanism of Photo-Oxidation of NH3 over TiO2: Fourier Transform Infrared Study of the Intermediate Species, J. Phys. Chem. C, 2007, 111, 11077–11085 CrossRef CAS.
- Q. Ma, L. Wang, B. Chu, J. Ma and H. He, Contrary Role of H2O and O2 in the Kinetics of Heterogeneous Photochemical Reactions of SO2 on TiO2, J. Phys. Chem. A, 2019, 123, 1311–1318 CrossRef CAS PubMed.
- C. E. Nanayakkara, J. Pettibone and V. H. Grassian, Sulfur dioxide adsorption and photooxidation on isotopically-labeled titanium dioxide nanoparticle surfaces: roles of surface hydroxyl groups and adsorbed water in the formation and stability of adsorbed sulfite and sulfate, Phys. Chem. Chem. Phys., 2012, 14, 6957–6966 RSC.
- Z. Zhang, L. Chen, Z. Li, P. Li, F. Yuan, X. Niu and Y. Zhu, Activity and SO2 resistance of amorphous CeaTiOx catalysts for the selective catalytic reduction of NO with NH3: in situ DRIFT studies, Catal. Sci. Technol., 2016, 6, 7151–7162 RSC.
- S. Yamazoe, T. Okumura and T. Tanaka, Photo-oxidation of NH3 over various TiO2, Catal. Today, 2007, 120, 220–225 CrossRef CAS.
- M. Chen, J. Ma, B. Zhang, F. Wang, Y. Li, C. Zhang and H. He, Facet-dependent performance of anatase TiO2 for photocatalytic oxidation of gaseous ammonia, Appl. Catal., B, 2018, 223, 209–215 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 abc,
Yaqi
Zhao
ab,
Peng
Zhang
*a,
Chang
Liu
abc,
Yaqi
Zhao
ab,
Peng
Zhang
*a,
Chang
Liu