
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ultra-stable reference electrode for scaled all-vanadium redox flow batteries†
Qian Huang *a,
Chaojie Songb,
Alasdair Crawfordc,
Zhengming Jiangb,
Alison Plattb,
Khalid Fatihb,
Christina Bockd and
David Reeda
*a,
Chaojie Songb,
Alasdair Crawfordc,
Zhengming Jiangb,
Alison Plattb,
Khalid Fatihb,
Christina Bockd and
David Reeda
aBattery Materials & Systems Group, Pacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: Qian.Huang@pnnl.gov
bEnergy, Mining & Environment, National Research Council Canada, Vancouver, BC V6T 1W5, Canada
cElectricity Infrastructure & Building Group, Pacific Northwest National Laboratory, Richland, WA 99352, USA
dEnergy, Mining & Environment, National Research Council Canada, Ottawa, ON K1A 0R6, Canada
First published on 9th November 2022
Abstract
Redox flow batteries (RFBs) have been investigated as a promising energy storage system (ESS) for grid applications over the past several decades due to their unique features, which include the separation of energy and power output, high safety, and long cycle life. It is therefore vital but still in severe deficiency to understand the reliability of RFBs, and the mechanisms that cause degradation with time. One of the primary challenges involves the unseparated contributions from individual electrodes due to the absence of a stable reference electrode (RE), particularly for long-term cycle testing in a scaled cell. Herein, we first develop an ultra-stable RE for scaled all-vanadium RFBs. The newly developed RE, based on a dynamic hydrogen electrode (DHE) with a novel design on the area (size) and surface roughness of platinum electrodes, demonstrates high accuracy and long-term stability that enables in situ monitoring of individual electrode potentials throughout 500 cycles. By introducing the RE approach to decouple the cathode and anode in conjunction with the measurement of voltage profiles, overpotentials and polarization curves, the reliability and degradation mechanism of a scaled all-vanadium RFB are further explored, revealing the diverse behaviors of individual electrodes. This exploratory work will benefit the future design and development of a stable RE for a scaled ESS, as well as the fundamental understanding of the RFB's reliability and degradation mechanism.
1. Introduction
Rapid expansion of renewable energies necessitates the development of efficient, cost-effective, and reliable electrochemical energy storage systems (ESSs). Recent calls for long duration energy storage technologies indicate a considerable demand for “big” ESSs in grid-level services.1 Because of their distinct characteristics, such as the separation of energy capacity and power output, high safety, long cycle life, and ease of manufacturing when compared to other rechargeable batteries,2,3 redox flow batteries have attracted growing interest in smart grid integration applications. Particularly, all-vanadium redox flow batteries (VRFBs), which use four different oxidation states of vanadium ions to form two soluble redox couples (VO2+/VO2+ and V2+/V3+) as catholyte and anolyte (see eqn (i)–(iii) of electrode and cell reactions), have demonstrated advantages on high electrochemical reversibility and high efficiencies, and are therefore promising candidates for stationary energy storage.4| Cathode: VO2+ + H2O ↔ VO2+ + 2H+ + e−, E° = 1.00 V | (i) |
| Anode: V3+ + e− ↔ V2+, E° = −0.25 V | (ii) |
| Full cell: VO2+ + V3+ + H2O ↔ VO2+ + V2+ + 2H+, E° = 1.25 V | (iii) |
However, one major technical obstacle haunting VRFB technology is the substantial capacity decay that occurs during long-term cycling. This is associated with complicated degradation mechanisms suffered inside the VRFB which includes (i) electrolyte crossover,4,5 (ii) electrolyte precipitation (as a strong function of temperature with different valences based vanadium ions),6 (iii) oxidation of carbon based electrodes (normally caused by electrolyte components or high potential),7,8 (iv) membrane degradation (mechanically or chemically),9–12 and (v) potential degradation from other inactive components (e.g. bipolar plate, gasket, and current collector).13,14 Therefore, it is essential to have a fundamental understanding of the reliability and degradation mechanisms of VRFBs, which is currently lacking but increasingly important for large-scaled applications.15 Moreover, the electrochemical performances of VRFBs have typically been tested in full cell mode, which makes it difficult to isolate contributions from individual electrodes and the cell degradation mechanism from each electrode challenging to identify. The creation of a reliable and stable reference electrode (RE) to decouple the cathode and anode and allow in situ monitoring of individual electrode signals to determine which electrode is causing performance degradation (i.e., which side is limiting the battery) during long-term cycling is in high demand.
The RE approaches that have been utilized for the research of RFBs are mostly leveraged from those in fuel cell applications.16,17 The REs can be classified into two types based on their positions: externally or internally located in the cell, as listed in Table S1.† The external REs, such as Ag/AgCl are inserted in the inlet or outlet tubing of the cell and normally show a relatively stable potential.18,19 However, the potential for chloride contamination and a large IR drop (associated with a large distance between the RE and electrode) are the primary issue for large-scaled applications. The internal REs, such as a single probe (Pt wire20,21 or carbon cloth/fiber22,23) or dynamic hydrogen electrode (DHE),24–26 is typically inserted between membranes and can decouple the cathode and anode potential drop and impedance. The internal REs, such as DHE, have been extensively used for the dynamic study of a VRFB.24,25 However, the potential of the internal RE continued to shift26 during cycling since the probe or the platinum electrodes (in DHE) inserted into the cell are easily influenced or contaminated by the surrounding environments inside the cell such as the continuous changing of the components (concentration and valence of vanadium ion) in the electrolytes.
Until now, there have been few studies on the design and development of REs specific for RFBs. Previous studies26 indicated that the main technical barrier is to maintain a stable and accurate RE potential in the presence of a dynamic electrolyte environment, including various redox couples (V2+, V3+, V4+, V5+) and supporting electrolyte (H+) with changes in concentration and valence of ions during cycling in a VRFB. More recently, the preliminary work on the reference electrode development for the reliability investigation of VRFB has been initiated in our group.27 The DHE based reference electrode was evaluated during long-term cycling, in which a couple of critical factors was found to influence the accuracy and stability of the DHE, including (i) the current flowing through the DHE, (ii) the contact area between DHE and cell membrane, and (iii) the proton concentration of the electrolytes. However, the stability is still a challenge for traditional DHEs, in particular when used in a scaled redox flow battery that in effect limits the use in practical applications.
In this work, a DHE based reference electrode with a novel design on the area and surface roughness of platinum electrode is first developed, which demonstrates a recorded high stability throughout 500 cycles for a scaled VRFB. The reliability and degradation mechanism of a scaled VRFB are further explored using this newly developed reference electrode to decouple the cathode and anode in voltage profiles, overpotentials, and polarization curves, revealing the different behavior of individual electrodes.
2. Experimental
2.1. DHE setup
A detailed setup of the DHE in this work was illustrated in Fig. 1a. Two Pt electrodes were inserted between membranes in a redox flow battery. The Pt electrodes (tips) were placed approx. 5 mm apart from the edge of the carbon felt electrodes of the VRFB to avoid any interference by the electric field generated between the two electrodes as well as for the ease of installation.26,28,29 These two Pt electrodes were connected to an external electric circuit with a 9 V battery and an optimized resistor of 1 MΩ.27 The Pt electrode that was directly connected to the negative terminal of the 9 V battery is employed as the DHE reference electrode where the hydrogen evolution reaction (HER) occurs and the current flowing through the cell is optimized using an adjustable resistor.26 Note that the distance between the two Pt electrodes (set at around 5 mm in our study) should have a negligible effect on the results because the adjustable resistor can probably compensate for the corresponding (electrolyte) resistance change.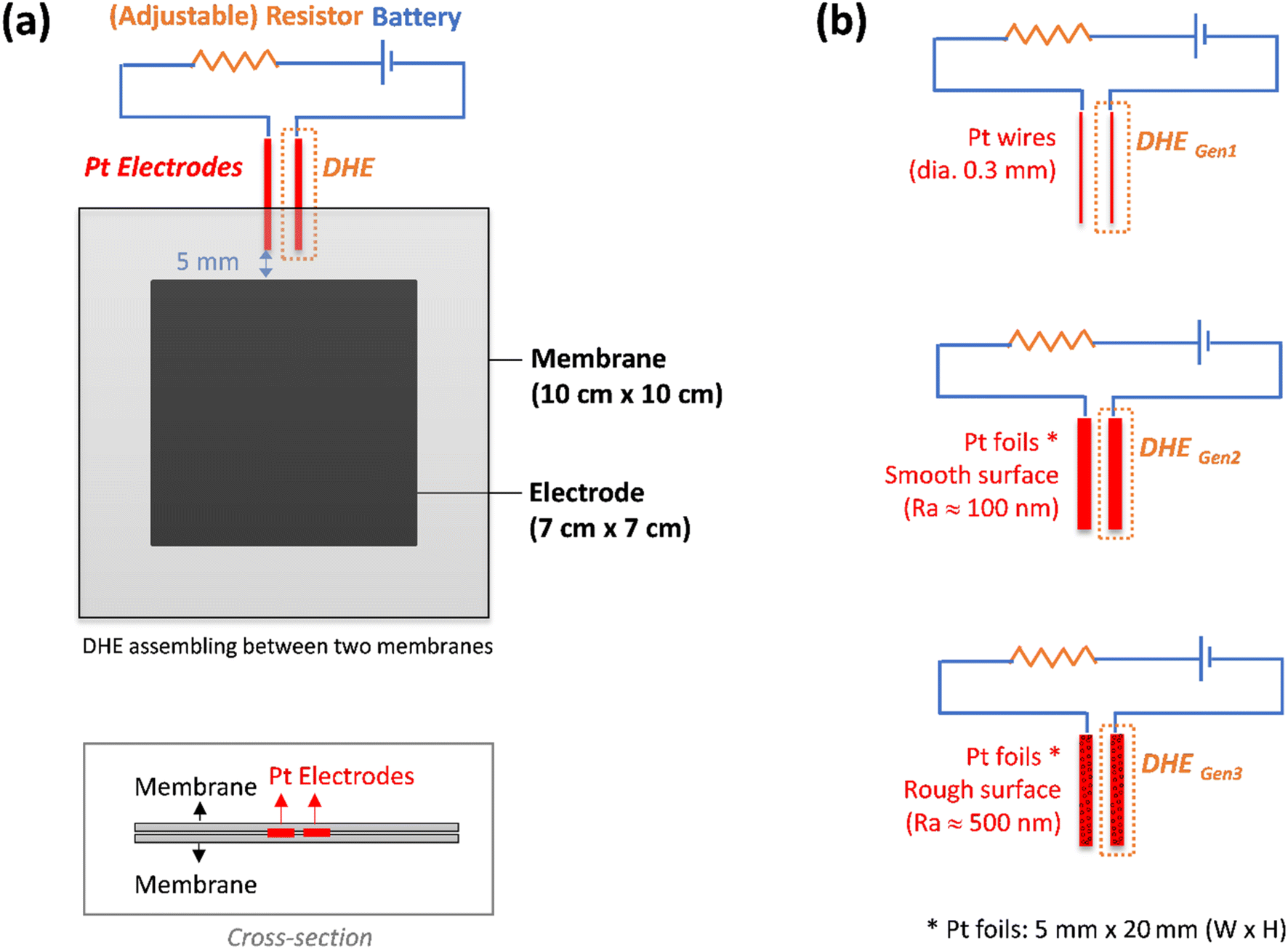 | ||
| Fig. 1 (a) A scheme of detailed design of DHE in an all-vanadium redox flow battery, and (b) DHE with three types of Pt electrodes: Pt wires (DHEGen1), Pt foils with smooth surface (DHEGen2), and Pt foils with rough surface (DHEGen3). | ||
In this study, three kinds of Pt electrodes with various shapes or surface roughness were introduced for the DHE setup respectively, as shown in Fig. 1b. They are (i) Pt wires (dia. 0.3 mm), (ii) Pt foils with the width of 5 mm and a smooth surface (surface roughness: Ra ≈ 100 nm), and (iii) Pt foils with the width of 5 mm and a rough surface (surface roughness: Ra ≈ 500 nm). The smooth surface of Pt foils in type (ii) was achieved by a general metal polishing process with fine sandpaper (1200 Grit) and diamond paste (3 micron/1 micron). The surface roughness of the Pt foils was measured by a profilometer, and the related results are shown in Fig. S1.†
2.2. Cell fabrication with internal DHE RE and external Ag/AgCl REs
A VRFB (49 cm2 active area, Standard Energy Co.) was fabricated by applying a pressure of 0.5 MPa to a stack consisting of a manifold frame, a current collector plate, a graphite bipolar plate (SIGRACET TF6, with a 0.025 inch thickness and flow-through type in no-flow pattern, SGL Group), a bipolar plate gasket, an internal flow frame (3 mm for thickness), a membrane gasket, a graphite felt electrode (GFD 4.6, SGL Group, 7 cm × 7 cm for active area), and a Nafion membrane (N212, Ion Power), for each half-cell (in order from exterior to interior). Prior to cell assembly, the graphite electrodes were thermally treated at 400 °C in air for 6 hours to increase its hydrophilicity. The DHE based internal RE was placed between both half cells. In addition, the two external REs of Ag/AgCl (with filling solution of 4 M KCl in AgCl, Pine Research Instrumentation) were placed in the inlet tubing of the catholyte and anolyte respectively, as shown in Fig. S2.†The vanadium electrolytes of 1.6 M V (V3+/V4+, 50/50, GfE)30 were used as received. The electrolytes were pumped from the electrolyte reservoirs (Pyrex graduated cylinders) to the flow cell compartments by using a peristaltic pump (Cole-Parmer, Masterflex L/S 7551) at a flow rate of 50 mL min−1 through Viton tubing. The reservoirs were bubbled with nitrogen for 10 min and sealed before testing.
2.3. Electrochemical testing and characterizations
The assembled flow cell was cycled in a charge–discharge process at room temperature with a voltage window between 1.6 and 0.8 V at a constant current density of 80 mA cm−2 using an Arbin cycler. Since the beginning electrolyte solution contains vanadium ions with a valence of 3.5 (a mixture of V3+/V4+, 50/50) for both catholyte and anolyte, the preparation of V4+ for catholyte and V3+ for anolyte was achieved by the electrochemical approach in the initial charging process of the cell, as illustrated in Fig. S3a.†Polarization curves were measured in a charged cell (charged to 1.6 V and then rested for 5 minutes, with an OCV of 1.45–1.5 V). E/i-measurements were carried out from 1.45 V to 0.75 V by reducing the potential every 40 or 30 s by 0.05 V (with 5 s’ rest after each potential measurement). The collected data points of current (i) were the value measured over the last few seconds of each potential step.
3. Results and discussion
3.1. A stable DHE reference electrode development for a scaled VRFB
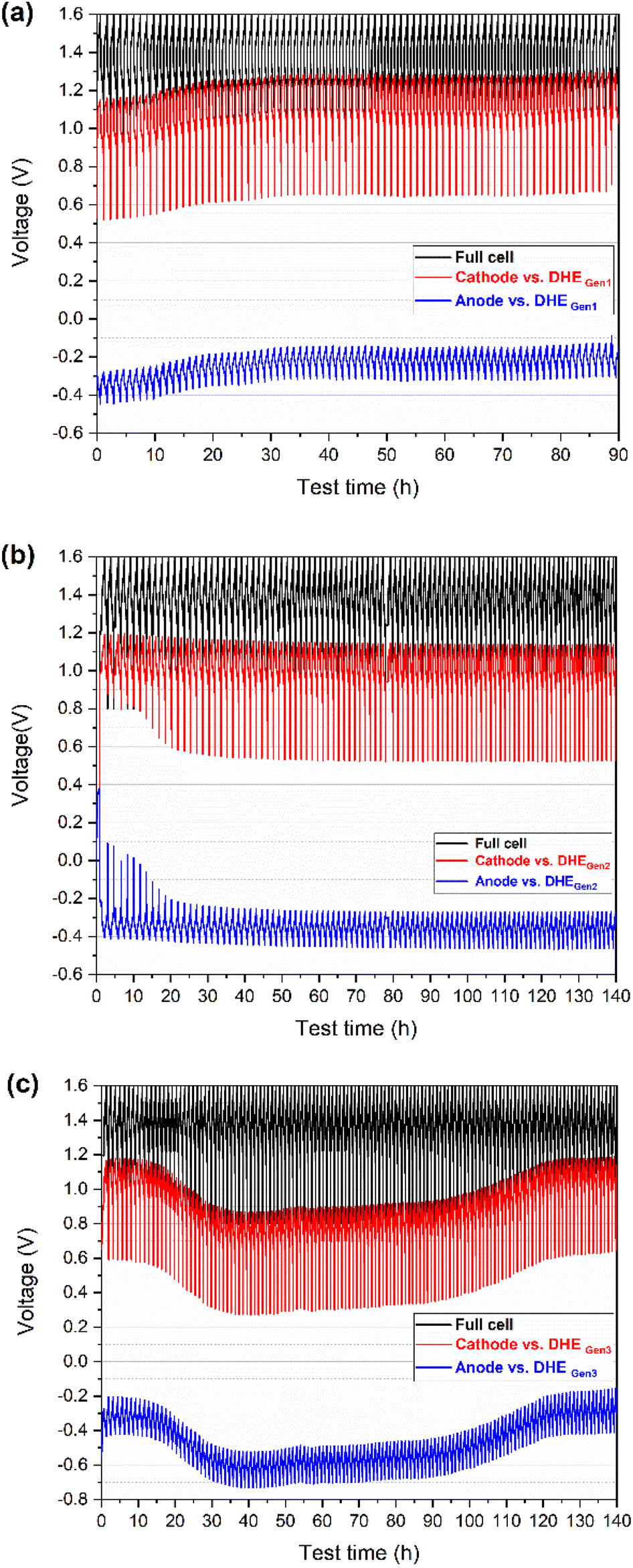 | ||
| Fig. 2 Charge–discharge voltage profiles (vs. time) of full cell (cathode vs. anode) and its individual electrode (cathode or anode) vs. DHE (with different Pt electrodes-based design) during long-term cycling of a scaled vanadium redox flow battery (49 cm2 in active area): (a) DHEGen1 (w/ Pt wires), (b) DHEGen2 (w/ Pt foils of smooth surface), and (c) DHEGen3 (w/ Pt foils of rough surface). | ||
As reported previously,26,27 a stable and reliable DHE reference electrode lies in the consistent existence of the hydrogen evolution reaction (HER) on the Pt electrode of DHE. To achieve a stable HER existence in a largely scaled flow cell, the design and optimization of the working (Pt) electrodes for DHE is proposed in this work. The hypothesis is the larger the area of the Pt electrode, the larger the contact area between the Pt electrode and the membrane. Therefore, the HER is more likely to take place on the Pt electrode yielding a more stable DHE potential. For further concept-approval of the hypothesis, the Pt foils with a larger electrode area (5 mm in width) instead of the Pt wires (dia. 0.3 mm) were designed as the DHE electrodes.
Firstly, the Pt foils with smooth surfaces (Ra ≈ 100 nm, achieved by polishing or pressing) were introduced for the DHE design (named DHEGen2, as shown in Fig. 1b). During the charging and discharging operation, the voltage curves of individual electrodes (vs. DHEGen2) were observed to be remarkably stable in Fig. 2b, indicating that a constant potential was achieved for the DHEGen2 by increasing the size of the Pt electrode. It is apparent that the charge–discharge curves of each electrode had somewhat distinct characteristics during the initial 10 cycles in which the discharge curves of the cathode and anode ended at a falling voltage as the cycling proceeds. This might be associated with notable changes in the open circuit voltage (OCV) and overpotential of the cathode and anode in the first few cycles, as indicated in Fig. 6. These results will be discussed in more details later. Here the consistent voltage of the cathode or the anode vs. DHEGen2 at the top of charge demonstrated the highly stable potential of DHE during cell cycling and this was achieved by the optimized design of Pt foil-based electrodes with a smooth surface.
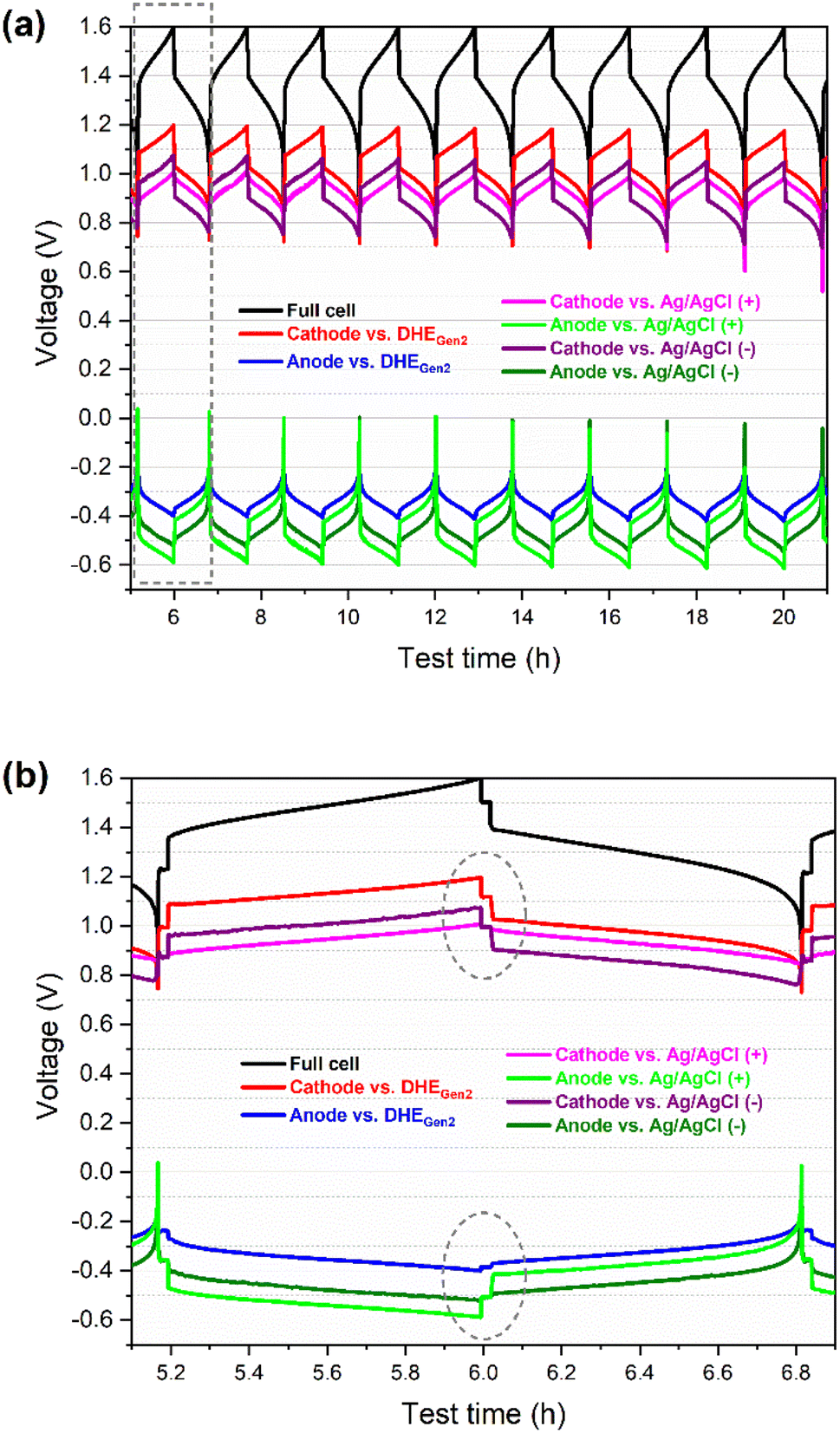 | ||
| Fig. 3 Charge–discharge voltage profiles (vs. time) of full cell and its individual electrode (cathode or anode) vs. RE (DHE, Ag/AgCl (+) or Ag/AgCl (−)) of a scaled vanadium redox flow battery (49 cm2 in active area): (a) for the initial 10 cycles, and (b) for the 2nd cycle, enlarged area highlighted in (a). Ag/AgCl (+) and Ag/AgCl (−) are the Ag/AgCl reference electrodes that are in the inlet of catholyte and anolyte respectively. | ||
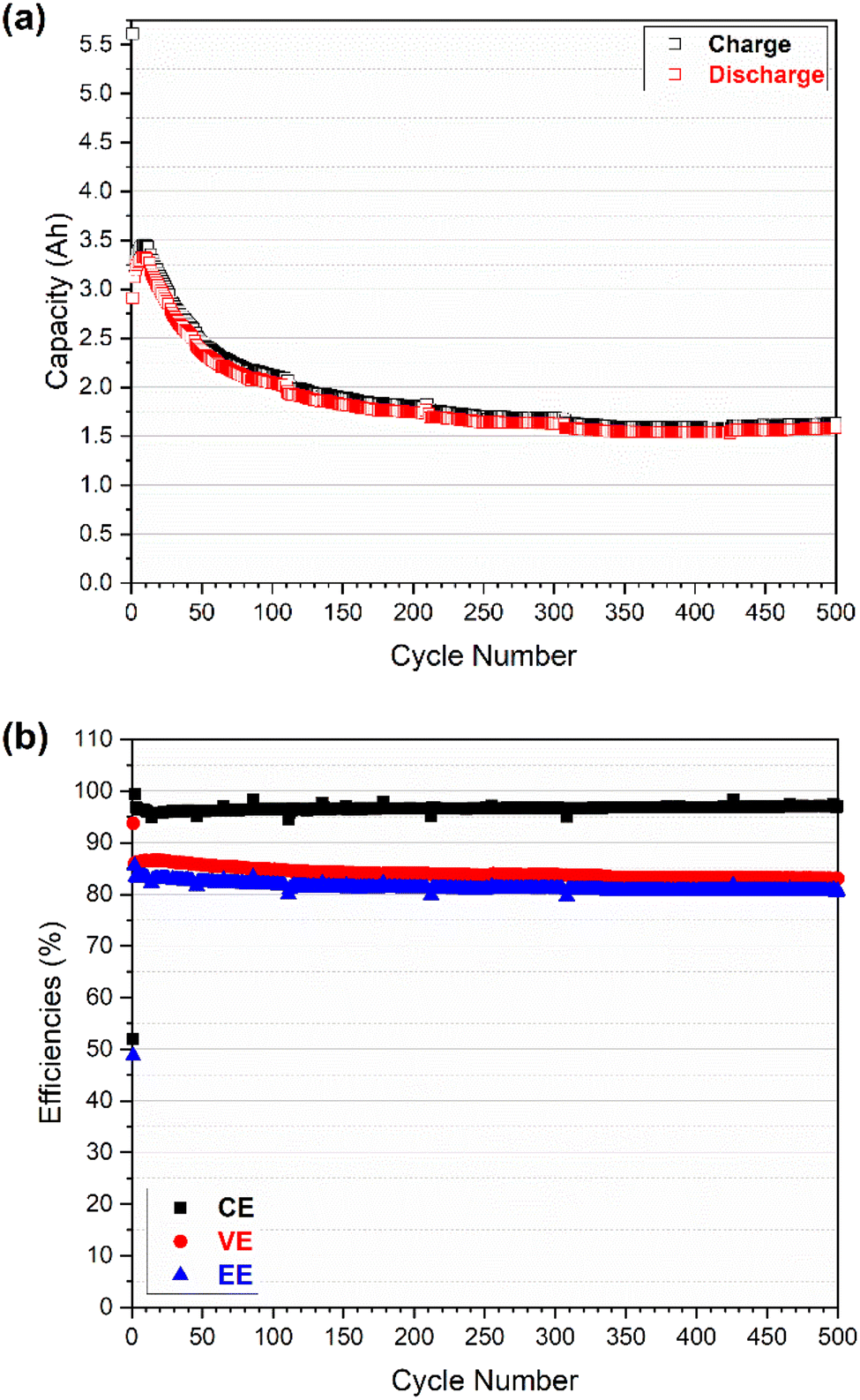 | ||
| Fig. 4 Cell performances of an all-vanadium RFB using GfE commercial electrolyte with V (1.6 M) and N212 double membranes: (a) charge and discharge capacities as a function of cycle number; and (b) coulombic efficiency (CE), voltage efficiency (VE), and energy efficiency (EE) as a function of cycle number. | ||
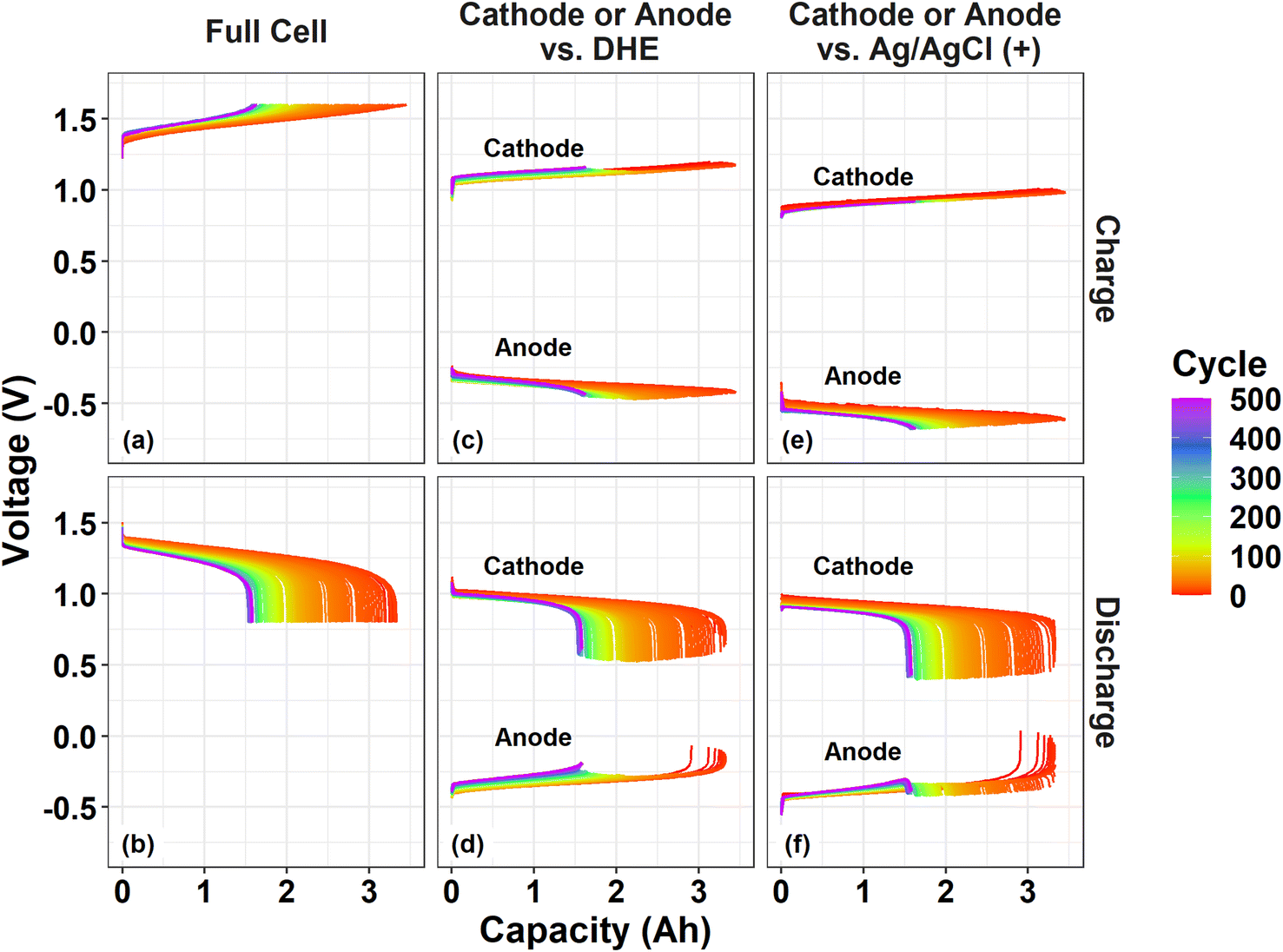 | ||
| Fig. 5 Charge and discharge voltage profile as a function of capacity throughout 500 cycles: a full cell (a and b), and individual electrodes vs. reference electrode of DHE (c and d) or Ag/AgCl (+) (e and f). | ||
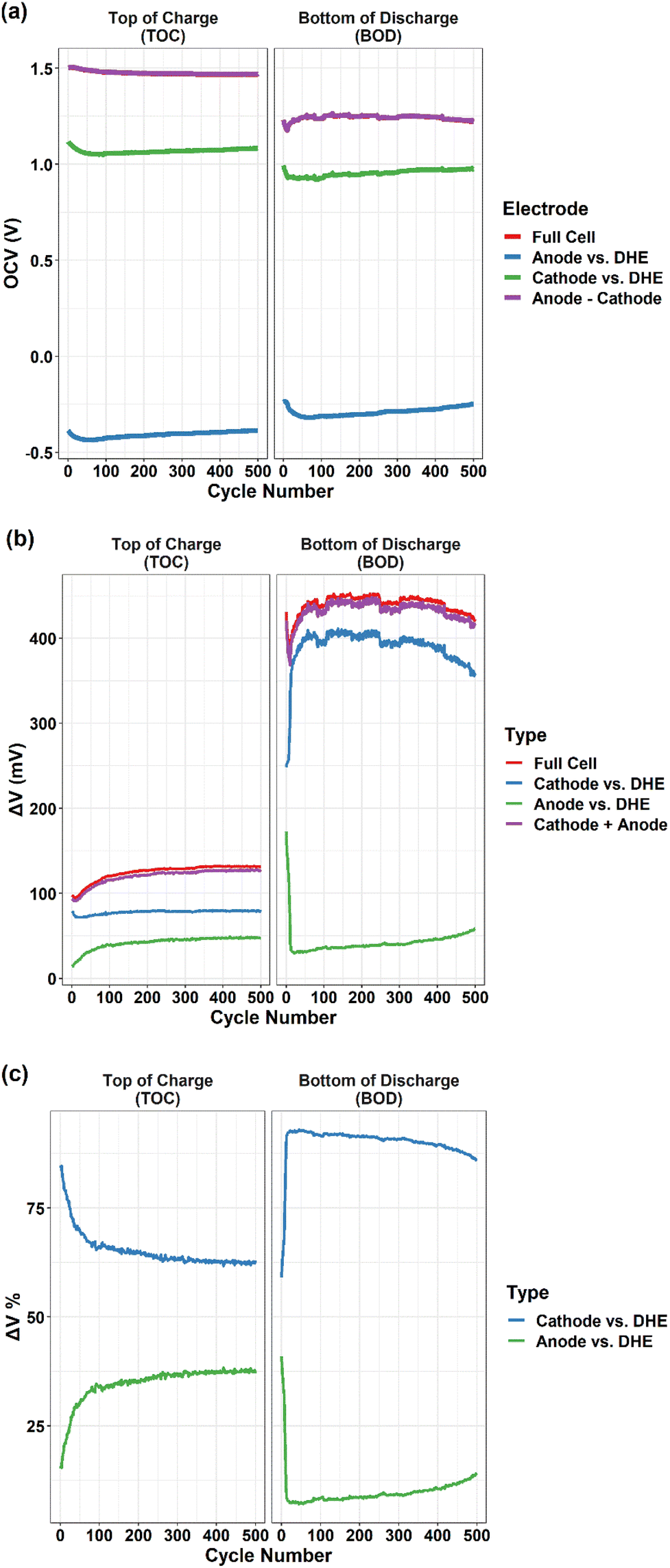 | ||
| Fig. 6 (a) OCV, (b) overpotentials (ΔV), and (c) the relative proportion of overpotential, at the top of charge (TOC) and bottom of discharge (BOD) as a function of cycle numbers for the full cell and individual electrode (cathode or anode vs. DHE). For (a), the purple line is the difference of OCV between cathode and anode. For (b), the purple line is the sum of overpotential of individual electrodes (cathode + anode). | ||
This phenomenon encouraged us to further think about the surface roughness of Pt electrode which might be another factor to influence the stability of DHE. Therefore, a rough surface (Ra ≈ 500 nm, achieved by using a rough sandpaper for polishing) was further introduced to the Pt foil (named DHEGen3, as shown in Fig. 1b) so as to create a higher surface area of the working electrode and thereby increase the opportunities of the HER resulting in the stability improvement of DHE. However, as shown in Fig. 2c, the cathode or anode voltage (vs. DHEGen3) shifted significantly more, indicating that the DHE with Pt foils with a rough surface was less stable than the normal Pt wires. In general, the cathode and anode voltages (vs. DHEGen3) were relatively stable in the initial 10 cycles, but their voltages dropped dramatically by ca. 0.3 V where they remained relatively stable for a prolonged period of time, and then followed by a gradual voltage increase returning to the initial value. In the initial 10 cycles or so, the cathode potential is approx. 1.1 V vs. DHE, close to the VO2+/VO2+ potential (E° = 1.00 V, eqn (i)), while the anode potential is approx. −0.3 V vs. DHE, close to the V3+/V2+ potential (E° = −0.25 V, eqn (ii)). Over the 30th to 80th cycles, the cathode potential decreased to 0.8 V vs. DHE, while the anode potential decreased to −0.6 V vs. DHE. After 120 cycles, both cathode and anode potential recovered to 0.8 V and −0.6 V vs. DHE. This phenomenon indicated that the potential of the DHE approached 0 V during the initial cycles, climbed progressively to 0.3 V, and then returned to 0 V.
As reported, there is co-existence of H+ and vanadium species in the electrolyte (nearby Pt electrodes),4,26 thus the competition between HER and vanadium redox reaction occurs on the DHE.
| 2H+ + 2e− → H2, E° = 0.00 V | (iv) |
| VO2+ + 2H+ + e− → V3+ + H2O, E° = 0.34 V | (v) |
According to eqn (iv) and (v), the DHE potential shifts from 0 V to around 0.3 V which indicates the competition between HER and vanadium redox reaction at the DHE; these two reactions switched with each other, leading to a dramatically changed DHE potential. The high surface area of the Pt foil working electrode in DHEGen3 accelerated the competition between HER and the vanadium redox reaction.
The results indicate that the high stability of the DHE reference electrode in a scaled cell can be achieved by optimizing the DHE design of tuning the size (area) and surface roughness of the Pt electrodes. The increase in the area of Pt electrode will benefit a stable HER and an improved stability of the DHE while a further increase in the surface area of the Pt electrode facilitates the competition of HER with other reactions inside the cell such as the vanadium redox reaction. The demonstrated highly stable DHEGen2 with an optimized design (based on Pt foils with a smooth surface) will serve as the reference electrode in the subsequent study.
The cathode (or anode) voltage curves vs. different REs (DHEGen2, Ag/AgCl (+), and Ag/AgCl (−)) show the same pattern and are almost parallel to each other. The gaps among the three voltage curves of each individual electrode (vs. three REs) includes the differences in: (a) the potential of REs (between DHE and Ag/AgCl electrodes), and (b) the overpotential from the membrane since these REs are placed in different positions of the flow cell (Fig. S2†).
The difference in the potential of the REs can be determined by the difference in the OCV of the individual electrode (cathode or anode) vs. different REs. As specified in the dashed circles in Fig. 3b, the OCV of the cathode (or anode) vs. external Ag/AgCl REs (at the state of rest) showed the same value, which is 1.00 V (or −0.51 V) while the OCV of cathode (or anode) vs. DHE RE is 1.12 V (or −0.39 V), resulting in 0.12 V for the difference in the potential of Ag/AgCl and DHE reference electrodes. The potential of Ag/AgCl (4 M KCl) is known as 0.199 Vvs. NHE at 25 °C.32 Therefore, the potential of the DHE is around 79 mVvs. NHE at 25 °C, as shown in eqn (vi)–(viii).
| EAg/AgCl (4 M KCl) − EDHE = 0.12 V | (vi) |
| EAg/AgCl (4 M KCl) = 0.199 Vvs. NHE at 25 °C | (vii) |
| EDHE = 0.079 Vvs. NHE at 25 °C | (viii) |
The hydrogen standard electrode potential (E°) is 0 V, thus the potential of the DHE in our cell (79 mV) probably reflects the internal environment of the cell such as the activity of the hydrogen ions (aH+) and the partial pressure of the hydrogen gas (pH2) surrounding the Pt electrodes of the DHE. This behavior can be explained according to the Nernst eqn (ix).33
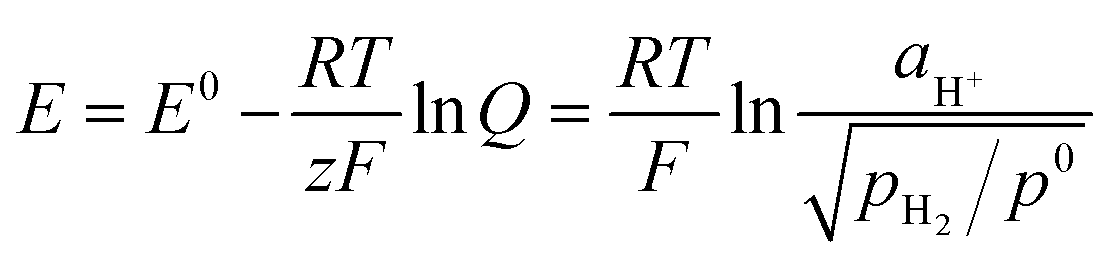 | (ix) |
Note that the Donnan potential (due to the proton concentration differences across the membrane) was included in a more accurate form of the Nernst equation for a VRFB.34 Here the two Pt electrodes in the DHE were on the same side of the membrane. As a result, the Donnan effect was not included in DHE's potential. However, when the potential of DHE (between two membrane) was compared with that of Ag/AgCl REs (in the inlet of the catholyte and anolyte), the Donnan effect may need to be taken into account for the potential comparison in eqn (vi) and (viii) since DHE and Ag/AgCl REs are on different sides of the membrane.
Since the DHE and the two Ag/AgCl REs are assembled in different positions of the cell (for example, between the membranes and in the inlet tubing of the catholyte or anolyte), the measured voltage of the individual electrode vs. the three REs during charging or discharging includes the overpotentials from the membrane. Since DHE was introduced between the two membranes, the electrode voltage vs. DHE contains the overpotential effect from one layer of the N212 membrane. Depending on the relative positions of the electrode and external Ag/AgCl RE, the electrode voltage vs. Ag/AgCl REs includes or excludes the overpotential effect from two layers of the membrane. For instance, the cathode (or anode) voltage vs. the RE placed in the inlet of catholyte (or anolyte) will not include the overpotential from the membrane, whereas the cathode (or anode) voltage versus the RE placed in the inlet of anolyte (or catholyte) will include the effect of the two layers of membranes. Despite the gaps between the voltage curves of each individual electrode, the consistent pattern of the curves for each electrode demonstrates the remarkable stability of the newly developed DHE in a scaled VRFB.
3.2. Reliability and degradation mechanism study of a scaled VRFB by newly developed DHE
As shown in Fig. 4a, the charge and discharge capacity continuously increased from 3.17 and 3.15 A h in the 2nd cycle to 3.32 and 3.20 A h in the 9th cycle respectively. This increase is probably associated with the sharp decrease in the overpotential of the cell in the initial cycles (as shown in Fig. 6b). Then the charge and discharge capacity decreased sharply to 2.32 and 2.24 A h in the 100th cycle followed by a slight decrease and appeared to be stable for the following 400 cycles; the capacity reached 1.84 and 1.80 A h in the 500th cycle. The discharge capacity retention is ca. 57.1% for a total of 500 cycles in which the average decrease rate of discharge capacity is 9.1 mA h per cycle for the initial 100 cycles and 1.1 mA h per cycle for the following 400 cycles. The more significant capacity fade observed in the initial 100 cycles is mostly associated with the imbalanced vanadium active species between the positive and negative half-cells that is induced by electrolyte (i.e., vanadium ion) crossover.4 As indicated in Fig. S4,† the total concentration of vanadium ions in the catholyte and anolyte (measured by ICP) kept changing particular during the initial 100 cycles where the total V concentration increased on the positive side and decreased on the negative side. The negative-to-positive transfer of vanadium ions (as well as water) during cycling is validated and in good agreement with a previous study.4 Once the concentration gap between the catholyte and anolyte reached the maximum at around 100 cycles, it remained constant during the following 400 cycles, and the discharge capacity faded due to the (vanadium ions) crossover being less significant.
Compared with the capacity, the efficiencies (CE-coulombic efficiency, VE-voltage efficiency, and EE-energy efficiency) in Fig. 4b exhibited much higher retention of 97.6, 96.6, and 94.3% for the CE, VE and EE, respectively. The low CE in the first cycle (51.9%) was due to the beginning electrolyte solution containing vanadium ions with a valence of 3.5 (a mixture of V3+/V4+, 50/50) for both catholyte and anolyte. As a result, the preparation of V4+ for catholyte and V3+ for anolyte was part of the initial charging process, leading to the low CE in the first cycle. The high CE (99.4%) in the second cycle could indicate that the system was still adjusting after the first cycle. After that the CE decreased slightly from 96.9% (in the 3rd cycle) to 96.2% (in the 10th cycle) and then increased gradually (to 96.5%) in the following 100 cycles. On the contrary, the VE increased slightly (from 86.0 to 86.5%) in the initial 10 cycles and then decreased gradually (to 84.8%) in the following 100 cycles. Both the CE and VE were relatively stable in the following 400 cycles, reaching 97.1 and 83.1% in the 500th cycle, respectively. The trends of CE and VE made the final EE decrease slightly in the initial 100 cycles (from 83.5% in the 3rd cycle to 81.8% in the 100th cycle) and then remained stable up to 500 cycles (80.6%). The crossover is assumed to be a primary factor to the considerable capacity fading and efficiency shifting in the first 100 cycles.4 As the crossover becomes less significant after 100 cycles (indicated by vanadium ion concentration of catholyte and anolyte vs. cycles in Fig. S4†), both capacity and efficiencies appear to be relatively steady. However, the capacity increase and efficiency shift observed in the initial 10 cycles (slight decrease and increase in CE and VE, respectively, accompanied by sharp drop in overpotential) might be also linked to the activation process of electrode, but the exact reason may require further investigation in future.
During the charge process, the cathode curves vs. DHE remain relatively consistent while the anode curves vs. DHE decreases by ∼0.05 V with cycling especially for the initial 100 cycles as shown in Fig. 5c. This trend aligns well with the behavior of the full cell in which the charge voltage rises by around 0.05 V in the initial 100 cycles. The phenomenon indicates that the performance degradation (overpotential increase) during charging of the cell is dominated by the anode. The shifting of the individual electrode voltages vs. Ag/AgCl (+) RE in Fig. 5e showed a similar trend when compared with the corresponding curves vs. DHE. This indicates that the influence of the membrane on the cell degradation upon charging is negligible.
During the discharge process, the voltage curve of the full cell decreased gradually by 0.05 V for the initial 100 cycles and by 0.03 V for the following 400 cycles (Fig. 5b), in which the cathode curve vs. DHE decreased by 0.05 V (for the initial 100 cycles) and increased slightly afterwards while the anode curve vs. DHE increased slightly in the initial 100 cycles and then more significantly (by 0.05 V) until 500 cycles (Fig. 5d). It can be deduced that the performance degradation during discharge is mostly derived from the cathode contribution in the initial 100 cycles but dominated by the anode in the following 400 cycles. The discharge curves for the individual electrode voltage vs. external RE Ag/AgCl (+) showed a somewhat different shifting trend in Fig. 5f, indicating a contribution from the membrane. The cathode curve vs. Ag/AgCl (+) decreased more significantly than that vs. DHE. The cathode curve vs. DHE includes the overpotential from one layer of the membrane but the one vs. Ag/AgCl (+) excludes the membrane effect, which suggests that the membrane plays a role in decreasing the overpotential of the discharge process. This effect was also validated by the anode curves during discharge. The anode curve vs. DHE (including one-layer of membrane) in Fig. 5d showed a more significant overpotential increase than that vs. Ag/AgCl (+) (including two-layer of membrane effect). The membrane induced overpotential decrease effect in the discharge process will be a subject of ongoing investigation in our laboratory.
In summary, the anode is primarily responsible for capacity fading (overpotential increase) during both the charge and discharge processes throughout 500 cycles except for the first 100 cycles of discharging where the cathode contributes more. In a prior work27 conducted in a smaller flow cell (almost 1/5 the active size of our scaled cell) with a different cell design, electrolyte composition (vanadium and acid concentration), and flow rate, it was observed that the cathode acted more dominantly for cell degradation. Several essential parameters, such as the size and design of the flow cell, cell components (electrolyte composition, electrode, and membrane), flow rate, and testing conditions, may influence the degradation mechanism of a VRFB, according to these studies. Even if the chemistry is identical, the cathode or anode in different cells may react differently. Future research will involve a more in-depth investigation of the mechanisms of degradation in various VRFBs.
The OCV of full cell decreased gradually at the TOC and increased at the BOD in the initial ∼100 cycles (excluding the first 10 cycles) and then tended to be relatively stable. As a strong indicator of the state of charge (SOC) of a VRFB,35 the OCV has an immediate relationship with the concentration of active materials (vanadium ions) in the catholyte and anolyte. The observed shift of OCV agreed well with the changes of vanadium ion concentration in the catholyte and anolyte due to the crossover of vanadium ion, as shown in Fig. S4† and discussed earlier. The results indicated that in our cell system, the crossover induced cell performance degradation mostly occurred in the first 100 cycles.
The OCV of the cathode and anode (vs. DHE) showed a similar trend that decreased in the initial 80–100 cycles and then increased gradually until 500 cycles. Considering the reverse feature of the cathode and anode (Vfull cell = Vcathode − Vanode), the consistent OCV curves of individual electrodes indicated the opposite roles of the cathode and anode in contributing to the overall degradation. The details will be further discussed in the overpotential (Fig. 6b and c) and polarization curve measurements (Fig. 7).
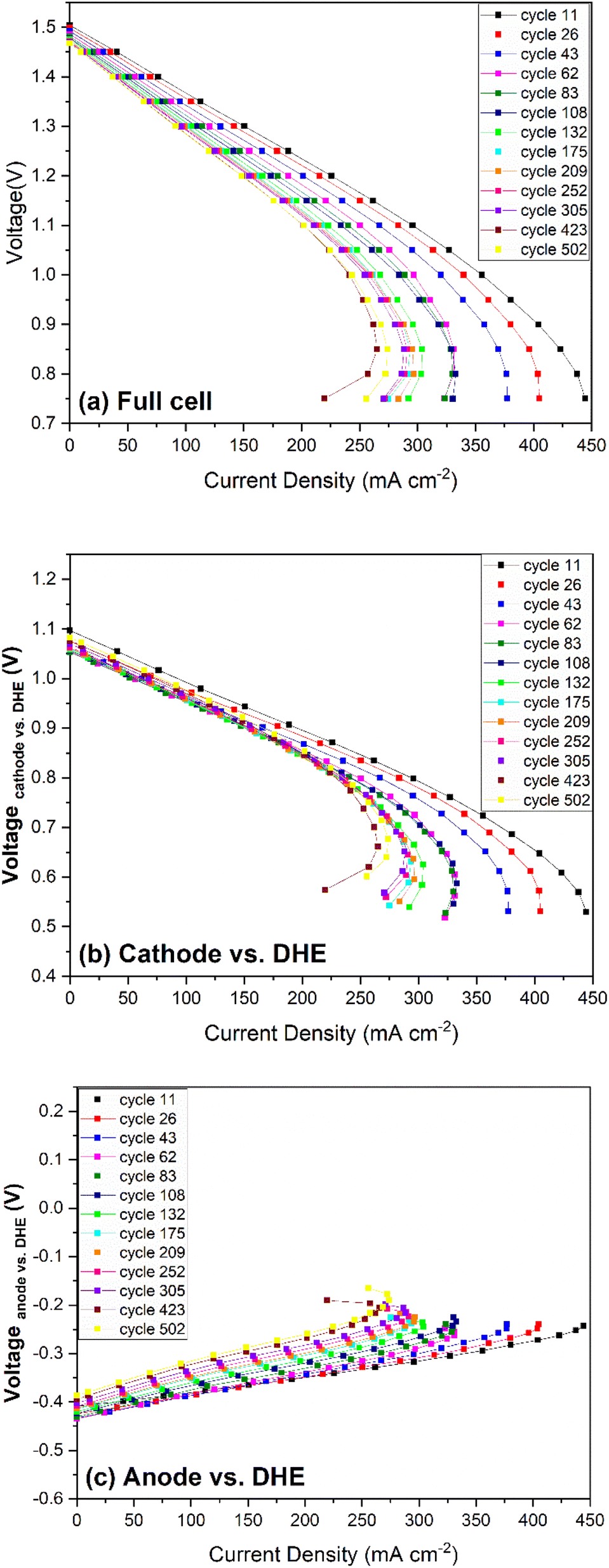 | ||
| Fig. 7 Polarization curves as a function of cycle numbers for (a) full cell, (b) cathode vs. DHE, and (c) anode vs. DHE. | ||
Fig. 6b shows the overpotentials at the TOC and BOD as a function of cycle numbers for the full cell, individual electrodes (cathode or anode vs. DHE), and the sum of individual electrodes (cathode + anode). The overpotential was the difference (absolute value) between the voltage at the operation current of 80 mA cm−2 (the last point of recorded voltage in the charge or discharge step) and the OCV (the recorded voltage at the 1.5 min of rest step after the charge or discharge step). In general, the overpotentials of full cell at the BOD (400–450 mV) are ∼3 times higher than those at the TOC (100–130 mV), which is mostly dominated by the cathode. Aligned well with those of the full cell, the overpotentials of cathode at the BOD (350–400 mV) are ∼3 times higher than at the TOC (75–80 mV), whereas the overpotentials of anode are kept stable and in a relatively lower value (20–50 mV) at both BOD and TOC (except for the initial 10 cycles).
In general, the largest change in the overpotential during cycling was observed in the initial 100 cycles for the full cell and the individual electrodes. This trend of an initial decrease (in the first 10 cycles) followed by an increase (until 100 cycles) was observed in the overpotentials of full cell at both the TOC and BOD; the trend is more significant at the BOD in comparison to the TOC. For the initial 10 cycles, the slight decrease in the overpotential at the TOC is mostly dominated by the cathode whereas the sharp decrease at the BOD is dominated by the anode. The increase in the overpotential between the 10th and 100th cycles of the full cell is mostly caused by the anode at the TOC and by the cathode at the BOD. The results indicated that the individual electrode showed quite different behavior in the overpotentials between the TOC and BOD. In addition, at the TOC or BOD, the cathode and anode contributed to the overpotential in an opposite way which is clearly indicated by the relative proportion of overpotentials for individual electrodes in Fig. 6c. This also aligned well with the OCV measurement discussed earlier. At the TOC, the largest difference in overpotentials between cathode and anode existed in the 1st cycle, with ∼85% and 15% ΔV for cathode and anode, respectively, as shown in Fig. 6c. The difference was dramatically decreased in the following 100 cycles, and then shifted slightly and remained relatively stable, leading to the smallest difference (∼62% and 38% ΔV for cathode and anode, respectively) near the end of 500 cycles. On the contrary, the smallest difference in overpotential (60% and 40% ΔV for cathode and anode) at the BOD was observed in the 1st cycle, and then the overpotentials of the cathode (up) and anode (down) went in the opposite direction sharply during the next 10 cycles and then reached the largest difference (88% and 12% ΔV for cathode and anode, respectively). The overpotentials then became stable status with the difference becoming gradually smaller particularly after 300 cycles.
It is known that the overpotential at the TOC or BOD plays a significant role in dominating the charge or discharge capacity of a VRFB,26,27 which has been verified in the present study where the shifting trend of overpotential during long-term cycling corresponds well with the capacity fading trend – the smaller overpotential, the higher capacity, and vice versa. It is noticeable that the charge and discharge capacity increase in the first 10 cycles of Fig. 4a is mostly attributed to the initial decrease in the overpotential of the full cell (dominated by the cathode at the TOC and anode at the BOD), as shown in Fig. 6b.
In summary, the cathode showed a much higher overpotential than the anode at both the TOC and BOD up to 500 cycles which indicates that the cathode reaction played a more significant role than the anode reaction in limiting the capacity particularly in the discharge process. However, the cell degradation has an overall larger contribution from the anode; the anode overpotential increased gradually during long-term cycling whereas the cathode showed the opposite contribution except for the initial 50 cycles where the cathode dominated at the BOD.
In addition, the difference in OCV between the cathode and anode (cathode − anode), and the sum of the overpotentials of the cathode and anode (cathode + anode) are plotted in Fig. 6a and b for both TOC and BOD. All curves aligned well with their corresponding full cell curves that validates high accuracy and long-term stability of the DHE throughout 500 cycles. To further compare the internal DHE RE with the external Ag/AgCl REs, the OCV of full cell and individual electrodes vs. different REs at the TOC and BOD are plotted in Fig. S5.† With different REs, the OCV curves of each electrode still show a consistent trend at the TOC or BOD over 500 cycles. The gap in the OCV value reflected the difference in the potential of REs (see eqn (vi)) as mentioned earlier and is consistent during 500 cycles. All these again validate the long-term stability of the DHE. It is noticeable that the unstable OCV curves of electrodes vs. Ag/AgCl (−) were observed in Fig. S5g and h,† indicating the unstable potential of Ag/AgCl (−) RE probably caused by bubbles blocking the tubing of anolyte starting at the ∼80th cycle. The behavior of Ag/AgCl (−) RE returned to normal once the bubble was removed, but the issue appeared again from the 200th cycle. The new DHE has a substantial advantage over the Ag/AgCl RE in terms of the stability of the potential during long-term cycling due to the reduced influence of gas generation, which should be accounted for in the external Ag/AgCl RE approach.
The polarization curves for the cathode (vs. DHE) in Fig. 7b showed significant decrease in OCV from 11th to 62nd cycles, and then the OCV remained relatively stable with a partial recovery during the remaining 400+ cycles. The change of ohmic loss was unobvious over 500 cycles, as indicated by the parallel feature of these curves at the current density below 200 mA cm−2. Similar to the full cell, a significant change in mass transport was observed in the first 100 cycles, after that, mass transport change became less significant. Owing to the reverse features of the anode when compared with those of the cathode (such as the voltage, Vfull cell = Vcathode − Vanode), it showed quite different polarization curves as shown in Fig. 7c. A significant increase in the OCV of the anode was observed after 100 cycles which indicates that the anode showed more degradation during long-term cycling. The ohmic loss increased slightly in the initial 100 cycles and then remained relatively constant. By further calculating the slope of the linear region in the polarization curves (Fig. 7b and c), it was found that the ohmic resistance of the cathode is much larger than that of anode which indicates that the ohmic loss of the full cell was mainly dominated by the cathode in the whole 500 cycles. In addition, a significant increase in the transport loss of anode was observed in the initial 100 cycles which is in good agreement with those trends in the full cell and cathode.
In general, the performance loss of the cell increased more significantly in the initial 100 cycles and then remained relatively stable up to 500 cycles. In terms of OCV (or overpotential at the TOC), the cell degradation is initially caused by the cathode but has a larger contribution from the anode up to 500 cycles. The ohmic loss of the full cell was mostly dominated by the cathode with insignificant changes throughout 500 cycles, but the initial slight increase in the ohmic loss is more induced by the anode. The mass transport loss, which shows a significant increase in the initial 100 cycles, is contributed by both the cathode and anode.
Lastly, the polarization curves of the cathode and anode vs. Ag/AgCl (+), shown in Fig. S6,† reflect the contribution from the membrane when compared with those of the cathode and anode vs. DHE (Fig. 7b and c). The cathode curves vs. Ag/AgCl (+) tend to more horizonal in direction (compared with Fig. 7b), indicating the lower ohmic loss due to the subtraction of the membrane contribution, while the anode curves vs. Ag/AgCl (+) present a steeper direction (compared with Fig. 7c), indicating the higher ohmic loss due to double layer membrane contribution.
It's worth noting that VRFB cell degradation is complicated by changes in the electrolyte composition caused by crossover as well as other cell components (electrode, membrane, etc.). To differentiate the detailed degradation sources (at least electrolyte changes from others) and gain a better understanding of degradation mechanism, further research using electrolyte remix and materials characterization before and after cycling, which is beyond the scope of this work, but will be the subject of our ongoing investigation in future.
4. Conclusions
The development of a stable reference electrode is critical for RFB reliability and degradation investigation. In this study, a reference electrode based on DHE with novel design on the area and surface roughness of platinum electrodes was developed for a scaled all-vanadium redox flow battery. The newly developed reference electrode demonstrated a recorded high accuracy and long-term stability throughout 500 cycles in a scaled vanadium RFB. By integrating the stable RE approach to decouple the cathode and anode in conjunction with voltage profiles, overpotentials, and polarization curve measurements, the degradation mechanism of a scaled vanadium RFB is further investigated. The performance fading (capacity and efficiencies) and losses (ohmic and transport loss) were found to occur dramatically in the initial 100 cycles, owing to the increase in cell polarization caused by electrolyte (vanadium ion) crossover. Relatively, the anode contributed more to overall cell degradation over long-term cycling, whereas the cathode reaction played a far more significant role than the anode response in limiting the capacity especially during discharge. In addition, the effect of the membrane on overpotential and performance losses was preliminarily evaluated by comparing individual electrodes to reference electrodes (DHE and Ag/AgCl) situated in various positions across the cell. A variety of crucial elements, including the size and design of the flow cell, cell components (electrolyte composition/concentration, electrode, and membrane), flow rate, and testing conditions, are likely to affect the degradation mechanism of a redox flow battery, according to our research. Future study may examine the implementation of this RE approach within an RFB stack. This exploratory effort will aid future design and development of a stable reference electrode for use in the practical application of scaled energy storage systems, as well as fundamental understanding of the RFB's reliability and degradation mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from the U.S. Department of Energy's (DOE) Office of Electricity (OE) (under Contract No. 57558). We also are grateful for insightful discussions with Dr Imre Gyuk of the DOE-OE Grid Storage Program. Pacific Northwest National Laboratory is a multi-program national laboratory operated by Battelle for DOE under Contract DE-AC05-76RL01830. The authors would thank Edwin C Thomsen, Hong (Amy) Qiao, and Anthony Guzman of the Pacific Northwest National Laboratory (PNNL) for the hardware setting of flow cell testing, profilometer measurement, and metal (platinum) polishing process development respectively.References
- DOE Long-Duration Energy Storage Workshop, March 9-10, 2021, https://www.sandia.gov/ess/LDES, accessed September 2022.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577 CrossRef CAS PubMed.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970 CrossRef CAS.
- Q. Luo, L. Li, W. Wang, Z. Nie, X. Wei, B. Li, B. Chen, Z. Yang and V. Sprenkle, ChemSusChem, 2013, 6, 268 CrossRef CAS PubMed.
- J. S. Lawton, A. Jones and T. Zawodzinski, J. Electrochem. Soc., 2013, 160, A697 CrossRef CAS.
- M. Skyllas-Kazacos, L. Cao, M. Kazacos, N. Kausar and A. Mousa, ChemSusChem, 2016, 9, 1521 CrossRef CAS PubMed.
- M. H. Chakrabarti, N. P. Brandon, S. A. Hajimolana, F. Tariq, V. Yufit, M. A. Hashim, M. A. Hussain, C. T. J. Low and P. V. Aravind, J. Power Sources, 2014, 253, 150 CrossRef CAS.
- I. Derr, M. Bruns, J. Langner, A. Fetyan, J. Melke and C. Roth, J. Power Sources, 2016, 325, 351 CrossRef CAS.
- S. Kim, T. B. Tighe, B. Schwenzer, J. Yan, J. Zhang, J. Liu, Z. Yang and M. A. Hickner, J. Appl. Electrochem., 2011, 41, 1201 CrossRef CAS.
- C. Fujimoto, S. Kim, R. Stains, X. Wei, L. Li and Z. Yang, Electrochem. Commun., 2012, 20, 48 CrossRef CAS.
- G. Qing, W. Guo, Y. Fan and B. Wang, CIESC J., 2013, 64, 427 CAS.
- D. Chen and M. A. Hickner, Phys. Chem. Chem. Phys., 2013, 15, 11299 RSC.
- S. Rudolph, U. Schröder, I. M. Bayanov and G. Pfeiffer, J. Electroanal. Chem., 2013, 709, 93 CrossRef CAS.
- A. Parasuramana, T. M. Lima, C. Menictas and M. Skyllas-Kazacos, Electrochim. Acta, 2013, 101, 27 CrossRef.
- T. Shigematsu, Curr. Opin. Electrochem., 2019, 18, 55 CrossRef CAS.
- S. Uhm, H. J. Lee and J. Lee, Phys. Chem. Chem. Phys., 2009, 11, 9326 RSC.
- S. Kaserer, C. Rakousky, J. Melke and C. Roth, J. Appl. Electrochem., 2013, 43, 1069 CrossRef CAS.
- J. Langner, J. Melke, H. Ehrenberg and C. Roth, ECS Trans., 2014, 58, 1 CrossRef.
- M. A. Miller, A. Bourke, N. Quill, J. S. Wainright, R. P. Lynch, D. N. Buckley and R. F. Savinell, J. Electrochem. Soc., 2016, 163, A2095 CrossRef CAS.
- Q. Liu, A. Turhan, T. A. Zawodzinski and M. M. Mench, Chem. Commun., 2013, 49, 6292 RSC.
- Y. A. Gandomi, D. S. Aaron, T. A. Zawodzinski and M. M. Mench, J. Electrochem. Soc., 2016, 163, A5188 CrossRef CAS.
- X. Wei, G. Xia, B. Kirby, E. Thomsen, B. Li, Z. Nie, G. G. Graff, J. Liu, V. Sprenkle and W. Wang, J. Electrochem. Soc., 2016, 163, A5150 CrossRef CAS.
- M. Becker, N. Bredemeyer, N. Tenhumberg and T. Turek, J. Power Sources, 2016, 307, 826 CrossRef CAS.
- C.-N. Sun, F. M. Delnick, D. S. Aaron, A. B. Papandrew, M. M. Mench and T. A. Zawodzinski, ECS Electrochem. Lett., 2013, 2, A43 CrossRef CAS.
- D. Aaron, C.-N. Sun, M. Bright, A. B. Papandrew, M. M. Mench and T. A. Zawodzinski, ECS Electrochem. Lett., 2013, 2, A29 CrossRef CAS.
- C. Choi, Y. Choi, S. Kim, H. Jung and H.-T. Kim, Electrochim. Acta, 2016, 213, 490 CrossRef CAS.
- Q. Huang, B. Li, C. Song, Z. Jiang, A. Platt, K. Fatih, C. Bock, D. Jang and D. Reed, J. Electrochem. Soc., 2020, 167, 160541 CrossRef CAS.
- G. Li and P. G. Pickup, Electrochim. Acta, 2004, 49, 4119 CrossRef CAS.
- S. B. Adler, B. T. Henderson, M. A. Wilson, D. M. Taylor and R. E. Richards, Solid State Ionics, 2000, 134, 35 CrossRef CAS.
- The GfE Vanadium Electrolyte Solution 1.6 M Online, https://www.gfe.com/02_produkte_loesungen/03_vanadium-chemikalien/PDB/Vanadium-Electrolyte-Solution-1.6-M-2012-114_V8.pdf, accessed September 2022 Search PubMed.
- G. Li and P. G. Pickup, Electrochem. Solid-State Lett., 2006, 9, A249 CrossRef CAS.
- The Standard Potential of Ag/AgCl Reference Electrode (4 M KCl) Online, https://pineresearch.com/shop/products/electrodes/reference-electrodes/standard-95mmod/silver-chloride-reference-electrode/#specifications, accessed September 2022 Search PubMed.
- The Nernst Equation and Standard Hydrogen Electrode in Wikipedia Online, https://en.wikipedia.org/wiki/Nernst_equation, and https://en.wikipedia.org/wiki/Standard_hydrogen_electrode, accessed September 2022 Search PubMed.
- K. W. Knehr and E. C. Kumbur, Electrochem. Commun., 2011, 13, 342 CrossRef CAS.
- M. R. Mohamed, H. Ahmad and M. N. Abu Seman, Elektron. Elektrotech., 2013, 19, 37 Search PubMed.
- D. Aaron, Z. Tang, A. B. Papandrew and T. A. Zawodzinski, J. Appl. Electrochem., 2011, 41, 1175 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05781f |
| This journal is © The Royal Society of Chemistry 2022 |