
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation vacancy driven efficient CoFe-LDH-based electrocatalysts for water splitting and Zn–air batteries†
Zhenyu
Kong‡
a,
Jingying
Chen‡
ab,
Xiaoxia
Wang
a,
Xiaojing
Long
 a,
Xilin
She
*a,
Daohao
Li
*a and
Dongjiang
Yang
a
a,
Xilin
She
*a,
Daohao
Li
*a and
Dongjiang
Yang
a
aState Key Laboratory of Bio-fibers and Eco-textiles, Shandong Collaborative Innovation Center of Marine Bio-based Fibers and Ecological Textiles, College of Environmental Science and Engineering, College of Materials Science and Engineering, Qingdao University, Qingdao 266071, P. R. China. E-mail: xlshe@qdu.edu.cn; lidaohao@qdu.edu.cn
bDepartment of Blood Transfusion, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao 266003, P. R. China
First published on 6th October 2021
Abstract
Rational design of multifunctional electrocatalysts can optimize the mutual conversion of water and oxygen. CoFe-LDH-based electrocatalysts with highly tunable electronic, component, and catalytic properties have attracted considerable attention. Herein, we report the synthesis of cation vacancy rich CoFe-LDH (CoFeV-LDH) and its trace Pt loaded material (Pt@CoFeV-LDH, Pt dosage: 2.85%), which can act as an efficient electrocatalyst toward the oxygen evolution reaction (OER), hydrogen evolution reaction (HER), and oxygen reduction reaction (ORR). The electrolyzer comprising the CoFeV-LDH cathode and Pt@CoFeV-LDH anode exhibited excellent water splitting performance with a small cell voltage of 1.57 V to reach 10 mA cm−2 and 99% efficiency after 8 h. Furthermore, the liquid-state ZAB using CoFeV-LDH//Pt@CoFeV-LDH as air electrodes shows satisfactory charge–discharge performance with excellent reversibility of 50 h at 50 mA cm−2.
1. Introduction
The exploitation and design of efficient catalysts for metal–air cells, fuel cells and water splitting devices has become a potential strategy to address the growing energy needs and environmental remediation of modern society.1–3 Among the feasible renewable energy electrocatalytic technologies, the oxygen evolution reaction (OER), hydrogen evolution reaction (HER), and oxygen reduction reaction (ORR) are widely researched in energy conversion processes owing to their internal interrelationship.4–6 The extensive application of energy conversion devices (such as fuel cells, zinc–air batteries and full water splitting devices) is impeded by the cost of noble metals.7–9 Hence, in consideration of the expense, activity and stability of the above-mentioned devices, catalysts should be designed legitimately in terms of efficiency and cost. Exploring highly efficient electrocatalysts with free or low-dosage noble metals for economical energy transformation and storage is mightily desired.Layered double hydroxides consisting of cheaper transition metals (TM-LDHs) have been typically considered as promising catalysts due to their high electrocatalytic performance and easily tailored structures.10–12 Selective alkali etching can specifically remove the target metal (such as Zn, Al) to induce cation vacancies in TM-LDHs (TMV-LDHs), which can modulate their chemical coordination and surface electronic states.13,14 The unsaturated sites created by the cation vacancies can effectively facilitate the OER process of the electrocatalyst.15 However, the HER and ORR catalytic activities of TMV-LDHs are limited due to the intrinsic nature of TM-LDHs as they do not have adequate active sites.16,17
Pt is one of the most common noble metals with convincing performance for the HER and ORR. However, due to the cost and low utilization rate of Pt, its application in energy conversion devices is limited and promising strategies are desired to replace it or reduce its dosage. Active site-support catalysts driven by cation vacancies have attracted wide attention due to the synergistic interaction between diverse components.18–20 The cation vacancy rich metal-based support can not only provide adhesion sites for the externally introduced other components by strong metal–support interaction (SMSI), but also optimize its internal electron distribution to enhance catalytic activities.21–23 Anchoring Pt on cation vacancy rich metal-based support through SMSI can facilitate the efficient utilization of Pt to reduce its dosage and enhance the intrinsic activity of Pt.24,25 Thus, trifle Pt coupled with vacancies within TMV-LDHs could show highly efficient HER and ORR performance, and it is meaningful to investigate the electrocatalytic activities of TMV-LDHs with trace Pt loading to achieve multiple energy conversions.
Herein, we report the cation vacancy rich CoFe-LDH (CoFeV-LDH) and its trace Pt loaded material (Pt@CoFeV-LDH, Pt dosage: 2.85%) for achieving highly efficient full water splitting and Zn–air batteries (ZABs). The CoFeV-LDH exhibits satisfactory OER performance with a 241 mV overpotential to drive 10 mA cm−2. Due to the synergistic effect of Pt and CoFeV-LDH support, the Pt@CoFeV-LDH exhibits potential HER and ORR performance. The HER activity of Pt@CoFeV-LDH is similar to that of 20 wt% Pt/C, which only requires 72 mV to supply 10 mA cm−2 current density. When the electrolyzer was composed of the CoFeV-LDH anode and Pt@CoFeV-LDH cathode, the current density of water splitting reaches 10 mA cm−2 at a low voltage of 1.57 V and remained at 99% after the 8 h long-term stability test. The Pt@CoFeV-LDH shows effective ORR performance in alkaline electrolytes (a half-wave potential of ∼0.819 V). The turnover frequency (TOF) value of Pt@CoFeV-LDH (0.2771 S−1) is about six-fold higher than that of Pt/C (0.042 S−1), indicating that the intrinsic activity of Pt is sufficiently enhanced by the CoFeV-LDH support. The liquid-state ZAB using CoFeV-LDH//Pt@CoFeV-LDH as air electrodes shows satisfactory charge–discharge performance with excellent reversibility.
2. Experimental section
2.1. Synthesis of CoFeZn layered double hydroxide (CoFeZn-LDH) nanosheets
CoFe-LDH was prepared according to a classical procedure. At room temperature, 3 mmol of cobalt nitrate hexahydrate, 1 mmol ferric nitrate nonahydrate, and 10 mmol urea were poured into water (70 mL) and stirred vigorously. The obtained mixture was transferred to a 100 mL stainless steel autoclave lined with polytetrafluoroethylene (PTFE) and heated at 100 °C for 12 hours. The obtained CoFe-LDH brown powder was washed three times with water/ethanol and dried at 60 °C for 6 hours in a vacuum oven. The Co2Fe1Zn1-LDH was prepared with 2 mmol cobalt nitrate hexahydrate, 1 mmol ferric nitrate nonahydrate, 1 mmol zinc nitrate hexahydrate and 10 mmol urea similarly to that described above. The Co2.5Fe1Zn0.5-LDH was prepared with 2.5 mmol cobalt nitrate hexahydrate, 1 mmol ferric nitrate nonahydrate, 0.5 mmol zinc nitrate hexahydrate and 10 mmol urea.2.2. Synthesis of CoFeV-LDH nanosheets
To take out the Zn2+, vacancies were created in CoFe-LDHs nanosheets, and the as-obtained CoFeZn-LDH nanosheet precursor was etched by using 6 M KOH solution (treatment time: 180 min) at 60 °C. The obtained product (CoFeV-LDH) was washed with water/ethanol three times and dried at 60 °C for 10 hours in a vacuum oven.2.3. Synthesis of Pt@CoFeV-LDH
Under UV (ultraviolet) irradiation, CoFeV-LDH was put into the H2PtCl6 aqueous solution. The photoreduction process was performed for 60 min. Then, the Pt@CoFeV-LDH powder was obtained after centrifugation and extraction. The sample was cleaned with deionized water and dried in a vacuum oven for 4 hours.2.4. Material characterization
X-ray diffraction (XRD) was performed with a DX2700 at 40 kV and 30 mA with Cu Kα radiation (λ = 1.5418 Å). The synthetic samples were investigated by using a FEI Magellan 400 field emission scanning electron microscope (FESEM) for obtaining their morphology. A JOEL JEM-2100F scanning transmission electron microscope (STEM) equipped with a Cs probe corrector at 200 Kv was employed to observe their structures. X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB 250XL electron spectrometer (Thermo Scientific Corporation) with monochromatic 150 W Al Kα radiation. Inductively coupled plasma (ICP) spectroscopy was performed using a Focused Photonics ICP-5000. The thickness of the nanosheets was recorded using an atomic force microscope (AFM) (Agilent 5400, America). The electrochemical performances were measured using a CHI760E electrochemical work station. Typical specific capacity and discharge curves of the zinc–air battery were investigated using a blue battery test system.2.5. Electrochemical measurements
The turnover frequency (TOF) value was computed according to the following equation:
| TOF = (J × A)/(4 × F × N) | (1) |
3. Results and discussion
The synthetic process of the cation vacancy rich Pt@CoFeV-LDHs is illustrated in Scheme 1. Primarily, a one-pot hydrothermal reaction described in the Experimental section was performed to grow pristine CoFeZn-LDHs. The atomic molar ratio of Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe within the original CoFe-LDH is 3:1. When the amount of zinc added is one-third of that of cobalt, it is denoted as Co2Fe1Zn1-LDH, and when the molar ratio of Co/Zn is 6, it is denoted as Co2.5Fe1Zn0.5-LDH. Based on the above-mentioned materials, alkali etching was performed to remove Zn species for creating cation vacancies to generate CoFe1/3V-LDH and CoFe1/6V-LDH. The OER performance of these catalyst was optimized due to the formation of unsaturated sites. Finally, cation vacancy rich CoFeV-LDH was impregnated in H2PtCl4 aqueous solution for photoreduction to form Pt@CoFe1/3v-LDH and Pt@CoFe1/6v-LDH.26 The cation vacancy driven CoFeV-LDH introduced Pt as an exogenous component, exhibiting efficient HER and ORR performances for full water splitting and zinc–air batteries.
:Fe within the original CoFe-LDH is 3:1. When the amount of zinc added is one-third of that of cobalt, it is denoted as Co2Fe1Zn1-LDH, and when the molar ratio of Co/Zn is 6, it is denoted as Co2.5Fe1Zn0.5-LDH. Based on the above-mentioned materials, alkali etching was performed to remove Zn species for creating cation vacancies to generate CoFe1/3V-LDH and CoFe1/6V-LDH. The OER performance of these catalyst was optimized due to the formation of unsaturated sites. Finally, cation vacancy rich CoFeV-LDH was impregnated in H2PtCl4 aqueous solution for photoreduction to form Pt@CoFe1/3v-LDH and Pt@CoFe1/6v-LDH.26 The cation vacancy driven CoFeV-LDH introduced Pt as an exogenous component, exhibiting efficient HER and ORR performances for full water splitting and zinc–air batteries.
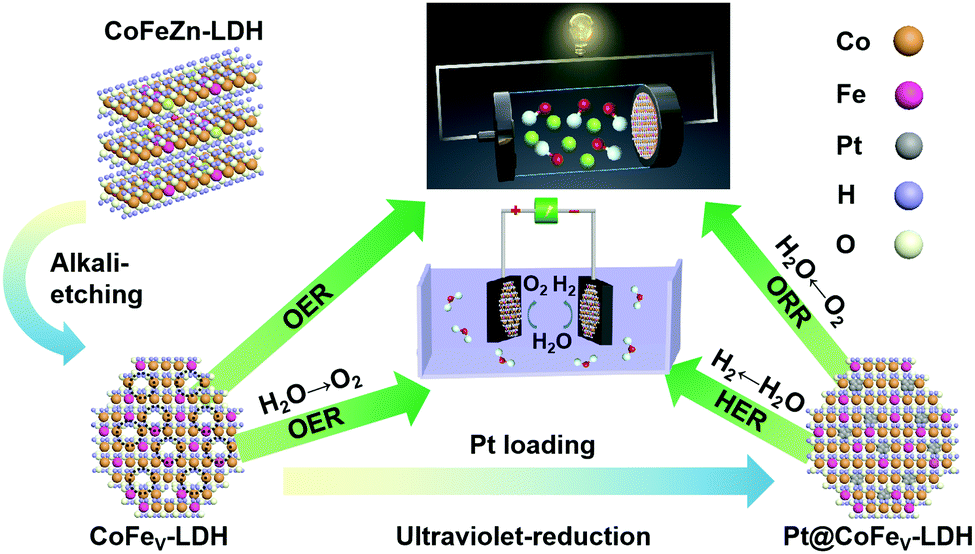 | ||
| Scheme 1 Schematic illustration of the synthesis process for multifunctional Pt@CoFeV-LDH. | ||
Fig. 1a shows the X-ray diffraction (XRD) patterns of CoFe-LDH, CoFeZn-LDH, CoFeV-LDH and Pt@CoFeV-LDH. The peaks located at 11.6°, 23.4°, 34.1°, 59° and 60.54° correspond to the (003), (006), (012), (110) and (113) planes, respectively, according to the JCPDS #50-0235.27 Although the vacancies created by the alkali etching process destroyed part of the structure, resulting in poor crystallinity, the crystalline image of CoFeV-LDH still remains. The characteristic peak of Pt is not obvious due to the trace amount of Pt injection, with the content of Pt being 2.85 wt% (detected by ICP). The ratio of Co:Fe:Pt in Pt@CoFe1/3V-LDH was calculated to be 1:21:7.3 by the ICP test, while that of the Pt@CoFe1/6V-LDH was 1:18:9.1. The Pt@CoFeV-LDH shows a typical 2 D nanosheet morphology (Fig. 1b).28–30 The lamellar 2D structure increases the number of active sites exposed. The SEM of CoFeV-LDH is shown in Fig. S2 (ESI†). By comparison, the morphology of the catalyst did not change before and after loading Pt. Transmission electron microscopy (TEM) images also confirmed the presence of hexagonal nanosheets (Fig. 1c). And the energy dispersive X-ray spectroscopy (EDS) mappings (inset of Fig. 1c) verified the presence of Co, Fe and Pt in equal amounts in the prepared nanosheet. Fig. 1d demonstrates that Pt is present in the form of very fine (5–7 nm) nanoparticles (NPs). The 0.226 nm lattice spacing corresponds to the (111) plane of Pt (inset of Fig. 1d).31 The surface of the CoFeV-LDH edge was observed by HRTEM (Fig. S1, ESI†), and parallel lattice stripes with an interplanar distance of 0.25 nm were assigned to the (012) plane of the crystalline CoFe-LDH (inset of Fig. S1, ESI†).32 The thickness of the CoFeV-LDH and Pt@CoFeV-LDH nanosheets was detected by AFM to be about 20 and 22 nm (Fig. 1e and f), indicating that the lamellar structure of CoFeV-LDH was not transformed after Pt loading.
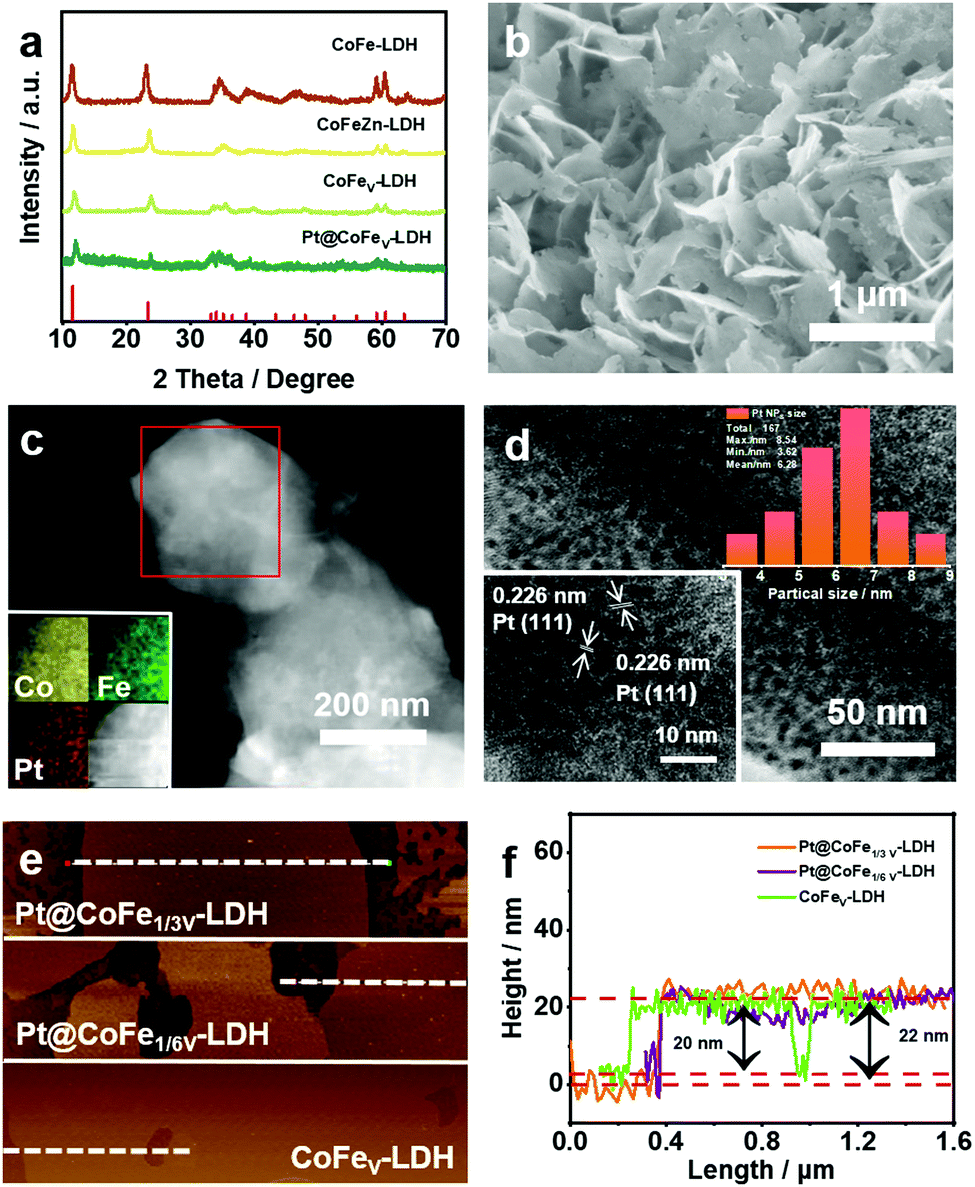 | ||
| Fig. 1 (a) XRD patterns of CoFe-LDH, CoFeZn-LDH, CoFeV-LDH and Pt@CoFeV-LDH. (b) FESEM image of the Pt@CoFeV-LDH nanosheet. (c) TEM and EDS mapping images, (d) HRTEM of Pt@CoFeV-LDH (inset: lattice space and size distribution of Pt NPs). (e and f) AFM images and curves of CoFeV-LDH and Pt@CoFeV-LDH. | ||
In order to analyse the surface chemical structure of the as-prepared samples, X-ray photoelectron spectroscopy (XPS) was performed to monitor the valence of metal elements on the surface of the CoFeV-LDH and Pt@CoFeV-LDH catalysts. As shown in Fig. 2a, the full XPS survey spectra of samples show strong signals of Co, Fe metal elements. The XPS spectrum of Pt@CoFeV-LDH shows two fresh tiny peaks around 71.6 eV and 74.7 eV, which are attributed to Pt 4f, indicating the successful loading of Pt on the CoFev-LDH support.33 Compared with the peak position of the sample before etching, the peaks of CoFeV-LDH are significantly negatively shifted, which is due to the decrease of the nearby metal valence state caused by the appearance of vacancies.34 The XPS spectra of Co 2p exhibited peaks located at 780 eV, 781.5 eV, 795.4 eV and 796.6 eV, which are assigned to Co 2p3/2 (+3, +2) and Co 2p1/2 (+3, +2) of Co within CoFeV-LDH, and the surrounding two satellite peaks located at 785.9 eV and 801.9 eV (Fig. 2b).35 The peaks of Co 2p3/2, Co 2p1/2 and satellite had a negative shift with different degrees (0.1–0.4 eV). The orbit of Fe 2p was linearly split into Fe 2p3/2 (+2, +3) at binding energies of 710.6 eV and 713 eV, and Fe 2p1/2 (+2, +3) peaks are located at 722.5 eV and 733 eV (Fig. 2c).36 Similar to the trend of the XPS peak shift within Co, Fe 2p and satellite peaks were negatively shifted. The negative shift of both Fe 2p and Co 2p implied that the effect of etching has not worked for single metals. After Pt loading, the peaks of Co 2p and Fe 2p are significantly positively shifted due to the loss of electrons from the metal in the carrier under SMSI, which leads to an increase in the energy band.37 The high resolution XPS spectrum of Pt 4f for Pt@CoFeV-LDH (Fig. 2d) displays the spin–orbit doublet with the peaks located at around 71.6 eV (Pt 4f7/2) and 74.8 eV (Pt 4f5/2), which indicates the subsistence of metallic Pt0 (predominant) and Pt2+ species in the composites.
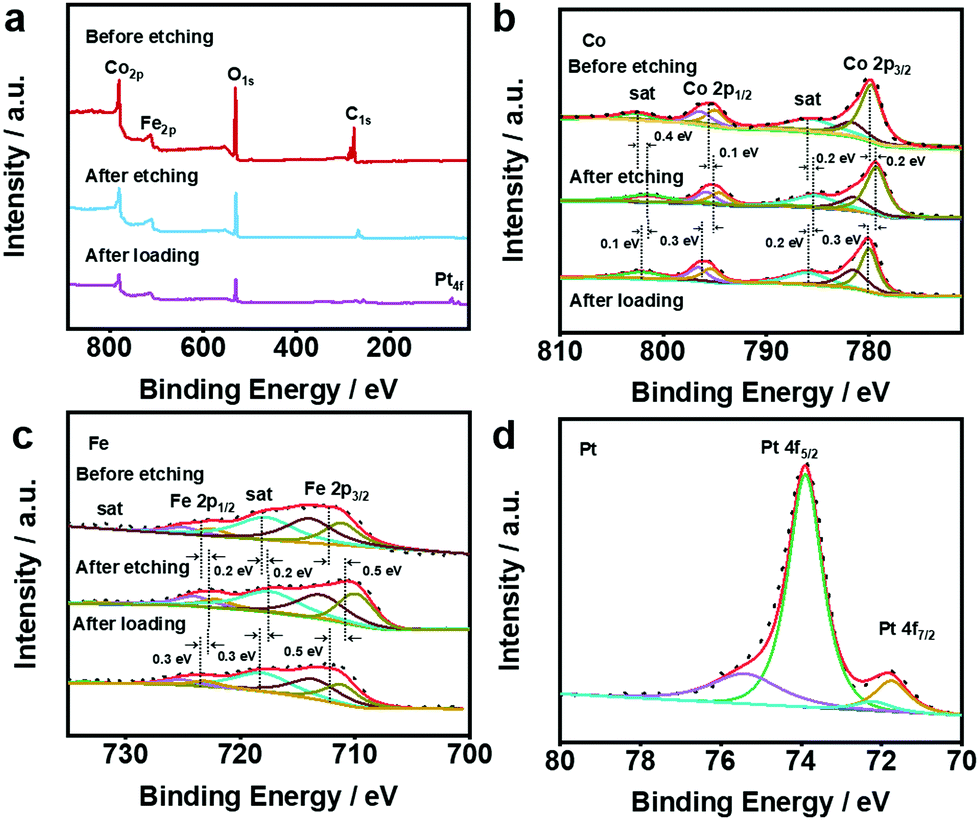 | ||
| Fig. 2 XPS spectra of (a) the full, (b) Co 2p and (c) Fe 2p within CoFeV-LDH and Pt@CoFeV-LDH. (d) XPS spectra of Pt within Pt@CoFeV-LDH. | ||
OER plays an important role as one of the two half-reactions of water splitting, thus it is necessary to consider its kinetic factors. The electrocatalytic properties of the as-synthesized CoFe-LDH, CoFeZn-LDH, CoFeV-LDH, and Pt@CoFeV-LDH nanosheets were evaluated by investigating the linear sweep voltammetry (LSV) curves in 1.0 M KOH solution. As shown in Fig. 3a, the fitted LSV curve for CoFe1/3V-LDH has a smaller starting potential relative to those of CoFeZn-LDH and CoFe-LDH. Obviously, the doped Zn occupied the original Co which was the active site, thus the OER performance of CoFe-LDH was inhibited. Moreover, the overpotential of CoFe1/3V-LDH is demonstrated to drive 241 mV at a current density of 10 mA cm−2 (CoFe1/6V-LDH, 267 mV) (Fig. 3b). The overpotential (317 mV) required for CoFe1/3V-LDH to supply 50 mA cm−2 current density was lower than that of Ir/C (330 mV), indicating that the kinetic advantage of CoFeV-LDH is shown with the increase of current density. The cation vacancies play a vital role in optimizing the OER performance by facilitating the adsorption of OER intermediates, which is consistent with the results reported by others.38 In addition, the OER performance of CoFe1/3V-LDH is better than that of CoFe1/6V-LDH, which reflects the importance for density regulation of vacancies. However, after Pt loading, the OER performance of Pt@CoFeV-LDH became slightly worse, as the active sites were provided by the cation vacancies (Fig. S3 and S4, ESI†). The Tafel slope of CoFe1/3V-LDH is 54 mV dec−1 (CoFe1/6V-LDH 61 mV dec−1), which is lower than that of Ir/C (65 mV dec−1) (Fig. S5, ESI†). The excellent Tafel slope proved the electron transfer ability of CoFeV-LDH which was optimized by etching. Fig. 3c shows that the current density of the CoFeV-LDH catalyst increases accordingly with different applied potentials (10, 50, 100, 150, and 200 mA cm−2), and the reverse process is consistent with the previous performance, indicating the reversible stability of the catalyst.39 In addition, compared to Pt loading before, the current density fitted curves of Pt@CoFeV-LDH became smoother in the reverse process (Fig. S6 and S7, ESI†). The linear scanning voltammograms (LSV) of Pt@CoFeV-LDH before and after 1000 cyclic voltammograms (CV) were unchanged (Fig. S8 and S9, ESI†). In long-term stability experiments for the OER (Fig. 3d), CoFeV-LDH (1/6 V, ∼93.1%; 1/3 V, ∼92.3%) and Pt@CoFeV-LDH (Fig. S10 and S11 (ESI†), 1/6 V, ∼99.1%; 1/3 V, ∼94.2%) maintained a stable current density after 18 h. The Pt@CoFeV-LDH showed perfectly reproducible performance in the reversible process and long-term stability experiments, demonstrating that Pt anchoring within the vacancies of CoFe-LDH by SMSI facilitates the structural stability of the catalyst with optimized electron distribution.
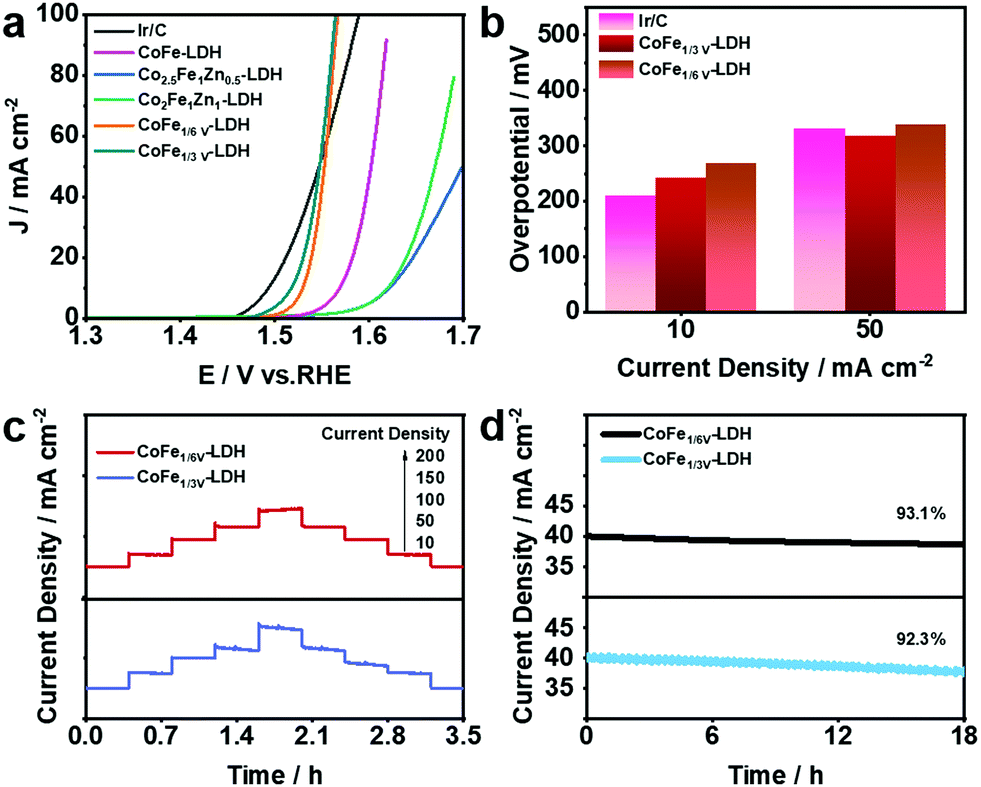 | ||
| Fig. 3 (a) LSV curves of Ir/C, CoFe-LDH, CoFeZn-LDH and CoFeV-LDH for catalyzing the OER. (b) Histogram of the overpotentials of the Ir/C and CoFeV-LDH to drive 10 mA cm−2 and 50 mA cm−2. (c) Multistep chronoamperometric curves of CoFeV-LDH at different applied potentials. (d) Long-term stability measurement of the CoFeV-LDH for over 18 h. | ||
The HER performance of the synthesized electrocatalysts and Pt/C was investigated with a three-electrode system in 1.0 M KOH. Fig. 4a shows the LSV curves of Pt/C, CoFe-LDH, CoFeZn-LDH, CoFeV-LDH, and Pt@CoFeV-LDH. The CoFe-LDH and support have poor electrocatalytic hydrogen release properties, and the coexistence of slight Pt active site loading promotes the intrinsic HER activity. Compared to Pt/C (45 mV), the Pt@CoFe1/3V-LDH exhibits a low overpotential of 72 mV (Pt@CoFe1/6V-LDH, 73 mV) to supply 10 mA cm−2. Also, the same trend for the overpotential is observed at 50 mA cm−2 (Fig. 4b). The Tafel slope of Pt@CoFe1/3V-LDH is 65 mV dec−1 (Pt@CoFe1/6V-LDH, 71 mV dec−1), which is higher than that of Pt/C (43 mV dec−1), which manifested that the HER process of the as-prepared samples abided by the combined Volmer–Heyrovsky pathway, whereas that of Pt/C followed the Tafel mechanism (Fig. 4c).40 The anchoring of trace Pt within the surface of CoFeV-LDH not only promotes the intrinsic activity of hydrogen evolution but also optimizes the stability, which is more beneficial for practical applications. After 24 hours of the uninterrupted chronopotentiometric (CA) test, Pt@CoFe1/3V-LDH showed ∼96.1% efficiency (Pt@CoFe1/6V-LDH, ∼92.4%) with no significant current decay, proving the long-term stability of the catalyst (Fig. 4d). In contrast, the performance of CoFe1/3V-LDH decayed to ∼82.5% significantly after the long stability test (CoFe1/6V-LDH, ∼83.3%) probably due to the transformation near the unsaturated sites that deformed the catalyst structure,41 demonstrating that Pt anchoring in vacancies facilitates the structural stability (Fig. S12 and S13, ESI†). The LSV curves of Pt@CoFeV-LDH before and after 1000 CV cycles have no significant change, thus demonstrating its excellent stability (Fig. S14 and S15, ESI†).
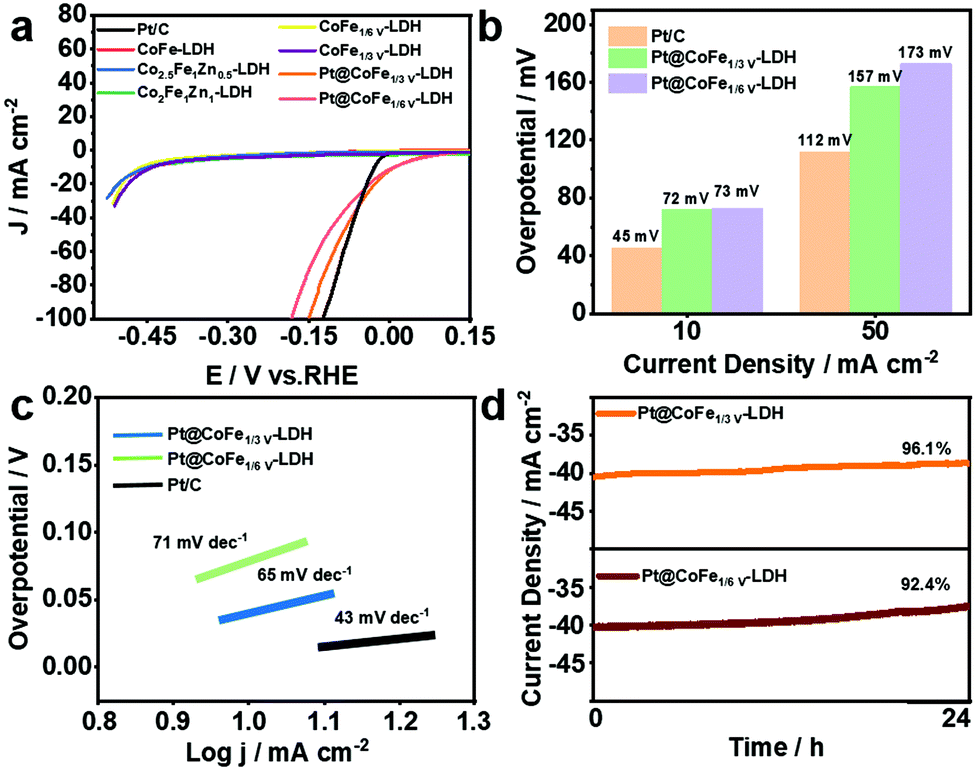 | ||
| Fig. 4 (a) LSV curves of Pt/C, CoFe-LDH, CoFeZn-LDH, CoFeV-LDH and Pt@CoFeV-LDH for catalyzing the HER in 1.0 M KOH. (b) Histogram of the overpotentials of the Pt/C and synthesized nanomaterials to drive 10 mA cm−2 and 50 mA cm−2. (c) Tafel slopes of Pt/C and Pt@CoFeV-LDH. (d) Long-term stability measurement of the Pt@CoFeV-LDH for over 24 h. | ||
To further enhance the versatility of the catalyst, the ORR performance of Pt@CoFeV-LDH was investigated in 0.1 M KOH solution (Fig. 5a). The LSV curves of the Pt@CoFe1/3V-LDH were also obtained in the range of 400 to 2500 rpm under O2-saturated alkaline conditions (Fig. 5b). As the rotation speed of the glassy carbon electrode increases, the corresponding fitted Koutecky–Levich (K–L) plots all show a good linear relationship (inset of Fig. 5b). The electron transfer number (n) = 3.89 ± 0.02 was calculated using the slope of the K–L curve, and R2 = 0.9998. The LSV curves recorded with a rotation speed of the electrode of 1600 rpm manifest that the ORR performance of Pt@CoFeV-LDH is close to that of 20% Pt/C (a half-wave potential (E1/2) of 0.819 V (vs. RHE) for Pt@CoFe1/3V-LDH and 0.801 V (vs. RHE) for Pt@CoFe1/6V-LDH), only 11 mV and 29 mV less than that of Pt/C (0.83 V) (Fig. 5c). The Tafel slope of Pt@CoFe1/3V-LDH is 58 mV dec−1, which is close to that of Pt/C (56 mV dec−1) and better than that of Pt@CoFe1/6V-LDH (65 mV dec−1) (Fig. 5d), further manifesting the fast ORR kinetics of Pt@CoFeV-LDH. The intrinsic competence of Pt@CoFeV-LDH was evaluated by the turnover frequency (TOF) over 0.5 V vs. RHE, which hypothesized that total Pt acted as active sites in the Pt@CoFeV-LDH. As shown in Fig. 5c, the TOF value of Pt@CoFe1/3V-LDH is 0.2771 S−1, indicating that active sites were more and sufficient to utilize compared to those of Pt@CoFe1/6V-LDH (0.2333 S−1).
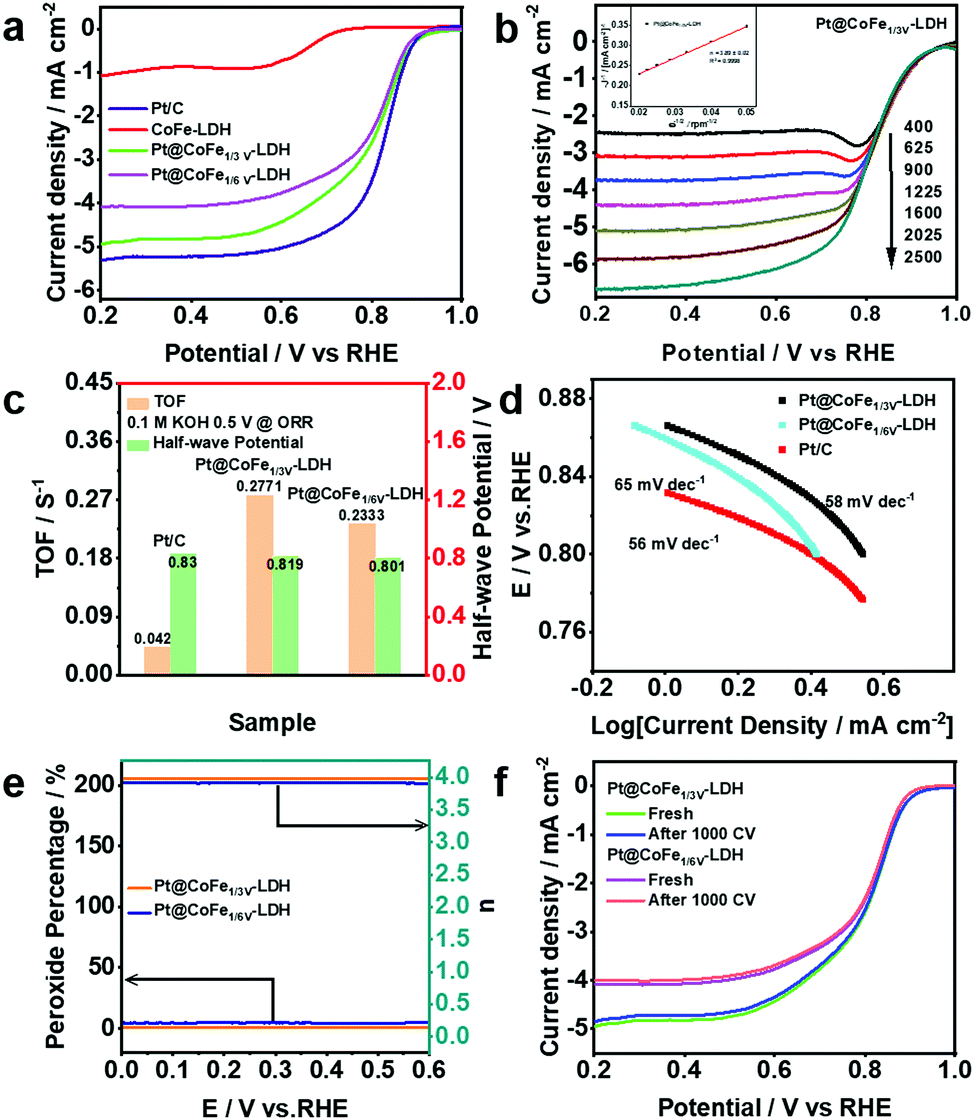 | ||
| Fig. 5 (a) LSV curves of Pt/C, CoFe-LDH and Pt@CoFeV-LDH. (b) LSV curves of Pt@CoFe1/3V-LDH at various rotation speeds (inset: K–L plots for Pt@CoFe1/3V-LDH at 0.2 V vs. RHE). (c) Turnover frequency (TOF) and half-wave potential of Pt/C and Pt@CoFeV-LDH. (d) Tafel plots. (e) Peroxide percentage, electron transfer number and (f) LSV curves before and after 1000 CVs of Pt@CoFeV-LDH. | ||
Rotating ring-disk electrode (RRDE) voltammograms of Pt@CoFeV-LDH were obtained in the 0.1 M KOH electrolyte with oxygen saturation. The electron-transfer number was calculated exactly with a 1600 rpm rotation rate. The electron-transfer numbers of Pt@CoFe1/3V-LDH and Pt@CoFe1/6V-LDH were calculated to be 3.92 and 3.87 over the potential range from 0 to 0.6 V (RHE), and the H2O2 yield remained below 2% (Fig. 5e). Moreover, the LSV curves of Pt@CoFeV-LDH before and after 1000 cycles of CV indicate that the ORR stability of Pt@CoFeV-LDH is appreciable (Fig. 5f). The trace Pt anchored within the cation vacancies led to an effective ORR performance with CoFeV-LDH in alkaline electrolytes, although it cannot match that of commercial Pt/C.
On the premise of the remarkable electrocatalytic activity of the synthesized CoFe-LDH-based catalysts towards the HER and OER, full water splitting reactions were performed in 1.0 M KOH using the Pt@CoFeV-LDH as the cathode and CoFeV-LDH as the anode for an electrolyzer (Fig. 6a). Only a small cell voltage of 1.57 V is needed to supply 10 mA cm−2 for the electrolyser containing CoFe1/3V-LDH//Pt@CoFe1/3V-LDH (CoFe1/6V-LDH//Pt@CoFe1/6V-LDH, 1.62 V) and the electrogenerated gas bubbles were clearly detected in Fig. 6a illustration. Moreover, CoFe1/3V-LDH//Pt@CoFe1/3V-LDH also demonstrated excellent stability at ∼99% (CoFe1/6V-LDH//Pt@CoFe1/6V-LDH, ∼94.7%) during the 8 h full water-splitting process (Fig. 6b).
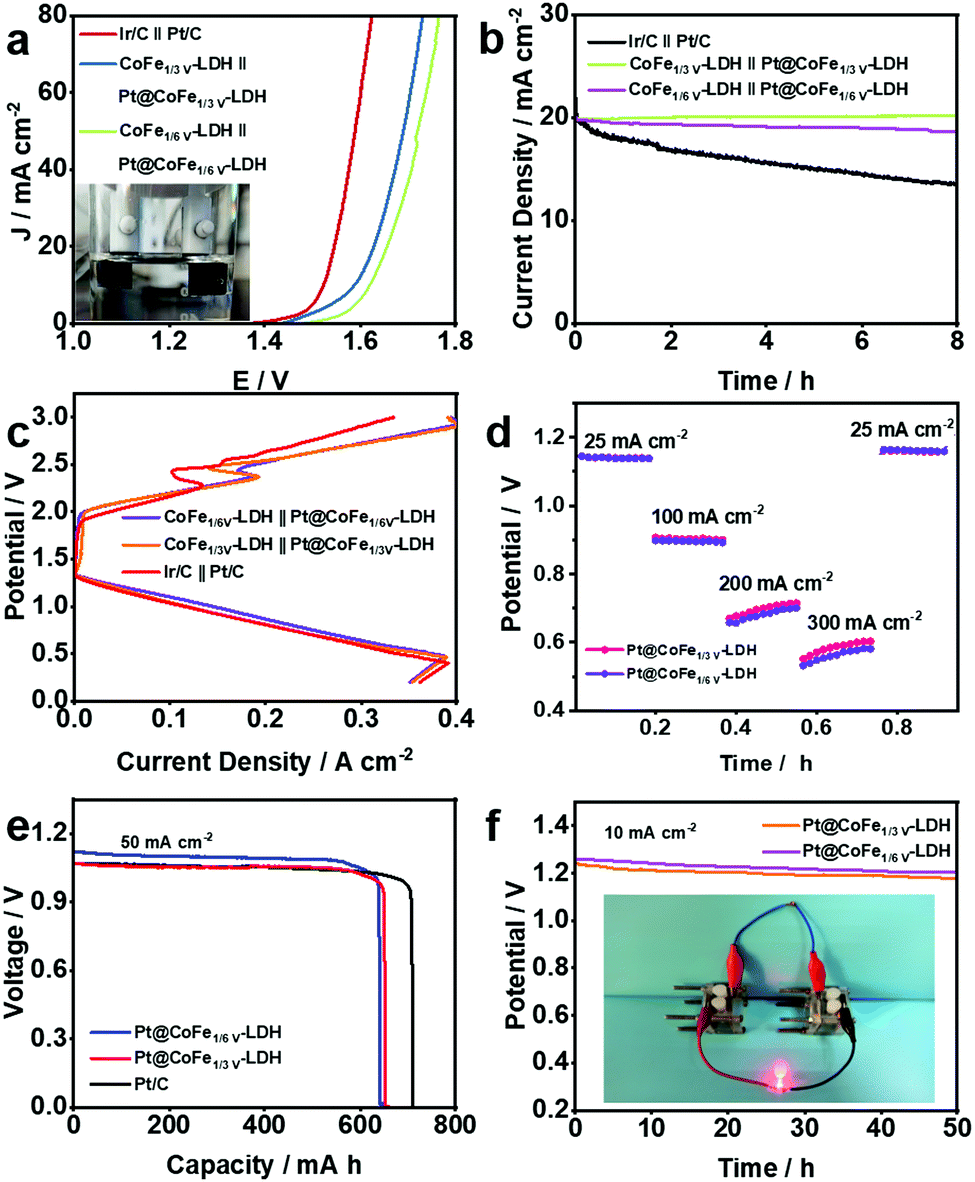 | ||
| Fig. 6 (a) LSV curves and (b) long-term stability test for full water splitting of the composited catalyst and Ir/C//Pt/C in 1 M KOH. (c) Discharge and charge polarization curves of the rechargeable battery using the CoFeV-LDH//Pt@CoFeV-LDH and Ir/C//Pt/C air electrodes. (d) Discharged curves from 25 to 300 mA cm−2. (e) Typical specific capacity (normalized to the mass of the zinc electrode) curves of the battery using the Pt@CoFeV-LDH air electrode at 50 mA cm−2 current densities. (f) The discharge curve of a Pt@CoFeV-LDH-based ZAB over 50 h and a picture of a LED bulb offering power by two ZABs in series. | ||
Distinctively, the kinetics in a rechargeable Zn–air battery (ZAB) is primarily confined by the oxygen reaction occurring in the cathode: O2 + 2H2O + 4e− ⇌ 4OH−.42 On account of the satisfactory ORR catalytic performance of the Pt@CoFeV-LDH, a liquid ZAB was also established, which broadens its potential for the manufacture of a practical device. As expected, the Pt@CoFe1/3V-LD-based ZAB displays an extreme power density of 200 mW cm−2 (Pt@CoFe1/6V-LDH, 190 mW cm−2) with a 350 mA cm−2 cut-off current density (Pt@CoFe1/6V-LDH, 390 mA cm−2) (Fig. 6c), which is relevant to 105.3% (Pt@CoFe1/6V-LDH, 100%) for the ZAB based on a commercial Pt/C catalyst (190 mW cm−2) (Fig. S16 and S17, ESI†).43 Discharge and charge polarization curves of the rechargeable battery using the CoFeV-LDH//Pt@CoFeV-LDH and Ir/C//Pt/C air electrodes are shown in Fig. 6c. The discharge and charge performances of the as-synthetic catalyst exhibit potential overpotentials, which are consistent with the OER and ORR performance of CoFeV-LDH and Pt@CoFeV-LDH. The Pt@CoFeV-LDH-based ZAB can be discharged from 25 to 300 mA cm−2 (Fig. 6d), suggesting that it can be operated in a wide range of current densities. Additionally, the specific capacity (normalized to the mass of the zinc electrode) of the Pt@CoFe1/3V-LDH-battery is ∼646.8 (Pt@CoFe1/6V-LDH, ∼641.3 mA h g−1) at 50 mA cm−2 current density (Fig. 6e). The potentials of Pt@CoFe1/3V-LDH and Pt@CoFe1/6V-LDH are 1.24 and 1.28 V, respectively, at 10 mA cm−2 after 50 h (Fig. 6f). Two series-connected Pt@CoFeV-LDH ZABs can light a LED bulb (Fig. 6f inset), which demonstrated that it can be practically applied in metal–air batteries.
4. Conclusions
In summary, we have synthesized high-efficiency cation vacancy rich CoFeV-LDH and Pt@CoFeV-LDH electrocatalysts, among which the CoFeV-LDH showed excellent OER performance due to cation vacancies providing affluent active sites and Pt@CoFeV-LDH, which is demonstrated for catalyzing the HER and ORR, exhibited efficient HER and ORR catalytic activities derived from the anchored Pt on cation vacancies of CoFeV-LDH. More importantly, the electrolyzer composed of the CoFeV-LDH anode and the Pt@CoFeV-LDH cathode showed excellent water splitting performance and stability. The liquid-state ZAB using CoFeV-LDH//Pt@CoFeV-LDH as air electrodes showed satisfactory charge–discharge performance. Moreover, Pt@CoFeV-LDH revealed sufficient potential and feasibility in liquid ZAB systems. This work provides a new strategy for the wide application of active sites-support catalysts in energy conversion and storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 51672143 and 51808303), the Natural Science Foundation of Shandong Province (ZR2017MEM018, ZR2018BEM002, ZR2019BEE027 and ZR2019BC007), the Taishan Scholars Program, Key Research and Development Program of Shandong Province (No. 2017GSF18128), and the State Key Laboratory of Bio-Fibers and Eco-Textiles, Qingdao University (No. ZKT10 and GZRC202006).Notes and references
- M. Ahsan, A. Santiago, Y. Hong, N. Zhang, M. Cano, E. Rodriguez-Castellon, L. Echegoyen, S. Sreenivasan and J. Noveron, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- J. Fu, F. Hassan, J. Li, D. Lee, A. Ghannoum, G. Lui, M. Hoque and Z. Chen, Adv. Mater., 2016, 28, 6420 CrossRef CAS PubMed.
- X. Long, D. Li, B. Wang, Z. Jiang, W. Xu, B. Wang, D. Yang and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 11369–11373 CrossRef CAS PubMed.
- H. Wang, J. Wang, Y. Pi, Q. Shao, Y. Tan and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 2316–2320 CrossRef CAS PubMed.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- H. Sun, M. Wang, S. Zhang, S. Liu, X. Shen, T. Qian, X. Niu, J. Xiong and C. Yan, Adv. Funct. Mater., 2021, 31, 2006533 CrossRef CAS.
- C. Zhang, Y. Cui, Y. Yang, L. Lu, S. Yu, Z. Meng, Y. Wu, Y. Li, Y. Wang, H. Tian and W. Zheng, Adv. Funct. Mater., 2021, 2105372, DOI:10.1002/adfm.202105372.
- Y. Zhou, S. Sun, C. Wei, Y. Sun, P. Xi, Z. Feng and Z. Xu, Adv. Mater., 2019, 31, 1902509 CrossRef CAS PubMed.
- Z. Cai, D. Zhou, M. Wang, S. Bak, Y. Wu, Z. Wu, Y. Tian, X. Xiong, Y. Li, W. Liu, S. Siahrostami, Y. Kuang, X. Yang, H. Duan, Z. Feng, H. Wang and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 9392–9396 CrossRef CAS PubMed.
- X. Zhang, Y. Zhao, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2019, 9, 1900881 CrossRef.
- A. Karmakar, K. Kannimuthu, S. Sam Sankar, K. Sangeetha, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC.
- Y. Zhao, L. Zheng, R. Shi, S. Zhang, X. Bian, F. Wu, X. Cao, G. Waterhouse and T. Zhang, Adv. Energy Mater., 2020, 10, 2002199 CrossRef CAS.
- Q. Xie, Z. Cai, P. Li, D. Zhou, Y. Bi, X. Xiong, E. Hu, Y. Li, Y. Kuang and X. Sun, Nano Res., 2018, 11, 4524–4534 CrossRef CAS.
- Y. Wang, M. Qiao, Y. Li and S. Wang, Small, 2018, 1800136 CrossRef PubMed.
- F. Wang, G. Xu, Y. He, Z. Liu, Z. Zhang, Q. Mao and Y. Huang, J. Energy Chem., 2020, 51, 101–104 CrossRef.
- G. Tomboc, J. Kim, Y. Wang, Y. Son, J. Li, J. Y. Kim and K. Lee, J. Mater. Chem. A, 2021, 9, 4528 RSC.
- J. Wu, P. Bag, Y. Xu, L. Gong, C. He, X. Chen and J. Zhang, Adv. Mater., 2021, 33, 2007368 CrossRef CAS.
- J. Zhang, H. Wang, L. Wang, S. Ali, C. Wang, L. Wang, X. Meng, B. Li, D. S. Su and F. S. Xiao, J. Am. Chem. Soc., 2019, 141, 2975–2983 CrossRef CAS PubMed.
- L. Hu, R. Xiao, X. Wang, X. Wang, C. Wang, J. Wen, W. Gu and C. Zhu, Appl. Catal., B, 2021, 298, 120599 CrossRef CAS.
- H. Chen, Z. Yang, X. Wang, F. Polo-Garzon, P. Halstenberg, T. Wang, X. Suo, S. Yang, H. Meyer III, Z. Wu and S. Dai, J. Am. Chem. Soc., 2021, 143, 8521–8526 CrossRef CAS PubMed.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C. Tung, J. Wu, H. Chen and J. Yu, Adv. Mater., 2021, 33, 2008599 CrossRef CAS PubMed.
- J. Yang, W. Li, S. Tan, K. Xu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19085–19091 CrossRef CAS.
- Y. Tang, Q. Liu, L. Dong, H. Wu and X. Yu, Appl. Catal., B, 2020, 266, 118627 CrossRef CAS.
- Y. Zhao, Y. Gao, Z. Chen, Z. Li, T. Ma, Z. Wu and L. Wang, Appl. Catal., B, 2021, 297, 120395 CrossRef CAS.
- L. Zhuang, Y. Jia, H. Liu, X. Wang, R. Hocking, H. Liu, J. Chen, L. Ge, L. Zhang, M. Li, C. Dong, Y. Huang, S. Shen, D. Yang, Z. Zhu and X. Yao, Adv. Mater., 2019, 31, 1805581 CrossRef PubMed.
- L. Wen, X. Zhang, J. Liu, X. Li, C. Xing, X. Lyu, W. Cai, W. Wang and Y. Li, Small, 2019, 15, 1902373 CrossRef PubMed.
- Z. Wang, W. Liu, Y. Hu, M. Guan, L. Xu, H. Li, J. Bao and H. Li, Appl. Catal., B, 2020, 272, 118959 CrossRef CAS.
- G. Tomboc, J. Kim, Y. Wang, Y. Son, J. Li, J. Y. Kim and K. Lee, J. Mater. Chem. A, 2021, 9, 4528 RSC.
- S. Liu, J. Zhu, M. Sun, Z. Ma, K. Hu, T. Nakajima, X. Liu, P. Schmuki and L. Wang, J. Mater. Chem. A, 2020, 8, 2490–2497 RSC.
- J. Chang, G. Wang and Y. Yang, Small Sci. DOI:10.1002/smsc.202100044.
- Q. Zhang, Y. Kuang, Y. Li, M. Jiang, Z. Cai, Y. Pang, Z. Chang and X. Sun, J. Mater. Chem. A, 2019, 7, 9517–9522 RSC.
- P. Wu, Y. Wu, L. Chen, J. He, M. Hua, F. Zhu, X. Chu, J. Xiong, M. He, W. Zhu and H. Li, Chem. Eng. J., 2020, 380, 122526 CrossRef CAS.
- Z. Chen, M. Chen, X. Yan, H. Jia and R. Wu, ACS Nano, 2020, 14, 6968–6979 CrossRef CAS PubMed.
- X. Zhou, X. Li, D. Chen, D. Zhao and X. Huang, J. Mater. Chem. A, 2018, 6, 24603–24613 RSC.
- K. Zhang, Y. Zhang, Q. Zhang, Z. Liang, L. Gu, W. Guo, B. Zhu, S. Guo and R. Zou, Carbon Energy, 2020, 2, 283–293 CrossRef CAS.
- X. Mao, A. Foucher, T. Montini, E. Stach, P. Fornasiero and R. Gorte, J. Am. Chem. Soc., 2020, 142, 10373–10382 CrossRef CAS.
- Y. Zhou, D. Yan, Q. Gu, S. Zhu, L. Wang, H. Peng and Y. Zhao, Appl. Catal., B, 2021, 285, 119792 CrossRef CAS.
- H. Sun, L. Chen, Y. Lian, W. Yang, L. Lin, Y. Chen, J. Xu, D. Wang, X. Yang, M. Rümmerli, J. Guo, J. Zhong, Z. Deng, Y. Jiao, Y. Peng and S. Qiao, Adv. Mater., 2020, 32, 2006784 CrossRef.
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Angew. Chem., Int. Ed., 2021, 60, 18981–19006 CrossRef CAS PubMed.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2020, 31, 2009070 CrossRef.
- X. Xie, L. Shang, R. Shi, G. I. N. Waterhouse, J. Zhao and T. Zhang, Nanoscale, 2020, 12, 13129–13136 RSC.
- C. Lv, Y. Zhu, W. Zhang, W. Xu, J. Ren, H. Liu, X. Yang, R. Cai, S. Jin, D. Li and D. Yang, Mater. Today Energy, 2021, 21, 100834 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00836f |
| ‡ Zhenyu Kong and Jingying Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |