DOI:
10.1039/D1MA00614B
(Paper)
Mater. Adv., 2021,
2, 7922-7931
Rationalization of excited state energy transfer in D–π–A porphyrin sensitizers enhancing efficiency in dye-sensitized solar cells†
Received
15th July 2021
, Accepted 30th September 2021
First published on 2nd October 2021
Abstract
Donor–π–acceptor (D–π–A) porphyrin based photo-sensitizers are extensively utilized in dye sensitized solar cells (DSSCs). However, investigation on how the intramolecular photoinduced energy/electron transfer influences the device performance is still limited. Herein, we report three new D–π–A porphyrin sensitizers (LG8, LG9 and LG10) comprising 3-ethynylfluoranthene as the donor and 2,1,3-benzothiadiazole (BTD), 2,3-diphenylquinoxaline (DPQ), and 2,3-di(thiophen-2-yl)quinoxaline (DTQ) as auxiliary acceptors. LG8 showed more efficient and red-shifted energy transfer between the donor and acceptors, which translates into a higher power conversion efficiency (PCE). UV-visible absorption and fluorescence spectra revealed a large bathochromic shift and a significant quenching of the donor emission confirming an efficient intramolecular energy transfer from the fluoranthene-linked porphyrin to the auxiliary acceptor benzothiadiazole in LG8. Density functional theory calculations showed that the LUMO of LG8 and LG10 destabilized and matched with the conduction band edge of the anatase TiO2 nanoparticles. As a result, LG8 and LG10 exhibited the best power conversion efficiency (η ≈ 3.0%) compared to LG9 (η = 1.50%) owing to faster electron injection from the dye excited states into the conduction band of TiO2. This work underlines that the modification of the electron donor and the acceptor in D–π–A porphyrin sensitizers has noticeable influence on the photo-physical properties of π-conjugated systems, thereby affecting device performances.
1. Introduction
The penetration of renewable energies into our energy mix is indispensable owing to the excess consumption of fossil fuels and the increased awareness of environmental consequences.1 In this context, solar energy received much attention amongst other green resources as an enormous amount of sunlight can be converted into electricity. Recently, organic photovoltaics have been in high demand to generate flexible, stable and low production cost solar cells in which dye-sensitized solar cells (DSSCs) are of significant interest due to ease of fabrication and excellent incident photon-to-electron conversion efficiency (IPCE) while being eco-friendly.2–8 In 1991, Grätzel et al. introduced the basic principle mechanism of nanocrystalline DSSCs.9 In this technology, the core is the photo-sensitizer that rules the two key basic principles of PV, light harvesting, and exciton formation and separation in contact with nanocrystalline TiO2 and a redox mediator in an electrolyte. Earlier, ruthenium-based DSSCs exhibited a certified PCE of over 11%. However, the low abundance of the Ru metal, purification of isomers and lack of absorption into the NIR region prompt the development of new alternatives to further improve stability, and efficiency and thus accelerate DSSCs commercialization.10–13 Therefore, the development of metal-free organic dyes allowed record PCEs exceeding 14% to be reached.14–21
Amongst all various photosensitizers developed, porphyrin dyes possess high molar extinction coefficients, a broad absorption spectrum from the visible to near-infrared (NIR) region and ease of structural modification at β-pyrrole and meso-positions to tune optical and electrochemical properties.22–29 Initially, meso-phenyl carboxy zinc porphyrin-based DSSCs reported an efficiency of 3.5%.29 Officer et al. improved the porphyrin sensitizer by substituting the carboxylic group at the β-pyrrole position, reaching a PCE of 7.1%.30,31 Subsequently, Grätzel et al. developed a D–π–A strategy using several porphyrin sensitizers YD2-O-C832 and SM31533 and achieved PCE values of 12.5% and 13%, respectively, along with stronger oxidizing Co-based redox couples. Furthermore, the porphyrin dye based device efficiency enhanced to 14.2% using either an organic co-sensitized or in a tandem fashion.34,35 Recently, we reported butterfly-shaped D–π–A porphyrin systems composed of phenothiazine as an electron donor and benzothiadiazole as an acceptor which result with the efficiency of 10.2%.36 Furthermore, the strong electron donating nature of phenanthroimidazole substituted in porphyrin D–π–A systems achieved an efficiency of 8.2% upon co-sensitisation with an organic dye.37 However, the device efficiency increase, induced by energy or electron transfer with the modifications of the electron and acceptor moieties in porphyrin D–π–A systems for DSSCs, is well documented in the literature.26–28 Therefore, we herein report a systematic investigation of fluoranthene as the energy donor and various auxiliary acceptor units such as benzothiadiazole/quinoxalines attached to the meso-position of porphyrins (LG8–LG10) and studied their optical, electrochemical and device properties. Detailed studies revealed that intramolecular energy transfer is more favoured in the case of LG8 leading to a higher efficiency compared to LG9 and LG10. These results suggest that the energy level matching between the electron donor and acceptor units affects the device performance and leads towards the design of diverse D–π–A systems for organic electronics and energy related applications.
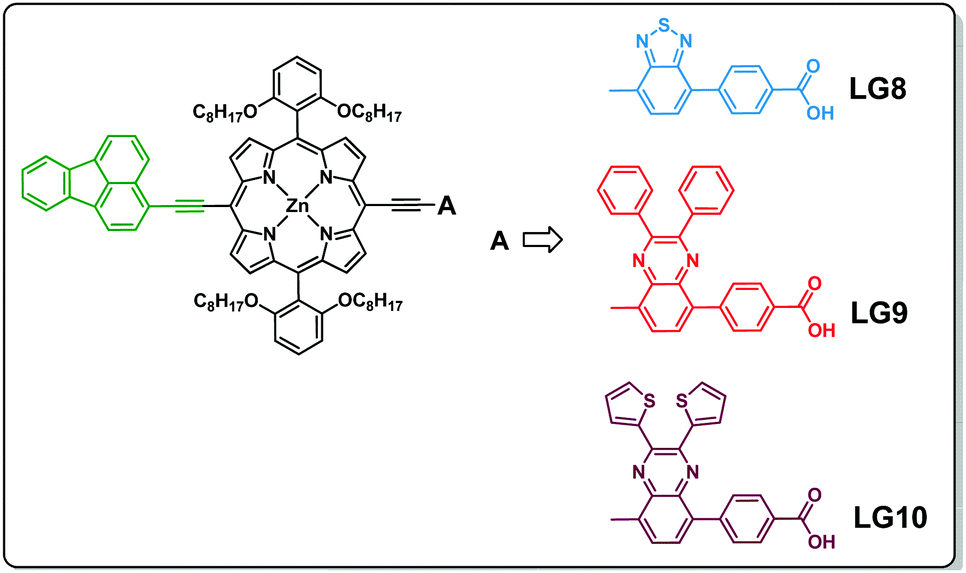
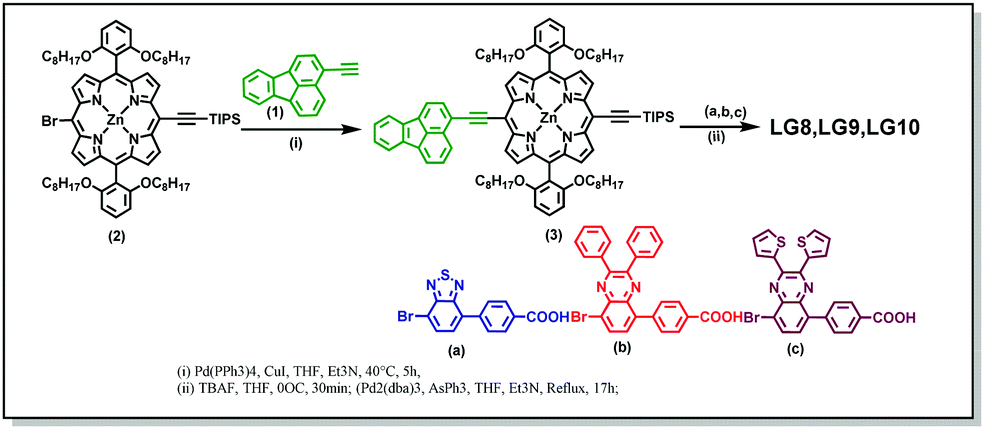
Three porphyrin D–π–A sensitizers (LG8–LG10) are herein designed, comprising fluoranthene as the electron donor unit tethered to porphyrin in its meso-position and three different auxiliary acceptor groups: benzothiadiazole (BTD) for LG8, 2,3-diphenylquinoxaline (DPQ) for LG9 and 2,3-di(thiophen-2-yl)quinoxaline (DTQ) for LG10, all three ending with a benzoic acid anchoring group. Fluoranthene is considered as one of the good energy donating units, which absorbs in the UV region, where energy transfer to the porphyrin macrocycle is possible to enhance charge separation and thus the device efficiency (Scheme 1).34,35 The detailed synthesis, characterization, energy transfer and device studies are herein presented and discussed.
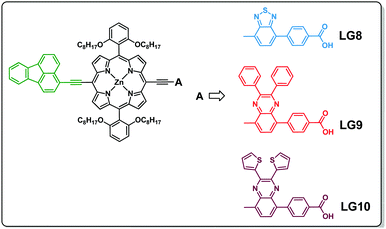 |
| | Scheme 1 Molecular structures of the three D–π–A porphyrin sensitizers (LG8, LG9 and LG10). | |
2. Experimental section
Materials
All the chemicals and reagents were purchased from Merck and Sigma-Aldrich and were used in reactions without further purification. Dry solvents were used for column chromatography and ACME silica gel (60–120 mesh) for the purification of compounds. Thin-layer chromatography was performed on Merck-pre-coated silica gel 60-F254 plates. All the reactions were carried out under a nitrogen or argon atmosphere using dry and degassed solvents.
Synthesis
3-Bromofluoranthene was used as received from TCI. 3-Ethynylfluoranthene (1), 4-(7-bromobenzo[c][1,2,5]thiadiazol-4-yl)benzoic acid (a), 4-(8-bromo-2,3-diphenylquinoxalin-5-yl)benzoic acid (b), and 4-(8-bromo-2,3-di(thiophen-2-yl)quinoxalin-5-yl)benzoic acid (c) and [5-bromo-15-(triisopropylsilyl)ethynyl-10,20-bis(2,6-di-octoxy phenyl)porphyrinato]zinc(II) (2) and 4-(7-bromobenzo[c][1,2,5]thiadiazol-4-yl)benzaldehyde were synthesized as reported in the literature.32,33,36–40
3-Ethynylfluoranthene.
3-Iodofluoranthene was dissolved in piperidine (15 mL) and THF (15 mL) followed by the addition of Pd(PPh3)2Cl2 (61.7 mg, 0.08 mmol), trimethylsilylacetylene (TMSA) (0.37 mL, 2.64 mmol), CuI (16 mg, 0.08 mmol), and PPh3 (23.08 mg, 0.08 mmol) and refluxed for 2 hours. The reaction mixture was brought to room-temperature while stirring and completion of the reaction was monitored by TLC. The solvent was removed by rotary evaporation. The residue was purified by column chromatography (ethyl acetate/n-hexanes = 1/10) to give compound 1. HRMS (APPI-FTMS) m/z [M]+ calcd for C18H10, 226.27; found 227. 1H NMR (400 MHz, CDCl3) δ: 8.32 (1H, d, J 8.3), 7.91 (1H, d, J 7.6), 7.85 (4H, m, J 8.4), 7.52 (1H, t, J 6.4), 4.38 (2H, t), 3.52 (1H, s).
[3-Ethynylfluoranthene-15-(triisopropylsilyl)ethynyl-10,20-bis(2,6-di-octoxy phenyl)porphyrinato] zinc(II) (3).
Compound 2 (270 mg, 0.196 mmol) and compound 1 (89.6 mg, 0.393 mmol) were dissolved in 30 mL of THF and 5 mL of triethylamine. To this, Pd(PPh3)4 (11.3 mg, 0.010 mmol) and CuI (1.8 mg, 0.010 mmol) were added under an inert atmosphere. The reaction mixture was stirred at 40 °C for 24 hours. The completion of the reaction was monitored by TLC. The solvent was removed by rotary evaporation. The residue was purified by column chromatography (THF/n-hexane = 1/10) to give compound 3 (yield = 30.8%) as a green solid. MALDI-TOF: m/z [M + H]+ calcd for C93H112N4O4SiZn, 1443.37; found 1444. 1H NMR (500 MHz, CDCl3 + pyridine-d5) 9.82 (2H, d, J 4.5), 9.65 (2H, J 4.4), 8.93 (2H, d, J 4.5), 8.87 (2H, d, J 4.5), 8.80 (1H, d, J 8.2), 8.33 (1H, d, J 7.1), 8.08 (1H, d, J 7.0), 8.03 (1H, d, J 6.7), 7.90–7.85 (4H, m), 7.69 (3H, t, J 8.5), 7.31–7.29 (2H, m), 7.00 (2H, d, J 6.7), 3.86 (8H, t, J 8.0), 2.20–2.15 (3H, s), 1.54–1.50 (16H, m), 1.43 (8H, s), 0.97 (12H, s), 0.77 (12H, d, J 7.4), 0.57 (4H, s), 0.51 (4H, s), 0.45 (14H, d, J 4.8), 0.41 (8H, s). FT-IR (neat, cm−1): 3446, 2923, 2856, 2130, 1633, 1585, 1453, 1379, 1334, 1293, 1244, 1203, 1159, 1094, 997, 882, 830, 783, 766, 711, 662, 467.
[5,15-Bis(2,6-dioctoxyphenyl)-10-(3-ethynyl fluoranthene)-20-(4-(7-benzo[c][1,2,5]thiadiazol-4-yl)carboxyphenylethynyl)porphyrinato]zinc(II) (LG8).
To a solution of compound 3 (200 mg, 0.138 mmol) in anhydrous THF (20 mL) was added TBAF (0.6 mL, 1 M in THF). The solution was stirred at 0 °C for 30 minutes under a N2 atmosphere. The mixture was quenched with H2O and then extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue (180 mg, 0.139 mmol) and 4-(7-bromobenzo[c][1,2,5]thiadiazol-4-yl)benzoic acid (111.5 mg, 0.401 mmol) were dissolved in a mixture of anhydrous THF (20 mL) and Et3N (8 mL) under a nitrogen atmosphere, and then Pd2(dba)3 (6.4 mg, 0.007 mmol) and AsPh3 (2.1 mg, 0.007 mmol) were added to the mixture. The solution was refluxed for 17 hours. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH = 20/1) and recrystallization from MeOH/ether to give dye LG8 (yield 59%) as a brownish green solid. Anal. calcd for C97H98N6O6SZn % (1541.30): C, 75.59; H, 6.41; N, 5.45; O, 6.23; S, 2.08; Zn, 4.24 found: C, 75.55; H, 6.36; N, 5.40; O, 6.18; S, 2.04; Zn, 4.18. MALDI-TOF: m/z [M − H]+ calcd for C97H98N6O6SZn % 1541.30; found, 1541. 1H NMR (300 MHz, CDCl3 + pyridine-d5) 9.99 (2H, s), 9.80 (2H, d, J 5.0), 8.97–8.92 (4H, dd, J 11.8), 8.81 (1H, d, J 8.0), 8.39 (2H, d, J 7.2), 8.29 (1H, d, J 5.5), 8.21 (4H, dd, J 13.0, 7.7), 8.08–8.02 (2H, m), 7.92 (4H, d, J 8.2), 7.87 (2H, t, J 6.9), 7.85 (3H, d, J 8.0), 7.06 (2H, d, J 5.5), 3.72 (8H, t, J 8.1), 1.26 (3H, d, J 6.5), 1.00–0.94 (8H, m), 0.89–0.84 (7H, m), 0.71 (6H, s), 0.58 (12H, m), 0.54 (8H, s), 0.52 (10H, m), 0.49 (6H, s). FT-IR (neat, cm−1): 3449, 2921, 2851, 2181, 1686, 1585, 1501, 1453, 1418, 1382, 1291, 1244, 1205, 1096, 997, 885, 830, 791, 770, 708, 653, 534, 509, 466.
[5,15-Bis(2,6-dioctoxyphenyl)-10-(3-ethynylfluoranthene)-20-(4-(8-(2,3 diphenylquinoxalin-5-yl))carboxyphenylethynyl)porphyrinato] zinc(II) (LG9).
We have adopted a similar procedure as for LG8, but instead of 4-(7-ethynylbenzo[c][1,2,5]thiadiazol-4-yl)benzoic acid, 4-(8-ethynyl-2,3-diphenylquinoxalin-5-yl)benzoic acid (170.2 mg, 0.344 mmol) was used. The crude compound was purified by silica gel column chromatography (CH2Cl2/MeOH = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), recrystallized from MeOH/ether to give dye LG9 (yield 57%) as a green solid. Anal. calcd for C111H108N6O6Zn % (1687.46): C, 79.01; H, 6.45; N, 4.98; O, 5.69; Zn, 3.87 found: C, 78.96; H, 6.36; N, 4.92; O, 5.62; Zn, 3.82. MALDI-TOF: m/z [M − H]+ calcd for C111H108N6O6Zn % 1687.46; found, 1687. 1H NMR (300 MHz, CDCl3 + pyridine-d5) 9.87 (2H, d, J 4.5), 9.02 (4H, dd, J 10.7, 4.4), 8.85 (2H, d, J 8.3), 8.78 (2H, d, J 4.6), 8.31 (4H, d, J 8.2), 8.12 (2H, d, J 7.8), 8.06 (2H, t, J 8.2), 7.96 (2H, d, J 2.9), 7.87 (3H, d, J 4.2), 7.72 (2H, m), 7.57 (3H, d, J 6.5), 7.49 (1H, s), 7.37 (3H, d, J 4.5), 7.32 (2H, s), 7.27 (3H, s), 7.05 (2H, d, 5.0), 3.72 (8H, t, J 7.9), 1.26 (4H, d, J 6.5), 0.90–0.86 (7H, m), 0.84 (7H, m), 0.77–0.74 (6H, s), 0.60–0.58 (12H, m), 0.57–0.55 (8H, s), 0.52 (10H, m), 0.54–0.49 (6H, s). FT-IR (neat, cm−1): 3448, 2921, 2853, 1683, 1595, 1451, 1243, 1093, 994, 767, 701.
:1, v/v), recrystallized from MeOH/ether to give dye LG9 (yield 57%) as a green solid. Anal. calcd for C111H108N6O6Zn % (1687.46): C, 79.01; H, 6.45; N, 4.98; O, 5.69; Zn, 3.87 found: C, 78.96; H, 6.36; N, 4.92; O, 5.62; Zn, 3.82. MALDI-TOF: m/z [M − H]+ calcd for C111H108N6O6Zn % 1687.46; found, 1687. 1H NMR (300 MHz, CDCl3 + pyridine-d5) 9.87 (2H, d, J 4.5), 9.02 (4H, dd, J 10.7, 4.4), 8.85 (2H, d, J 8.3), 8.78 (2H, d, J 4.6), 8.31 (4H, d, J 8.2), 8.12 (2H, d, J 7.8), 8.06 (2H, t, J 8.2), 7.96 (2H, d, J 2.9), 7.87 (3H, d, J 4.2), 7.72 (2H, m), 7.57 (3H, d, J 6.5), 7.49 (1H, s), 7.37 (3H, d, J 4.5), 7.32 (2H, s), 7.27 (3H, s), 7.05 (2H, d, 5.0), 3.72 (8H, t, J 7.9), 1.26 (4H, d, J 6.5), 0.90–0.86 (7H, m), 0.84 (7H, m), 0.77–0.74 (6H, s), 0.60–0.58 (12H, m), 0.57–0.55 (8H, s), 0.52 (10H, m), 0.54–0.49 (6H, s). FT-IR (neat, cm−1): 3448, 2921, 2853, 1683, 1595, 1451, 1243, 1093, 994, 767, 701.
[5,15-Bis(2,6-dioctoxyphenyl)-10-(3-ethynylfluoranthene)-20-(4-(8-(2,3-di(thiophen-2-yl)quinoxalin-5-yl))carboxyphenylethynyl)porphyrinato] zinc(II) (LG10).
We have adopted a similar procedure as for LG8, but used 4-(7-ethynylbenzo[c][1,2,5]thiadiazol-4-yl)benzoic acid and 4-(8-ethynyl-2,3-di(thiophen-2-yl)quinoxalin-5-yl)benzoic acid (176.5 mg, 0.357 mmol). The crude compound was purified by silica gel column chromatography (CH2Cl2/MeOH = 20:1, v/v), recrystallized from MeOH/ether to give dye LG10 (yield 69%) as a greenish-brown solid. Anal. calcd for C107H104N6O6S2Zn % (1699.52): C, 75.62; H, 6.17; N, 4.94; O, 5.65; S, 3.77; Zn, 3.85 found: C, 75.55; H, 6.11; N, 4.90; O, 5.62; S, 3.73; Zn, 3.82. MALDI-TOF: m/z [M − H]+ calcd for C107H104N6O6S2Zn % 1699.52; found, 1698. 1H NMR (300 MHz, CDCl3 + pyridine-d5) 10.12 (2H, d, J 4.5), 9.81 (2H, d, J 4.5), 8.97 (2H, d, J 4.4), 8.90 (2H, d, J 4.5), 8.81 (1H, s), 8.42 (3H, s), 8.37 (2H, s), 8.28 (1H, s), 8.04 (3H, s), 7.95 (2H, s), 7.88–7.86 (2H, m), 7.81–7.79 (1H, d, J 4.4), 7.75–7.74 (1H, m), 7.69 (2H, s), 7.53 (3H, d, J 5.0), 7.43 (2H, s), 7.39–7.38 (2H, m), 7.10 (2H, d, J 5.0), 3.93 (8H, t, J 7.3), 1.27 (4H, s), 1.19 (8H, m), 1.16–1.13 (6H, s), 0.98–0.86 (6H, m), 0.60–0.58 (12H, m), 0.53 (8H, m), 0.50 (10H, s), 0.48 (6H, s). FT-IR (neat, cm−1): 3446, 2922, 2851, 2180, 1682, 1588, 1455, 1420, 1382, 1294, 1241, 1095, 997, 953, 842, 775, 705, 545, 466.
Methods and instrumentation
MALDI-MS spectra were recorded using a TO-4X KOMPACT SEQ, KARTOS, UK, mass spectrometer. Major fragmentations are given as percentages relative to the base peak intensity. 1H NMR spectra were obtained at 400 MHz using a Bruker 400 Advance NMR spectrometer with X-WIN NMR software and all the 1H NMRs were referenced to trimethylsilane (TMS). The elemental vario MICRO CUBE analyzer was used to perform elemental analysis. Electrochemical studies were performed using a PC-controlled CH instruments model CHI 620C electrochemical analyzer. 1 mM solutions in THF solvent were used for cyclic and differential pulse voltammetry studies. The supporting electrolyte was 0.1 M tetrabutylammonium perchlorate (TBAP) at a scan rate of 100 mV s−1 using glassy carbon, the saturated calomel electrode (SCE), and platinum wire as working, reference and auxiliary electrodes, respectively. A first cyclic voltammogram (CV) was recorded, and then ferrocene was added before the measurement of a second cyclic voltammogram to determine the potential of both HOMO and LUMO levels. Spectro-electrochemical experiments were performed using a CH instruments model CHI 620C electrochemical analyzer utilizing a three-electrode configuration of a thin layer quartz spectro-electrochemical cell at 25 °C. The working electrode was a Pt gauze, the Pt wire counter electrode and the SCE reference electrode were separated from the solution by a double bridge. Thermogravimetric measurements were carried out on the Mettler Toledo TGA/SDTA 851e instrument at a heating rate of 10 °C min−1 with 10 mg of sample.
Steady-state absorption and time-resolved fluorescence measurements
The optical absorption spectra were recorded using a Shimadzu (Model UV-3600) spectrophotometer with solutions of concentration 1 × 10−6 M for the Soret band and 1 × 10−5 M for the Q band in THF solvent. Steady-state fluorescence spectra were recorded (Spex model Fluorolog-3) for solutions having an optical density of ca. 0.2 at the excitation wavelength. Excitation–emission map and time-correlated single photon counting (TCSPC) experiments were carried out using an Edinburgh Instrument FLS980 spectrometer. For steady-state, the setup is composed of a continuous Xe light source associated with double excitation/emission monochromators. TCSPC experiments were carried out using a 70 ps pulse width laser diode at 480 nm as an excitation source (20 MHz repetition rate, 50 pJ cm−2 average energy, instrumental response of 90 ps FWHM) and a microchannel plate photomultiplier tube (MCP-PMT) Hamamatsu detector set after the first emission monochromator. A 510 nm long-pass filter was used in emission to avoid light scattering from the glass. The numerical analysis of the excited-state lifetime has been determined after reconvolution of the photoluminescence decay considering the instrumental response function (IRF) determined using Ludox solution.
DSSC device fabrication
Unless specified, all devices were fabricated with a standard mesoporous 8 μm thick TiO2 film sensitized with either LG8, LG9 or LG10 dye in a tert-butanol:acetonitrile (1:1 in volume) solvent mixture, including an optimized concentration of 2 mM of chenodeoxycholic acid (CDCA) to avoid the formation of molecular aggregation in both solution and at the surface of the nanocrystals. TiO2 paste is commercially available (Greatcell solar materials 18 NRT). Optimized electrolyte composition is based on a high volatile acetonitrile solvent including 0.6 M 1,3 dimethylimidazolium iodide (DMII), 0.05 M iodine, 0.1 M LiI and 0.5 M tert-butyl pyridine (TBP).
Picosecond transient absorption spectroscopy (TAS) measurements
The picosecond pump–probe set-up for TAS measurements with a streak camera detector was used to investigate regeneration and geminate recombination kinetics in the LG8–LG10 based DSSCs. The set-up is explained in detail in our previous report.41 Dye regeneration and recombination kinetics are measured after pump excitation at 430 nm. A 700 μJ cm−2 pump fluence was used. A 455 nm long-pass filter (Thorlabs) was placed before the spectrograph to remove scattered light from the pump. The transient absorption is calculated from four streak images according to the equation:
| ΔA = −log((data − emission)/(monitor − dark)) |
where data represent the intensity of the image obtained when both pump and probe are on, emission when only pump is on, monitor when only probe is on, and dark when both pump and probe are off in order to subtract the contribution of the stray light. Each of these four images consisted of N integrated camera images. Measurements are performed in sequence: data, emission, monitor, dark and this sequence is repeated M times. In order to obtain a satisfactory signal-to-noise ratio, N = 500 and M = 5–10.
DFT studies
The attained optimized structures for LG8–LG10 were stable and confirmed by means of genuine global minimum energy, which was carried out by Density Functional Theory (DFT) and B3LYP hybrid functional.42 The 6-31G(d,p) basis set43 was used as an input for further calculations. All the calculations were carried out using the Gaussian 09 package on a personal computer.44 Energy-minimized structures and frontier molecular orbitals (FMOs) were executed ground state properties processed by density functional theory (DFT) in the gas phase. The excited state properties, such as the percentage of molecular contribution, oscillatory strength, and singlet transition energy, were investigated by employing the time-dependent density functional theory (TD-DFT) in THF. The optimized geometries were used to determine the frontier molecular orbitals (FMOs) and subjected to single-point TD-DFT studies (first 20 vertical singlet–singlet transitions) to obtain the UV–vis spectra of these dyes. M06-2x function and the solvation of the dyes in tetrahydrofuran were described by the integral equation formalism polarizable continuum model (PCM)45,46 within the self-consistent reaction field (SCRF) theory in the TD-DFT calculations. To interpret the nature of electron transitions, foremost portions of the absorption spectra were simulated by GaussSum 2.2.5 software.47,48 The percentage contribution of individual moieties present in the dyes was calculated using their respective molecular orbitals.
3. Results and discussion
Synthesis
Scheme 2 illustrates the synthesis of new porphyrin dyes LG8–LG10 using Sonogashira coupling reactions. The precursors of three sensitizers, i.e., TIPS derivatives (3) were synthesized from compound 1 and 2 in the presence of the Pd(PPh3)4 catalyst. Subsequently, deprotection of TIPS-acetylene of 3 treated with bromoaromatic acids (a–c) using AsPh3 and Pd2(dba)3 followed by column chromatography results in the LG8–LG10 sensitizers. All these sensitizers are characterized by spectroscopic techniques such as 1H NMR, MALDI-TOF-MS and FT-IR. Furthermore, optical and electrochemical properties of LG8–LG10 were recorded using UV-visible absorption, transient absorption, fluorescence and cyclic voltammetry. These results suggest that the modification of the acceptor unit in D–π–A porphyrin sensitizers affects energy levels of D–A linear π-conjugated systems and promote energy transfer from fluoranthene-linked porphyrin to auxiliary acceptor BTD, thus improving the solar-to-electrical power conversion efficiency. Preliminary characterizations of all three sensitizers were carried out by elemental analysis, MALDI-MS, and 1H NMR and FT-IR spectroscopies (see the Experimental section and Fig. S1–S10, ESI†).
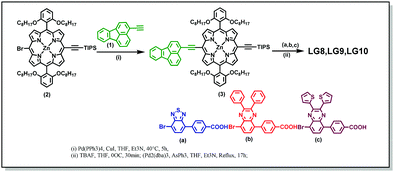 |
| | Scheme 2 Synthetic route for the LG8–LG10 dyes. | |
Optical properties
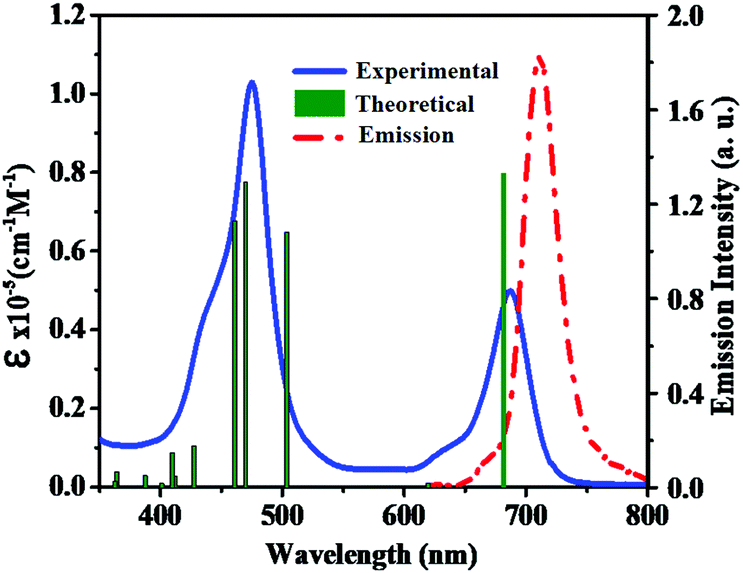
To examine the ground-state electronic properties of fluoranthene and BTD, DPQ, and DTQ containing linear D–π–A porphyrin sensitizers LG8–LG10, UV–visible optical absorption spectra are recorded in tetrahydrofuran (THF) at a concentration of 1 × 10−5 M at 25 °C. The two absorption bands at 400–500 nm and 600–750 nm of the LG8 are attributed to the Soret and the Q-band, respectively (Fig. 1), whereas LG9 and LG10 exhibit similar spectral features (Fig. S11, ESI†). However, the Q-band of LG8 at 687 nm showed a significant red-shift when compared to LG9 (643 nm) and LG10 (683 nm). It reveals that the incorporation of the benzo[c][1,2,5]thiadiazole (BTD) moiety in auxiliary position influenced the ground state vibrational transitions responsible for this bathochromic shift in LG8 absorption maxima.23 Subsequently, calculated absorption maxima (λmax) and molar extinction coefficient (ε) of three porphyrin sensitizers are included to support the changes in experimental absorption data via modifications of auxiliary acceptors in fluoranthene appended D–π–A porphyrin sensitizers as shown in Table 1. In addition, the heteroaromatic units in the D–π–A systems are bathochromically shifted, which indicates that the excited state is more polar than the ground state such as BTD and thiophene units as π-spacers.49
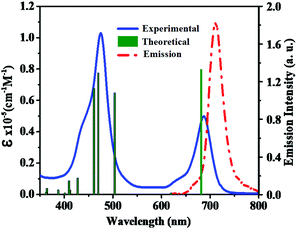 |
| | Fig. 1 Absorption (left, solid line) and emission (right, dashed line) spectra of porphyrin sensitizer LG8 in the THF solvent. Simulated absorption bands are shown as vertical bars. | |
Table 1 Photophysical and electrochemical data of LG8–LG10 dyes
| Sensitizer |
Absorption λmax (nm), logε, (M−1 cm−1)a |
Emission λem, nm, (ϕ)b |
τ, ns (A%)c |
E
0–0
(eV) |
E
OX (V)/NHEe |
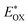
(V) |
|
λ
ex = 357 nm |
λ
ex = 470 nm |
Solvent: THF, error limits: λmax, ±1 nm, ε, ±10%.
Error limits: λem, ±1 nm, ϕ, ±10%.
Error limits: τ ∼ 10%.
E
0–0 was determined from the intersection of absorption and emission spectra, error limit ±10%.
Solvent: THF, error limits: EOX, ±0.03 V, 0.1 M TBAP.
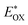 was determined as EOX − E0–0. was determined as EOX − E0–0.
|
|
LG8
|
475 (5.43) |
636 (4.38) |
687 (5.06) |
45855 (0.01) |
70256 (0.25) |
1.20 (100) |
1.80 |
1.05 |
−0.75 |
|
LG9
|
464 (5.54) |
578 (4.39) |
643 (4.90) |
459 (0.02) |
660 (0.19) |
1.52 (100) |
1.90 |
0.95 |
−0.95 |
|
LG10
|
473 (5.30) |
645 (4.42) |
685 (4.78) |
458 (0.01) |
698 (0.24) |
1.48 (100) |
1.78 |
1.03 |
−0.75 |
Emission studies in solution
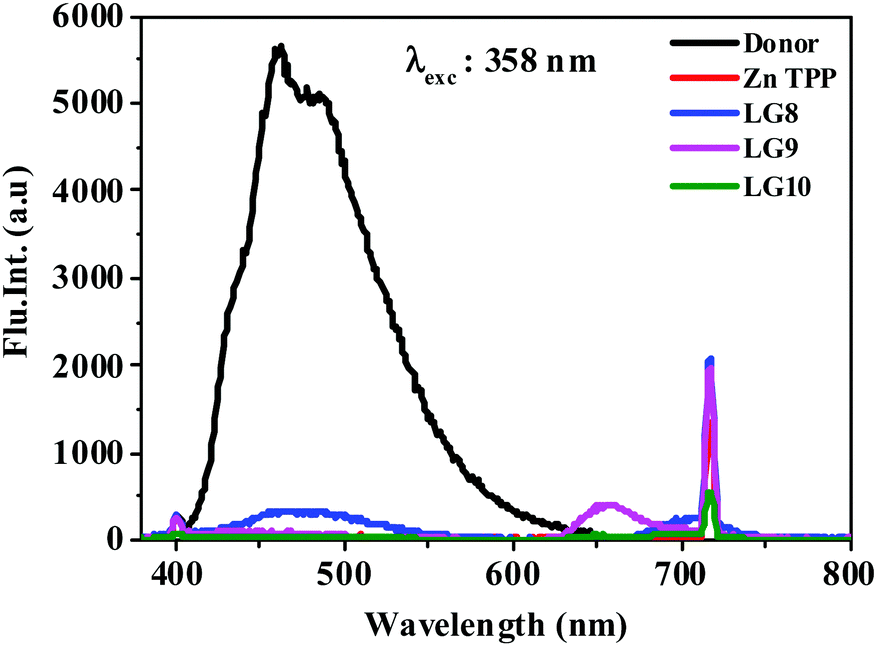
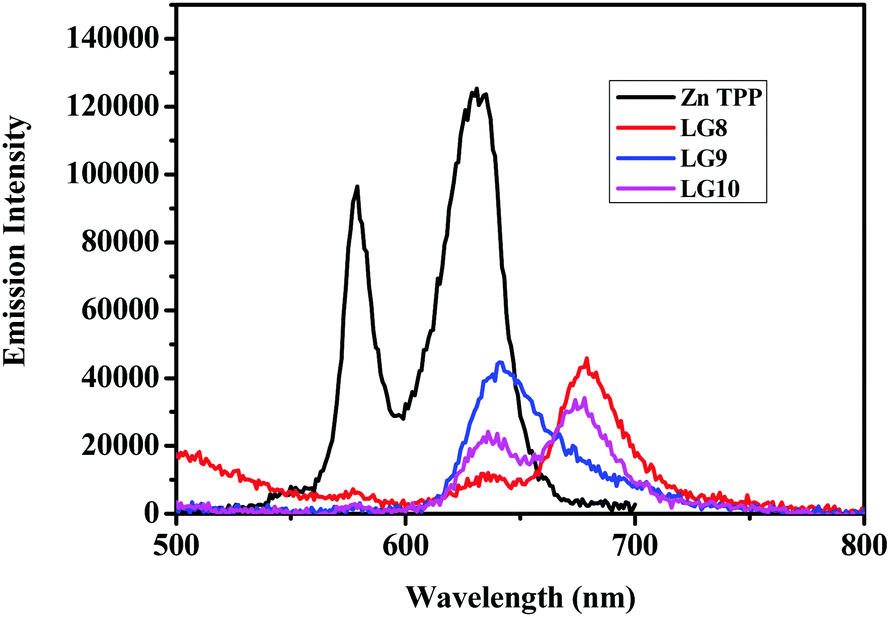
The photo-induced intramolecular excited state energy/electron transfer process between the donor and the acceptor in D–π–A porphyrin sensitizers LG8–LG10 is characterized by steady-state emission studies measured in non-coordinating solvents such as DCM and O-DCB and coordinating solvent THF, at λexc = 358 nm, where donor fluoranthene absorbs predominantly and is compared with the emission of the individual constituent 3-ethynylfluoranthene (FLU). The emission intensity of donor fluoranthene is quenched relative to pristine compound 3-ethynylfluoranthene at room-temperature. Interestingly, the emission of fluoranthene overlaps with the absorption of the porphyrin π-spacer in D–π–A sensitizers as shown in Fig. 1 and 2. In addition, the appearence of porphyrin emission at ca. 700 nm suggests that the quenching of FLU emission intensity is due to intramolecular excitational energy transfer from the singlet state of FLU to porphyrin. Furthermore, the excitation spectrum of the three sensitizers at the porphyrin emission maximum of 680 nm showed characteristic fluoranthene absorption bands, suggesting the evidence of intramolecular excited energy transfer in D–π–A sensitizers. In addition, upon excitation at ca. 470 nm, where the porphyrin π-spacer absorbs predominantly and respective emission maxima with quantum yields are presented in Table 1. The emission maxima of all three sensitizers are bathochromically shifted when compared to their pristine compound zinc tetraphenyl porphyrin (ZnTPP) (Fig. 3). From Fig. 2 and Table 1, the results suggest that the emission quenching of the donor fluoranthene is more pronounced in LG8 sensitizers compared to the two other sensitizers. This is further supported by the quantum yield data presented in Table 1. As a result, one can expect higher power conversion efficiency for the LG8 sensitizer.
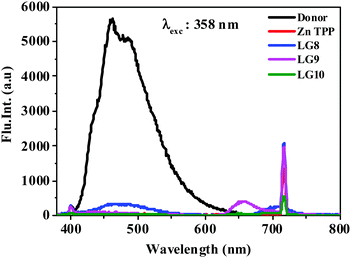 |
| | Fig. 2 Emission spectra of equi-absorbing solutions of donor, ZnTPP, LG8–LG10 in the THF solvent at 358 nm. | |
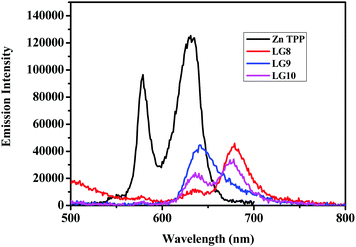 |
| | Fig. 3 Emission spectra of equi-absorbing solutions of ZnTPP and LG8–LG10 dyes in THF solvent. | |
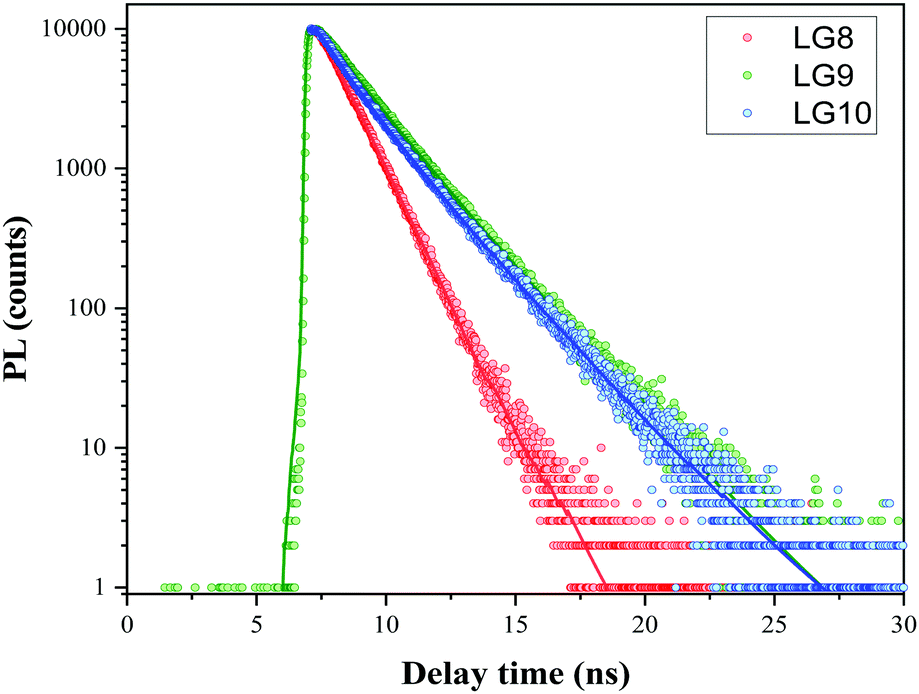
Time-Correlated Single Photon Counting (TCSPC) experiments were carried out in solution to determine the dynamics of the excited-state in solution for LG8–LG10 (Fig. 4). Regardless of the dye, the photoluminescence (PL) decay shows a single component feature. The excited states lifetime is faster for LG8 (τ = 1.20 ns) bearing the benzothiadazole compared to the quinoxaline-based LG9 (τ = 1.52 ns) and LG10 (τ = 1.48 ns), and for the latter, the excited states lifetime is very similar. We have determined singlet state energies (bandgap, E0–0) from the absorption and emission spectra, in the range of 1.78–1.90 eV. This is comparatively lower than the Zn Por counterpart (2.05 eV). Finally, we recorded absorption spectra of all three sensitizers when anchored onto a 6 μm thick TiO2 layer.
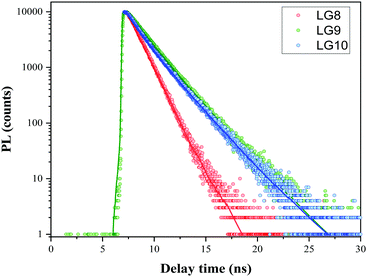 |
| | Fig. 4 Comparison of PL decay using the TCSPC technique at 480 nm excitation wavelength LG8–LG10 dyes in the tert-butanol:acetonitrile mixture including 2 mM CDCA (solution O.D. = 0.5). Fit after reconvolution from the IRF is represented by plain lines. | |
The absorption spectra of all the three sensitizers are broadened with a red-shift in the maxima. This is due to molecular aggregation in the self-assembled monolayer and the electronic coupling of the dye anchoring group with the TiO2 nanoparticles, respectively.
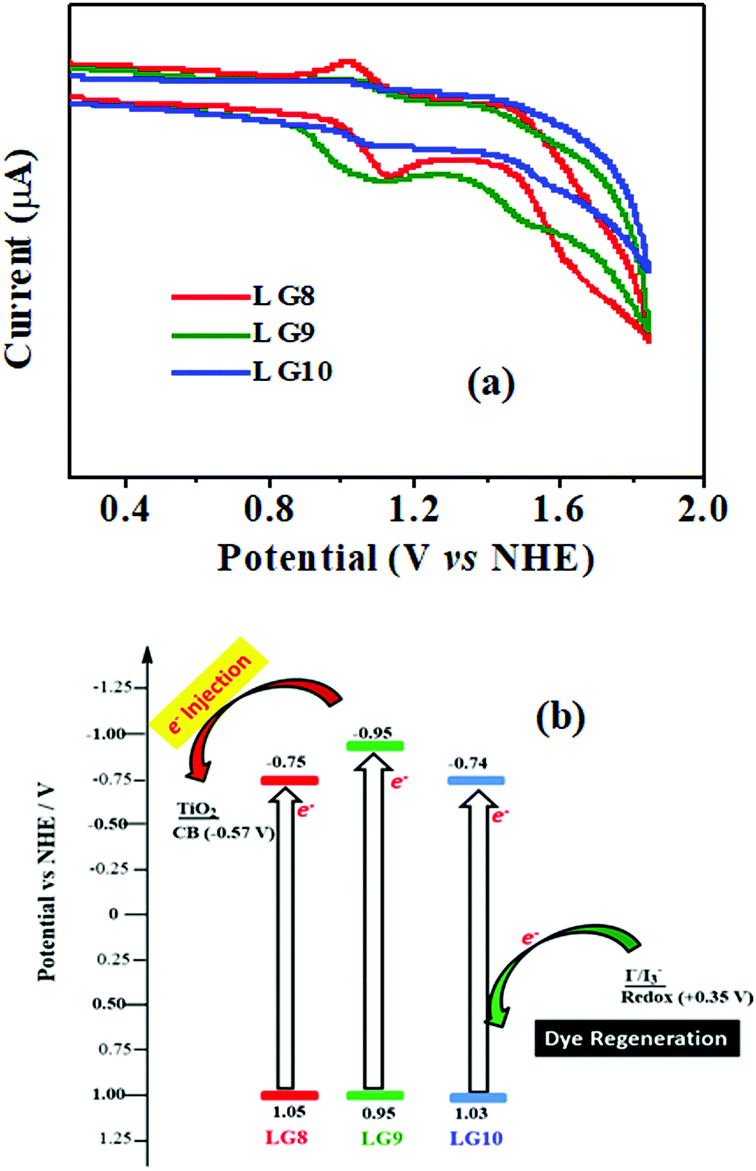
Electrochemical studies
Cyclic voltammetry is employed for the electrochemical analysis of the LG8–LG10 dyes as shown in Fig. 5a and the corresponding data are reported in Table 1. The ground state oxidation potentials (EOX) of LG8–LG10 are found at +1.05 V, +0.95 V and +1.03 V (vs. NHE), respectively. The excited state oxidation potential 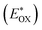 of LG8–LG10 is obtained from the following expression
of LG8–LG10 is obtained from the following expression 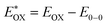 . The resulting values are −0.75 V, −0.95 V and −0.75 V (vs. NHE), respectively. However, LG8 and LG10 showed a decrease in energy gap compared to LG9, which leads to a red-shifted absorption tail and the relatively positive
. The resulting values are −0.75 V, −0.95 V and −0.75 V (vs. NHE), respectively. However, LG8 and LG10 showed a decrease in energy gap compared to LG9, which leads to a red-shifted absorption tail and the relatively positive 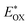 level suggests that BTD and DTQ moieties play a crucial role in LG8 and LG10. On the other hand, the excited state oxidation potential of LG8–LG10 sensitizers showed more reductive values than the conduction band edge of TiO2 (−0.57 V vs. NHE)50 and the ground state oxidation potential is more positive than the redox potential of I3−/I− (+0.35 V vs. NHE), which will afford an efficient electron injection and dye regeneration in the complete devices (Fig. 5b).
level suggests that BTD and DTQ moieties play a crucial role in LG8 and LG10. On the other hand, the excited state oxidation potential of LG8–LG10 sensitizers showed more reductive values than the conduction band edge of TiO2 (−0.57 V vs. NHE)50 and the ground state oxidation potential is more positive than the redox potential of I3−/I− (+0.35 V vs. NHE), which will afford an efficient electron injection and dye regeneration in the complete devices (Fig. 5b).
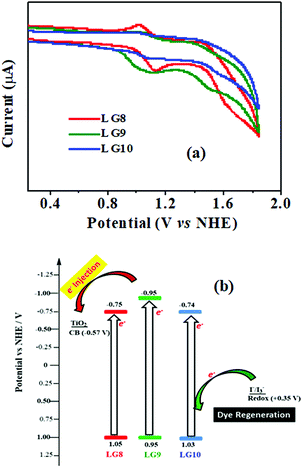 |
| | Fig. 5 (a) Cyclic voltammograms of LG8–10 dyes in THF (0.1 M TBAPF6, a glassy carbon electrode as the working electrode, Pt as the counter electrode and a saturated calomel electrode as the reference electrode were used, scan rate: 100 mV s−1). (b) An energy-level diagram of the LG8–LG10 porphyrins, redox potential from I3−/I− redox couple and TiO2 conduction band edge; E(HOMO) = Eoxd and E(LUMO) = E(HOMO) − E0–0. | |
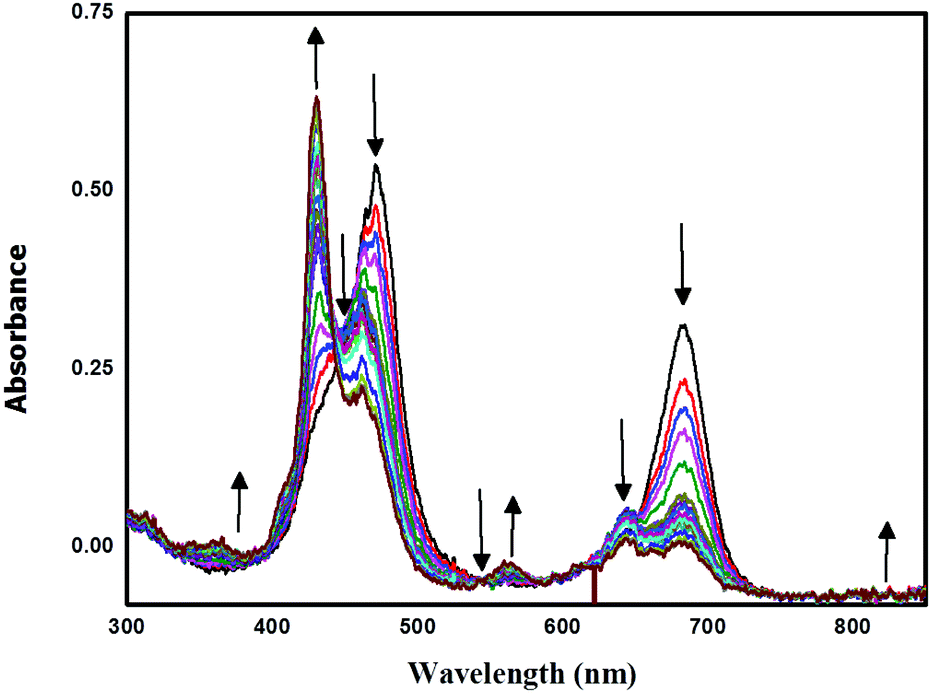
Spectro-electrochemical studies
Spectro-electrochemical studies of LG8–LG10 were carried out at a controlled oxidation potential of +1.10 V vs. NHE to determine the optical fingerprint of the oxidized dye and its stability. From the evolution of the UV-vis absorption spectra of LG8 (Fig. 6), upon oxidation, a decrease in the intensity of the absorption bands is observed at 475 nm and 687 nm, corresponding to the Soret and Q bands, respectively. This evolution is concomitant with the formation of a new band at 430 nm attributed to the dye radical cation. Three isobestic points at 444 nm, 543 nm and 735 nm were observed during this process. Likewise, LG9 and LG10 porphyrin sensitizers exhibit the same spectral features as observed in LG8 (Fig. S9, ESI†). Therefore, a higher stability of the oxidized species of LG8–LG10 porphyrins than in Ru-sensitizer N719 under similar conditions helps to improve the performance of solar cells.51
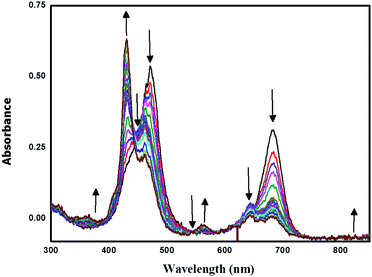 |
| | Fig. 6 Oxidative OTTLE studies of LG8 in 0.3 M TBAP/THF with an applied potential of +1.10 V (vs. NHE/KCl). | |
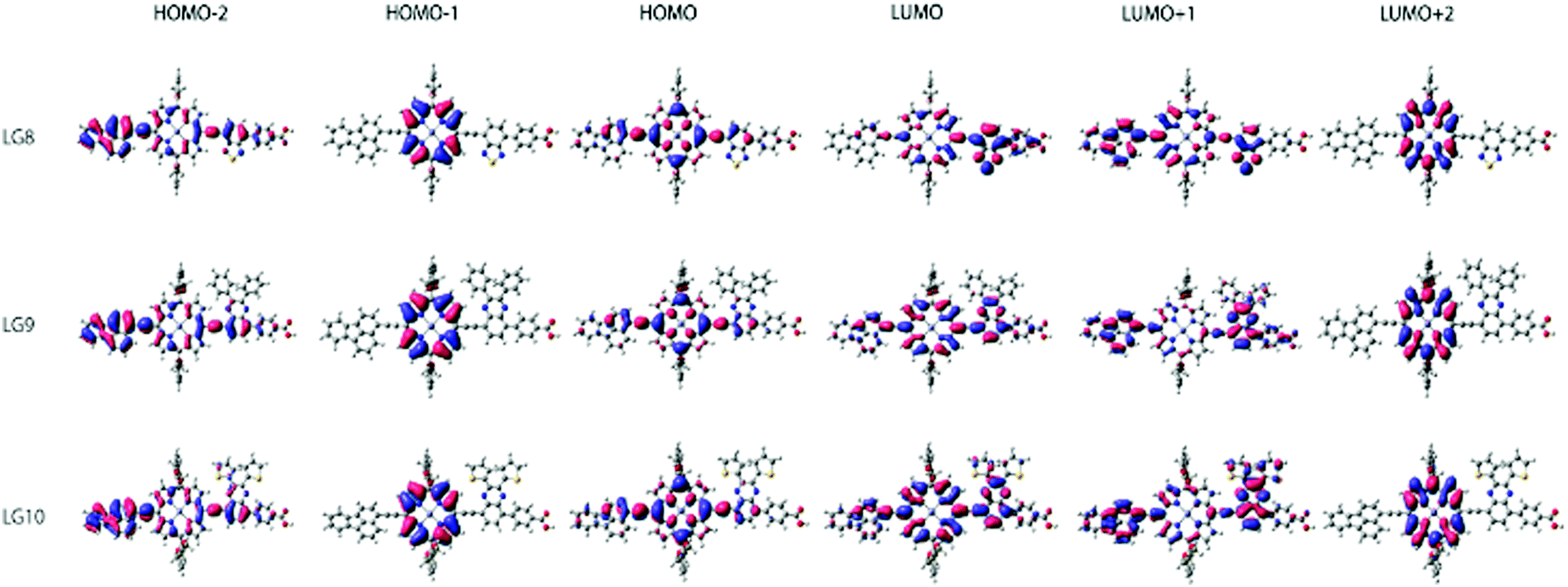
Theoretical studies
Theoretical DFT and TD-DFT calculations with the functional basis set of B3LYP/6-31G(d,p) were performed to find the match between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of LG8–LG10 porphyrin sensitizers with the TiO2 nanoparticles and liquid electrolytes to avoid the charge recombination process in DSSCs. The ground state optimized structures of LG8–LG10 (Fig. S15, ESI†) suggest that functionalization with auxiliary groups (BDT, DPQ, DTQ) between the porphyrin ring and phenyl carboxylic acid reduces the aggregation on the TiO2 surface and improves the overall performance of the device when compared to previous reported dyes.36,37 The optimized structure of LG8–LG10 showed that HOMO levels are delocalized on both the ethynylfluoranthrene and the porphyrin part while the LUMO level is located on the auxiliary acceptor unit and the phenyl carboxylic acid anchoring group to afford the intramolecular donor–acceptor energy transfer mechanism. Subsequently, HOMO−1 exists on porphyrin and LUMO+1 on the donor and partially on the porphyrin ring and auxiliary groups while exhibiting an efficient energy transfer, stable oxidation potentials and suitable HOMO–LUMO levels. LG8–LG10 based DSSCs are prepared according to the experimental section and following previously reported literature.51–53 HOMO−2 and LUMO+2 electrons are present on the donor and the porphyrin core, respectively (Fig. 7). Later, excited-state transitions of LG8–LG10 were calculated using the polarizable continuum model (PCM) in THF with the M06-2X function using the B3LYP/6-31G(d,p) basis set. The resulting calculated vertical excitation energies for singlet and oscillator strengths match with the experimental data. Therefore, the incorporation of electron-deficient auxiliary acceptor units adjacent to the phenyl group aids to delocalize the LUMO orbitals of the sensitizer and promote electron transfer from the porphyrin moiety to the acceptor unit, thus facilitating light harvesting and charge separation to achieve the long-lived excited state. As a whole, the electron distribution of the LUMO of LG8 is more concentrated on acceptors or anchoring groups and one can expect better device efficiency, when compared to LG9 and LG10 sensitizers.
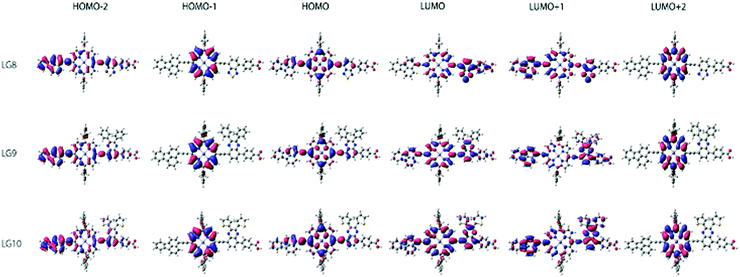 |
| | Fig. 7 Isodensity (0.02) plots of FMOs by using the B3LYP method 6-31G(d,p). | |
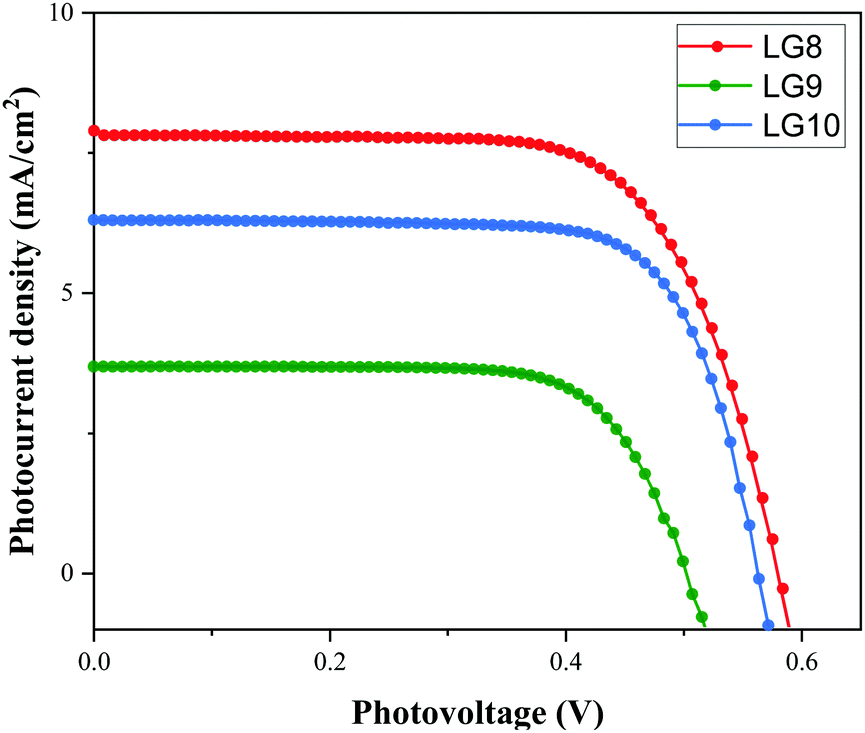
Device studies
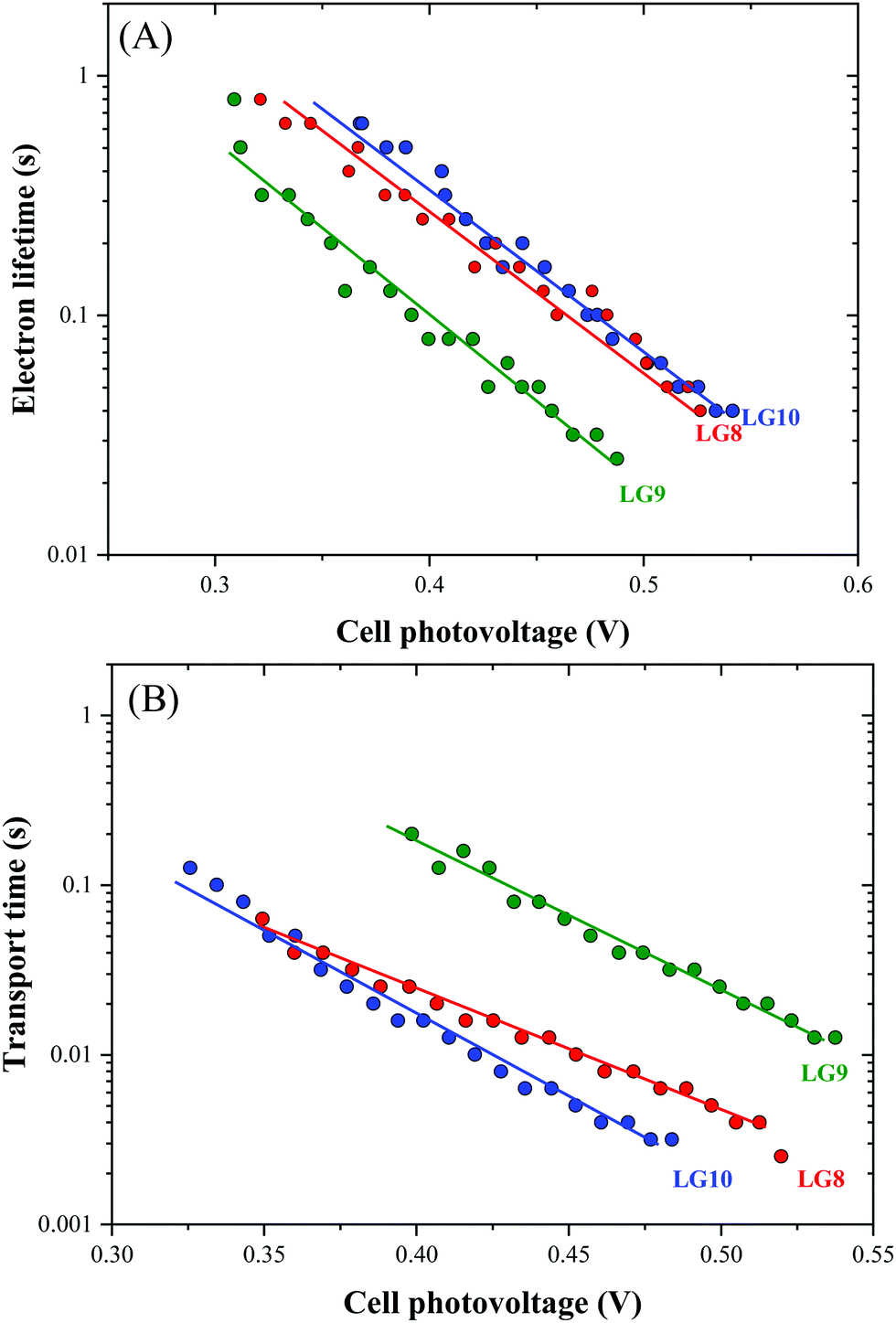
Optical, electrochemical and theoretical studies suggested that fluoranthene appended porphyrin sensitizers in LG8–LG10.Fig. 8 shows the evolution of current density–voltage (J–V) characteristics of the devices under standard A.M.1.5G irradiation conditions (100 mW cm−2). LG8 and LG10 show the highest PCE value of 3.11% and 2.86%, respectively (Table 2). LG9 exhibits a PCE value of only 1.50% under the same conditions. The lower efficiency of LG9 is attributed to the 2,3-diphenylquinoxaline (DPQ) as the auxiliary group compared to benzothiadiazole (BTD) and 2,3-di(thiophen-2-yl)quinoxaline (DTQ) in LG8 and LG10. The LUMO is more destabilized in the LG9 dye and is almost similar for both LG8 and LG10 dyes. The photovoltaic characteristics are for LG8 (JSC = 7.8 mA cm−2, VOC = 0.58 V, FF = 68.3%), LG9 (JSC = 3.53 mA cm−2, VOC = 0.51 V, FF = 72.0%) and LG10 (JSC = 6.28 mA cm−2, VOC = 0.56 V, FF = 73.0%). Thus, the incorporation of auxiliary acceptors like BTD and heteroaromatic substituents on quinoxaline moieties affords to improve the dye performances compared to phenyl groups on the quinoxaline unit. IMVS/IMPS analysis shows that changing the acceptor unit of the new porphyrin dyes affects both the non-geminate recombination (i.e. interfacial electron transfer with the redox mediator) and the electron transport time (Fig. 9). While benzothiadiazole and 2,3-di-thiophen-2-yl quinoxaline are providing very similar properties, the introduction of 2,3 diphenylquinoxaline in LG9 dyes is accelerated by ca. two times the non-geminate recombination and is affected by a similar order of the electron transport, signifying that charge collection efficiency in the LG9 dye is lower than in its LG8 and LG10 counterparts. This provides a first explanation on why LG9 suffers from lower photovoltage and photocurrent density in full devices. These results stress that the shorter excited state lifetime of the LG8 dye does not compromise the device performance.
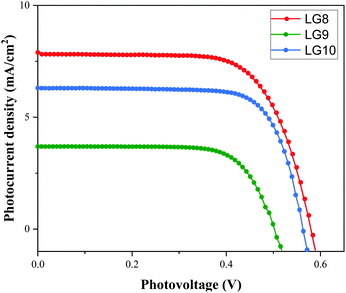 |
| | Fig. 8 Current–voltage characteristics of LG8–LG10 sensitizers under A.M.1.5G conditions. | |
Table 2 Photovoltaic performance parameters of LG8–LG10 sensitizers
| Dye |
TBP (mM) |
J
SC
(mA cm−2) |
V
OC (V) |
FFa |
η (%) |
|
Error limits: JSC = ±0.20 mA cm−2, VOC = ±0.30 mV, and FF = ±0.03.
|
|
LG8
|
0.5 M |
7.8 |
0.58 |
68.3 |
3.11 |
|
LG9
|
0.5 M |
3.53 |
0.51 |
72 |
1.50 |
|
LG10
|
0.5 M |
6.28 |
0.56 |
73 |
2.86 |
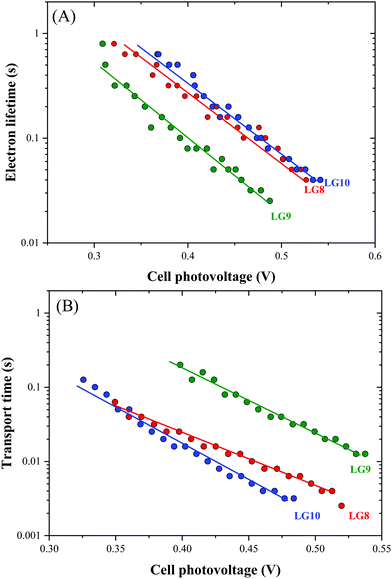 |
| | Fig. 9 Comparison of (A) electron lifetime and (B) transport time determined by IMVS and IMPS, respectively, for LG8–LG10-based DSSCs. | |
Kinetic studies
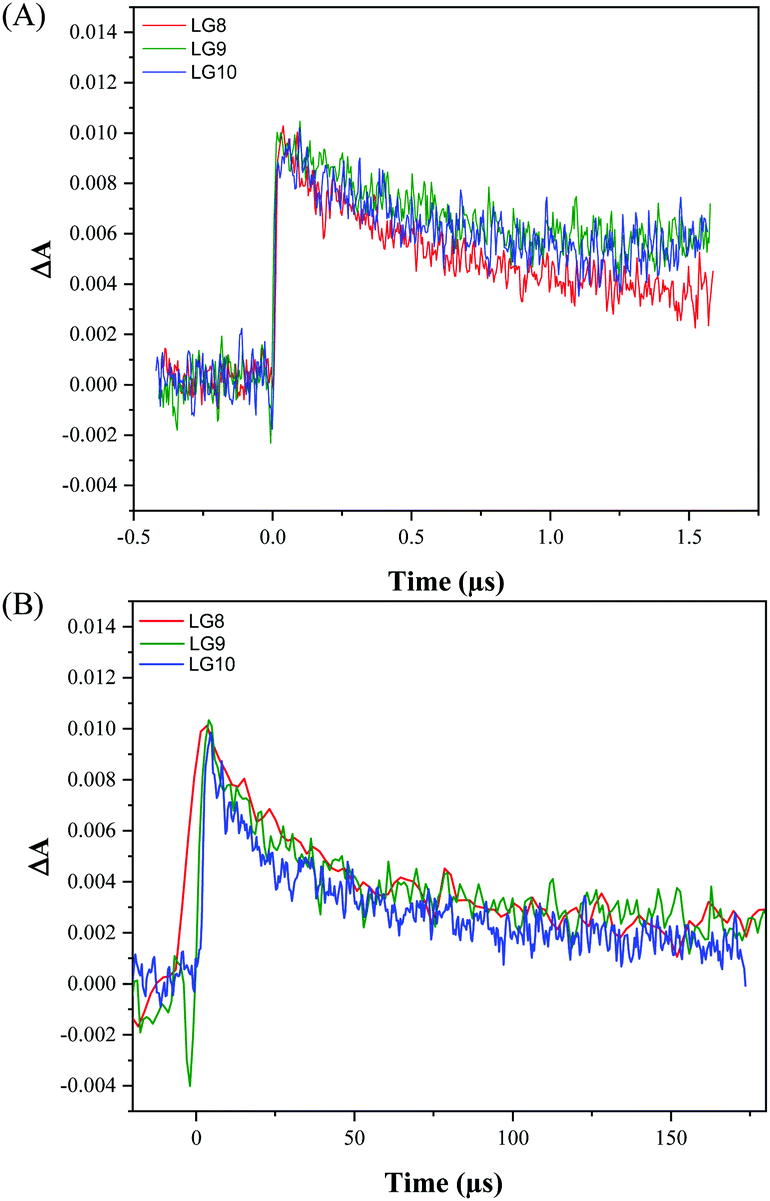
Transient absorption spectroscopy using a Streak camera in the detection of both spectral and temporal resolution was employed to investigate LG8–LG10 dye regeneration and geminate recombination kinetics (Fig. S18, ESI†). The transient dye radical cation absorption spectrum for the different dyes is presented in Fig. S19 (ESI†) and the corresponding kinetics in Fig. 10.
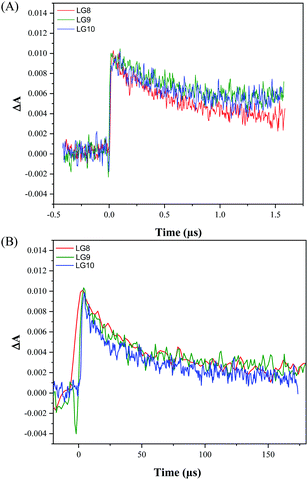 |
| | Fig. 10 Comparison of transient absorption kinetics for LG8–LG10-based DSSCs (A) including the I3−/I− redox mediator in the electrolyte, (B) without redox couple in the electrolyte. The devices were pumped at 430 nm (10 Hz) with an energy of 700 μJ cm−2. | |
Dye radical cations of all dyes show in a broad spectral range two bands, one at 550–620 nm and another at 750–800 nm. This is in good agreement with the spectral evolution upon oxidation obtained by spectro-electrochemistry in TBAP/THF (Fig. 6). The LG8–LG10 dye radical cation formed upon 30 ps pump at 430 nm depopulates within 1.5 μs in a process of dye regeneration where the transfer of an electron from the redox couple regenerates the original dye oxidation state. The unwanted and competitive process to the dye regeneration step is the back electron transfer from TiO2 to the dye radical cation. The lifetime of this geminate recombination process is determined to be 60 μs, which is sufficiently longer than regeneration. Interestingly, both regeneration and non-geminate recombination are independent of the type of acceptor unit. This result combined with TD-DFT calculations confirms that these two kinetic processes are mainly governed by the fluoranthene unit attached to the porphyrin core. Based on these two numbers, one can estimate that the yield of dye regeneration is 97.5%, thus suggesting that dye regeneration is not a limiting step in the device performance.
Finally, we have carried out thermogravimetric analysis of all three new sensitizers to characterize the dye's thermal stability, a characteristic essential for the commercialization of the technology. The thermal behaviour of the three sensitizers suggests that these dyes are stable up to 200 °C and the initial weight loss in between 200 °C and 250 °C is due to decarboxylation of the cyanoacrylic group (Fig. S21, ESI†).54 Therefore, the results revealed that LG8–LG10 based porphyrin sensitizers are highly thermally stable and the values associated with them are close to the corresponding values of the previously reported porphyrin sensitizers.
4. Conclusions
In summary, fluoranthene D–π–A based porphyrin sensitizers alter the photovoltaic performance upon varying the acceptor moieties such as benzothiadiazole (BTD), 2,3-diphenylquinoxaline (DPQ) and 2,3-di(thiophen-2-yl)quinoxaline for LG8, LG9 and LG10, respectively. Optical properties suggest that the presence of the BDT acceptor group in the LG8 sensitizer shifts the absorption maxima towards the red region. Emission, electrochemical and theoretical studies indicate an efficient intramolecular excited energy transfer mechanism and the energy transfer efficiency is higher in LG8 than in LG9 and LG10 sensitizers. Spectro-electrochemical studies revealed that the ease of oxidation in porphyrins to form radical cations occurs at an applied potential of +1.10 V vs. NHE in LG8. Furthermore, the DFT calculations supported the energy transfer mechanism while HOMO levels localized on porphyrin and fluoranthene while LUMO levels localized on the acceptor moieties. Device performance of LG8 exhibits a PCE of 3.11%, which suggests that the novel design of the diverse electron and acceptor moieties appended D–π–A planar system may find applications not only in DSSCs but also in broader perspectives for organic electronics.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Department of Science & Technology (DST), under DST-CERI (DST/TM/CERI/C228 & DST/TMD/CERI D46) for the financial support to carry out this work. JVSK acknowledges the UGC for research fellowships. I. D. and F. S. thank the Conseil Régional des Hauts-de-France for the financial research fellowship support of I. D. We thank the CSIR-IICT director for the support (IICT/Pubs./2021/206).
Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- C. Dragonetti and A. Colombo, Molecules, 2021, 26, 2461 CrossRef CAS PubMed.
- M. Kokkonen, P. Talebi, J. Zhou, S. Asgari, S. A. Soomro, F. Elsehrawy, J. Halme, S. Ahmad, A. Hagfeldt and S. G. Hashmi, J. Mater. Chem. A, 2021, 9, 10527–10545 RSC.
- K. S. Srivishnu, S. Prasanthkumar and L. Giribabu, Mater. Adv., 2021, 2, 1229–1247 RSC.
- A. Abate, M. Planells, D. J. Hollman, S. D. Stranks, A. Petrozza, A. R. S. Kanada, Y. Vaynzof, S. K. Pathak, N. Robertzon and H. J. Snaith, Adv. Energy Mater., 2014, 4, 1400116 CrossRef.
- M.-E. Ragoussi, M. Ince and T. Torres, Eur. J. Org. Chem., 2013, 6475–6489 CrossRef CAS.
- L. Giribabu and R. K. Kanaparthi, Curr. Sci., 2013, 104, 847855 Search PubMed.
- A. Hagfeld, G. Boschlo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- V. K. Singh, R. K. Kanaparthi and L. Giribabu, RSC Adv., 2014, 4, 6970–6984 RSC.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- F. Gao, Y. Wang, D. Shi, J. Zhang, M. Wang, X. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720–10728 CrossRef CAS PubMed.
- C. Chen, M. Wang, J. Li, N. Pootrakulchote, L. Alibabaei, C. Ngoc-le, J. Decoppet, J. Tsai, C. Grätzel, C. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS PubMed.
- P. S. Gangadhar, A. Jagadeesh, A. S. George, G. Reddy, S. Prasanthkumar, S. Soman and L. Giribabu, Mol. Syst. Des. Eng., 2021, 6, 779–789 RSC.
- M. V. Vinayak, T. M. Lakshmykanth, M. Yoosuf, S. Soman and K. R. Gopidas, Sol. Energy, 2016, 124, 227–241 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J.-I. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- L. Giribabu, R. K. Kanaparthi and V. Velkannan, Chem. Rec., 2012, 12, 306–328 CrossRef CAS PubMed.
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 CrossRef CAS PubMed.
- Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224–7230 CrossRef CAS.
- S. Ferrere and B. A. Greg, New J. Chem., 2002, 26, 1155 RSC.
- T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036–3037 RSC.
- K. Devulapally, G. Reddy, S. Prasanthkumar, A. Jagadeesh, S. Soman and L. Giribabu, J. Porphyrins Phthalocyanines, 2021, 25, 407–417 CrossRef CAS.
- J. V. S. Krishna, D. Koteshwar, T. H. Chowdhury, S. P. Singh, I. Bedja, A. Islam and L. Giribabu, J. Mater. Chem. C, 2019, 7, 13594–13605 RSC.
- V. Nikalou, A. Charisiadis, S. Chalkiadaki, I. Alexandropolous, S. C. Pradhan, S. Soman, M. K. Panda and A. G. Coustolelos, Polyhedron, 2018, 140, 9–18 CrossRef.
- V. K. Narra, H. Ullah, V. K. Singh, L. Giribabu, S. Senthilarasu, S. Z. Karazhanov, A. A. Tahir, T. K. Mallick and H. M. Upadhyaya, Polyhedron, 2015, 100, 313–320 CrossRef CAS.
- K. Zheng, Z. Tong, L. Ma, W.-H. Zhu, W. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1617–1657 RSC.
- T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- S. Cherian and C. C. Wamser, J. Phys. Chem. B, 2000, 104, 3624–3629 CrossRef CAS.
- L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang and M. Gratzel, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS.
- A. Yella, H.-W. Lee, N. Tsao, C. H. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Cur-chod, N. Ashari-Astani, I. Tavernelli, U. Rothlis-berger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- J.-M. Ji, H. Zhou, Y. K. Eom, C. H. Kim and H. K. Kim, Adv. Energy Mater., 2020, 10, 2000124 CrossRef CAS.
- Y. K. Eom, S. H. Kang, I. T. Choi, Y. Yoo, J. Kim and H. K. Kim, J. Mater. Chem. A, 2017, 5, 2297–2308 RSC.
- N. V. Krishna, J. V. S. Krishna, S. P. Singh, L. Giribabu, L. Han, I. Bedja, R. K. Gupta and A. Islam, J. Phys. Chem. C, 2017, 121, 6464–6477 CrossRef CAS.
- N. V. Krishna, J. V. S. Krishna, S. P. Singh, L. Giribabu, A. Islam and I. Bedja, J. Phys. Chem. C, 2017, 121, 25691–25704 CrossRef.
- S. Kumar and S. Patil, J. Phys. Chem. C, 2015, 119, 19297–19304 CrossRef CAS.
- S. Kumar, D. Kumar, Y. Patil and S. Patil, J. Mater. Chem. C, 2015, 4, 193–200 RSC.
- F. Lu, X. Wang, Y. Zhao, G. Yang, J. Zhang, B. Zhang and Y. Feng, J. Power Sources, 2016, 333, 1–9 CrossRef CAS.
- W. Naim, V. Novelli, I. Nikolinakos, N. Barbero, I. Dzeba, F. Grifoni, Y. Ren, T. Alnasser, A. Velardo, R. Boreelli, S. Haacke, S. M. Zakeeruddin, M. Grätzel, C. Barolo and F. Sauvage, J. Am. Chem. Soc., 2021, 1, 409–426 CAS.
- D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- G. A. Peterson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. Robb, A. J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain and O. Farkas, et al., Gaussian 09, Gaussian, Inc., Pittsburgh PA, 2009 Search PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117–129 CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- N. M. O’Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
-
R. Dennington, T. Keith and J. Millam, GaussView, version 5, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
-
L. R. Milgrom, The Colours of Life: an Introduction to the Chemistry of Porphyrins and Related Compounds, Oxford University Press, New York, 1997 Search PubMed.
- A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- L. Giribabu, N. Duvva, S. Prasanthkumar, S. P. Singh, L. Han, I. Bedja, R. K. Gupta and A. Islam, Sustainable Energy Fuels, 2017, 1, 345–353 RSC.
- Y.-S. Chen, C. Zeng, Z.-H. Li, W.-B. Wang, X.-S. Wang and B.-W. Zhang, J. Mater. Chem., 2005, 15, 1654–1661 RSC.
- Y. S. Yen, J. S. Ni, T. Y. Lin, W. I. Hung, J. T. Lin and M. C. P. Yeh, Eur. J. Org. Chem., 2015, 7367–7377 CrossRef CAS.
- X. Wei, X. Du, D. Chen and Z. Chen, Thermochim. Acta, 2006, 440, 181–187 CrossRef CAS.
- Quantum yields are calculated with reference to fluoranthene. K.-M. Bark and R. K. Force, Spectrochim. Acta, 1993, 49A, 1605–1611 CrossRef CAS.
- Quantum yields are calculated with reference to Zn Por. L. Giribabu, T. A. Rao and B. G. Maiya, Inorg. Chem., 1999, 38, 4971–4980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00614b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Lingamallu
Giribabu
*c and
Lingamallu
Giribabu
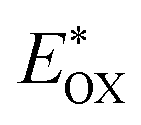 was determined as EOX − E0–0.
was determined as EOX − E0–0.
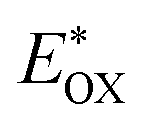
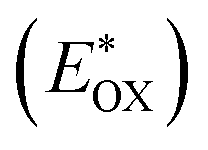 of LG8–LG10 is obtained from the following expression
of LG8–LG10 is obtained from the following expression 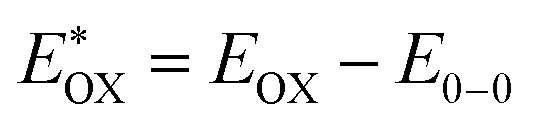 . The resulting values are −0.75 V, −0.95 V and −0.75 V (vs. NHE), respectively. However, LG8 and LG10 showed a decrease in energy gap compared to LG9, which leads to a red-shifted absorption tail and the relatively positive
. The resulting values are −0.75 V, −0.95 V and −0.75 V (vs. NHE), respectively. However, LG8 and LG10 showed a decrease in energy gap compared to LG9, which leads to a red-shifted absorption tail and the relatively positive 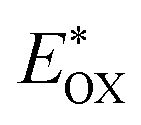 level suggests that BTD and DTQ moieties play a crucial role in LG8 and LG10. On the other hand, the excited state oxidation potential of LG8–LG10 sensitizers showed more reductive values than the conduction band edge of TiO2 (−0.57 V vs. NHE)50 and the ground state oxidation potential is more positive than the redox potential of I3−/I− (+0.35 V vs. NHE), which will afford an efficient electron injection and dye regeneration in the complete devices (Fig. 5b).
level suggests that BTD and DTQ moieties play a crucial role in LG8 and LG10. On the other hand, the excited state oxidation potential of LG8–LG10 sensitizers showed more reductive values than the conduction band edge of TiO2 (−0.57 V vs. NHE)50 and the ground state oxidation potential is more positive than the redox potential of I3−/I− (+0.35 V vs. NHE), which will afford an efficient electron injection and dye regeneration in the complete devices (Fig. 5b).