
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceATP-fuelled self-assembly to regulate chemical reactivity in the time domain†
Maria A.
Cardona
 and
Leonard J.
Prins
*
and
Leonard J.
Prins
*
Department of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: leonard.prins@unipd.it
First published on 18th December 2019
Abstract
Here, we exploit a small biomolecule – ATP – to gain temporal control over chemical reactivity in a synthetic system composed of small self-assembling molecules and reactants. The approach relies on the capacity of ATP to template the formation of amphiphile-based assemblies. The presence of the enzyme alkaline phosphatase causes a gradual decrease in the ATP-concentration in time and, consequently, a spontaneous dissociation of the assemblies. The uptake of apolar reactants in the hydrophobic domain of the assemblies leads to an enhancement of the reaction rate. It is shown that ATP-triggered self-assembly causes the selective upregulation of one out of two hydrazone-bond formation reactions that take place concurrently in the system. This leads to an inversion in the product ratio, which, however, is transient in nature because the upregulated reaction spontaneously reverts to its basal low reaction rate once the ATP has been consumed by the enzyme. Overall, the results demonstrate the potential of chemically-fuelled self-assembly under dissipative conditions to gain temporal control over reactivity in complex chemical systems.
Introduction
The bioregulatory system is mediated by small molecules, which can efficiently up- or downregulate biochemical pathways through allosterism.1 The high selectivity of the interactions between these molecules and their biochemical targets permits control over reactivity in a chemically highly complex mixture. The transient availability of small molecules provides temporal control and ensures that the system returns to a basal state once the trigger has disappeared.2 Only recently, chemists have started to appreciate chemically-fuelled dissipative processes as a tool to actively control the properties of synthetic systems.3–5 Within a short time span experimental studies have already demonstrated that the obtained materials, catalysts, and molecular machinery have unique properties when compared to analogous systems that operate under equilibrium conditions.6–10 One important novelty is that the functional properties of a system can be temporally controlled by regulating the concentration of chemical fuel, just as in nature. This implies that the time domain has now become accessible as a design parameter for the development of functional chemical systems.11–14 Whereas the majority of these studies have been dedicated to the development of materials and a study of their properties, the exploitation of chemically-fuelled driven self-assembly for the purpose of controlling chemical reactivity has received much less attention.15–25 Here, we exploit adenosine triphosphate (ATP)-fuelled self-assembly16,26–29 to control the reaction kinetics in a reaction mixture in which two reactions take place concurrently (Fig. 1). We demonstrate that the addition of ATP causes the selective transient upregulation of one of the two chemical reactions, which translates in a temporary inversion of the product ratio in the system as long as the chemical fuel is present.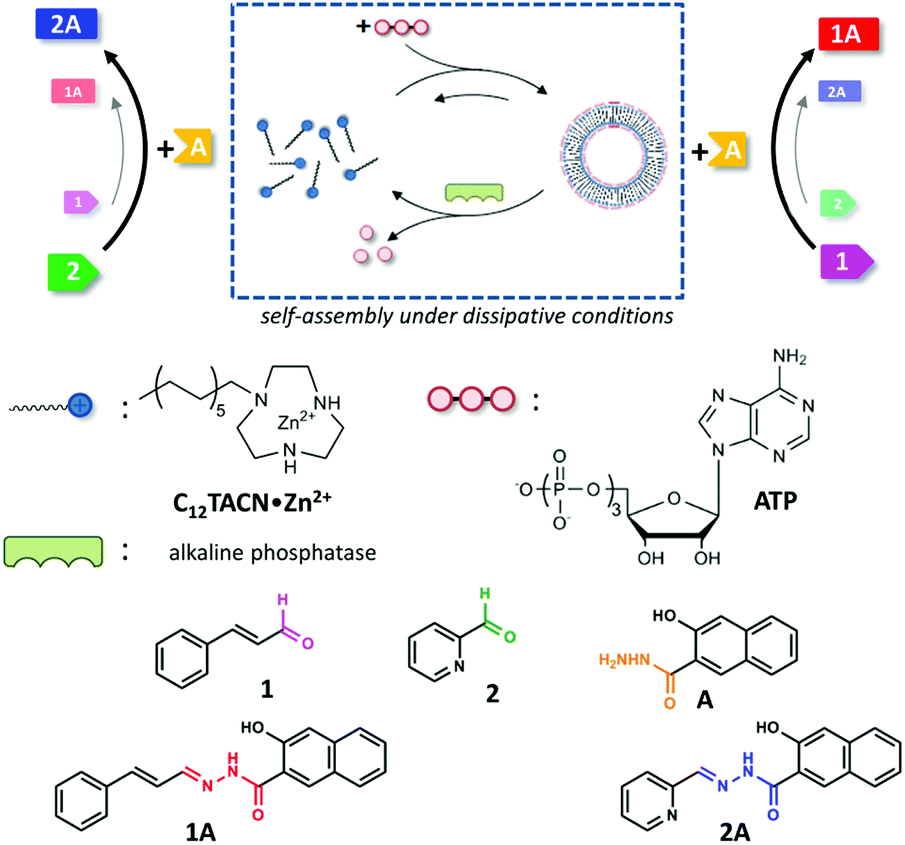 | ||
| Fig. 1 Schematic representation of the ATP-templated assembly of C12TACN·Zn2+ into spherical assemblies. It is noted that throughout the paper we use vesicle-like structures to represent the assembled state. Although the data indicate that the assemblies have a similar nature as those reported before,16 we could not contain conclusive evidence that indeed vesicles are formed. A disassembly pathway is installed in the presence of the ATP-cleaving enzyme alkaline phosphatase. In the unassembled state the reaction between 2 and A occurs at a higher rate compared to the reaction between 1 and A. In the assembled state the situation is reversed and the reaction between 1 and A is the faster one. | ||
Results
Synthesis and characterisation
We synthesized the new amphiphile C12TACN, which contains the same 1,4,7-triazacyclononane (TACN) head group as the amphiphile C16TACN used in previous studies but attached to a shorter apolar chain (C12vs. C16) (Fig. 1).16,30 We expected that the lower intrinsic stability of the resulting assemblies would lead to a larger difference in critical aggregation concentration (CAC) in the presence and absence of templates. This would allow us to work at higher amphiphile-concentrations without having to worry about spontaneous, non-templated self-assembly. This difference was indeed confirmed by self-assembly studies carried out by titrating C12TACN·Zn2+, the Zn2+-complex of C12TACN, to a solution of an apolar fluorophore, such as DPH (1,6-diphenyl-1,3,5-hexatriene) or laurdan dye, in aqueous buffer ([HEPES] = 5 mM, pH = 7.0). These probes are insoluble in the solution, but spontaneously partition in the apolar domain of structures formed by self-assembly of the amphiphiles causing an increase in fluorescence intensity. The onset of fluorescence intensity is thus an indicator for the CAC. For C12TACN·Zn2+ the fluorescence intensity of DPH started to increase slightly only at a concentration of around 5 mM, indicating that the CAC lies around this concentration (Fig. 2a). This marks a strong difference with the CAC of around 100 μM previously determined for C16TACN·Zn2+ (Fig. 2a).16 This result was confirmed by surface tension measurements (Fig. 2a).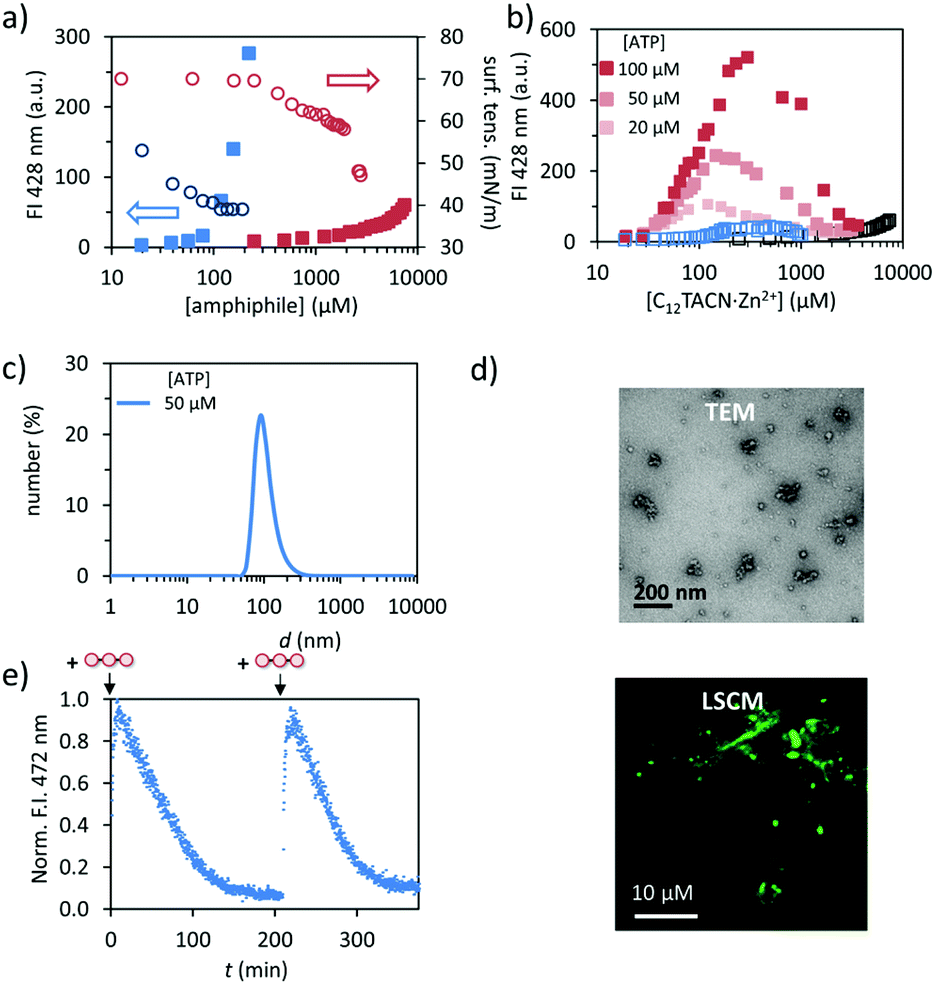 | ||
| Fig. 2 (a) Fluorescence intensity (FI) at 428 nm (filled squares) and surface tension (empty circles) as a function of the concentration of C12TACN·Zn2+ (red) and C16TACN·Zn2+ (blue) in the presence of DPH (2.5 μM). (b) Fluorescence intensity (FI) at 428 nm as a function of the concentration of C12TACN·Zn2+ in the presence of different concentrations of ATP (20, 50, 100 μM). The black trace indicates the fluorescence response of a sample without ATP present, whereas the blue trace indicates the response in the presence of adenosine (200 μM) and Pi (600 μM). (c) Hydrodynamic diameter of assemblies formed in the presence of 50 μM ATP as measured by DLS. (d) TEM and LSCM images of aggregates with [C12TACN·Zn2+] = 100 μM and [ATP] = 50 μM. TEM images are stained with 2% uranyl acetate solution. (e) Fluorescence intensity (FI) at 472 nm following repetitive additions of ATP (50 μM) to a solution of C12TACN·Zn2+ (100 μM) and laurdan dye (2 μM) in the presence of alkaline phosphatase (0.5 U). General experimental conditions: [HEPES] = 5 mM, pH 7.0, T = 25 °C. | ||
We then studied the effect of ATP (adenosine triphosphate) on the CAC of C12TACN·Zn2+ by repeating the titrations in the presence of different amounts of ATP (20–100 μM) (Fig. 2b). In all cases the fluorescence intensity started to increase at a C12TACN·Zn2+-concentration of around 30 μM indicating that ATP templated the formation of assemblies at concentrations well below the CAC. All curves exhibited a maximum which implied that at elevated concentrations of C12TACN·Zn2+ the amount of ATP is not sufficient to stabilize the assemblies. The ATP-templated formation of assemblies with nanometer-sized dimensions was confirmed by dynamic light scattering (DLS), transmission electron microscopy (TEM) and laser scanning confocal microscopy (LSCM) measurements (Fig. 2c and d). The presence of an aqueous inner phase in these structures, as evidenced from the possibility to encapsulate the water-soluble cationic dye rhodamine 6G (Fig. S3, ESI†), suggests that the assemblies have a vesicular structure similar to those observed previously for C16TACN·Zn2+.16,30 Altogether, the data show that ATP templates the self-assembly of C12TACN·Zn2+ in spherical assemblies. Similar to previous studies,16,30 transient assembly formation was observed under dissipative conditions installed by the presence of the enzyme alkaline phosphatase (Fig. 2e). The waste products (adenosine and inorganic phosphate) obtained from enzymatic hydrolysis of ATP are unable to stabilize the assemblies (Fig. 2b) and spontaneous dissociation takes place after enzymatic ATP hydrolysis. Full reversibility of this process was demonstrated by an experiment in which the same solution was fuelled with a second batch of ATP (50 μM) (Fig. 2e).
Transient regulation of chemical reactivity
Previously we have shown that transiently stable vesicular nanoreactors can upregulate a nucleophilic aromatic substitution reaction by concentrating the apolar reactants in the hydrophobic bilayer.16 We were interested in expanding the reaction scope to hydrazone-bond formation, because this reaction is widely applied for the development of molecular receptors, materials and catalysts.31,32 Furthermore, we wanted to demonstrate that a chemical trigger can be used to selectively and transiently upregulate one chemical reaction in a system of higher complexity.After a preliminary screening of a set of hydrazides and aldehydes, we selected aldehydes 1 (trans-cinnamaldehyde) and 2 (2-pyridinecarboxaldehyde) and hydrazide A (3-hydroxy-2-napthoic hydrazide) for a detailed study (Fig. 1 and 3a). The formation of hydrazone 1A from 1 and A could be conveniently detected using UV-vis spectroscopy because the conjugated structure of 1A gives rise to a new absorption band with a maximum at 332 nm in aqueous buffer (Fig. S18, ESI†). The identity of the product and the kinetics of product formation were independently confirmed by LC-MS (Fig. S20–S25†). In aqueous buffer the formation of 1A after mixing 1 (5 μM) and A (20 μM) is slow and just trace amounts of product are observed after several hours (Fig. 3b). Performing the reaction in the presence of just C12TACN·Zn2+ (100 μM) caused a minor 3-fold increase in initial rate from 6.1 × 10−5 to 1.8 × 10−4 μM min−1 (Fig. 3b). On the other hand, when the reagents were added to a solution containing both C12TACN·Zn2+ (100 μM) and ATP (200 μM) the reaction rate proceeded at a 25-fold higher rate (1.5 × 10−3 μM min−1) (Fig. 3b). TEM images confirmed that the presence of the reagents did not affect the assembly process (Fig. S19, ESI†). A direct visualization of the correlation between the formation of ATP-templated assemblies and the increase in reaction rate was obtained from a control experiment in which ATP was added 12 hours after mixing C12TACN·Zn2+ and reagents 1 and A (Fig. 3c). While the reaction proceeded slowly in the absence of ATP, the addition of ATP caused an immediate strong increase in reaction rate. A control experiment in which ATP was added to a solution of the reactants in buffer in the absence of C12TACN·Zn2+ did not result in any rate increase (Fig. S25, ESI†).
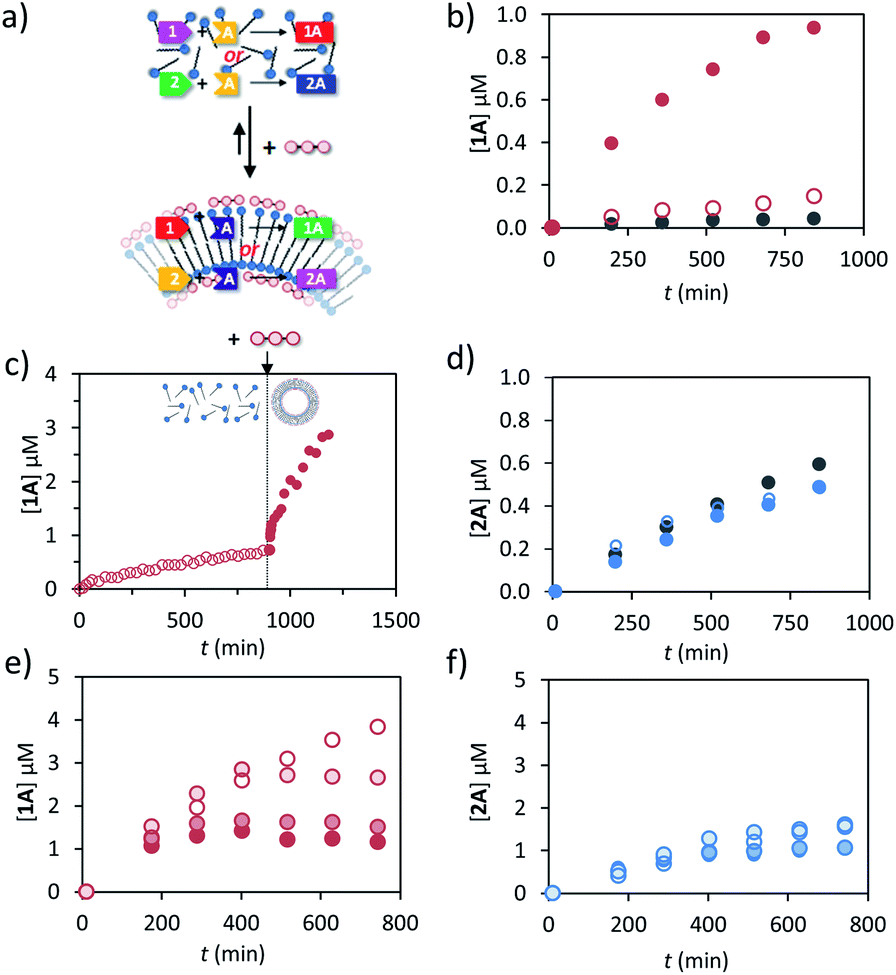 | ||
| Fig. 3 (a) Schematic representation of the reactions between 1, 2 and A to give hydrazones 1A and 2A in the unassembled and assembled state. (b) Concentration of 1A as a function of time as determined by HPLC when 1 (20 μM) and A (5 μM) were added to aqueous buffer at pH 7.0 (grey), a solution of C12TACN·Zn2+ (100 μM) (empty red circles) or a solution of C12TACN·Zn2+ (100 μM) and ATP (200 μM) (filled red circles). (c) Concentration of 1A as a function of time as determined by HPLC when 1 and A (20 μM) were added to a solution of C12TACN·Zn2+ (100 μM) and to which ATP (200 μM) was added after 900 minutes. (d) Concentration of 2A as a function of time when 2 (20 μM) and A (5 μM) were added to aqueous buffer at pH 7.0 (grey), a solution of C12TACN·Zn2+ (100 μM) (empty blue circles) or a solution of C12TACN·Zn2+ (100 μM) and ATP (200 μM) (filled blue circles). (e and f). Concentration of 1A (e)/2A (f) as a function of time when 1 (e)/2 (f) (20 μM) and A (20 μM) were reacted together in the presence of C12TACN·Zn2+ (100 μM), ATP (200 μM) and increasing concentrations of alkaline phosphatase (0–2 U). General experimental conditions: [HEPES] = 5 mM, pH 7.0, T = 25 °C. | ||
On the other hand, the reaction between aldehyde 2 and hydrazide A to form hydrazone 2A was not affected by the presence of either C12TACN·Zn2+ or ATP-templated assemblies (Fig. 3d). In aqueous buffer, the reaction proceeds with a significant rate (7.9 × 10−4 μM min−1), attributed to the higher reactivity of the aldehyde-moiety in 2 compared to 1 caused by the pyridine-moiety.33,34 Similar rates were observed in the presence of either C12TACN·Zn2+ (100 μM) or C12TACN·Zn2+ (100 μM) and ATP (200 μM) (ν0 = 9.9 × 10−4 and 6.4 × 10−4 μM, respectively). The slight decrease in rate observed in the presence of assemblies is tentatively ascribed to an uptake of hydrazide A in the hydrophobic part of the assemblies, which reduces its concentration in the aqueous medium.
The fact that the reaction between 1 and A is mediated by the assemblies creates – from a kinetic point of view –an interesting difference with the same reaction carried out in bulk. Although ATP does not participate in the reaction, its concentration enters in the rate equation because it determines the availability of reactor volume. Indeed, kinetic studies revealed that the reaction rate was of order 0.9 in the concentration of ATP (Fig. S21, ESI†). The order in ATP indicates the efficiency at which ATP creates an apolar reaction environment in the system. Interestingly, it was observed that the addition of ATP under dissipative conditions, i.e. in the presence of enzyme, caused an increase in rate, but only for a limited amount of time depending on the enzyme concentration (Fig. 3e). The final concentrations of 1A (at t = 800 min) were inversely related to the enzyme concentration. This is caused by the reduction in life time of the assemblies upon an increase in enzyme concentration which provides less time for the reactions to occur. Importantly, for the experiments that will be described below, the formation of product 2A was not affected by the presence of enzyme (Fig. 3f). The transientness of ATP-induced upregulation emerged from an experiment in which 3 batches of ATP (50 μM) were repetitively added to a solution of reactants and C12TACN·Zn2+ in the presence of enzyme (0.5 U). For 1 and A each addition of ATP resulted in a rapid increase in rate which gradually levelled off as ATP was hydrolysed, whereas for 1 and B the addition of ATP had no effect on the reaction rate (Fig. S26, ESI†).
Regulation of chemical reactivity in the time domain
The different dependence of the two reactions on the presence of templated assemblies prompted us to investigate reactivity in a system in which both reactions take place simultaneously (Fig. 4a). In line with the observations reported above, in a solution containing all reactants 1, 2, and A and just C12TACN·Zn2+ the reaction producing 2A occurred at a significantly higher rate and after 350 minutes hydrazone 2A was the dominant product in the reaction mixture (Fig. 4b). However, the product distribution in the system changed completely after the addition of ATP (200 μM). The ATP-templated formation of assemblies led to a strong acceleration of the reaction leading to 1A, without significantly affecting the rate of formation of 2A. Consequently, the concentration of 1A started to increase rapidly resulting in an equimolar concentration of hydrazones 1A and 2A at around 300 minutes after addition of ATP. After that time, hydrazone 1A became the dominant product in the reaction mixture. A similar inversion of product composition was observed when C12TACN·Zn2+ was added to a reaction mixture which initially contained the reactants and just ATP (Fig. S27, ESI†).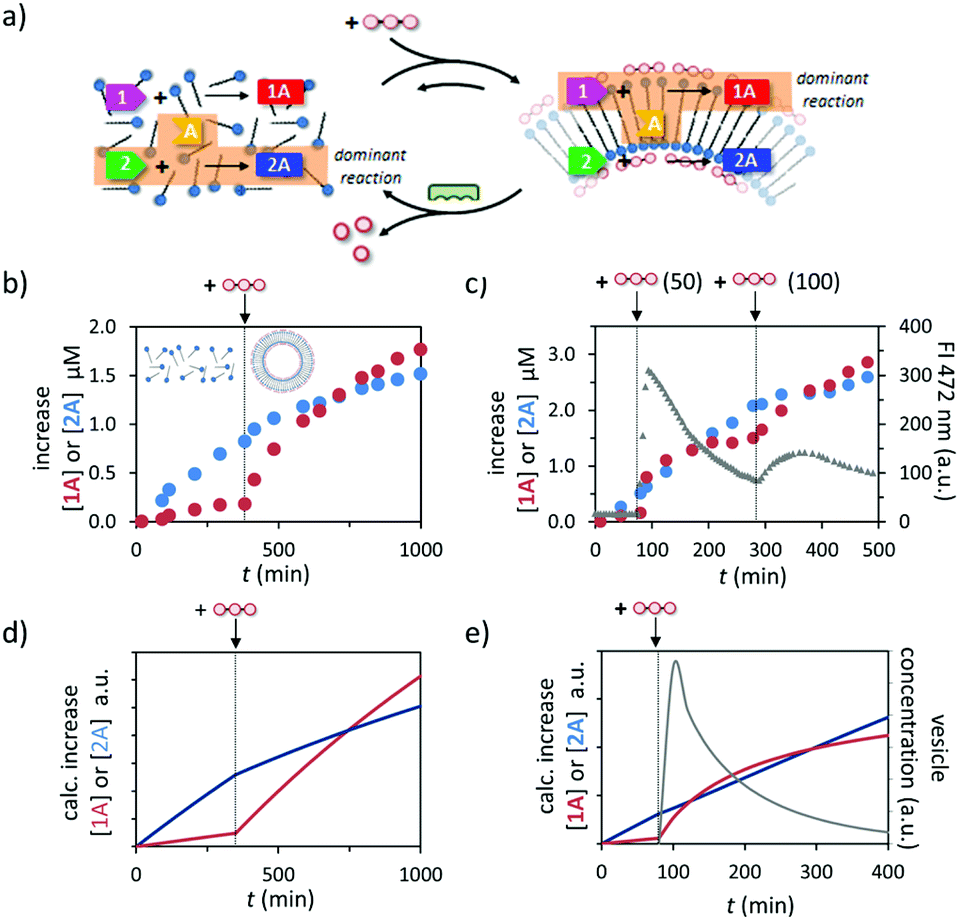 | ||
| Fig. 4 (a) Schematic representation of the change in dominant reaction as the system transits between the unassembled and the assembled state under dissipative conditions. (b) Increase in the concentration of 1A and 2A over time when a mixture of 1, 2 and A (20 μM) were added to a solution of C12TACN·Zn2+ (100 μM) and to which ATP (200 μM) was added after 350 minutes. (c) Increase in the concentration of 1A and 2A over time when a mixture of 1 (20 μM), 2 (20 μM) and A (40 μM) were added to a solution of C12TACN·Zn2+ (100 μM) and alkaline phosphatase (0.5 U) and to which ATP (50 μM) was added at t = 80 min and a new batch (100 μM) at t = 300 min. Overlaid on the same graph is the change in FI at 472 nm over time upon the addition of the same amounts of ATP to a solution containing C12TACN·Zn2+ (100 μM) and laurdan dye (2 μM) under the same experimental conditions. General experimental conditions: [HEPES] = 5 mM, pH 7.0, T = 25 °C. (d) Simulation of the experiment described in (b) using a kinetic model (ESI†). (e) Simulation of the experiment described in (c) (first cycle) using a kinetic model (ESI†). | ||
Finally, temporal control over the product distribution in the system could be obtained exploiting the fact that the upregulation of 1A formation is controlled by ATP. This is illustrated by the following experiment in which ATP was added to a solution containing reagents 1, 2, and A, C12TACN·Zn2+ and the enzyme alkaline phosphatase under optimized conditions in terms of concentrations. The formation of reaction product was monitored by UPLC, which revealed a higher rate of formation of 2A prior to addition of ATP (50 μM), as expected based on the results described above (Fig. 4c and S28, ESI†). The addition of ATP at t = 80 min resulted in a rapid increase in rate of formation of 1A and at t = 100 min hydrazone 1A became the dominant product in the mixture. However, under the dissipative conditions installed by the presence of enzyme, the assemblies have a limited lifetime, which could be determined independently by monitoring the fluorescence intensity of the apolar probe laurdan dye under the same experimental conditions. It was observed that after a rapid rise the FI dropped rapidly over the time course of around 200 minutes. Importantly, in the same time interval the increase in concentration of 1A slowed down significantly reaching a nearly complete stop in concomitance with completion of the disassembly process. However, over this period the concentration of 2A continued to increase steadily because this reaction is not affected by the presence – or absence – of the assemblies. As a result, from t = 200 min onwards, hydrazone 2A became again the dominant product in the reaction mixture. To unequivocally demonstrate that the product composition can be temporally controlled by ATP, we added a new batch of ATP (100 μM) at t = 300 min to start a new cycle. A rapid increase in concentration of 1A was again observed and from t = 400 min onwards hydrazone 1A became again the major product in the mixture.
A kinetic model was developed to improve the understanding how the different processes occurring in the system – self-assembly, reactivity, energy dissipation – affect the output (ESI†). The model includes the reaction rates for the formation of hydrazones 1A and 2A in the absence and presence of assemblies, ATP-induced assembly formation, and the enzyme-catalysed degradation of ATP. The simulated addition of ATP to a mixture in the absence of enzyme indeed leads to the experimentally observed inversion of product selectivity (compare Fig. 4d to b) and under dissipative conditions the calculated trace for the increase in concentration of 1A compares well to the experimentally observed data. The model is thus able to capture the essential kinetic features of the system in the correct time frame. The possibility to capture the system in a relatively simple kinetic scheme will facilitate the design of systems of higher complexity.
Conclusions
In conclusion, we have shown that chemically-fuelled self-assembly under dissipative conditions permits the transient upregulation of a chemical reaction. This changes the product selectivity in the reaction mixture for a limited amount of time depending on the availability of ATP. Kinetic reaction profiles of the kind demonstrated here are not possible to obtain using conventional approaches. Overall, the results indicate a novel way of controlling chemical reactivity in reaction mixtures, which will serve to program reaction pathways in systems of higher complexity.35Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the European Union Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement 642793. We are grateful to Dr Ilaria Fortunati for the LSCM measurements and to Dr Subhabrata Maiti for advice and stimulating discussions.Notes and references
- M. Perutz, Mechanisms of Cooperativity and Allosteric Regulation in Proteins, Cambridge University Press, 1990 Search PubMed.
- P. Goloubinoff, A. S. Sassi, B. Fauvet, A. Barducci and P. De los Rios, Nat. Chem. Biol., 2018, 14, 388–395 CrossRef CAS PubMed.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- F. della Sala, S. Neri, S. Maiti, J. L. Y. Chen and L. J. Prins, Curr. Opin. Biotechnol., 2017, 46, 27–33 CrossRef CAS PubMed.
- S. De and R. Klajn, Adv. Mater., 2018, 30, 1706750 CrossRef PubMed.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- B. A. Grzybowski and W. T. S. Huck, Nat. Nanotechnol., 2016, 11, 584–591 CrossRef PubMed.
- R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- T. Heuser, A. K. Steppert, C. M. Lopez, B. L. Zhu and A. Walther, Nano Lett., 2015, 15, 2213–2219 CrossRef CAS PubMed.
- S. Dhiman, A. Jain, M. Kumar and S. J. George, J. Am. Chem. Soc., 2017, 139, 16568–16575 CrossRef CAS PubMed.
- C. Pezzato, C. Y. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491–5507 RSC.
- J. Leira-Iglesias, A. Sorrenti, A. Sato, P. A. Dunne and T. M. Hermans, Chem. Commun., 2016, 52, 9009–9012 RSC.
- H. Fanlo-Virgos, A. N. R. Alba, S. Hamieh, M. Colomb-Delsuc and S. Otto, Angew. Chem., Int. Ed., 2014, 53, 11346–11350 CrossRef CAS PubMed.
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed.
- Y. Okamoto and T. R. Ward, Angew. Chem., Int. Ed., 2017, 56, 10156–10160 CrossRef CAS PubMed.
- H. L. Che, B. C. Buddingh and J. C. M. van Hest, Angew. Chem., Int. Ed., 2017, 56, 12581–12585 CrossRef CAS PubMed.
- H. L. Che, S. P. Cao and J. C. M. van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef CAS PubMed.
- P. S. Munana, G. Ragazzon, J. Dupont, C. Z. J. Ren, L. J. Prins and J. L. Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 16469–16474 CrossRef PubMed.
- I. Colomer, S. M. Morrow and S. P. Fletcher, Nat. Commun., 2018, 9, 2239 CrossRef PubMed.
- S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS PubMed.
- H. Wang, Y. Wang, B. Shen, X. Liu and M. Lee, J. Am. Chem. Soc., 2019, 141, 4182–4185 CrossRef CAS PubMed.
- C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 CrossRef CAS PubMed.
- S. P. Afrose, S. Bal, A. Chatterjee, K. Das and D. Das, Angew. Chem., Int. Ed., 2019, 58, 15783–15787 CrossRef CAS PubMed.
- S. Dhiman, A. Jain and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 1329–1333 CrossRef CAS PubMed.
- X. Hao, W. Sang, J. Hu and Q. Yan, ACS Macro Lett., 2017, 6, 1151–1155 CrossRef CAS.
- A. Sorrenti, J. Leira-Iglesias, A. Sato and T. M. Hermans, Nat. Commun., 2017, 8, 15899 CrossRef CAS PubMed.
- L. Heinen and A. Walther, Sci. Adv., 2019, 5, eaaw0590 CrossRef PubMed.
- J. L. Y. Chen, S. Maiti, I. Fortunati, C. Ferrante and L. J. Prins, Chem.–Eur. J., 2017, 23, 11549–11559 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- E. T. Kool, P. Crisalli and K. M. Chan, Org. Lett., 2014, 16, 1454–1457 CrossRef CAS PubMed.
- E. T. Kool, D. H. Park and P. Crisalli, J. Am. Chem. Soc., 2013, 135, 17663–17666 CrossRef CAS PubMed.
- G. Vantomme and E. W. Meijer, Science, 2019, 363, 1396–1397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05188k |
| This journal is © The Royal Society of Chemistry 2020 |