
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase segregated Cu2−xSe/Ni3Se4 bimetallic selenide nanocrystals formed through the cation exchange reaction for active water oxidation precatalysts†
Sungwon
Kim
a,
Hiroki
Mizuno
a,
Masaki
Saruyama
*b,
Masanori
Sakamoto
 b,
Mitsutaka
Haruta
b,
Hiroki
Kurata
b,
Taro
Yamada
c,
Kazunari
Domen
c and
Toshiharu
Teranishi
*b
b,
Mitsutaka
Haruta
b,
Hiroki
Kurata
b,
Taro
Yamada
c,
Kazunari
Domen
c and
Toshiharu
Teranishi
*b
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
bInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: saruyama@scl.kyoto-u.ac.jp; teranisi@scl.kyoto-u.ac.jp
cDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 19th December 2019
Abstract
Control over the composition and nanostructure of solid electrocatalysts is quite important for drastic improvement of their performance. The cation exchange reaction of nanocrystals (NCs) has been reported as the way to provide metastable crystal structures and complicated functional nanostructures that are not accessible by conventional synthetic methods. Herein we demonstrate the cation exchange-derived formation of metastable spinel Ni3Se4 NCs (sp-Ni3Se4) and phase segregated berzelianite Cu2−xSe (ber-Cu2−xSe)/sp-Ni3Se4 heterostructured NCs as active oxygen evolution reaction (OER) catalysts. A rare sp-Ni3Se4 phase was formed by cation exchange of ber-Cu2−xSe NCs with Ni2+ ions, because both phases have the face-centered cubic (fcc) Se anion sublattice. Tuning the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu molar ratio leads to the formation of Janus-type ber-Cu2−xSe/sp-Ni3Se4 heterostructured NCs. The NCs of sp-Ni3Se4 and ber-Cu2−xSe/sp-Ni3Se4 heterostructures exhibited high catalytic activities in the OER with small overpotentials of 250 and 230 mV at 10 mA cm−2 in 0.1 M KOH, respectively. They were electrochemically oxidized during the OER to give hydroxides as the real active species. We anticipate that the cation exchange reaction could have enormous potential for the creation of novel heterostructured NCs showing superior catalytic performance.
:Cu molar ratio leads to the formation of Janus-type ber-Cu2−xSe/sp-Ni3Se4 heterostructured NCs. The NCs of sp-Ni3Se4 and ber-Cu2−xSe/sp-Ni3Se4 heterostructures exhibited high catalytic activities in the OER with small overpotentials of 250 and 230 mV at 10 mA cm−2 in 0.1 M KOH, respectively. They were electrochemically oxidized during the OER to give hydroxides as the real active species. We anticipate that the cation exchange reaction could have enormous potential for the creation of novel heterostructured NCs showing superior catalytic performance.
Introduction
Electrochemical water splitting, a combination of two half-cell reactions consisting of the hydrogen evolution reaction at the cathode and the oxygen evolution reaction (OER) at the anode, provides an attractive path in various energy conversion1–5 and storage systems.6,7 However, electrochemical water splitting usually suffers from significant efficiency loss and high overpotential in the oxidative half-cell reaction. The OER is complex and sluggish because the generation of one oxygen molecule involves the transfer of 4e− and removal of 4H+ from water, which hinders practical application of overall water splitting.8–10 To date, the most efficient OER catalysts have been IrO2 and RuO2;11–13 however, the scarcity of these noble metals has further hindered practical application of water splitting on a large scale. Therefore, it is imperative to develop an earth-abundant and cost-effective OER catalyst with excellent electrocatalytic activity to make water splitting a viable energy conversion system.Transition metal-based chalcogenides represent an alternative to the use of noble metals in water electrolysis.14 They exhibit diverse properties that depend on their composition and crystalline structures, leading to enhanced catalytic activity in the water splitting reaction. In particular, Ni-based electrocatalysts show great potential as alternative catalysts for the OER due to their unique 3d electron number and special eg orbitals.15,16 Recently, catalysts of the nickel selenide group have gained attention because of their earth-abundance and high activity for water splitting. They have various crystal structures, such as hexagonal NiSe,17 cubic NiSe2 (ref. 18–20) and rhombohedral Ni3Se2 (ref. 21), mediated by the slight difference in electronegativity between Ni and Se. Although nickel selenide systems have shown a favorably low onset potential and overpotential for the OER in alkaline media, they have practical limitations associated with their bulk-like morphologies. In this context, catalytic systems based on nanocrystals (NCs) are highly promising because of their large specific surface areas. In addition, homogeneous dispersions of catalyst NCs in a solvent can be used to modify various electrodes and photocatalysts by simple deposition or coating methods.22–24 However, it remains challenging to synthesize uniformly structured nickel selenide NCs with high electrocatalytic activity.
One of the ultimate goals of functional NC synthesis is to access arbitrarily defined structures with desired compositions. The cation exchange reaction has become a growing area in NC research, because the substitution of the original cations with other species enables us to synthesize metastable NCs that are not accessible by conventional one-pot synthesis.25–29 Furthermore, if specific procedures are employed in cation exchange reactions, the residual cations can be removed selectively and/or partially, representing a new strategy for accessing structurally novel and multicomponent NCs. Recently, cation exchange reactions of Sn4+, Sn2+, In3+, Cd2+, Zn2+, Hg2+ and Pb2+ cations, using parent Cu2−xSe NCs, have been developed.26,30–32 The exchange reactions of these cations give rise to ternary alloys with distinct nanoheterostructures, including core@shell, sandwich and Janus structures, making them potentially useful materials for solar energy harvesting and catalysis.33,34
Here we report the synthesis of a monodisperse spinel-type nickel selenide (sp-Ni3Se4) NC based OER electrocatalyst using the cation exchange reaction. In this work, we successfully replaced Cu+ of berzelianite copper selenide (ber-Cu2−xSe) as starting NCs with Ni2+ (Scheme 1). The formation of rare and metastable sp-Ni3Se4 is feasible because both ber-Cu2−xSe and sp-Ni3Se4 phases contain an fcc Se anion sublattice, which is retained during the cation exchange reaction. We also achieved fine tuning of the Ni/Cu molar ratio in the NCs, resulting in the formation of partially cation-exchanged NCs with a Janus-type heterostructure composed of ber-Cu2−xSe and sp-Ni3Se4 domains arising from the large lattice mismatch, anisotropic reaction process and immiscibility of the two phases. These Ni-cation-exchanged ber-Cu2−xSe NCs showed efficient electrocatalytic water oxidation activity. Specifically, the heterostructured ber-Cu2−xSe/sp-Ni3Se4 NCs exhibited a lower overpotential (230 mV@10 mA cm−2 in 0.1 M KOH) than pure sp-Ni3Se4 NCs. The characterization of NCs after the OER revealed that the NCs were electrochemically oxidized to hydroxides to serve as the real active species. Thus, these selenide NCs were found to behave as precatalysts for efficient water oxidation. Our work not only provides synthetic pathways to these novel-phased NCs but also supplements the existing modification methods for constructing high-performance catalysts.
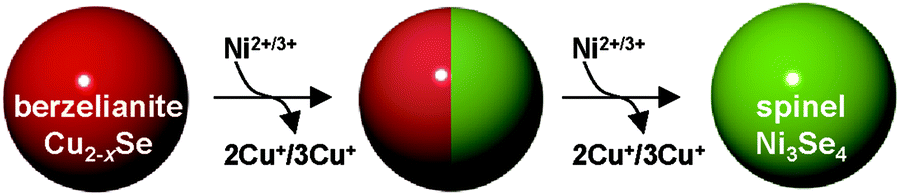 | ||
| Scheme 1 Schematic of the cation exchange reaction of ber-Cu2−xSe NCs with Ni cations. | ||
Results and discussion
To obtain nickel selenide NCs by the cation exchange reaction, we chose ber-Cu2−xSe NCs as the parent material. The ber-Cu2−xSe NCs were synthesized using the procedure reported by Shen et al. with minor modifications (see the ESI for details†).35 A transmission electron microscope (TEM) image of the resulting NCs shows the formation of monodisperse and cuboctahedral NCs with a size of 16.2 ± 1.4 nm (Fig. 1a). The X-ray diffraction (XRD) pattern of the NCs was well matched with that of the ber-Cu2−xSe crystal structure (JCPDS 00-006-0680, Fig. 1e). The elemental analysis of the NCs using X-ray fluorescence spectroscopy (XRF) gave a Cu:Se molar ratio of 64:36, indicating the sub-stoichiometry of Cu in the ber-Cu2−xSe NCs (x = 0.22). These ber-Cu2−xSe NCs exhibited an intense near-infrared (NIR) absorption peak at around 1000 nm related to localized surface plasmon resonance (LSPR) arising from the Cu deficiency in their composition (Fig. S1†).36 A high density of Cu vacancies in sub-stoichiometric copper chalcogenide NCs is generally expected to enhance the cation exchange reaction by vacancy diffusion.37
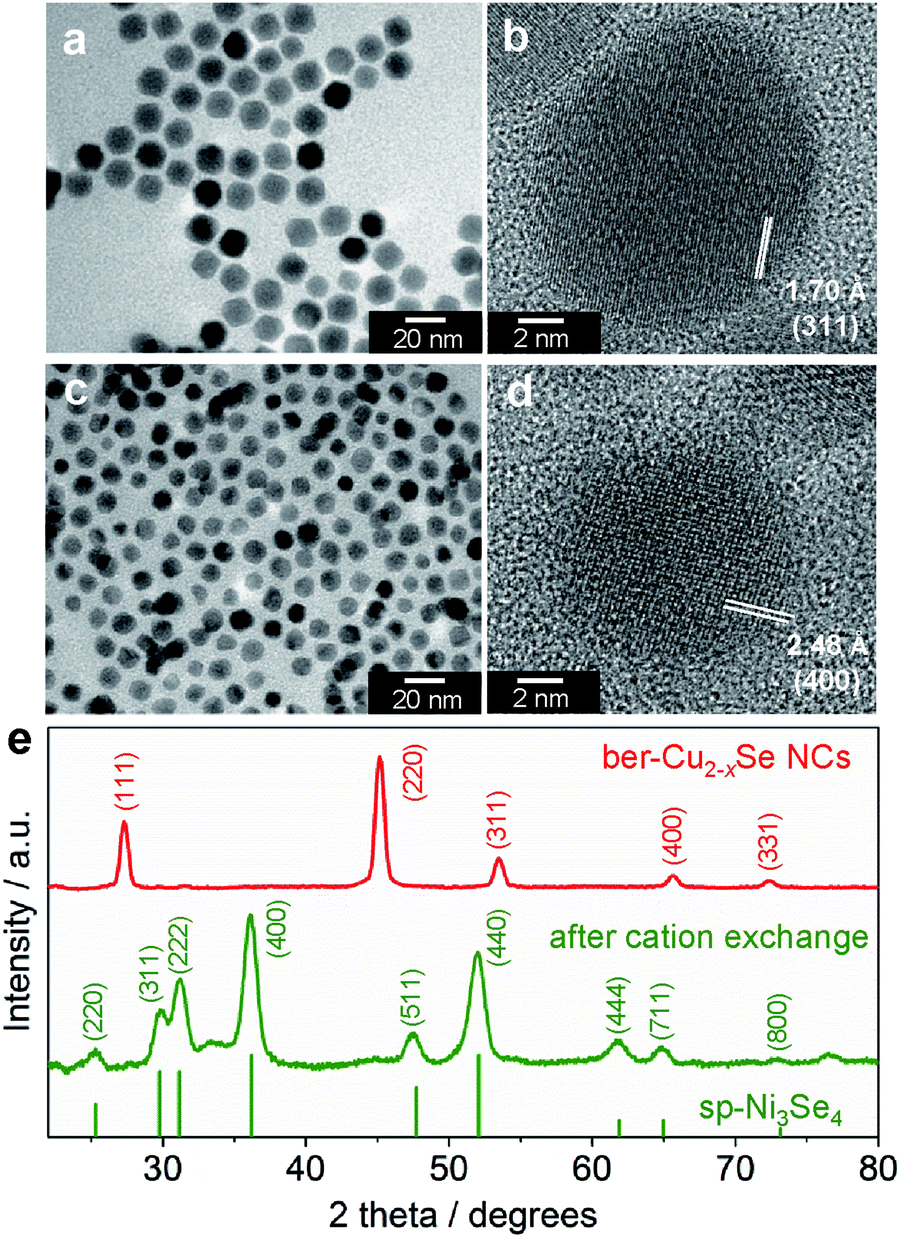 | ||
| Fig. 1 (a and c) TEM and (b and d) HRTEM images of (a and b) ber-Cu2−xSe NCs and (c and d) sp-Ni3Se4 NCs. (e) XRD patterns of ber-Cu2−xSe NCs and sp-Ni3Se4 NCs. The reference XRD pattern shows sp-Ni3Se4 (JCPDS 18-0889). | ||
Then, the as-synthesized ber-Cu2−xSe NCs underwent cation exchange with Ni acetate in the presence of tri-n-octylphosphine (TOP) at 140 °C. For Cu+ → Ni2+ exchange, we used half the amount of Ni2+ relative to Cu+ in the ber-Cu2−xSe NCs, as the reaction involves the replacement of two Cu+ cations with one Ni2+. In the ber-Cu2−xSe NCs, Cu+ (chemical hardness: η = 6.3 eV) can be easily exchanged with harder metal cations when employing soft Lewis bases like TOP (η = 6 eV) to promote Cu+ dissolution into the solvent.38 Finally, we obtained monodisperse NCs of 11.7 ± 1.3 nm size, i.e., smaller than the original ber-Cu2−xSe NCs (Fig. 1c).
When we monitored the change in the molar ratio of Cu, Ni and Se in the NCs during the cation exchange reaction by XRF, it was found that the cation exchange proceeded quite rapidly and was completed within 15 min (Table S1 and Fig. S2†). The rapid replacement of Cu+ with Ni2+ was also confirmed from the UV-vis-NIR absorption spectra, which showed that the LSPR peak arising from the ber-Cu2−xSe phase completely disappeared within 15 min (Fig. S1†). In the XRD analyses, the diffraction pattern of ber-Cu2−xSe NCs also completely disappeared within 15 min, while a new set of peaks assignable to the sp-Ni3Se4 structure emerged (Fig. 1e). This sp-Ni3Se4 phase was maintained when prolonging the reaction time up to 60 min (Fig. S3†). In the high-resolution TEM (HRTEM) images in Fig. 1b and d, the ber-Cu2−xSe NC seeds show a lattice spacing of 1.70 Å, which is consistent with the (311) lattice spacing of ber-Cu2−xSe, while the cation-exchanged NCs exhibit a lattice spacing of 2.48 Å, which can be assigned to the (400) family of planes of sp-Ni3Se4 (additional HRTEM images of sp-Ni3Se4 NCs are shown in Fig. S4†).
Sp-Ni3Se4 was first discovered in 1964 as a natural mineral in Finland and named trüstedtite.39 The crystal structure resembles that of polydymite, spinel Ni3S4, with a larger lattice constant (sp-Ni3Se4: a = 9.94 Å and Ni3S4: a = 9.48 Å, Table S2 and Fig. S5†). Although many researchers have reported the synthesis of nickel selenide crystals, most of them were rhombohedral Ni3Se2,40,41 hexagonal NiSe,42,43 monoclinic Ni3Se4 (ref. 44 and 45) or cubic NiSe2 (ref. 46 and 47) (Table S3†), which are thermodynamically stable phases in the phase diagram of the nickel selenide series at RT.48 On the other hand, spinel Ni3Se4 has been artificially obtained only under extreme conditions.49
We compared the crystal structure of ber-Cu2−xSe and sp-Ni3Se4. Ber-Cu2−xSe has a lattice constant of 5.69 Å and two kinds of Cu sites: Cu1 occupying every tetrahedral coordination site and Cu2 occupying nontetrahedral coordination sites. On the other hand, sp-Ni3Se4 also contains two kinds of Ni sites. While the first of these, Ni1, occupies one-eighth of tetrahedral coordination sites, the other, Ni2, occupies half of the octahedral coordination sites (Table S2†). Focusing on the Se arrangement, ber-Cu2−xSe has an fcc Se anionic sublattice with a lattice constant of 5.69 Å (same as that of a unit cell), while sp-Ni3Se4 has a distorted quasi-fcc Se sublattice with a lattice constant of ∼4.97 Å on average (half that of a unit cell) as shown in Fig. 2b and S6.† This indicates that the fcc Se sublattice is maintained with a slight distortion during the cation exchange process. In a fcc Se unit cell, sp-Ni3Se4 has one Ni1 and two Ni2 sites on average, indicating that one-eighth of Cu1 sites are replaced with Ni cations, and seven-eighth of Cu1 sites and all of Cu2 sites become empty after cation exchange (Table S4†). Further Ni cations newly occupy the octahedral Ni2 sites (Fig. S7†). As a typical spinel structure, Ni2 is located at only a half of the octahedral sites in the lattice, which disproportionates the size of Se octahedral units (Fig. S8†). As a result, forceful insertion of Ni2 sites induces the local distortion of the Se configuration in perfect cubic ber-Cu2−xSe, while the comprehensive fcc Se sublattice is preserved.
Replacement of Cu+ with half the amount of smaller Ni2+ to transform the ber-Cu2−xSe lattice into the sp-Ni3Se4 lattice requires a volume shrinkage of 33%. Based on the comparative TEM images, the actual volume shrinkage of the NCs was calculated to be 48%. This further volume reduction can be explained by etching with TOP and OLAm during the reaction, because the size of the ber-Cu2−xSe NCs decreased from 16 ± 1.3 nm to 10 ± 1.6 nm in the reaction even without Ni acetate. (Fig. S9†).
According to a few number of reports of sp-Ni3Se4 synthesis, we speculate that sp-Ni3Se4 is metastable. To better understand the stability of the nickel selenide phases, we monitored the transition of the crystal phases under annealing in an Ar atmosphere (Fig. S10†). Although the spinel structure of the Ni3Se4 NCs was retained at 100 °C, annealing at >200 °C caused the phase transition to the hexagonal NiSe phase, which confirms that sp-Ni3Se4 is the less stable nickel selenide species.
To reveal the reason why such a rare and metastable sp-Ni3Se4 phase formed, we compared the Se sublattice structure with other common Ni selenides (Fig. 2c). Rhombohedral Ni3Se2 and hexagonal NiSe have bcc and hcp Se sublattices, respectively. Although monoclinic Ni3Se4 has the same stoichiometry as sp-Ni3Se4, its Se sublattice has an hcp configuration with a small distortion. Cubic NiSe2 has an fcc conformation; however, it is constructed from Se–Se dimer components, which is totally different from the usual fcc Se sublattice. This comparison shows that only sp-Ni3Se4 has an fcc Se sublattice which is the same as that of ber-Cu2−xSe. This indicates that Se sublattice preservation preferentially occurs during cation exchange rather than the formation of a more thermodynamically preferable structure, even if the product (sp-Ni3Se4) is less stable.
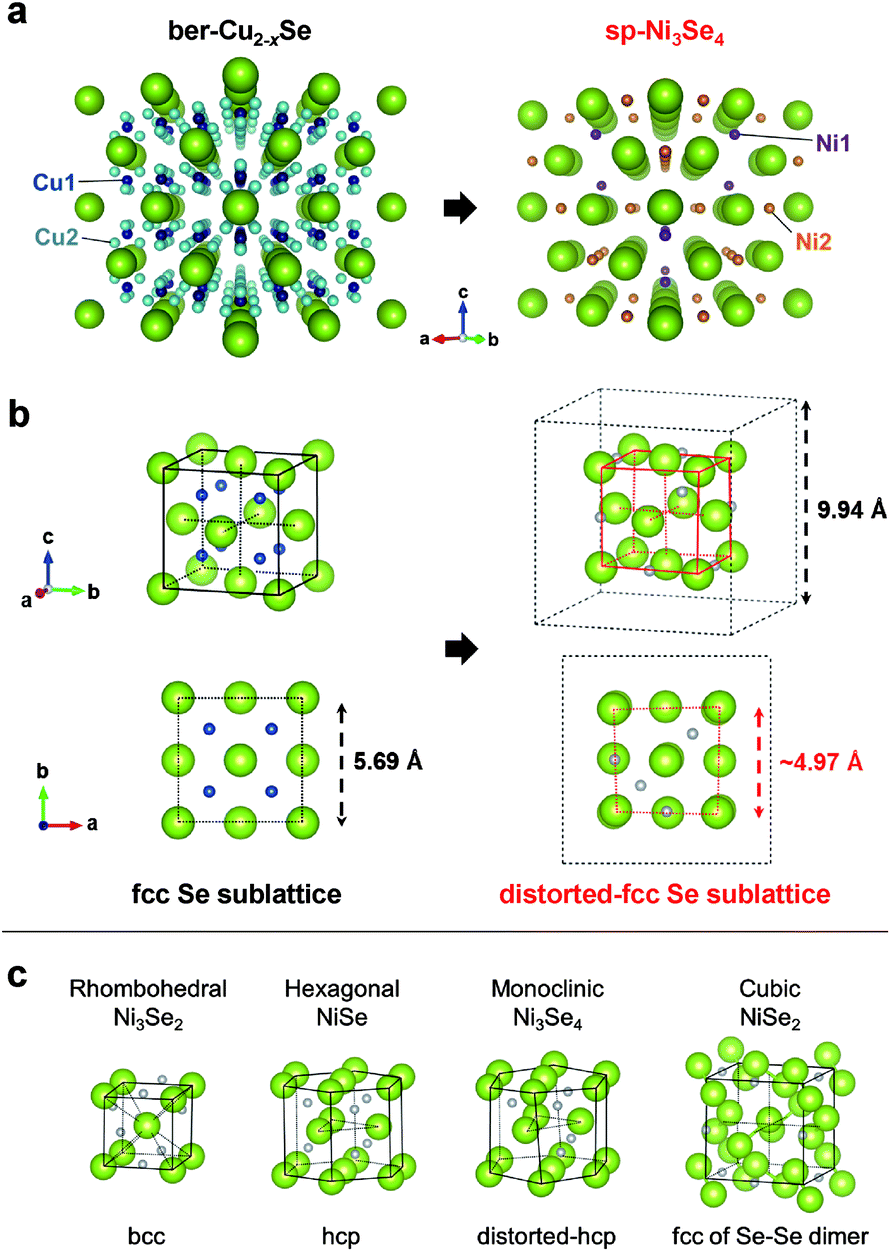 | ||
| Fig. 2 (a) Perspective images of ber-Cu2−xSe (left, 2 × 2 × 2 = 8 unit cells) and sp-Ni3Se4 (right, 1 unit cell) along the <110> zone axis. Cu1, Cu2, Ni1 and Ni2 sites are coloured in dark blue, light blue, purple and orange, respectively. (b) Se sublattice (pseudo) unit cell of ber-Cu2−xSe and sp-Ni3Se4. Only Cu1 sites in ber-Cu2−xSe are shown for simplification. Black dashed lines of sp-Ni3Se4 highlight the size of the sp-Ni3Se4 unit cell. (c) Se pseudo-unit cell of a common nickel selenide system. | ||
Binary transition metal selenides often exceed monometallic selenides in OER activity due to the synergetic effect between two metals. The cation exchange method can facilely provide NCs with bimetallic composition at the intermediate stage of the reaction. However, in general, the cation exchange reaction proceeds too fast to obtain such partially exchanged NCs. Because the stability constant of Ni(acac)2 (logK = 6.1) is larger than that of Ni acetate (logK = 1.8),50 using Ni(acac)2 as a Ni2+ precursor was expected to slow down the cation exchange kinetics. TEM images of the NCs obtained by cation exchange using Ni(acac)2 at different reaction times showed monodisperse 11 nm NCs, which is similar to the case of Ni acetate. The Ni/Cu molar ratio during the reaction increased much more slowly than the case of Ni acetate as expected (Table S5 and Fig. S11†). Thanks to this slow reaction, we succeeded in obtaining partially cation-exchanged NCs with tunable Ni:Cu molar ratios from 10:50 to 40:20 (Table S5 and Fig. S11†). Fig. 3e shows the XRD patterns of partially cation-exchanged NCs after different reaction times. The patterns show a combination of the ber-Cu2−xSe and the sp-Ni3Se4 phases without other CuxNiySez-related solid solution phases. As the reaction proceeded, the peak intensities of the sp-Ni3Se4 phase increased, whereas those of ber-Cu2−xSe decreased and almost disappeared at 60 min. This slow cation exchange also induced the gradual disappearance of LSPR absorption of the ber-Cu2−xSe phase (Fig. S12†).
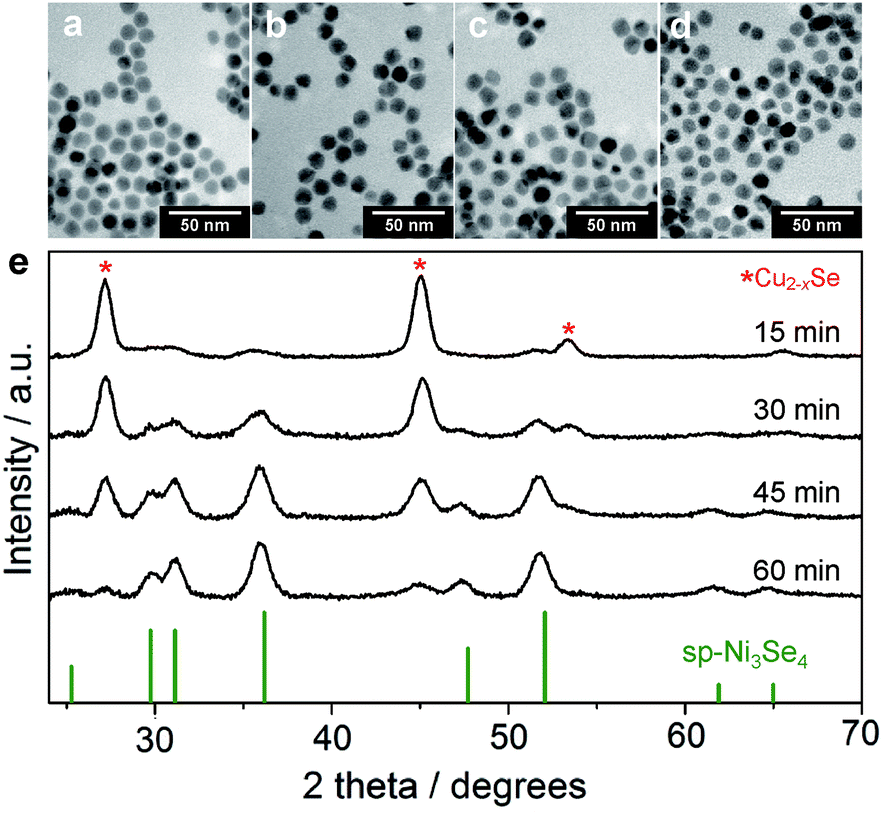 | ||
| Fig. 3 (a–d) TEM images of ber-Cu2−xSe/sp-Ni3Se4 HNCs at different cation exchange reaction times of (a) 15 min, (b) 30 min, (c) 45 min and (d) 60 min. (e) XRD patterns of ber-Cu2−xSe/sp-Ni3Se4 HNCs at different cation exchange reaction times. The red-star symbols and reference XRD pattern indicate ber-Cu2−xSe and sp-Ni3Se4 (JCPDS 18-0889), respectively. | ||
Scanning TEM (STEM)–energy-dispersive X-ray spectroscopy (EDS) elemental mapping was carried out to reveal the spatial distributions of the elements Cu, Ni and Se in the partially cation-exchanged NCs (Fig. 4a–d). Cu and Ni were concentrated at specific regions in each individual NC, while Se was distributed throughout each NC. This means that the ber-Cu2−xSe and sp-Ni3Se4 phases were anisotropically segregated to form Janus-type heterostructured NCs (HNCs), which is consistent with the XRD results. In the HRTEM image of partially exchanged NCs (Fig. 4e), we can see a clear boundary between domains with lattice spacings of ber-Cu2−xSe ({002}, 2.86 Å) and sp-Ni3Se4 ({004}, 2.48 Å). The fact that these two planes are parallel indicates the epitaxial relationship between ber-Cu2−xSe and sp-Ni3Se4 with a large lattice mismatch of ∼13% (Fig. 4f). Additional HRTEM images (Fig. S13†) also exhibit an epitaxial relationship between two phases. These observations indicate that these two phases share a continuous Se sublattice and the Se framework is maintained during the cation exchange reaction.
 | ||
| Fig. 4 (a–d) STEM-EDS elemental mapping for each element and (e) HRTEM image of ber-Cu2−xSe/sp-Ni3Se4 HNCs (Ni:Cu:Se = 22:37:41 mol% in XRF). (f) Schematic atomic arrangement of the heterointerface between ber-Cu2−xSe and sp-Ni3Se4 phases along with the [1−10] zone axis. Cu1, Ni1 and Ni2 sites are coloured in dark blue, purple and orange, respectively. | ||
The formation of Janus-type HNCs through the cation exchange of copper chalcogenide NCs has been recently reported.51 Typical examples are the formation of Cu2−xSe/CdSe and Cu2−xSe/ZnSe HNCs,37 which are structurally similar to the as-synthesized ber-Cu2−xSe/sp-Ni3Se4 HNCs. The phase segregation can be explained as a consequence of the preferential starting sites and propagation paths for cation exchange within the NCs. The cation exchange reaction observed herein is also likely to have been initiated at the most energetically favored positions, like Cu vacancies. In addition, considering that no data for the Cu–Ni–Se ternary alloy can be found in the phase diagrams,52 the ber-Cu2−xSe and sp-Ni3Se4 phases seem to be immiscible, which thermodynamically drives their phase segregation. As an exception, spinel CuNi2Se4 was very recently found as a natural mineral in 2017 (ref. 53) in the Andes in Bolivia and named Nickeltyrrellite.54 However, it took several millions of years to mineralize CuNi2Se4 under extreme conditions of continuous volcanic hydrothermal activity with repeated pressure and heating. Considering that there is almost no other literature of Cu–Ni–Se ternary systems, it can be concluded that ber-Cu2−xSe and sp-Ni3Se4 are basically immiscible. Such a large lattice mismatch, preferable reaction site and pathway, and immiscibility of ber-Cu2−xSe and sp-Ni3Se4 phases result in the formation of a Janus-type heterostructure with a single interface which is favorable to minimize the lattice strain and the interfacial area.
The sp-Ni3Se4 based NCs produced by the cation exchange reaction are expected to have high OER catalytic activity, as observed in bulk transition metal selenides electrocatalysts. The OER activity of the ber-Cu2−xSe NCs, sp-Ni3Se4 NCs and ber-Cu2−xSe/sp-Ni3Se4 (Ni:Cu:Se = 34:31:35 mol% in XRF) HNCs was evaluated in 0.1 M KOH electrolyte (pH 13). The working electrodes were prepared by the simple deposition of NCs on carbon paper. Fig. 5a shows the iR-corrected polarization curves. While pure ber-Cu2−xSe NCs showed a high overpotential of 568 mV at 10 mA cm−2, the fully cation-exchanged sp-Ni3Se4 NCs exhibited a much lower overpotential of 250 mV at 10 mA cm−2, which is comparable to the reported values for metal chalcogenide systems (Table S6†). Interestingly, the ber-Cu2−xSe/sp-Ni3Se4 HNCs showed an even better catalytic activity with a lower overpotential of 230 mV at 10 mA cm−2. For comparison, a simple mixture of ber-Cu2−xSe NCs and sp-Ni3Se4 NCs was also applied to the OER; however, it has lower activity than sp-Ni3Se4 NCs. A scanning electron microscopy (SEM) image of the electrode of mixed NCs showed segregation of each kind of NCs (Fig. S14†), suggesting that the homogeneous distribution of ber-Cu2−xSe and sp-Ni3Se4 phases contributes to the synergetic improvement of catalysis. Fig. 5b shows the Tafel slopes of the catalysts. It is generally accepted that a smaller Tafel slope indicates faster OER kinetics.55 The ber-Cu2−xSe NCs had a high Tafel slope of 949 mV dec−1 (not shown), implying their poor intrinsic OER kinetics. The sp-Ni3Se4 NCs and ber-Cu2−xSe/sp-Ni3Se4 HNCs exhibited Tafel slopes of 57 and 48 mV dec−1, respectively, revealing that the latter had the fastest OER kinetics.
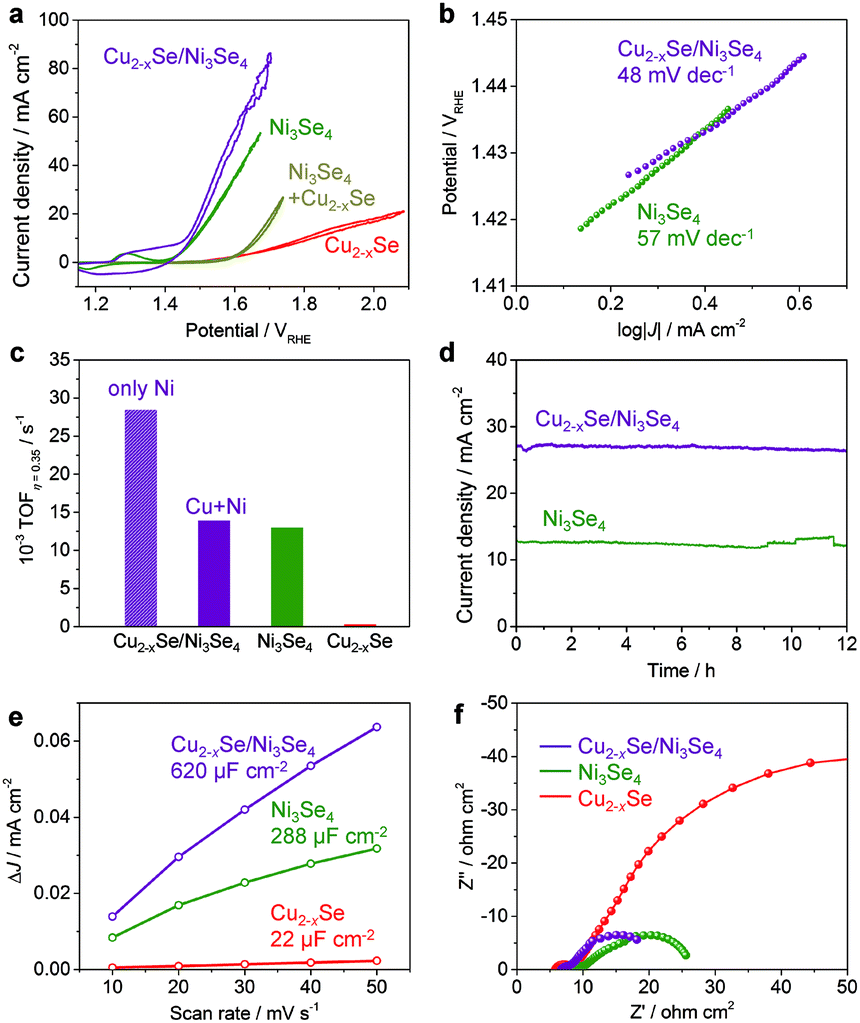 | ||
| Fig. 5 (a) Cyclic voltammograms of ber-Cu2−xSe, sp-Ni3Se4, the mixture of ber-Cu2−xSe and sp-Ni3Se4 NCs and ber-Cu2−xSe/sp-Ni3Se4 HNCs in 0.1 M KOH. (b) Tafel plots of sp-Ni3Se4 NCs and ber-Cu2−xSe/sp-Ni3Se4 HNCs. (c) TOF calculations at an overpotential of 350 mV and (d) long-term durable operation with a constant potential of 1.5 V vs. RHE. (e) Capacitive current density versus scan rate and (f) Nyquist plots of the ber-Cu2−xSe NC, sp-Ni3Se4 NC and ber-Cu2−xSe/sp-Ni3Se4 HNC-loaded electrodes. | ||
To further estimate the inherent OER activity of the ber-Cu2−xSe/sp-Ni3Se4 HNCs, the turnover frequency (TOF) at an overpotential of 0.35 V was calculated under the assumption that all the metal content in the NCs was catalytically active (Fig. 5c). While the TOF was 0.32 × 10−3 s−1 for the ber-Cu2−xSe NCs, this was enhanced by ∼40 times to 12.98 × 10−3 s−1 and 13.91 × 10−3 s−1 for sp-Ni3Se4 NCs and ber-Cu2−xSe/sp-Ni3Se4 HNCs, respectively, which are comparable to or even surpass the TOFs reported for metal chalcogenide catalysts.56–58 Alternatively, assuming that only the Ni ions work as active sites, the TOF of the ber-Cu2−xSe/sp-Ni3Se4 HNCs was calculated to be 27 × 10−3 s−1, which is twice that of pure sp-Ni3Se4 NCs. After 500 cyclic voltammetry (CV) sweeps, the overpotential at 10 mA cm−2 increased from the initial sweep by only 22 mV (from 230 to 252 mV) for the ber-Cu2−xSe/sp-Ni3Se4 HNCs and by 15 mV (from 250 to 265 mV) for the sp-Ni3Se4 NCs (Fig. S15†), which illustrated their high durability as catalysts. Moreover, the sp-Ni3Se4 NCs and ber-Cu2−xSe/sp-Ni3Se4 HNCs also exhibited excellent stability in long-term continuous operation at 1.5 vs. the reversible hydrogen electrode (RHE), in which the current densities were retained at more than 10 mA cm−2 for 12 h without obvious degradation (Fig. 5d).
The electrochemically active surface area (ECSA) was determined from the electrochemical double-layer capacitance (Cdl) in the potential range in which the current response was non-faradic. The ber-Cu2−xSe/sp-Ni3Se4 HNCs and sp-Ni3Se4 NCs showed 6-times and 3-times higher Cdl values than the ber-Cu2−xSe NCs, respectively, as shown in Fig. 5e and S16.† To clarify the reason for the different ECSA values of these NCs, the differences in NC morphologies before and after the OER were microscopically investigated (Fig. S17†). The SEM images showed that the ber-Cu2−xSe NCs on carbon paper were transformed into larger solid particles of 50–100 nm size after the OER. The sp-Ni3Se4 NCs were also fused into aggregates after the OER. For the ber-Cu2−xSe/sp-Ni3Se4 HNCs, the NC size was maintained, with only slight contact among NCs, during the OER, providing the largest surface area. The reason for this shape retention is still not fully understood. We speculate that the immiscibility of the ber-Cu2−xSe and sp-Ni3Se4 phases prevents both species of metal ions from migrating to form larger agglomerates. XRF analyses of each NC catalyst after the OER demonstrated that the molar proportion of Se decreased, compared with the as-synthesized NCs, suggesting that Se dissolution occurred during OER operation (Fig. S17†). Surface oxidation of the bulk metal selenide catalyst during the OER has often been reported recently, resulting in the formation of micropores to increase the number of active sites.59 For NCs, however, surface oxidation triggers the fusion of NCs, decreasing the merit of the large relative surface area. Therefore, suppressing such a fusion to maintain the nanoparticulate form is apparently advantageous to improve the overall activity.
The catalytic enhancement was further confirmed by electrochemical impedance spectroscopy (EIS) analyses, as shown in Fig. 5f. We fitted Nyquist and Bode plots using the equivalent circuit model which contains three kinds of resistances to reproduce the multistep kinetics in the OER (Fig. S18 and Table S7†).60 For each catalyst, Rads (resistance related to the ease of reaction intermediate adsorption on the surface) is much larger than RCT (charge transfer resistance), which suggests that OER catalysis is dominated by the formation of chemical intermediates. Ni3Se4 and ber-Cu2−xSe/sp-Ni3Se4 HNCs have much smaller Rads than ber-Cu2−xSe, illustrating drastic improvement of OER activity after Ni cation exchange. Ber-Cu2−xSe/sp-Ni3Se4 HNCs have not only the smallest Rads but also the largest capacitance (Cads), which suggests that ber-Cu2−xSe/sp-Ni3Se4 HNCs possess a larger number of preferential active sites for OER intermediates.
From the above results, the coexistence of ber-Cu2−xSe and sp-Ni3Se4 phases in a single NC seems to cause a synergetic effect to promote the OER. In the cyclic voltammogram, the ber-Cu2−xSe/sp-Ni3Se4 HNCs have a larger Ni redox current area around 1.3 V vs. RHE than the pure sp-Ni3Se4 NCs (Fig. 5a), meaning that more Ni3+ species were generated than in the sp-Ni3Se4 NCs, probably because of their larger ECSA. It is well known that in higher valence states, Ni acts as a more efficient OER active site.61 Thus, the presence of the ber-Cu2−xSe phase supports the generation of both a larger ECSA and larger number of Ni3+ species in the sp-Ni3Se4 phase.
To further reveal the origin of the activity difference among the three kinds of NCs, X-ray photoluminescence spectroscopy (XPS) measurements were performed. First, we investigated the XPS data of the NCs before the OER (Fig. 6a–d). In the Cu 2p3/2 region, the ber-Cu2−xSe NCs showed a peak at 932.3 eV assigned to Cu+ in ber-Cu2−xSe, with a minor peak at higher binding energy assigned to Cu2+ in CuO (Fig. 6a). After the cation exchange, Ni 2p3/2 peaks of Ni2+/3+ in sp-Ni3Se4 and Ni2+ from NiO and Ni(OH)2 newly emerged (Fig. 6b). Simultaneously, new Se 3d peaks appeared at ∼58.6 eV resulting from SeOx formation (Fig. 6c), and O 1s peaks also become strong after cation exchange (Fig. 6d). These results indicate that the cation exchange reaction caused surface oxidation of NCs, probably during the purification step in air. We would like to claim that the main phases are selenides, because all XRD peaks are assigned to selenides (Fig. 3e), and no oxide layers were observed in HRTEM images. Therefore, the amorphous oxide layers are considered to be thin on the NCs. Second, we further conducted XPS measurements of all NCs after the OER (after 500 cycles CV, Fig. 6e–h). In all samples, the Se 3d peaks completely disappeared because of the Se dissolution during the OER (Fig. 6g), which is consistent with the decreased Se molar ratio after the OER (Fig. S17†). For the ber-Cu2−xSe NCs, the Cu 2p3/2 peak was shifted from 932.3 eV (before the OER) to 933.9 eV, suggesting the oxidation of Cu+ to Cu2+ and the formation of CuO (Fig. 6e). The sp-Ni3Se4 NCs showed a positive shift of the Ni 2p3/2 peak to 857.0 eV which can be assigned to the formation of Ni3+ in NiOOH (Fig. 6f). In the ber-Cu2−xSe/sp-Ni3Se4 HNCs, the Cu 2p3/2 peak at around ∼935.0 eV suggests the formation of CuO and Cu(OH)2, and Ni 2p3/2 peak at 856.8 eV and O 1s peak at 531.3 eV arise from NiOOH. The ber-Cu2−xSe/sp-Ni3Se4 HNCs have higher binding energy of the Cu 2p3/2 peak (934.3 eV) than pure Cu2−xSe NCs (933.9 eV), implying that the higher valence state of Cu was stabilized by the coexistence of NiOOH species. It has been reported that highly oxidized Cu species can act as efficient OER catalysts,62 which suggests that the high valence state of Cu in (Cu/Ni)OOH might also catalyze the OER. However, additional experiments are required to confirm this hypothesis. Selected area electron diffraction (SAED) measurements of ber-Cu2−xSe/sp-Ni3Se4 NCs were also carried out to further confirm the change of crystal structures during the OER (Fig. S19†). Immediately after immersing the NCs in 0.1 M KOH, the crystalline feature of ber-Cu2−xSe and sp-Ni3Se4 was retained. However, the NCs become less crystalline after 500 CV cycles, indicating that the oxidation of NCs is electrochemically induced. Such a process can be considered as the activation step, as we can see an increase of current density in the first 50 CV cycles (Fig. S20†).
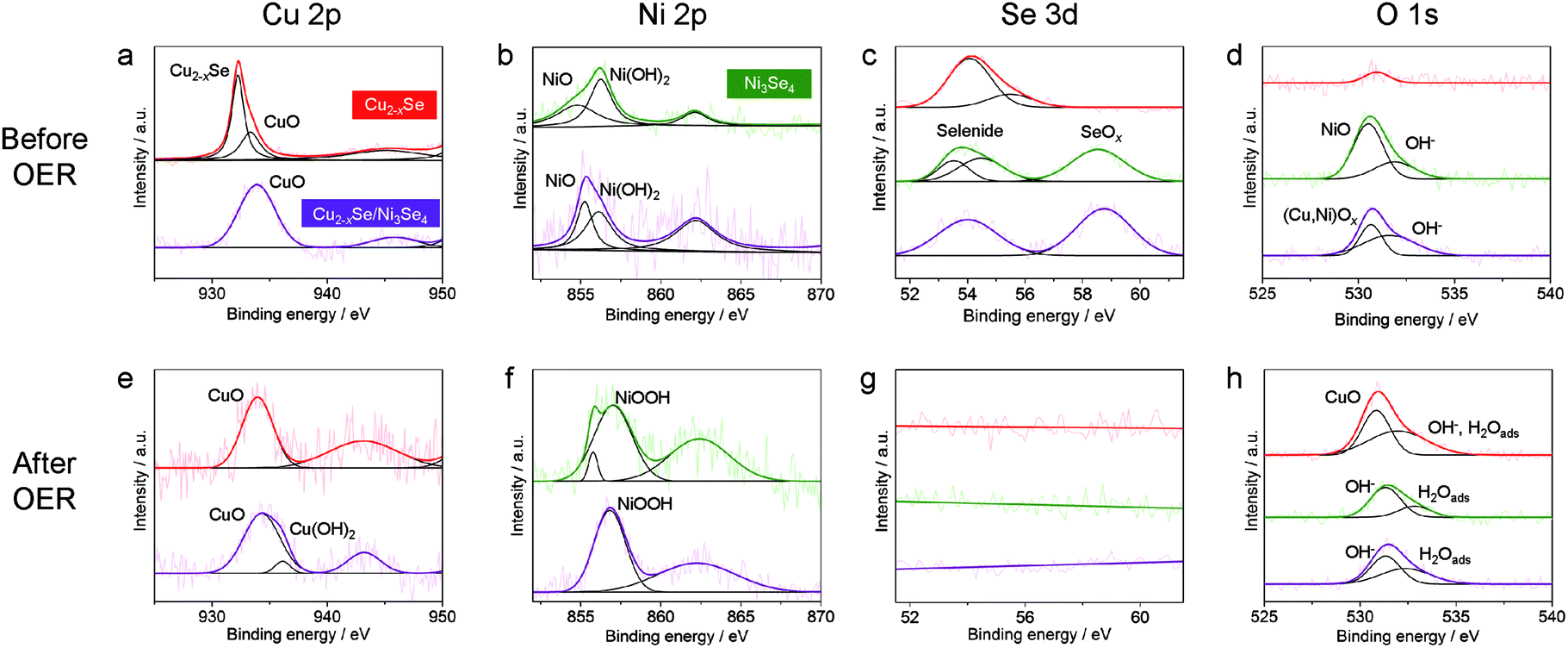 | ||
| Fig. 6 XPS spectra of NCs (a–d) before and (e–h) after the OER. XPS spectra of ber-Cu2−xSe NCs (red line), sp-Ni3Se4 NCs (green line) and ber-Cu2−xSe/sp-Ni3Se4 HNCs (purple line) loaded on carbon paper electrodes before and after OER experiments; (a and e) Cu 2p3/2, (band f) Ni 2p3/2, (c and g) Se 3d and (d and h) O1s regions. | ||
Conclusions
We have developed the selective synthesis of sp-Ni3Se4 NCs and novel Janus-type ber-Cu2−xSe/sp-Ni3Se4 HNCs by the cation exchange reaction, and demonstrated their high OER catalytic activity. A rare sp-Ni3Se4 phase was selectively formed because of the preservation of the fcc Se sublattice of ber-Cu2−xSe. Careful control of the progress of the cation exchange reaction selectively provided the Janus-like heterostructure as a result of lattice mismatch, anisotropic reaction process and immiscibility of the two phases. The ber-Cu2−xSe/sp-Ni3Se4 HNCs exhibited exceptional OER activity, manifesting a low overpotential of 230 mV at 10 mA cm−2, even better than that of the sp-Ni3Se4 NCs (250 mV). This outperforms the values for other Ni–Se systems and the most commonly reported solid-solution catalysts. The NCs were activated by in situ electrochemical oxidation, and their hydroxide species work as the real active species. Such an enhanced activity of the HNCs arose from the coexistence of ber-Cu2−xSe and sp-Ni3Se4 phases in each individual NC, which provided synergistic benefits by increasing the ECSA and generating highly active Cu species. We believe that the utility of the cation exchange reaction might extend to other transition metal-based materials for highly active next-generation electrocatalysts. We also anticipate that the novel heterostructured NCs might be exploited in other materials systems for fabricating nanostructures with crystal phases that are not easily accessed by direct chemical synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Artificial Photosynthesis Project of the New Energy and Industrial Technology Development Organization (NEDO) of Japan. This work was partially supported by JSPS KAKENHI for Scientific Research S (Grant No. JP19H0563), Scientific Research B (Grant No. JP16H03826), Scientific Research on Innovative Areas (Grant No. JP16H06520 (Coordination Asymmetry)) (T. T.) and Young Scientists B (Grant No. 17K14081) (M. S.).References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. DeGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocerea, D. M. Tiede, H. Dau, W. Hillier, L. Wang and S. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 RSC.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Zhao, J. R. Swierk, J. D. Megiatto Jr, B. Sherman, J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- M. E. G. Lyons and S. Floquet, Phys. Chem. Chem. Phys., 2011, 13, 5314–5335 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L. Z. Wu, C. H. Tung, D. O'Hare and T. R. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS PubMed.
- S. Kukunuri, M. R. Krishnan and S. Sampath, Phys. Chem. Chem. Phys., 2015, 17, 23448–23459 RSC.
- F. Wang, Y. Li, T. A. Shifa, K. Liu, F. Wang, Z. Wang, P. Xu, Q. Wang and J. He, Angew. Chem., Int. Ed., 2016, 55, 6919–6924 CrossRef CAS PubMed.
- A. T. Swesi, J. Jasud, W. P. R. Liyanage, S. Umapathi, E. Bohannan, J. Medvedeva and M. Nath, Sci. Rep., 2017, 7, 2401 CrossRef PubMed.
- G. Nagaraju, S. M. Cha, C. Sekhar and J. S. Yu, Adv. Energy Mater., 2017, 7, 1601362 CrossRef.
- Z. Zhuang, Q. Peng, J. Zhuang, X. Wang and Y. Li, Chem.–Eur. J., 2006, 12, 211–217 CrossRef CAS PubMed.
- M. Saruyama, S. Kim, T. Nishino, M. Sakamoto, M. Haruta, H. Kurata, S. Akiyama, T. Yamada, K. Domen and T. Teranishi, Chem. Sci., 2018, 9, 4830–4836 RSC.
- T. Yoshinaga, M. Saruyama, A. Xiong, Y. Ham, Y. Kuang, R. Niishiro, S. Akiyama, M. Sakamoto, T. Hisatomi, K. Domen and T. Teranishi, Nanoscale, 2018, 10, 10420–10427 RSC.
- S. Kim, T. Nishino, M. Saruyama, M. Sakamoto, H. Kobayashi, S. Akiyama, T. Yamada, K. Domen and T. Teranishi, ChemNanoMat, 2017, 3, 764–771 CrossRef CAS.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- H. Li, M. Zanella, A. Genovese, M. Povia, A. Falqui, C. Giannini and L. Manna, Nano Lett., 2011, 11, 4964–4970 CrossRef CAS PubMed.
- J. L. Fenton and R. E. Schaak, Angew. Chem., Int. Ed., 2017, 56, 6464–6467 CrossRef CAS PubMed.
- A. E. Powell, J. M. Hodges and R. E. Schaak, J. Am. Chem. Soc., 2016, 138, 471–474 CrossRef CAS PubMed.
- H.-L. Wu, R. Sato, A. Yamaguchi, M. Kimura, M. Haruta, H. Kurata and T. Teranishi, Science, 2016, 351, 1306–1310 CrossRef CAS PubMed.
- L. D. Trizio, H. Li, A. Casu, A. Genovese, A. Sathya, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2014, 136, 16277–16284 CrossRef PubMed.
- A. C. Berends, W. Stam, Q. A. Akkerman, J. D. Meeldijk, J. Lit and C. M. Donega, Chem. Mater., 2018, 30, 3836–3846 CrossRef CAS PubMed.
- R. Tu, Y. Xie, G. Bertoni, A. Lak, R. Gaspari, A. Rapallo, A. Cavalli, L. D. Trizio and L. Manna, J. Am. Chem. Soc., 2016, 138, 7082–7090 CrossRef CAS PubMed.
- H. Tahara, M. Sakamoto, T. Teranishi and Y. Kanemitsu, Phys. Rev. Lett., 2017, 119, 247401 CrossRef PubMed.
- Z. Lian, M. Sakamoto, J. J. M. Vequizo, C. S. K. Ranasinghe, A. Yamakata, T. Nagai, K. Kimoto, Y. Kobayashi, N. Tamai and T. Teranishi, J. Am. Chem. Soc., 2019, 141, 2446–2450 CrossRef CAS PubMed.
- H. Shen, H. Wang, H. Yuan, L. Ma and L. S. Li, CrystEngComm, 2012, 14, 555–560 RSC.
- D. Dorfs, T. Jartling, K. Miszta, N. C. Bigall, M. E. Kim, A. Genovese, A. Falqui, M. Povia and L. Manna, J. Am. Chem. Soc., 2011, 133, 11175–11180 CrossRef CAS PubMed.
- V. Lesnyak, R. Brescia, G. C. Messina and L. Manna, J. Am. Chem. Soc., 2015, 137, 9315–9323 CrossRef CAS PubMed.
- J. Gui, M. Ji, J. Liu, M. Xu, J. Zhang and H. Zhu, Angew. Chem., Int. Ed., 2015, 127, 3754–3758 CrossRef.
- Y. Vuorelainen, A. Huhma and A. Hakli, Bull. Comm. Geol. Finl., 1964, 215, 114–125 Search PubMed.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 RSC.
- K. Xu, H. Ding, H. Lv, S. Tao, P. Chen, X. Wu, W. Chu, C. Wu and Y. Xie, ACS Catal., 2016, 7, 310–315 CrossRef.
- X. Li, G. Han, Y. Liu, B. Dong, W. Hu, X. Shang, Y. Chai and C. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 20057–20066 CrossRef CAS PubMed.
- S. Kukunuri, M. R. Krishnan and S. Sampath, Phys. Chem. Chem. Phys., 2015, 17, 23448–23459 RSC.
- S. Ananthara, J. Kennedy and S. Kundu, ACS Appl. Mater. Interfaces, 2017, 9, 8714–8728 CrossRef PubMed.
- J. Y. Zhang, X. Tian, T. He, S. Zaman, M. Miao, Y. Yan, K. Qi, Z. Dong, H. Liu and B. Y. Xia, J. Mater. Chem. A, 2018, 6, 15653–15658 RSC.
- Z. Pu, Y. Luo, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 4718–4723 CrossRef CAS PubMed.
- H. Fan, H. Yu, X. Wu, Z. Yu, Y. Zhang, Z. Luo, H. Wang, Y. Guo, S. Madhavi and Q. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25261–25267 CrossRef CAS PubMed.
- H. Baker, ASM Handbook, Alloy phase diagrams, ASM International, 1992 Search PubMed.
- G. Wang, S. K. Lok, G. K. L. Wong and I. K. Sou, Small, 2011, 7, 1546–1551 CrossRef CAS PubMed.
- M. Misono, E. Ochiai, Y. Saito and Y. Yoneda, J. Inorg. Nucl. Chem., 1967, 29, 2685–2691 CrossRef CAS.
- L. D. Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef PubMed.
- P. Vilars, A. Prince and H. Okamoto, Handbook of Ternary Alloy Phase Diagrams, ASM International, 1995 Search PubMed.
- G. Grundmann and H. J. Föster, Minerals, 2017, 7, 68 CrossRef.
- H. J. Förster, C. Ma, G. Grundmann, L. Bindi and C. J. Stanley, Eur. J. Mineral., 2019, 31, 197–202 CrossRef.
- L. Liang, H. Cheng, F. Lei, J. Han, S. Gao, C. Wang and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 12004–12008 CrossRef CAS.
- L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shem, J. Guo and S. J. Sun, J. Am. Chem. Soc., 2015, 137, 7071–7074 CrossRef CAS PubMed.
- Y. Shi, Y. Zhou, D. Yang, W. Xu, C. Wang, F. Wang, J. Xu, X. Xia and H. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 CrossRef CAS PubMed.
- Y. H. Fang and Z. P. Liu, ACS Catal., 2014, 4, 4364–4376 CrossRef CAS.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CrossRef CAS.
- M. Yoshida, Y. Mitsutomi, T. Mineo, M. Nagasaka, H. Yuzawa, N. Kosugi and H. Kondoh, J. Phys. Chem. C, 2015, 119, 19279–19286 CrossRef CAS.
- S. Cui, X. Liu, Z. Sun and P. Du, ACS Sustainable Chem. Eng., 2016, 4, 2593–2600 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional characterization and results. See DOI: 10.1039/c9sc04371c |
| This journal is © The Royal Society of Chemistry 2020 |