
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Al(ORF)3 (RF = C(CF3)3) activated silica: a well-defined weakly coordinating surface anion†
Damien B.
Culver
a,
Amrit
Venkatesh
 b,
Winn
Huynh
a,
Aaron J.
Rossini
b and
Matthew P.
Conley
*a
b,
Winn
Huynh
a,
Aaron J.
Rossini
b and
Matthew P.
Conley
*a
aDepartment of Chemistry, University of California, Riverside, California 92521, USA. E-mail: matthew.conley@ucr.edu
bDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA
First published on 19th December 2019
Abstract
Weakly Coordinating Anions (WCAs) containing electron deficient delocalized anionic fragments that are reasonably inert allow for the isolation of strong electrophiles. Perfluorinated borates, perfluorinated aluminum alkoxides, and halogenated carborane anions are a few families of WCAs that are commonly used in synthesis. Application of similar design strategies to oxide surfaces is challenging. This paper describes the reaction of Al(ORF)3*PhF (RF = C(CF3)3) with silica partially dehydroxylated at 700 °C (SiO2-700) to form the bridging silanol ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH⋯Al(ORF)3 (1). DFT calculations using small clusters to model 1 show that the gas phase acidity (GPA) of the bridging silanol is 43.2 kcal mol−1 lower than the GPA of H2SO4, but higher than the strongest carborane acids, suggesting that deprotonated 1 would be a WCA. Reactions of 1 with NOct3 show that 1 forms weaker ion-pairs than classical WCAs, but stronger ion-pairs than carborane or borate anions. Though 1 forms stronger ion-pairs than these state-of-the-art WCAs, 1 reacts with alkylsilanes to form silylium type surface species. To the best of our knowledge, this is the first example of a silylium supported on derivatized silica.
Si–OH⋯Al(ORF)3 (1). DFT calculations using small clusters to model 1 show that the gas phase acidity (GPA) of the bridging silanol is 43.2 kcal mol−1 lower than the GPA of H2SO4, but higher than the strongest carborane acids, suggesting that deprotonated 1 would be a WCA. Reactions of 1 with NOct3 show that 1 forms weaker ion-pairs than classical WCAs, but stronger ion-pairs than carborane or borate anions. Though 1 forms stronger ion-pairs than these state-of-the-art WCAs, 1 reacts with alkylsilanes to form silylium type surface species. To the best of our knowledge, this is the first example of a silylium supported on derivatized silica.
Introduction
The development of inert Weakly Coordinating Anions (WCAs) was critical to isolate very reactive electrophilic species.1 Studies of superacid media resulted in the first generation of WCAs (CF3SO3−, PF6−, SbF6−, etc., Fig. 1).2 The first generation WCAs continue to find broad applications in the synthetic community, but these anions are too reactive or coordinating to stabilize highly reactive cations. For example, organometallic Zr(IV) cations, key 14-electron intermediates in the synthesis of polyolefins, are incompatible with first generation WCAs.3 These anions are also not sufficiently weakly coordinating to form R3Si+ cations.4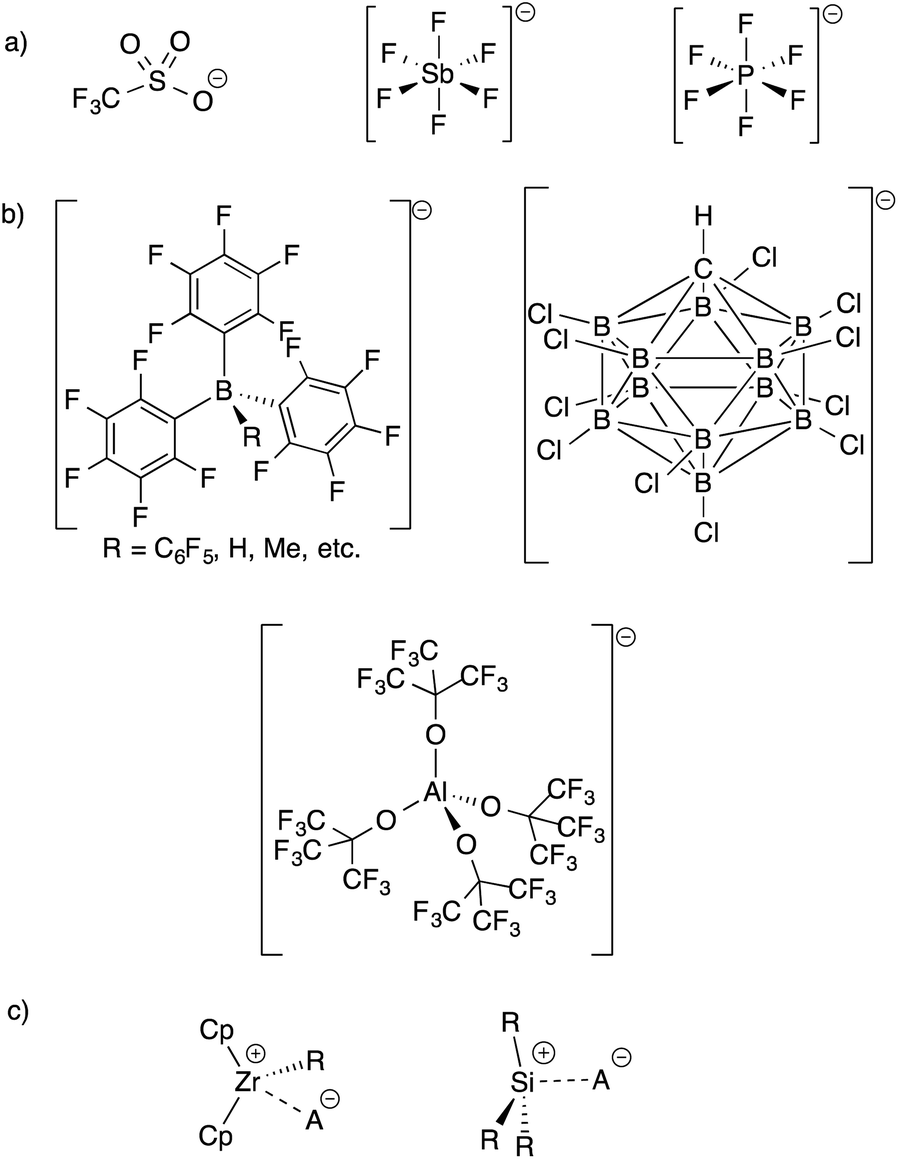 | ||
| Fig. 1 Structures of first generation WCAs (a) and bulky inert WCAs (b); very strong electrophiles that are too reactive to form with first generation WCAs (c). | ||
Fluorinated borates (e.g.−B(C6F5)4, −B(3,5-(CF3)2–C6H3)4),5 aluminates (e.g.−Al(OC(CF3)3)4),6 or carborane anions (e.g.−CHB11H6X5, −CHB11X11; X = halide),7 shown in Fig. 1, stabilize organometallic Zr(IV) cations or R3Si+. The anions are designed to delocalize charge throughout the structure of the WCA, which results in low basicity. The conjugate acids of the WCAs shown in Fig. 1, when isolable, are the strongest known Brønsted acids.8 The strong C–F or B–X bonds in these WCAs also provide some degree of chemical inertness, which is important in reactions involving the strong electrophiles mentioned above.
Direct translation of these concepts to well-defined heterogeneous catalysts is more challenging. Well-defined heterogeneous catalysts are desirable because the molecular structure of a catalytically active site can be determined using spectroscopic methods,9–11 which provides opportunities to optimize the properties of these catalysts based on the structure of the active site. The largest class of well-defined heterogeneous catalysts are supported on SiO2 partially dehydroxylated at 700 °C. Well-defined sites supported on SiO2 generally do not form ion-pairs but rather SiO–MLn. For example, the 29Si Cross Polarization Magic Angle Spinning (CPMAS) NMR spectrum of alkylsilane functionalized silica (SiO–SiMe3, Fig. 2a) contains a signal at 14 ppm for the alkylsilane fragment, which is inconsistent with formation of a Me3Si+ species on the silica surface.12–15 Similarly, Cp*ZrMe3 (Cp* = pentamethylcyclopentadienyl) reacts with partially dehydroxylated SiO2 to form SiO–Zr(Cp*)Me2 (Fig. 2a),16 which is inactive in the polymerization of ethylene. However, SiO–Zr(Cp*)Me2 does react with B(C6F5)3 to form electrophilic ion-pairs that are active in the polymerization of ethylene.17 Silica surfaces can also form strong ion-pars with between surface siloxide anions and tetraalkylphosphonium groups.18,19
 | ||
| Fig. 2 Formation of R3Si–Ox and Cp*Zr(Me)2–Ox (Ox = surface oxygen) on partially dehydroxylated SiO2 (a); reaction of organometallics with partially dehydroxylated Al2O3 (* = Lewis acid site), minor product are ion-pairs (b); formation of electrophilic ion pairs on sulfated oxides (c). | ||
Partially dehydroxylated Al2O3 contains a very small quantity of tri-coordinate Al Lewis-acid sites20 that react with organometallic complexes to form electrophilic ion-pairs, Fig. 2b.21,22 However, the surface coverage of the –OH sites is much higher than the surface coverage of Lewis sites, resulting in low active site loadings in these well-defined catalysts.23
The trends in WCAs described above suggest that oxides containing more acidic –OH sites may be more weakly coordinating. Zeolites contain –OH sites that are more acidic than –OH sites on SiO2, and can support organometallic species.24 Studies of well-defined organometallics are limited to small molecules because SiO2/Al2O3 zeolite materials have small pore sizes. Oxides treated with sulfuric acid, sulfated oxides, were claimed to contain superacid Brønsted acid sites.25 This relates sulfated oxides to first generation WCAs, and several studies showed that sulfated oxides form electrophilic ion pairs with organometallics (Fig. 2c).26–31 However, titrations of the –OH sites on sulfated oxides with phosphines are inconsistent with superacid –OH sites.32 This data is consistent with DFT calculations showing that sulfated oxides are weaker acids than zeolites.33 Detailed studies showed that sulfated oxides also contain significant amounts of oxidative pyrosulfate sites, which can result in undesirable side reactions with organometallic substrates.34
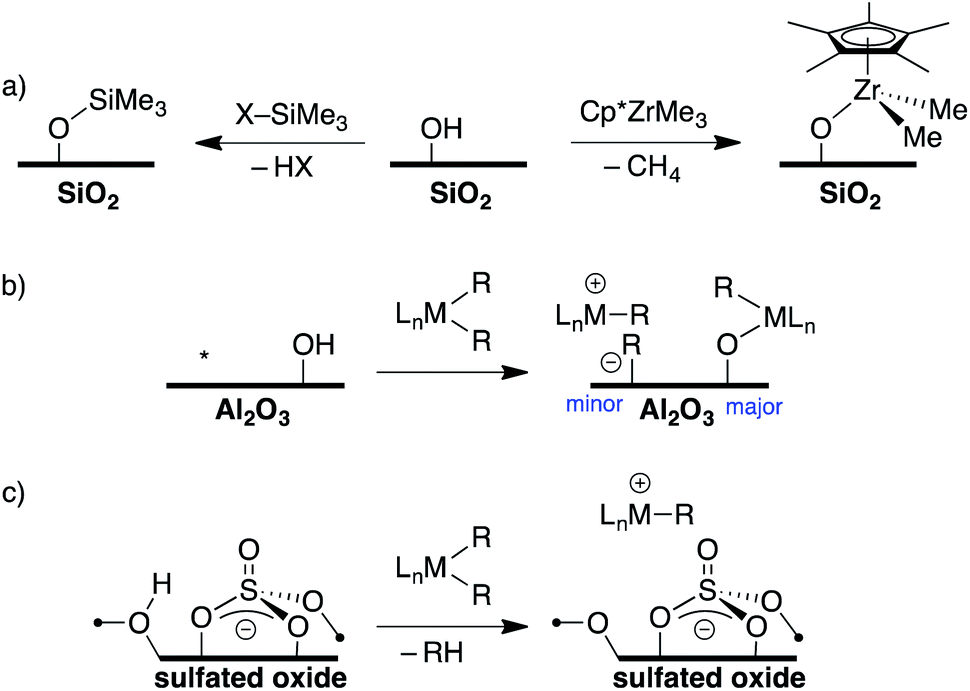
The reaction of a strong Lewis acid and a Si–OH on partially dehydroxylated silica should form a strong Brønsted acid site (Fig. 3a).35 Deprotonation of the strong Brønsted acid should result in a weakly coordinating anion that may stabilize electrophilic surface species that would not typically form on SiO2 surfaces, and also translate solution WCA concepts to heterogeneous supports. Contacting dehydroxylated silica with AlCl3 forms strong Brønsted acid sites, but also results in various side reactions leading to strong Lewis sites on the silica surface,36 which is common in this class of functionalized oxides.37 Redox inactive strong Lewis acids, such as B(C6F5)3, are not sufficiently Lewis acidic to form stable bridging silanols with silica (Fig. 3b).38 However, B(C6F5)3 reacts with silica and aniline bases to form ion-pairs that are capable of activating organometallic species,39,40 or with exogenous H2O to form grafted species on the SiO2 surface.41 This paper describes the reaction of Al(ORF)3*PhF (R = C(CF3)3)42 with silica partially dehydroxylated at 700 °C (SiO2-700) to generate Si–OH⋯Al(ORF)3 (1, Fig. 3c). Calculated gas phase acidity (GPA) of 1 shows that the activated silanols are very strong Brønsted acids. Reactions of 1 with silane reagents result in the formation of [R3Si][Si–O⋯Al(ORF)3], a rare example of a silylium supported on SiO2.
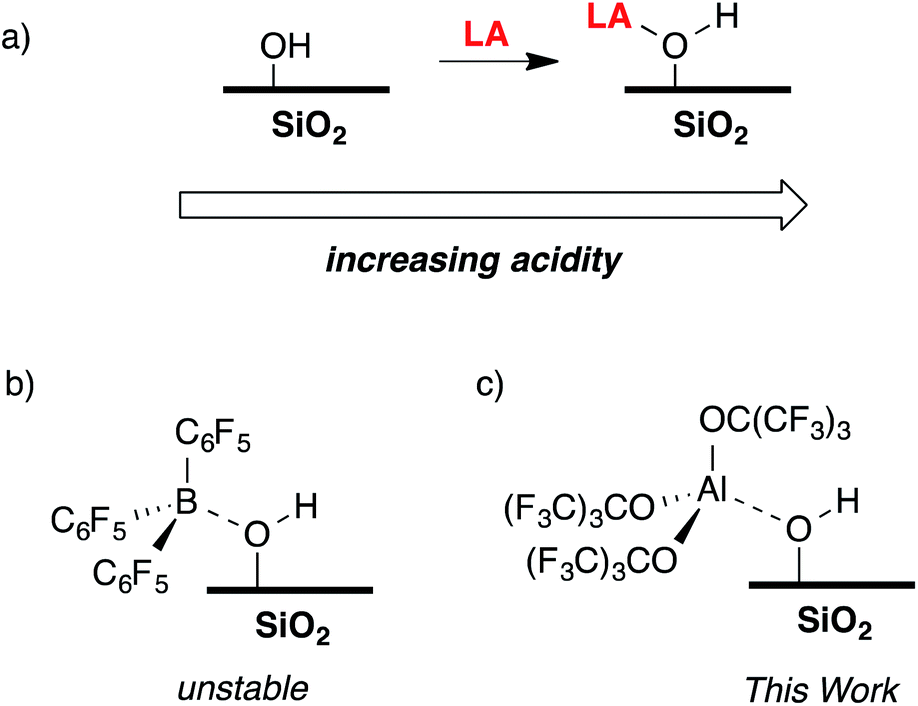 | ||
| Fig. 3 The reaction of a Lewis acid with silica to form a bridging silanol (a); B(C6F5)3 reacts with silica to form unstable bridging silanols (b); the focus of this work, generation of Si–OH⋯Al(ORF)3 (c). | ||
Results and discussion
Reaction of Al(ORF)3*PhF with partially dehydroxylated SiO2
A perfluorohexane slurry of SiO2-700 (0.26 mmol OH g−1) reacts with Al(ORF)3*PhF to formSi–OH⋯Al(ORF)3 (1, Fig. 4a). ICP-OES analysis shows that 0.24 mmol g−1 is present in 1, indicating that most of the silanols in SiO2-700 are coordinated to Al(ORF)3. The FTIR spectrum of 1, shown in Fig. 4b, contains a new red-shifted νOH at 3542 cm−1 that is typical of bridging silanols in silica–alumina materials. This spectrum also contains a νOH corresponding to silanols that do not form adducts with Al(ORF)3. Weak sp2–νCH and νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C are also present, suggesting that some fluorobenzene remains adsorbed to 1. Consistent with this observation, 19F{1H} NMR measurements of 1 suspended in CD3CN show that 0.045 ± 0.004 mmol g−1 of PhF leaches off the silica surface (Fig. S19†).
C are also present, suggesting that some fluorobenzene remains adsorbed to 1. Consistent with this observation, 19F{1H} NMR measurements of 1 suspended in CD3CN show that 0.045 ± 0.004 mmol g−1 of PhF leaches off the silica surface (Fig. S19†).
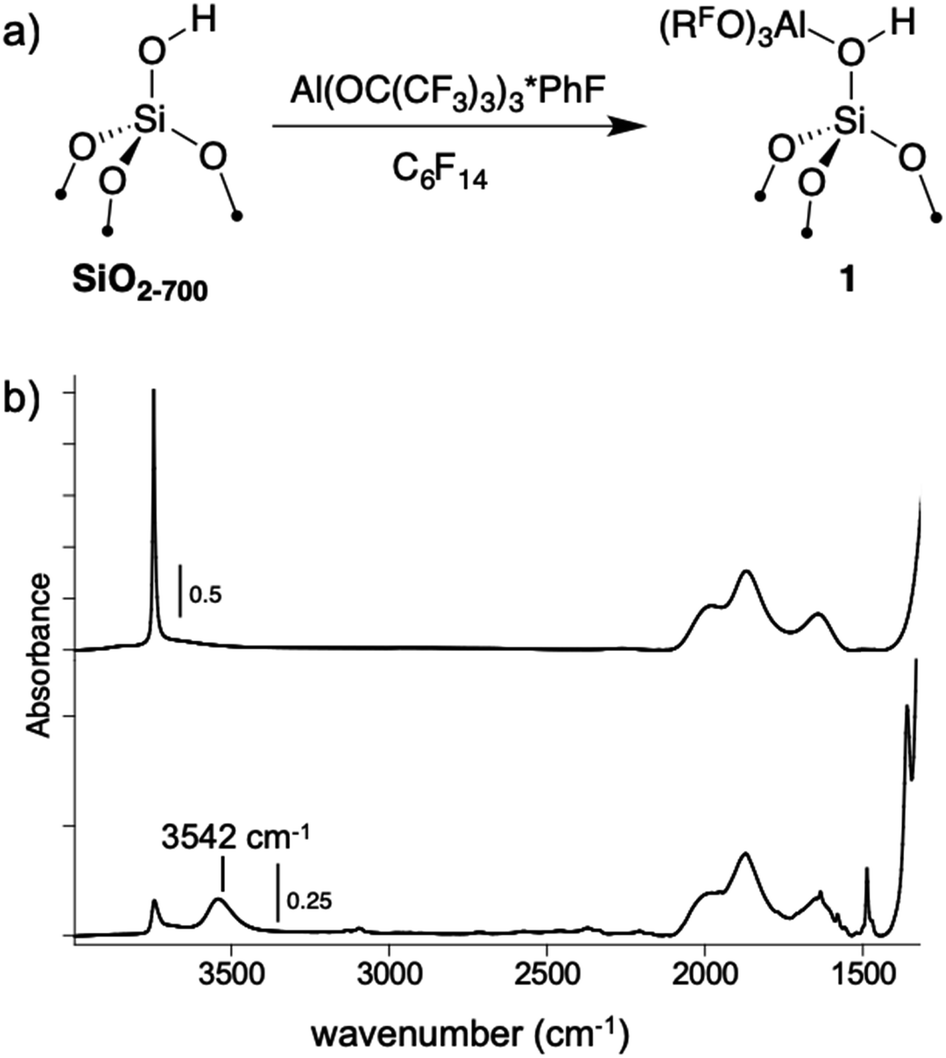 | ||
| Fig. 4 The reaction of Al(ORF)3*PhF with SiO2-700 in perfluorohexane (a); FTIR spectra of SiO2-700 (top) and 1 (bottom, b). | ||
The static 27Al NMR spectrum of 1 contains a typical quadrupolar powder pattern that can be simulated with a single site (Fig. 5a).43 The isotropic chemical shift (δiso = 43 ppm) and large quadrupolar coupling constant (CQ = 14.6 MHz) is consistent with a highly distorted tetrahedral Al coordination environment. These values are in agreement with those obtained from 27Al MAS measurements of 1 (Fig. S4†). The 1H magic angle spinning (MAS) NMR spectrum of 1 contains signals at 7.1 (adsorbed PhF), 5.0 (Si–OH⋯Al(ORF)3), and 2.3 (Si–OH) ppm (Fig. 5b, top trace). A 1H dipolar double-quantum single-quantum (DQ-SQ) spectrum does not show crosspeaks between adsorbed PhF and the bridging silanol (see the ESI, Fig. S5†), suggesting that adsorbed PhF is distant from the acidic silanol in 1. A 2D 1H{27Al} D-RINEPT spectrum shows that the 27Al signal in 1 correlates to the acidic silanol (Si–OH⋯Al(ORF)3) signal at 5.0 ppm (Fig. S7†), supporting these assignments.
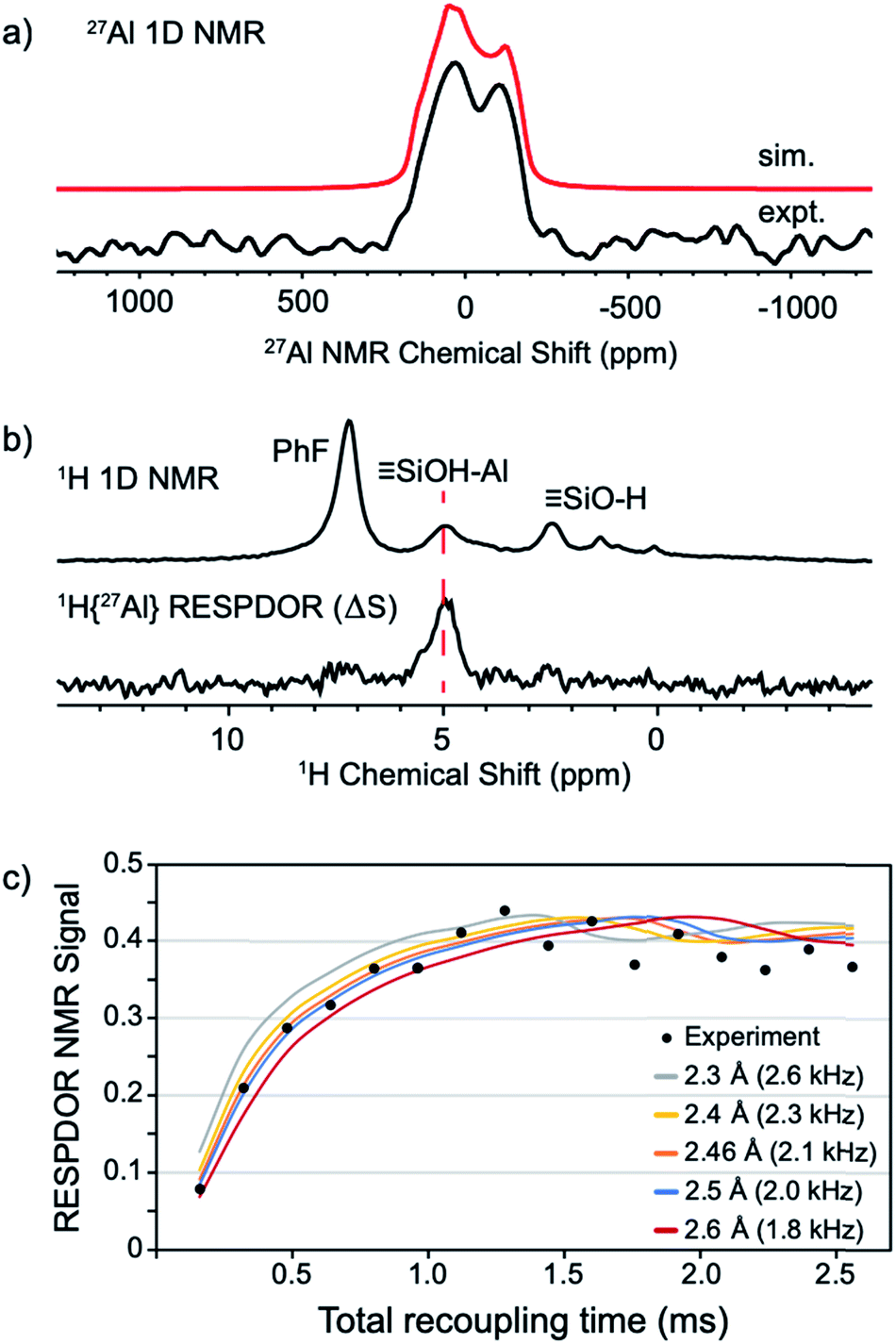 | ||
| Fig. 5 Static 27Al solid-state NMR spectrum of 1 acquired at 14.1 T (a); solid-state 1D 1H MAS NMR (top) and 1H{27Al} RESPDOR difference spectrum of 1 (middle, b); fit of RESPDOR dipolar dephasing curve to measure dipolar 1H–27Al dipolar coupling present in 1 (bottom, c). See the ESI† for Experimental details. | ||
The 1H{27Al} Resonance-Echo Saturation-Pulse Double-Resonance (RESPDOR)44,45 NMR experiment allows measurement of the dipolar coupling constant for 1H and 27Al spins. The 1H–27Al dipolar coupling constant is inversely proportional to the cube of the inter-atomic distance, so only 1H and 27Al spins that are in close spatial proximity (<5 Å) will be affected in this experiment. The 1H{27Al}-RESPDOR difference NMR spectrum (ΔS) is shown in Fig. 5b (bottom) and contains a single 1H NMR signal at 5.0 ppm (Si–OH⋯Al(ORF)3). This result indicates that the bridging silanol is close to the aluminum in Al(ORF)3, and that the signals at 7.1 ppm (PhF) and 2.3 ppm (Si–OH) are from protons distant from aluminum, as expected. Variation of recoupling times in the 1H{27Al}-RESPDOR pulse sequence, and numerical simulation of the RESPDOR dipolar dephasing curve allows the 1H–27Al dipolar coupling constant to be determined. These data are given in Fig. 5c, and show that the 1H–27Al dipolar coupling is ∼2.0–2.3 kHz, which corresponds to Al–OH distances in the range of 2.4–2.5 Å. This distance is in good agreement with structural models predicted by DFT (see below).
DFT studies of small cluster models of 1
1 was modeled using Al(ORF)3 and the –SiH3 capped polysesquisiloxane cluster46 at the B3LYP/6-31G(d,p) level of theory. The cluster 1-DFT is shown in Fig. 6. Al(ORF)3 in 1-DFT coordinates to the isolated silanol in the cluster and not Si–O–Si bridges. The aluminum fragment in 1-DFT adopts a distorted tetrahedral geometry, and the Al–OH distance in 1-DFT is 1.91 Å. The terminal Si–O distance is 1.70 Å, slightly longer than the average Si–O distances (1.62 Å) in the cluster. These observations are similar to those obtained for alcohol adducts of Al(ORF)3.47 The predicted Al–OH distance is 2.46 Å, and is in good agreement with an estimated Al–OH distances determined with the 1H{27Al} RESPDOR experiment.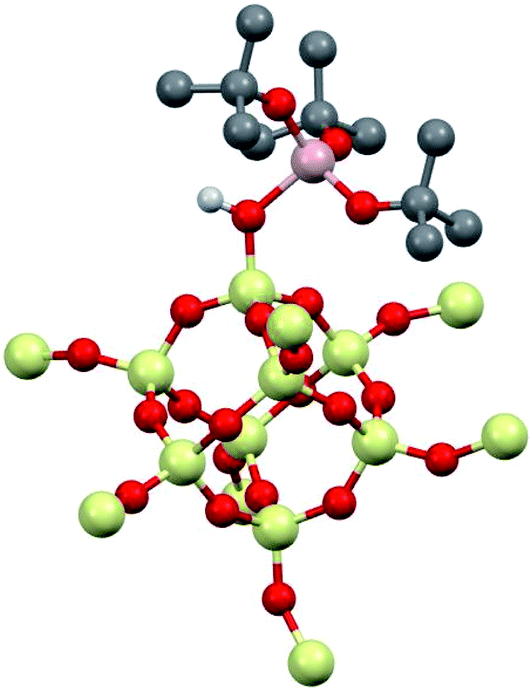 | ||
| Fig. 6 1-DFT with selected hydrogens and fluorines hidden for clarity. Selected distances (Å) and angles (deg.): O–H (0.98 Å), Al–OH (1.91 Å), Al–OH (2.46 Å) Al–ORF (1.74 Å), Si–OH (1.70 Å), Si–Ocluster (1.60–1.63 Å), H–O–Al 113°, H–O–Si 116°, Si–O–Al 128°, O–Si–O(H) 105°. | ||
The calculated IR spectrum of 1-DFT predicts a νOH at 3550 cm−1 (expt. νOH = 3542 cm−1). NMR calculations at the M06L/Al(6-311G(d,p)), 6-31G(d,p) level of theory predict that the acidic proton appears at 5.1 ppm, and that the 27Al CQ is 15.3 MHz. These values agree well with those obtained experimentally for 1, and are similar to those obtained for molecular H[Al(OC(CF3)3)4].47
Quantitative measurement of Brønsted acidity on oxides is challenging.48–50 Gas-phase acidity (GPA) can be calculated using DFT methods, and is reasonably accurate for small molecules. Table 1 gives the GPA of various mineral acids at BP86/def2-TZVP to calibrate the accuracy of this level of theory. The GPA of HCl is 334.5 kcal mol−1, which is very close to the experimental value (333.6 kcal mol−1). In general, we find good agreement between experimental and calculated values. The calculated deprotonation energy of 1-DFT is 262.7 kcal mol−1. For comparison, small clusters of Si–OH⋯Al(OMe)3, simplified models for bridging silanols in SiO2/Al2O3, were also calculated at this level of theory and have deprotonation energy of 279–299 kcal mol−1 (see the ESI† for details). These values are similar to those calculated for more complex models of zeolities,51–54 indicating that 1-DFT is more acidic than bridging silanols in silica/aluminas. However, 1-DFT is clearly a weaker acid than H[Al(OC(CF3)3)4] (GPA = 262.7 kcal mol−1) or the H[CHB11Cl11] carborane acid (GPA = 239.0 kcal mol−1). The strong Brønsted acidity of 1 suggests that the conjugate base of the bridging silanol may behave as a weakly coordinating anion.
| Acid | Expt. GPA (kcal mol−1) | Calc'd GPA (kcal mol−1) |
|---|---|---|
| HCl | 336.2 | 334.5 |
| HBr | 318.3 | 321.6 |
| HI | 309.2 | 305.9 |
| H2SO4 | 302.2 | 305.9 |
| HSO3F | 299.8 | 294.6 |
| Zeolite | — | 279–299 |
| HSO3CF3 | 299.5 | 293.3 |
| 1-DFT | — | 262.7 |
| H[Al(OC(CF3)3)4] | — | 248.8 (ref. 47) |
| H[CHB11Cl11] | — | 239.1 |
Formation of ion-pairs with 1
The most common experimental method to assess the ion-pairing on a solid involves adsorption of a probe molecule to the solid and measuring the change in a spectroscopic observable, usually Δν by FTIR or Δδ by NMR spectroscopy. However, solution 19F{1H} NMR studies indicate adsorption of common probes (pyridine or triethylphosphine oxide) or heteroatom containing solvents (CD3CN, Et2O, or CH2Cl2) to 1 results in desorption of solvated Al(ORF)3 from the silica surface.Reed and co-workers described the properties of [Oct3NH][X] contact ion pairs in CCl4 solution.55 The νNH stretch from FTIR measurements provides information about ion-pairing in [Oct3NH][X]. In a H-bonded contact ion-pair, weaker NH⋯X interactions will result in higher νNH stretching frequencies. The νNH values for selected [Oct3NH][X] contact ion pairs in CCl4 solution are given in Table 2.
The reaction of 1 with 0.95 equiv NOct3 in C6H12 at room temperature results in the formation of [Oct3NH][(RFO)3Al–OSi] (2, eqn (1)). Solution 19F NMR spectra of this mixture indicates that desorption of Al(ORF)3 or decomposition of 1 does not occur under these conditions. 2 was characterized by multinuclear solid-state NMR spectroscopy (see the ESI† for details).
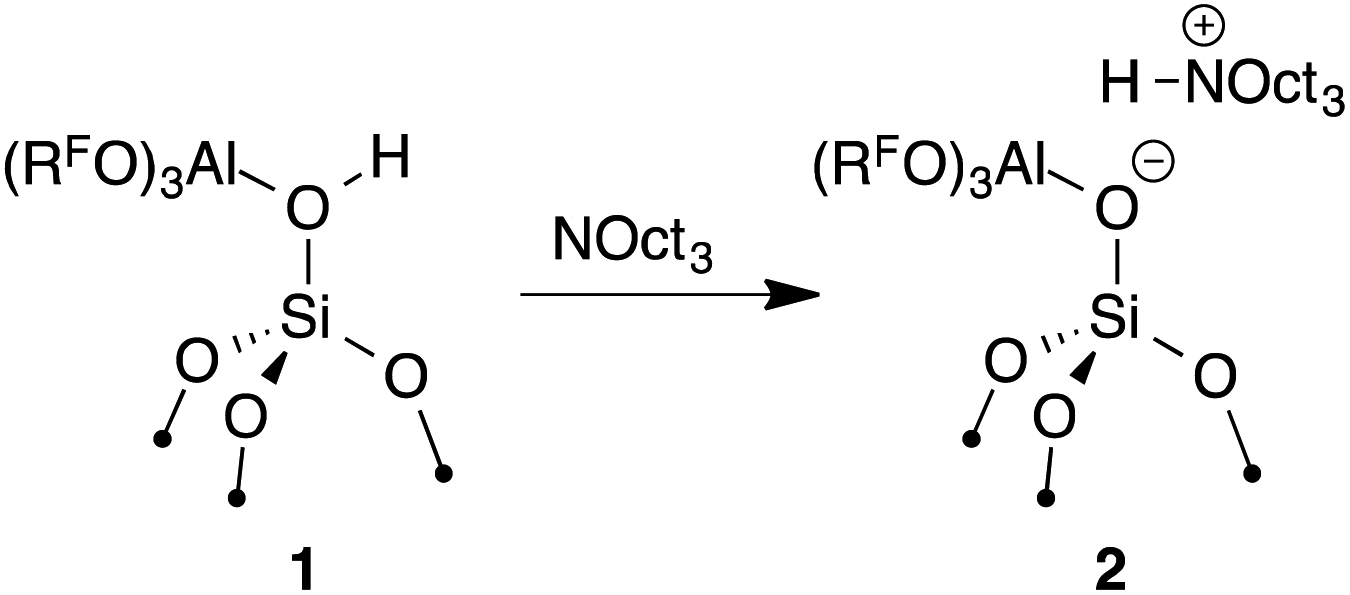 | (1) |
The FTIR spectrum of 2 shows νNH at 3070 cm−1. This result indicates 2 forms a weaker ion pair than [Oct3NH][SO3CF3] (νNH = 2939 cm−1) or [Oct3NH][ClO4] (νNH = 3049 cm−1), common first generation WCAs. This experimental data is consistent with the calculated GPA showing that 2 is a very strong Brønsted acid because strong acids form weak ion pairs. However, 2 forms stronger ion-pairs with [Oct3NH] than carborane or [B(C6F5)4] anions.
Formation of [iPr3Si][(RFO)3Al–OSi![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) ] (3)
] (3)
As mentioned above, R3Si+ ions are not stable in the presence of first generation WCAs because these WCAs either react or bind to the silylium ions. The characteristics of 1 suggest that R3Si+ species may be stable on this surface. The reaction of allyltriisopropylsilane and 1 results in the formation of [iPr3Si][(RFO)3Al–OSi] and small amounts of SiOSiiPr3 (3, Fig. 7a). The FTIR of 3 lacks the strong νOH for the bridging silanol observed in 1 (Fig. 7b). The 29Si CPMAS NMR spectrum of 3 contains a minor signal at 4.0 ppm, which is commonly observed in alkylsilane functionalized silica, and is consistent with the formation of SiOSiiPr3. The major signal in the 29Si CPMAS NMR spectrum is at 70 ppm (Fig. 7c), and is assigned to 3. This chemical shift is typical of R3Si+ fragments interacting with weak ligands. The 29Si chemical shift of [iPr3Si(SO2)][CH6B11Br6] appears at 85 ppm,56 and [Et3Si(toluene)][B(C6F5)4] appears at 94 ppm. Solvents that form stronger complexes with R3Si+ fragments appear at lower chemical shift values. For example, the 29Si chemical shift of [tBu3Si(OH2)][CH6B11Br6] is 46.7 ppm,57 and [iPr3Si(NCCH3)] [CH6B11Br6] appears at 37.2 ppm.58 These results suggest that the iPr3Si+ fragment in 3 is bound to a weaker ligand than MeCN or H2O, but a stronger ligand than toluene or SO2.
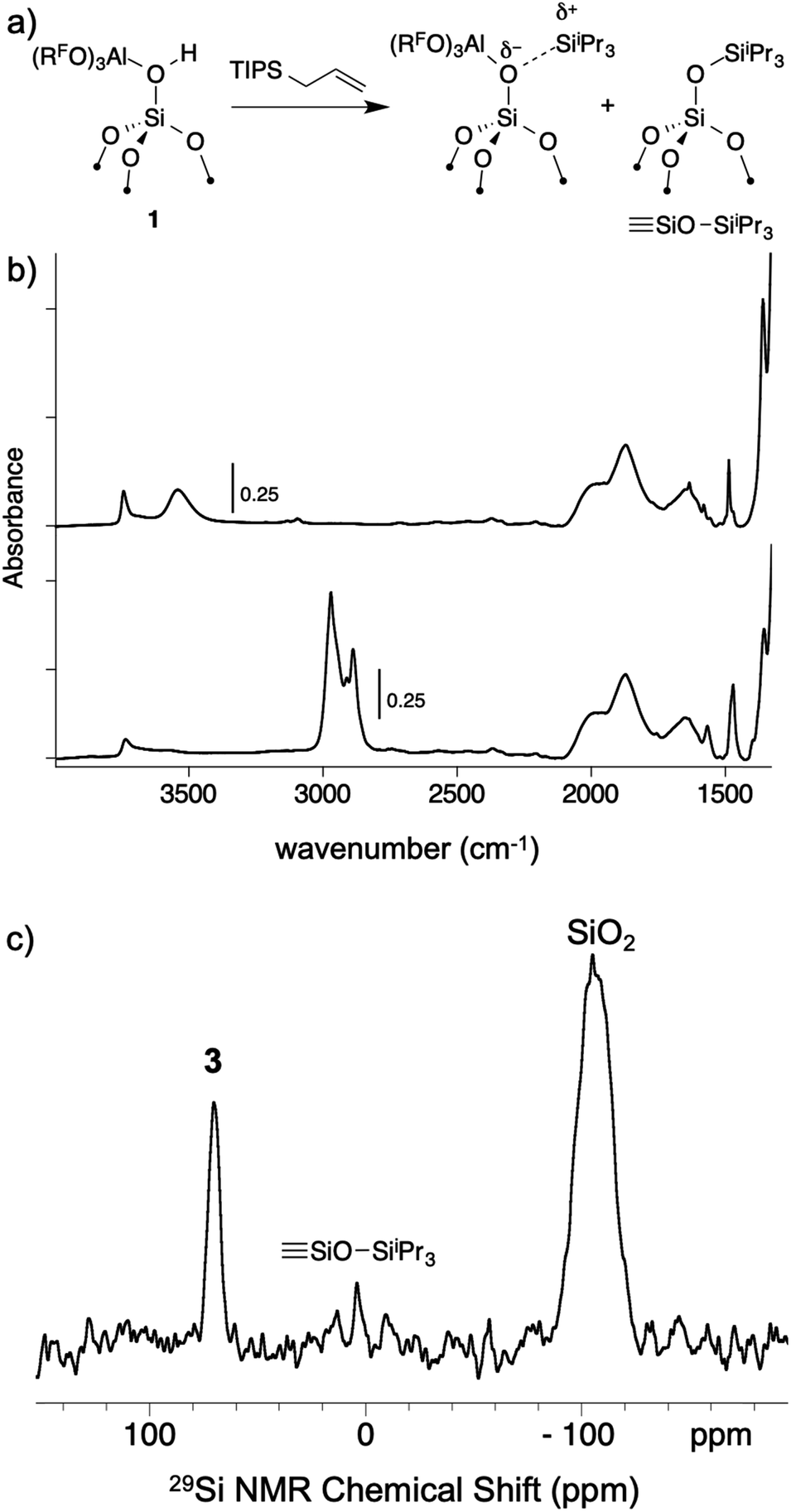 | ||
| Fig. 7 Reactivity of 1 with allyltriisopropylsilane to form 3 and small amounts of SiOSiiPr3 (a); FTIR spectrum of 1 (top) and 3 (bottom, b); 29Si CPMAS NMR spectrum of 3 (c). | ||
The structure of 3 was studied using DFT methods. The optimized structure of the [iPr3Si][(RFO)3Al–OSi] ion pair (3-DFT) at the B3LYP/6-31G(d,p) level of theory is shown in Fig. 8. The calculated 29Si NMR chemical shift of 3-DFT at the M06L/Al(6-311G(d,p)),6-31G(d,p) level of theory is 67 ppm, in good agreement with experimental data. The iPr3Si+ fragment coordinates to the most sterically open Si–O–Si bridge in the polysesquisiloxane model, and does not interact with the C–F bonds on the anionic (RFO)3Al–OSi fragment. The Si–O distance in 3-DFT is 1.86 Å, which is ∼0.1 Å longer than the Si–O bond in [tBu3Si(OH2)][CH6B11Br6].57 The Si is displaced from the plane defined by the three carbon atoms by 0.57 Å, a larger value than typically observed for silylium ions containing carborane anions (∼0.3–0.4 Å).
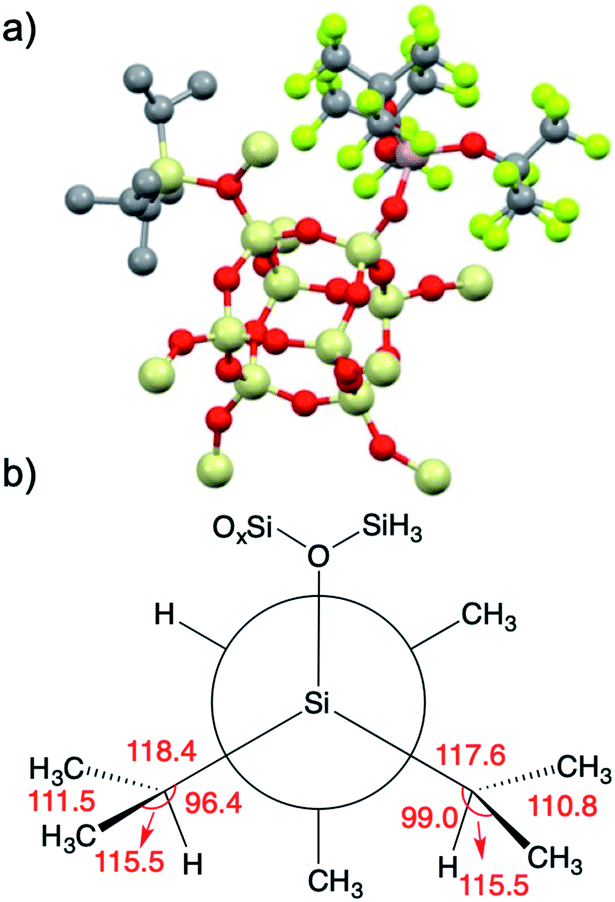 | ||
| Fig. 8 3-DFT with hydrogens hidden for clarity (a); Newman projection of the iPr3Si+ fragment in 3 (b). Selected angles (deg) are given in red and referred to in the text. | ||
iPr3Si+ salts contain σC–H/3p hyperconjugation interactions between the methine C–H group of an isopropyl group and the empty 3pz hybrid orbital on Si.59 The presence of hyperconjugation results in bond angles that deviate from those expected for sp3 geometries. A Newman projection showing the [iPr3Si] fragment in 3 is shown in Fig. 8b. The Si–C–H bond angles in two of the iPr units are 96.4° and 99.0°, respectively. These values are lower than the expected 109.5° expected for sp3 carbon, and is suggestive of σC–H/3p hyperconjugative interactions in 3-DFT. The sum of bond angles around these isopropyl carbons (ΣC–C–X; X = C or Si) are 345.4° and 343.9°, respectively. Similar trends in bond angles were observed in the solid-state structure of [iPr3Si][CH6B11Br6].59 The third isopropyl has bond angles closer to those expected for sp3 carbon (Si–C–H = 103.4°; ΣC–C–X = 339°).
A scale of 29Si NMR chemical shift for selected iPr3Si–X species is shown in Fig. 9. The 29Si NMR chemical shift of triisopropylsilane is 11 ppm, while triisopropylsilyltriflate has a chemical shift of 41 ppm. R3Si+ salts containing carborane anions are more deshielded with respect to these species, appearing between 97 ppm for [iPr3Si][CH6B11I6] and 115 ppm for [iPr3Si][CH6B11Cl6].60 [Me3Si][EtCB11F11] contains a more weakly coordinating carborane anion and has a 29Si NMR chemical shift of 138 ppm,61 similar to silylium zwitterions.62
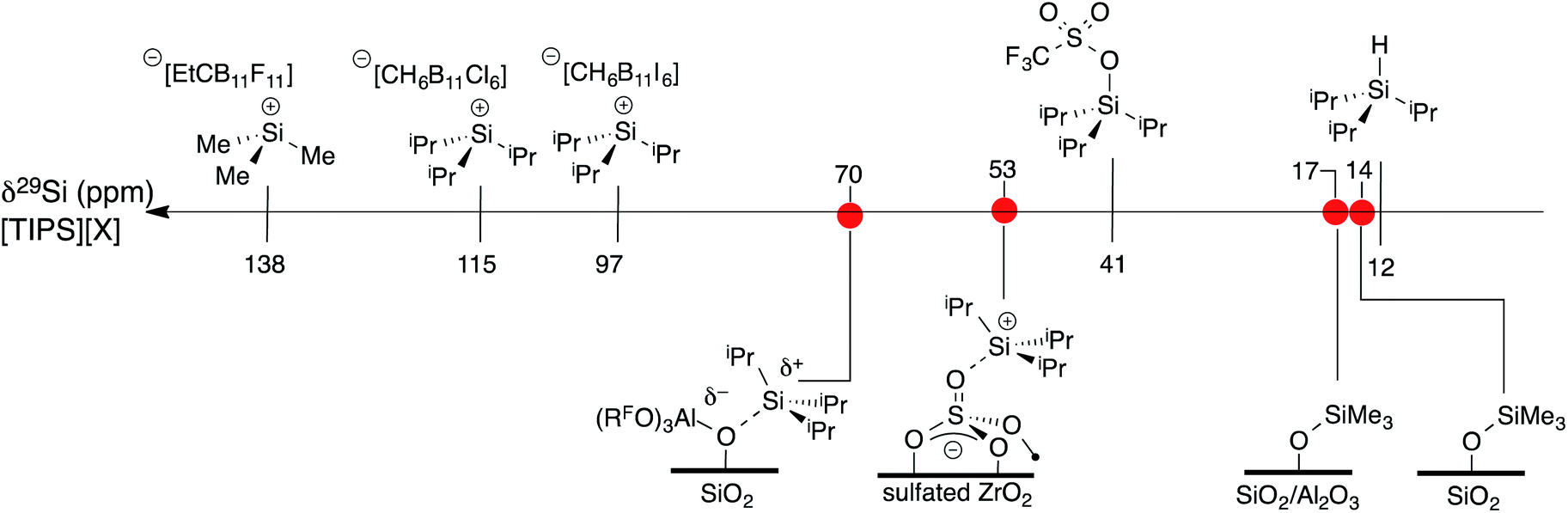 | ||
| Fig. 9 A scale relating buildup of positive charge on silicon to 29Si NMR chemical shift for various WCAs and oxides. | ||
Typical 29Si NMR chemical shifts for alkylsilanes on oxides are also included in Fig. 9. The 29Si CPMAS NMR spectrum of partially dehydroxylated silica containing –OSiMe3 groups contains a 29Si NMR signal at 14 ppm.12–15 The 29Si NMR chemical shift of trimethylsilyl functionalized zeolites appear at 17 ppm.63 These results are inconsistent with a silylium character in these materials. To the best of our knowledge, the only [R3Si][oxide] type species is iPr3Si+ supported on sulfated zirconia (δ29Si = 53 ppm).64 The 29Si NMR chemical shift of 3 is 17 ppm more downfield than that of [iPr3Si][sulfated zirconia].
The data in Fig. 9 indicates that the isotropic 29Si NMR chemical shift of R3Si–X relate to the electronics at silicon.60 A clear comparison is iPr3Si–OTf (δ29Si = 41 ppm) and [iPr3Si][CH6B11Cl6] (δ29Si = 115 ppm). Triflate anions bind to iPr3Si fragments stronger than electron deficient carborane anions, which modulates the Lewis acidity of the iPr3Si-fragment in these compounds because silicon is more positively charged in carborane salts than triflates. This is also reflected in the geometry of the iPr3Si-fragment, which becomes more planar in carborane salts than typical sp3 organosilanes. Less clear was if this trend would also apply to alkylsilanes supported on oxides. The available 29Si chemical shift values for R3Si-supported on silica and silica–alumina suggested that alkylsilanes do not form R3Si+ sites.12–15,63 This is a result of formation of SiO–SiR3 sites on these material surfaces.
Sulfated zirconium oxide and 1 are more acidic than silica or silica alumina based on proton affinity calculations.33 This suggests that these iPr3Si-functionalized materials would contain 29Si NMR chemical shifts more downfield than R3Si-functionalized silica or silica alumina. The 29Si chemical shift of [iPr3Si][sulfated zirconia] (δ29Si = 53 ppm) and 3 (δ29Si = 70 ppm) are consistent with formation of species with R3Si+ character. However, these chemical shifts are far from those of iPr3Si+ carborane salts. These data indicate that 29Si NMR chemical shift trends for molecular R3Si–X also apply to surface species. This implies that the 29Si NMR chemical shift on R3Si-functionalized surfaces gives information about ion-pairing on surfaces sites, which could be important in designing catalytic sites on these weakly coordinating surfaces.
Silylium ions are strong Lewis acids that catalyze or mediate numerous chemical reactions.65–67 Silylium ions activate C–F bonds to form R3Si–F and carbocation intermediates,68–72 which are rapidly quenched in the presence of excess silane to form C–H bonds. 3 activates C–F bonds in 1-adamantylfluoride in the presence of Et3SiH at 0 °C to give adamantane (TON = 18). This reactivity is consistent with silylium character in the iPr3Si+ fragment in 3. However, 3 is less stable than iPr3Si+ sites supported on sulfated zirconia, which gives 160 turnovers in this reaction.64 Solution 19F NMR spectra monitored during the C–F bond activation reaction contain signals for Al(ORF)3 and HOC(CF3)3, indicating that the low stability of 3 is probably related to decomposition reactions of the surface aluminum anion under these conditions (Fig. S20†).
Conclusions
This study shows that design strategies for WCAs in solution can be applied to generate well-defined surface WCAs. The reaction of SiO2-700 with Al(ORF)3*PhF in perfluorohexane formsSi–OH⋯Al(ORF)3 (1) and contains strong Brønsted acid sites based on GPA calculations. Experimental evaluation of the νNH stretch in [Oct3NH][(RFO)3Al–OSi)] (2) shows that this material forms weaker ion-pairs than typical first generation WCAs. 1 reacts with allyltriisopropylsilane to generate [iPr3Si][(RFO)3AlOSi] (3), a rare example of a silylium-like Lewis acid supported on an oxide, and to the best of our knowledge the only example supported on derivatized silica. Though the νNH stretch of 2 and 29Si NMR chemical shift of 3 show that 1 does fulfill the prerequisites to form WCAs, these data also suggest that ion-pairing on these surface sites is stronger than carborane or [B(C6F5)4] anions. Neutral Lewis acids stronger than Al(ORF)3 are necessary to form strong Brønsted acids with partially dehydroxylated silica to form weaker coordinating anions than 1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. C. is a member of the UCR Center for Catalysis. This work was supported by NSF CHE-1800561 (M. P. C.). Solid-state NMR measurements were recorded on an instrument supported by NSF CHE-1626673. A. J. R. acknowledges the Donors of the American Chemical Society Petroleum Research Fund (58627-DNI6) for supporting solid-state NMR measurements. Computations were performed using the computer clusters and data storage resources of the HPCC at UC Riverside, which were funded by grants from NSF (MRI-1429826) and NIH (1S10OD016290-01A1).References
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- G. A. Olah, G. K. Surya Prakash, Á. Molnár and J. Sommer, in Superacid Chemistry, John Wiley & Sons, Inc., 2008, ch. 1, pp. 1–34, DOI:10.1002/9780470421604.
- R. F. Jordan, W. E. Dasher and S. F. Echols, J. Am. Chem. Soc., 1986, 108, 1718–1719 CrossRef CAS.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 325–332 CrossRef CAS.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- A. B. A. Rupp and I. Krossing, Acc. Chem. Res., 2015, 48, 2537–2546 CrossRef CAS PubMed.
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed.
- C. A. Reed, Acc. Chem. Res., 2013, 46, 2567–2575 CrossRef CAS PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- S. L. Wegener, T. J. Marks and P. C. Stair, Acc. Chem. Res., 2011, 45, 206–214 CrossRef PubMed.
- M. P. Conley, C. Copéret and C. Thieuleux, ACS Catal., 2014, 4, 1458–1469 CrossRef CAS.
- C. Zapilko, M. Widenmeyer, I. Nagl, F. Estler, R. Anwander, G. Raudaschl-Sieber, O. Groeger and G. Engelhardt, J. Am. Chem. Soc., 2006, 128, 16266–16276 CrossRef CAS.
- J. Bluemel, J. Am. Chem. Soc., 1995, 117, 2112–2113 CrossRef CAS.
- K. D. Behringer and J. Blümel, J. Liq. Chromatogr. Relat. Technol., 1996, 19, 2753–2765 CrossRef CAS.
- M. Jezequel, V. r. Dufaud, M. J. Ruiz-Garcia, F. Carrillo-Hermosilla, U. Neugebauer, G. P. Niccolai, F. d. r. Lefebvre, F. β. Bayard, J. Corker, S. Fiddy, J. Evans, J.-P. Broyer, J. Malinge and J.-M. Basset, J. Am. Chem. Soc., 2001, 123, 3520–3540 CrossRef CAS PubMed.
- N. Millot, S. Soignier, C. C. Santini, A. Baudouin and J.-M. Basset, J. Am. Chem. Soc., 2006, 128, 9361–9370 CrossRef CAS PubMed.
- Y. Yang, B. Beele and J. Blümel, J. Am. Chem. Soc., 2008, 130, 3771–3773 CrossRef CAS PubMed.
- J. Sommer, Y. Yang, D. Rambow and J. Blümel, Inorg. Chem., 2004, 43, 7561–7563 CrossRef CAS PubMed.
- R. Wischert, P. Laurent, C. Copéret, F. Delbecq and P. Sautet, J. Am. Chem. Soc., 2012, 134, 14430–14449 CrossRef CAS PubMed.
- J. Joubert, F. Delbecq, P. Sautet, E. L. Roux, M. Taoufik, C. Thieuleux, F. Blanc, C. Copéret, J. Thivolle-Cazat and J.-M. Basset, J. Am. Chem. Soc., 2006, 128, 9157–9169 CrossRef CAS.
- P. J. Toscano and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 653–659 CrossRef CAS.
- T. J. Marks, Acc. Chem. Res., 1992, 25, 57–65 CrossRef CAS.
- J. Guzman and B. C. Gates, Dalton Trans., 2003, 3303–3318, 10.1039/b303285j.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1980, 851–852, 10.1039/c39800000851.
- D. B. Culver, H. Tafazolian and M. P. Conley, Organometallics, 2018, 37, 1001–1006 CrossRef CAS.
- H. Tafazolian, D. B. Culver and M. P. Conley, Organometallics, 2017, 36, 2385–2388 CrossRef CAS.
- D. M. Kaphan, R. C. Klet, F. A. Perras, M. Pruski, C. Yang, A. J. Kropf and M. Delferro, ACS Catal., 2018, 8, 5363–5373 CrossRef CAS.
- M. Stalzer, M. Delferro and T. Marks, Catal. Lett., 2015, 145, 3–14 CrossRef CAS.
- W. Gu, M. M. Stalzer, C. P. Nicholas, A. Bhattacharyya, A. Motta, J. R. Gallagher, G. Zhang, J. T. Miller, T. Kobayashi, M. Pruski, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 6770–6780 CrossRef CAS PubMed.
- Z. H. Syed, D. M. Kaphan, F. A. Perras, M. Pruski, M. S. Ferrandon, E. C. Wegener, G. Celik, J. Wen, C. Liu, F. Dogan, K. I. Goldberg and M. Delferro, J. Am. Chem. Soc., 2019, 141, 6325–6337 CrossRef CAS PubMed.
- J. Rodriguez, D. B. Culver and M. P. Conley, J. Am. Chem. Soc., 2019, 141, 1484–1488 CrossRef CAS PubMed.
- F. Haase and J. Sauer, J. Am. Chem. Soc., 1998, 120, 13503–13512 CrossRef CAS.
- R. C. Klet, D. M. Kaphan, C. Liu, C. Yang, A. J. Kropf, F. A. Perras, M. Pruski, A. S. Hock and M. Delferro, J. Am. Chem. Soc., 2018, 140, 6308–6316 CrossRef CAS PubMed.
- J. Sauer, J. Mol. Catal., 1989, 54, 312–323 CrossRef CAS.
- T. Xu, N. Kob, R. S. Drago, J. B. Nicholas and J. F. Haw, J. Am. Chem. Soc., 1997, 119, 12231–12239 CrossRef CAS.
- G. A. Olah, G. K. S. Prakash, J. Sommer and A. Molnar, Superacid Chemistry, John Wiley & Sons, 2009 Search PubMed.
- N. Millot, C. C. Santini, F. d. r. Lefebvre and J.-M. Basset, C. R. Chim., 2004, 7, 725–736 CrossRef CAS.
- J. F. Walzer, US Pat., 5,643,847, 1995.
- N. Millot, C. C. Santini, A. Baudouin and J.-M. Basset, Chem. Commun., 2003, 2034–2035, 10.1039/b304047j.
- Y.-J. Wanglee, J. Hu, R. E. White, M.-Y. Lee, S. M. Stewart, P. Perrotin and S. L. Scott, J. Am. Chem. Soc., 2012, 134, 355–366 CrossRef CAS PubMed.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed.
- S. E. Ashbrook and S. Sneddon, J. Am. Chem. Soc., 2014, 136, 15440–15456 CrossRef CAS PubMed.
- Z. Gan, Chem. Commun., 2006, 4712–4714, 10.1039/b611447d.
- L. Chen, Q. Wang, B. Hu, O. Lafon, J. Trébosc, F. Deng and J.-P. Amoureux, Phys. Chem. Chem. Phys., 2010, 12, 9395–9405 RSC.
- I. Del Rosal, I. C. Gerber, R. Poteau and L. Maron, J. Phys. Chem. A, 2010, 114, 6322–6330 CrossRef CAS PubMed.
- A. Kraft, J. Beck, G. Steinfeld, H. Scherer, D. Himmel and I. Krossing, Organometallics, 2012, 31, 7485–7491 CrossRef CAS.
- W. E. Farneth and R. J. Gorte, Chem. Rev., 1995, 95, 615–635 CrossRef CAS.
- J. F. Haw, Phys. Chem. Chem. Phys., 2002, 4, 5431–5441 RSC.
- A. Zheng, S.-B. Liu and F. Deng, Chem. Rev., 2017, 117, 12475–12531 CrossRef CAS PubMed.
- M. Rybicki and J. Sauer, Catal. Today, 2019, 323, 86–93 CrossRef CAS.
- A. J. Jones and E. Iglesia, ACS Catal., 2015, 5, 5741–5755 CrossRef CAS.
- H. Fang, A. Zheng, Y. Chu and F. Deng, J. Phys. Chem. C, 2010, 114, 12711–12718 CrossRef CAS.
- A. Zheng, H. Zhang, X. Lu, S.-B. Liu and F. Deng, J. Phys. Chem. B, 2008, 112, 4496–4505 CrossRef CAS PubMed.
- E. S. Stoyanov, K.-C. Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 8500–8508 CrossRef CAS PubMed.
- S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767–769, 10.1039/b511344j.
- Z. Xie, R. Bau and C. A. Reed, J. Chem. Soc., Chem. Commun., 1994, 2519–2520, 10.1039/c39940002519.
- Z. Xie, D. J. Liston, T. Jelínek, V. Mitro, R. Bau and C. A. Reed, J. Chem. Soc., Chem. Commun., 1993, 384–386, 10.1039/c39930000384.
- Z. Xie, R. Bau, A. Benesi and C. A. Reed, Organometallics, 1995, 14, 3933–3941 CrossRef CAS.
- Z. Xie, J. Manning, R. W. Reed, R. Mathur, P. D. W. Boyd, A. Benesi and C. A. Reed, J. Am. Chem. Soc., 1996, 118, 2922–2928 CrossRef CAS.
- T. Küppers, E. Bernhardt, R. Eujen, H. Willner and C. W. Lehmann, Angew. Chem., Int. Ed., 2007, 46, 6346–6349 CrossRef.
- R. Ramírez-Contreras, N. Bhuvanesh, J. Zhou and O. V. Ozerov, Angew. Chem., Int. Ed., 2013, 52, 10313–10315 CrossRef PubMed.
- W. Song, D. M. Marcus, S. M. Abubakar, E. Jani and J. F. Haw, J. Am. Chem. Soc., 2003, 125, 13964–13965 CrossRef CAS PubMed.
- D. B. Culver and M. P. Conley, Angew. Chem., Int. Ed., 2018, 57, 14902–14905 CrossRef CAS PubMed.
- H. F. T. Klare, ACS Catal., 2017, 7, 6999–7002 CrossRef CAS.
- H. F. T. Klare and M. Oestreich, Dalton Trans., 2010, 39, 9176–9184 RSC.
- Y. Seo and M. R. Gagné, ACS Catal., 2018, 8, 6993–6999 CrossRef CAS.
- C. Douvris, C. M. Nagaraja, C. H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed.
- V. J. Scott, R. Celenligil-Cetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852–2853 CrossRef CAS PubMed.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- S. Duttwyler, C. Douvris, N. L. P. Fackler, F. S. Tham, C. A. Reed, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2010, 49, 7519–7522 CrossRef CAS PubMed.
- B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05904k |
| This journal is © The Royal Society of Chemistry 2020 |