
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-inspired design: bulk iron–nickel sulfide allows for efficient solvent-dependent CO2 reduction†
Stefan
Piontek
a,
Kai
junge Puring
 ab,
Daniel
Siegmund
b,
Mathias
Smialkowski
a,
Ilya
Sinev
c,
David
Tetzlaff
a,
Beatriz
Roldan Cuenya
d and
Ulf-Peter
Apfel
*ab
ab,
Daniel
Siegmund
b,
Mathias
Smialkowski
a,
Ilya
Sinev
c,
David
Tetzlaff
a,
Beatriz
Roldan Cuenya
d and
Ulf-Peter
Apfel
*ab
aInorganic Chemistry I, Ruhr-University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany. E-mail: ulf.apfel@rub.de
bFraunhofer UMSICHT, Osterfelder Straße 3, 46047 Oberhausen, Germany
cDepartment of Physics, Ruhr-University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany
dDepartment of Interface Science, Fritz-Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
First published on 6th November 2018
Abstract
The electrocatalytic reduction of carbon dioxide (CO2RR) to valuable bulk chemicals is set to become a vital factor in the prevention of environmental pollution and the selective storage of sustainable energy. Inspired by structural analogues to the active site of the enzyme CODHNi, we envisioned that bulk Fe/Ni sulfides would enable the efficient reduction of CO2. By careful adjustment of the process conditions, we demonstrate that pentlandite (Fe4.5Ni4.5S8) electrodes, in addition to HER, also support the CO2RR reaching a peak faradaic efficiency of 87% and 13% for the formation of CO and methane, respectively at 3 mA cm−2. The choice of solvent, the presence of water/protons and CO2 solubility are identified as key-properties to adjust the balance between HER and CO2RR in favour of the latter. Such experiments can thus serve as model reactions to elucidate a potential catalyst within gas diffusion electrodes.
Introduction
Energy production from fossil fuels results in critical environmental contaminations due to the emission of the greenhouse gas CO2 into earth's atmosphere.1 While CO2 acts as a pollutant, it is also a cheap and abundant C1-feedstock for carbon-based chemicals if efficient pathways for its conversion are established.2 Especially, the development of sustainable production processes by means of electrocatalysis are a promising pathway to efficiently activate CO2 and allow for the utilization of electricity from renewable sources like wind or photovoltaics.3 While electrochemical CO2 reduction was previously shown, the hampered mass transport and low water solubility of CO2 render this process still inefficient and instead facilitate the formation of H2.4,5 In addition, the metal–substrate binding strength (H+vs. CO2), and electron transfer kinetics at the material surface, are among the key factors for the selectivity of the electrocatalytic CO2 reduction.6,7 While low-coordinated Cu-nanoparticle sites facilitate HER, up to a certain extent, nano-structuring of Cu particles was shown to favor CO2 reduction over HER.8–11 General design principles leading to more efficient electrochemical CO2 reduction catalysts, however, are not yet available.12 Notably, the capability of a catalyst to show either HER or CO2RR activity usually depends on the process conditions applied. E.g. Cu and pyrite (FeS2) were both reported to facilitate H2 generation as well as CO2 reduction at various conditions and further support the Janus-faced character of both processes.13–17Contrary to artificial catalysts,18–20 nature has developed sophisticated machineries to allow for an efficient CO2 reduction. E.g. the carbon monoxide dehydrogenase21 allows for selective reduction of CO2 to CO at −0.52 V vs. NHE and ambient pressure with turn over frequencies of up to 700 molecules per h.22–24
The efficient enzymatic reactivity is related to its unique [Ni–4Fe–5S] cluster (Fig. 1a), a finely enzyme regulated amount of protons/water at the reactive center, as well as a directed CO2 access to the low valent active sites. Furthermore, the protein scaffold of the enzyme allows for a synchronized influx of electrons and substrates enabling high enzymatic selectivity. We and others recently showed that efficient electrocatalysts can be designed following natural principles.24–31 In particular, the mineral Fe4.5Ni4.5S8 (Fig. 1b) acted as a functional mimic for [NiFe] hydrogenase and allowed for H+ reduction at overpotentials of 190 mV at 10 mA cm−2 with high stability and without the need of any artificial nano-structuring. The distinct material composition of the electrode surface and its high conductivity was shown to be responsible for its activity and supported by operando phonon spectroscopy as well as scanning electrochemical cell microscopy.32,33 Following our previous design concepts for electrode materials and our knowledge on the surface and bulk structural composition,26,32,33 we reasoned that due to close structural analogy with the enzyme CO dehydrogenase (CODHNi), Fe4.5Ni4.5S8 should likewise act as a potential electrocatalyst for the reduction of CO2. Here, comparable to the CODHNi, Fe4.5Ni4.5S8 possesses distinct [Fe,Ni] sulfur clusters and reveals Fe–Ni sites with an M–M bond of around 2.6 Å [ca. 2.8 Å in CODHNi]21 that is bridged by labile sulfur atoms. This bio-inspired design attempt is further supported by the hydrothermal hydrogenation of CO2 observed in hydrothermal vents and facilitated by Fe2Ni7S8 possessing a pentlandite structure.34 Considering the observed natural performance and the close resemblance of FeNi sulfides to the enzyme CO dehydrogenase, it is surprising that up to now only little is known about the performance of such iron- and nickel-sulfide materials for CO2RR.24 Markedly, solely pyrite (FeS2)13 and greigite (Fe3S4)35–37 were previously reported to allow electrochemical reduction of CO2 in water but with low overall faradaic efficiencies (FE) for CO2 reduction (<10%) with low stability. We herein show that Fe4.5Ni4.5S8 bulk electrodes can efficiently facilitate CO2RR with high faradaic efficiency towards CO formation (87 ± 4%), at moderate current densities (3 mA cm−2) and potentials (−1.8 V vs. NHE). We furthermore reveal the electrochemical behavior of common C1-reduction products like formaldehyde, CO, or formic acid explaining the observed product distribution.
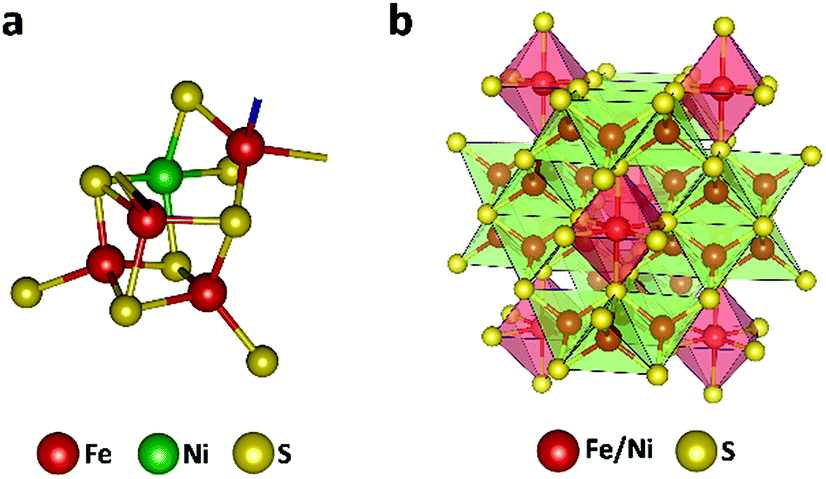 | ||
| Fig. 1 Active site in the nickel-containing CO dehydrogenase (pdb: 1su6) (a) and crystal structure of the Fe4.5Ni4.5S8 (b). | ||
Results and discussion
Solvent dependency of CO2RR with pentlandite
Fe4.5Ni4.5S8 was synthesized via a high-temperature route at 1100 °C following literature procedures.25,38 Pellet electrodes were prepared from the as-synthesized material, mounted into a Teflon casing.33 Their performance was subsequently tested in different organic and aqueous electrolytes (Fig. 2a and b). While performing linear sweep voltammetry (LSV) from 0 to −2.2 V vs. NHE and controlled potential coulometry (CPC) at various potentials, the electrolyte solution was continuously purged with CO2 and the gas flow analyzed by online gas chromatography.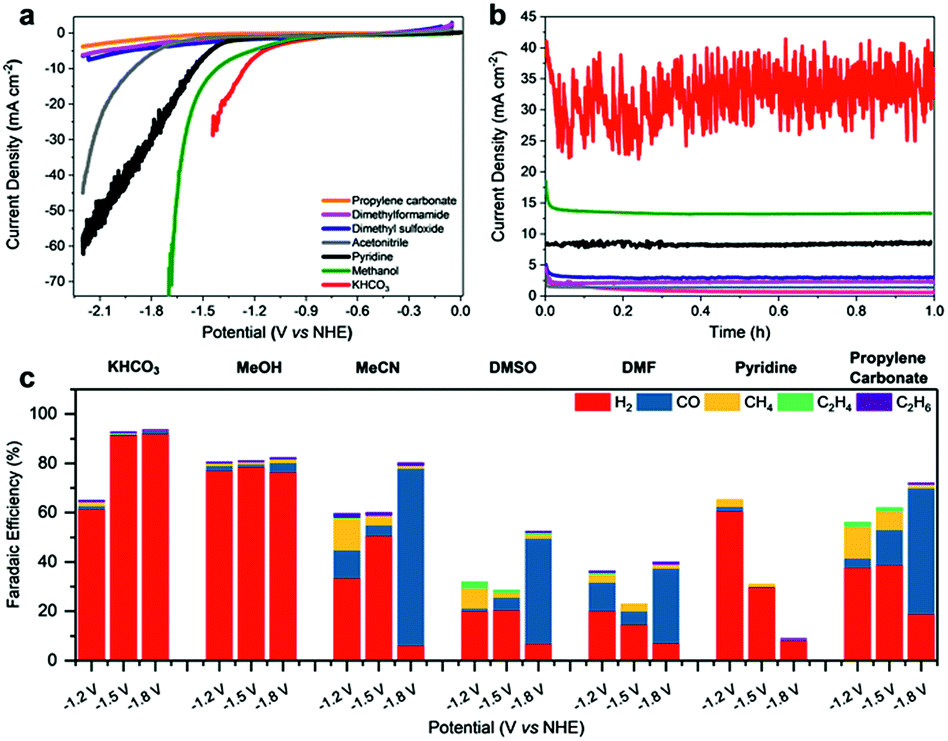 | ||
| Fig. 2 (a) Linear sweep voltammograms recorded at 5 mV s−1 in saturated CO2 solutions. (b) Controlled potential coulometry (CPC) was performed in different electrolytes (0.1 M TBAPF6) at −1.80 V vs. NHE and (c) the composition of the CO2 reduction products analyzed by on-line gas chromatography after 1 h. Notably, formation of soluble reduction products was not observed in additionally performed GC and HPLC experiments. | ||
Likewise, the liquid phase was analyzed via HPLC analysis to reveal any potentially soluble reduction products. However, neither alcohols, carboxylic acids or aldehydes were detected. In aqueous KHCO3 (0.1 M), the bulk electrode shows a catalytic current density of 20 mA cm−2 at −1.3 V. In contrast, in methanol, pyridine/water (0.01%) and acetonitrile (113 ppm H2O; 0.01%), comparable catalytic currents were observed at significantly more negative potentials of −1.6, −1.7 and −2.1 V, respectively. In dimethylformamide (DMF), dimethyl sulfoxide (DMSO) or propylene carbonate (PC) containing 0.1 M TBAPF6, current densities above 10 mA cm−2 were not observed reaching only maximum values of ca. 7.6 mA cm−2 at −2.2 V.
While certainly, the lower conductivity and with it the higher charge transfer resistance of the organic solvents will have an influence on the currents, the higher proton concentration in the more protic solvents also facilitates the H2 generation (Fig. 2c). Notably, in aqueous KHCO3, a faradaic efficiency (FE) for H2 formation of up to 93% was observed at −1.8 V vs. NHE which is in line with the previously reported high activity of this material towards HER. The contribution of the CO2RR is small and CO, CH4 as well as C2H6 were observed with low faradaic efficiencies of 1.0%, 0.2% and 0.1%, respectively. While at −1.2 V vs. NHE in KHCO3 HER is still the dominating reaction, the contribution of the CO2RR is slightly improved revealing faradaic efficiencies of 1.2% (CO), 1.8% (CH4), 0.3% (C2H6) and 61.5% (H2). The observed performance of Fe4.5Ni4.5S8 is in good agreement with the reports on greigite and pyrite in aqueous conditions.13,14,35
Although water is indeed the preferred electrolyte to perform CO2RR, the obvious CO2 reduction, led us to further investigate the role of other solvents in this process. The utilization of organic solvents is furthermore accompanied by a firm control of the proton concentration in the electrocatalytic double layer leading to analogous conditions that are usually found in gas diffusion electrode layers.39 In dry methanol, HER is still the dominating process (FE 78%). However, the fraction of CO2RR on the total faradaic efficiency is increased showing that the lower acidity of methanol, and thereby the lower proton availability, facilitates CO2 reduction (Table S1†). The overall impact of CO2RR in MeOH likewise slightly increases at more negative potentials with total faradaic efficiencies for all detected carbon products of 3.2% at −1.2 V to 5.7% at −1.8 V vs. NHE. In pyridine, HER is significantly suppressed showing faradaic efficiencies for H2 of solely 60, 30 and 8% at −1.2, −1.5 and −1.8 V. In addition, a clear increase of the FE for CH4 from solely 0.3 to 3.0% at −1.2 V was observed. In contrast, in MeCN, DMSO, DMF and PC, Fe4.5Ni4.5S8 electrodes reveal a significantly altered behavior. In these solvents, the faradaic yields for HER are significantly reduced and an increased amount of CO2RR products is observed. It is noteworthy that in MeCN and PC methane (FE 12.8% in MeCN, 8.3% in DMSO and 13.0% in PC) and ethylene (FE 0.5% in MeCN, 2.2% in DMSO and 1.4% in PC) are the major products at −1.2 V vs. NHE which corroborates the finding of pentlandite being capable to hydrothermally hydrogenate CO2.34
At very negative potentials, the CO formation capability of the material is generally increased reaching FE as high as 71% in MeCN, 42% in DMSO and 51% in PC at potentials of −1.8 V. Surprisingly, the product distribution in DMF is less dependent on the potential applied showing steady methane formation (FE 23%) and smaller variations in CO formation (FE 11–30%) as compared to MeCN, DMSO and PC. While CH4 and CO are the main products from CO2 reduction, it is notable that especially in aprotic solvents C2H4 and C2H6 are also observed (Fig. 2c). E.g. FE for C2H4 of 2.2% (DMSO), 0.7% (DMF) and 0.5% (MeCN) were observed. Likewise, C2H6 was observed in the range of 1–1.3%, respectively. The faradaic efficiencies below 100% observed within the non-protic solvents can most-likely be assigned to high capacitive currents and poor conductivity leading to e.g. heat formation, gas leakage, as well as a potential partial re-oxidation of CO2 reduction products and are not necessarily an effect of the electrode behavior in the different solvents.40 However, these amounts are among the highest ever reported for non-Cu containing materials (Table S4†).41 We conclude that beside the proton availability and the solvent chosen, the CO2 solubility in the various solvents has a major influence on the CO2RR by enhancing the effective concentration of CO2 at the electrode surface.
The solubility of CO2 in acetonitrile (0.27 M) was reported to be significantly higher compared to water (0.04 M) and methanol (0.16 M).42 Likewise, an increased CO2 solubility is observed for DMF (0.18 M)43 and PC (6.97 M)43 which encourages the CO2 reduction in these solvents (Table S5†). However, the solubility of CO2 in DMSO is just 0.02 M and contradicts this hypothesis. In this regard, the proton availability which is commonly displayed by the acidity of water in a particular electrolyte or the solvent itself will influence CO2RR and is highlighted by increased HER activity in aqueous KHCO3 (pKa 10.3) compared to MeOH (pKa 16).
Dependency on water/proton concentration
The efficiency of CO2RR seems to be a compromise between altered proton concentration at the electrode surface (generally caused by the water content in a given solvent) and the capability of a certain electrolyte to efficiently absorb CO2. We thus opted to determine the influence of the water amount on the activity of Fe4.5Ni4.5S8 electrodes. Hence, we examined the selectivity towards CO2RR using MeCN electrolyte solutions with varying water concentrations (24–30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) (Table S3†) by performing LSV (Fig. 3a).
000 ppm) (Table S3†) by performing LSV (Fig. 3a).
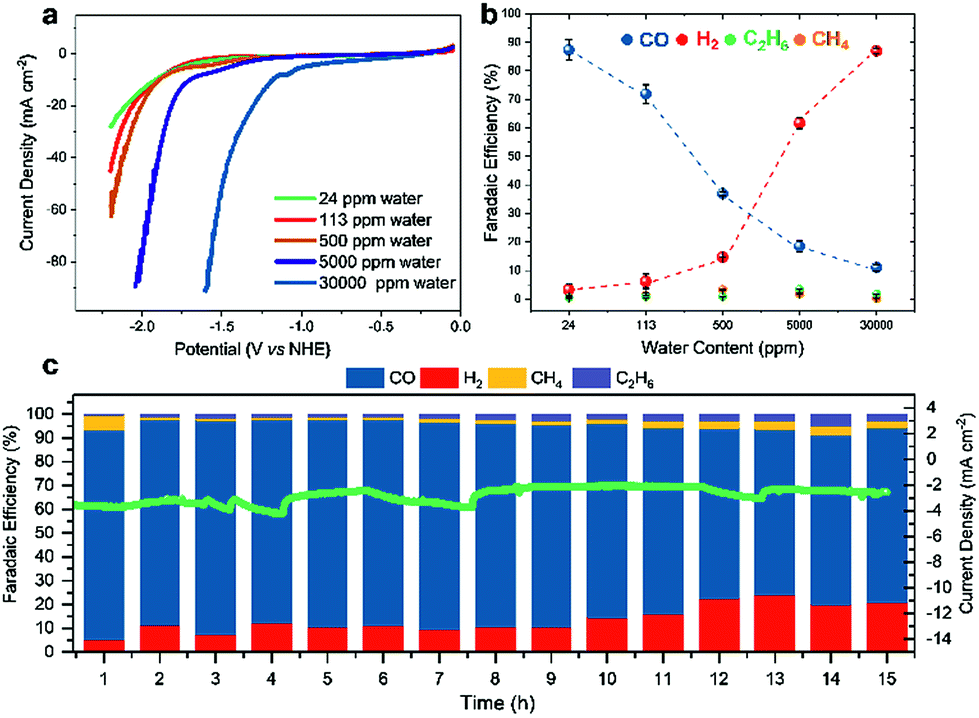 | ||
| Fig. 3 Linear sweep voltammograms recorded at 5 mV s−1 in MeCN with different water content and CO2 purge (a), faradaic efficiencies of CO2 reduction products in the gas phase at a constant potential of −1.8 V vs. NHE from MeCN with various amounts of water (b). Long term electrolysis for 15 h at −1.8 V vs. NHE with 24 ppm water including the observed faradaic efficiencies (c). | ||
A shift of the potential necessary to achieve a catalytic current of −20 mA cm−2 to lower potentials (−2.1 V; 24 ppm H2O vs. −1.3 V; 30000 ppm) is observed as the water concentration in the electrolyte increases. However, gas analyses after 1 h CPC at −1.8 V clearly shows that at higher water concentrations HER is facilitated and CO2RR is suppressed (Fig. 3b). While for high water concentrations in the electrolyte, FEs of 87% (30000 ppm) and 62% (5000 ppm) are observed for hydrogen, only 11% and 19% FE, respectively, were found for CO. Contrary, in the presence of lower water concentrations, CO2RR becomes the dominant reaction. Here, FE for CO significantly increases to 37% (500 ppm H2O) and 87% (24 ppm H2O), respectively, concomitant with a decrease of the FE for HER to 14% and 3%, respectively.
In control experiments using Ar purged electrolytes containing 24 ppm H2O, no formation of CO2RR was observed (Fig. S6†). In addition, to rule out a suppressed oxidation on the anode side due to an imbalanced amount of charges passed through the system, we added conc. sulfuric acid to the acetonitrile mixture at the cathodes chamber. This allows for a fast HER reaction and allows for a comparable oxidation reaction as observed under CO2RR catalytic condition. Similarly, no increased formation of CO2 reduction products (Fig. S7†) was observed proving the dominant reduction of CO2 rather than electrolyte decomposition. Similar results were observed when using DMSO or DMF under otherwise identical conditions. To confirm the origin of the various C-products observed, we performed a mass-selective product analysis utilizing 13CO2 in acetonitrile under otherwise identical electrochemical conditions. While the observed mass peak of 29 is indicative for the formation of 13CO from 13CO2 (Fig. S9†), the unaltered mass pattern of ethane suggests a different origin (Fig. S10†). We thus repeated the CO2 reduction experiment in the presence of CD3CN. The resulting mass pattern and the observed mass peak at 33 clearly indicate the formation of C2D3H3 and suggest that ethane originates from the hydrogenation of acetonitrile rather than reduction of CO2 (Fig. S10†). Likewise, we analyzed the origin of CH4 that was observed within our experiments via the same technique. While the MS pattern of CH4 overlaps with those of the reaction matrix under the applied instrumental conditions, increase of the peak intensity at m/z = 17 and the missing higher mass peaks for CD3H and derivatives upon performing the experiments in CD3CN indicate that likewise CH4 is formed from CO2 (Fig. S11†).
Notably, fine-tuning of the water concentration in the electrolysis cell and the applied potential allows for the tailored electrochemical production of syngas (Table S4†). Markedly, for CH4 and C2H6 that were likewise found as reduction products, FE is independent of the water concentration (Fig. S1†) suggesting a different catalytic mechanism. The results clearly show that HER and CO2RR are both catalyzed by Fe4.5Ni4.5S8 electrodes and the outcome of the reduction can be tuned by the process conditions rather than by further catalyst modification.
Durability and potential intermediates
To further investigate the Fe4.5Ni4.5S8 bulk electrodes for its capability to act as a potent electrocatalyst in CO2RR, we performed stability tests in acetonitrile (24 ppm water H2O) at an applied potential of −1.8 V over 15 h (Fig. 3c). During the electrolysis, the system operated at a constant current density of around 3 mA cm−2. While in the beginning of the controlled potential electrolysis, high faradaic efficiencies of up to 87% CO are observed, it decreases slowly with electrolysis time to 75%. In equal measure, the FE for H2 increases from 8 to 21%. This change in the CO:H2 selectivity can be caused by either an incremental change of the catalyst surface and hence its reactivity or an increase in water concentration over the course of the electrolysis. Notably, SEM-EDX showed only carbon deposition. The Fe:Ni:S content, however, is not affected, indicating high stability of the material. We subsequently quantified the water content after 15 h electrolysis and observed an elevated water concentration, responsible for the hampered catalyst performance. This increase most likely originates from traces of water present in the CO2 stream causing accumulation of water in the electrolyte over time. To further evaluate the stability of Fe4.5Ni4.5S8, we performed long-time stability experiments in the presence of various amounts of water. Noteworthy, after conducting electrolysis at −1.8 V in a 5000 ppm water acetonitrile solution, the electrode is covered with a black film and correlates with the observed decrease of CO2RR activity. Energy dispersive X-ray spectroscopy (EDX) revealed a high carbon content of up to 80% at the electrode surface (Fig. S2†). While similar results were previously observed for Cu electrodes,44 the exact reason of this electrode passivation, however, is not well understood and remains subject of future investigations.Ex situ X-ray photoelectron spectroscopy (XPS) measurements were carried out to reveal the composition and chemical state of the pentlandite catalysts as-prepared and after CO2RR. Both show a significant content of oxygen as it can be seen from the survey scans (Fig. S12†). Moreover, the sample after reaction bears considerably high amounts of the electrolyte residuals as indicated by intense F 1s, N 1s, and C 1s peaks. A closer investigation of the high-resolution regions of the metallic components (Fig. 4a and b) and sulfur (Fig. 5) confirms severe oxidation of the catalyst surface before and after the reaction and subsequent exposure to air. A spectral deconvolution of the Fe 2p3/2 photoemission line using an Fe(III) multiplet structure45 and a single FeSx peak (Fig. 4c) indicates only 18 at% of the latter.
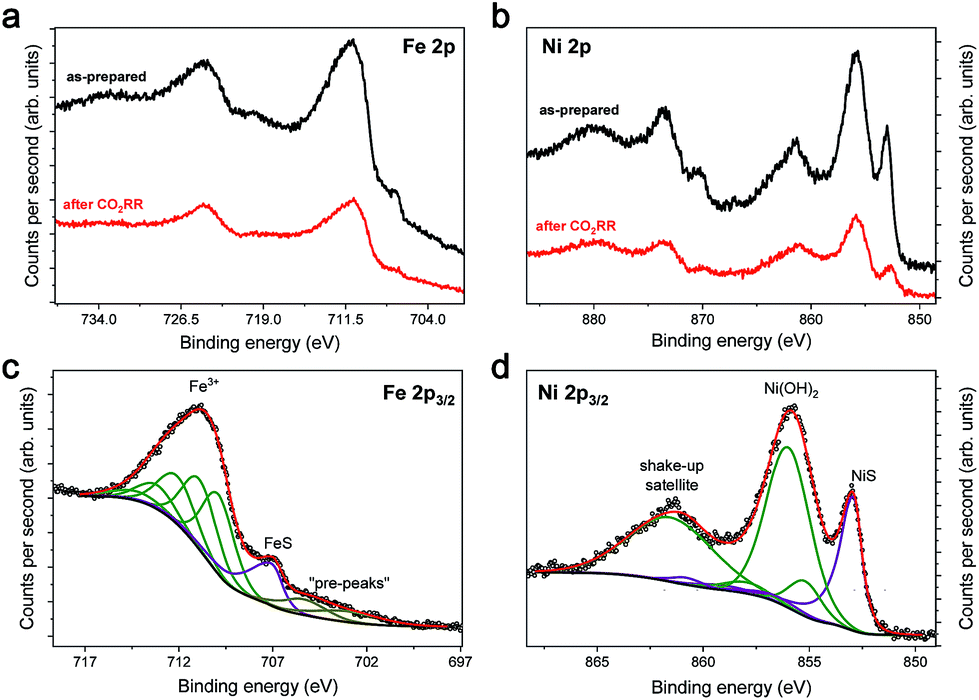 | ||
| Fig. 4 (a) Fe 2p and (b) Ni 2p XPS spectra of pentlandite before and after CO2RR. Examples of the spectral deconvolution fit for the as-prepared sample are shown in (c) for the Fe 2p3/2 core level and (d) for Ni 2p3/2. | ||
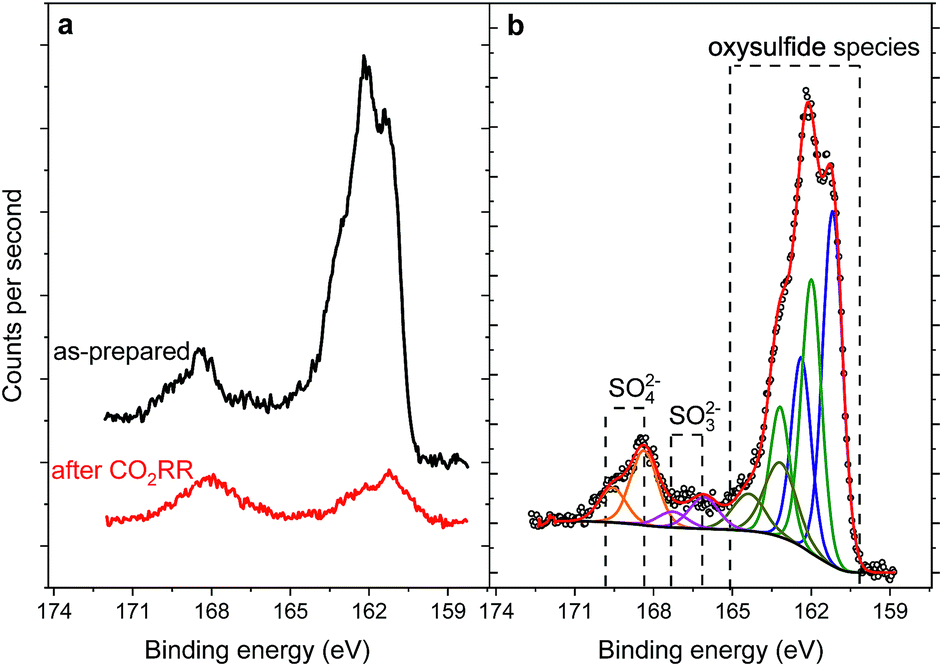 | ||
| Fig. 5 S 2p XPS spectra of the pentlandite before and after CO2RR (a) and spectral deconvolution exemplified for the as-prepared sample (b). The decrease in the intensity of the S signal observed after reaction is at least partially related to the overlayer resulting from electrolyte residues. | ||
Similarly, the Ni 2p3/2 region is dominated by the Ni(OH)2 structure46 represented by a photoemission peak at 855.9 eV and a characteristic shake-up satellite at 861.4 eV (Fig. 4d). The peak at 853.0 eV represents a sulfide species and corresponds to ∼23% of the total Ni content. XPS analysis of the sample after reaction does not display a significant change in the chemical state of Fe and Ni, although one needs to consider that both samples were measured ex situ and that at least partial catalyst reduction is expected under reaction conditions that can be further re-oxidized upon ex situ sample transfer in air. Moreover, calculation of the Fe:Ni ratio for the sulfide species, shows enrichment with nickel before and after reaction. The S 2p region of both samples shows a shape typical for FexNi9−xS8 materials25 with a structure between 160–165 eV, characteristic of oxysulfide films,47 and a mildly oxidized iron pyrite surface.48,49 The structure at higher binding energy is deconvoluted in two doublets, at 168.4–169.7 and 166.1–167.3 eV, representing sulfate and sulfite species correspondingly.50 The structure at higher binding energy is deconvoluted in two doublets, at 168.4–169.7 and 166.1–167.3 eV, representing sulfate and sulfite species, correspondingly. Remarkably, the amount of oxidic sulfur species increases from 17 to 50 at% after the reaction. Despite the high amount of oxidized species observed by XPS, we do not indent to identify those as active in the CO2RR. Both Fe and Ni are well known to form native oxide overlayers upon exposure to air. However, the increased amount of oxidic sulfur species is striking. We attribute it to the formation of S defect sites under reaction conditions, which are then more prone to oxidation when exposed to air. Notably, such [NiFe]-oxido-species are also found in the natural archetype Ni-dehydrogenases and are discussed as key intermediates in the enzymatic CO2 reduction.51,52
As copper is one of the benchmark materials in CO2RR, we decided to compare Fe4.5Ni4.5S8 and a copper disc electrode under the same experimental conditions. CPC experiments at −1.2, −1.5 and −1.8 V vs. NHE were subsequently performed in acetonitrile (113 ppm H2O) (Fig. S3†). While we observed current densities of −0.14, −0.73 and −3.2 mA cm−2 at −1.2, −1.5, and −1.8 V, respectively, for Fe4.5Ni4.5S8, Cu bulk electrodes showed significantly smaller current densities of −0.01, −0.10 and −0.28 mA cm−2 under otherwise identical conditions (Fig. S3†). Analysis of the gaseous products after 1 hour at −1.2 V and −1.5 V, revealed significant higher HER activity of Fe4.5Ni4.5S8 with FEs of 34 and 51%, respectively, for H2 in comparison to those observed for Cu bulk electrodes (0 and 6%). In lieu, Cu revealed higher efficiency towards methane formation with FE of 20 and 9% at −1.2 V and −1.5 V respectively versus the observed FE of 13 and 4% for Fe4.5Ni4.5S8 electrodes under identical conditions. Likewise, the efficiency for CO at Cu electrodes (FE 39%, −1.2 V; FE 9%, −1.5 V) is higher than observed for Fe4.5Ni4.5S8 (FE 11.4%, −1.2 V; FE 4%, −1.5 V). Notably, these differences are only observed at low current densities/low potentials. At −1.8 V, there is only a small difference in the product distribution. Both, Fe4.5Ni4.5S8 and Cu electrodes reveal suppressed H2 formation and facilitate the CO2 reduction to mainly CO (72% FE for both electrodes) and traces of methane (1% FE for Fe4.5Ni4.5S8 and 6% FE for Cu). Notably, at such high potential conditions, Fe4.5Ni4.5S8 generally reveals higher current densities suggesting faster conversion kinetics and revealing its potential (Fig. S3†).
The overall narrow product distribution of Fe4.5Ni4.5S8 pointed our attention towards potential intermediates of the CO2RR and their subsequent conversion on Fe4.5Ni4.5S8 bulk electrodes. We thus tested the reactivity of various potential CO2 reaction products, such as carbonate, CO, formaldehyde, formic acid and oxalic acid under electrocatalytic conditions (at −1.8 V vs. NHE) in acetonitrile containing 24 ppm water.
CPC in saturated Cs2CO3 solutions (3.31 mM) in MeCN yielded exclusively CO as the major product (42% FE, Fig. 6a) and suggests that likewise, carbonates can serve as a CO2 source. In the presence of oxalic acid, the major reduction product is CO (FE 3.9%) and only trace amounts of hydrogenation to afford C2H4 (FE 0.05%), C2H6 (FE 1.60%) is also detected. Additionally, CO poisoning experiments were performed since Fe and Ni strongly adsorb CO and usually undergo surface deactivation. CPC experiments at −1.8 V vs. NHE in a solution of MeCN (24 ppm H2O, Fig. S4 and S5†) applying a continuously applied CO stream showed no obvious influence on the catalytic current and suggests no CO poisoning. Furthermore, neither gaseous nor liquid CO conversion products were detected, underlining the inertness of the Fe4.5Ni4.5S8 electrodes towards further CO conversion. Under same conditions, a 37% formaldehyde solution afforded high amounts of CH4 (FE 4.2%) and traces of C2H4 (FE 0.13%) and C2H6 (FE 0.10%) (Fig. 6b). Notably, only negligible amounts of CO were observed herein. It is worth to highlight that C2-reduction products are observed in the presence of oxalic or formic acid (Fig. 6c and d) although with low efficiency (FE < 1%). Our results clearly show that although the formation of alternative reduction products seems possible, they will be converted to mainly CO and methane with a minor amount of C–C coupling products.
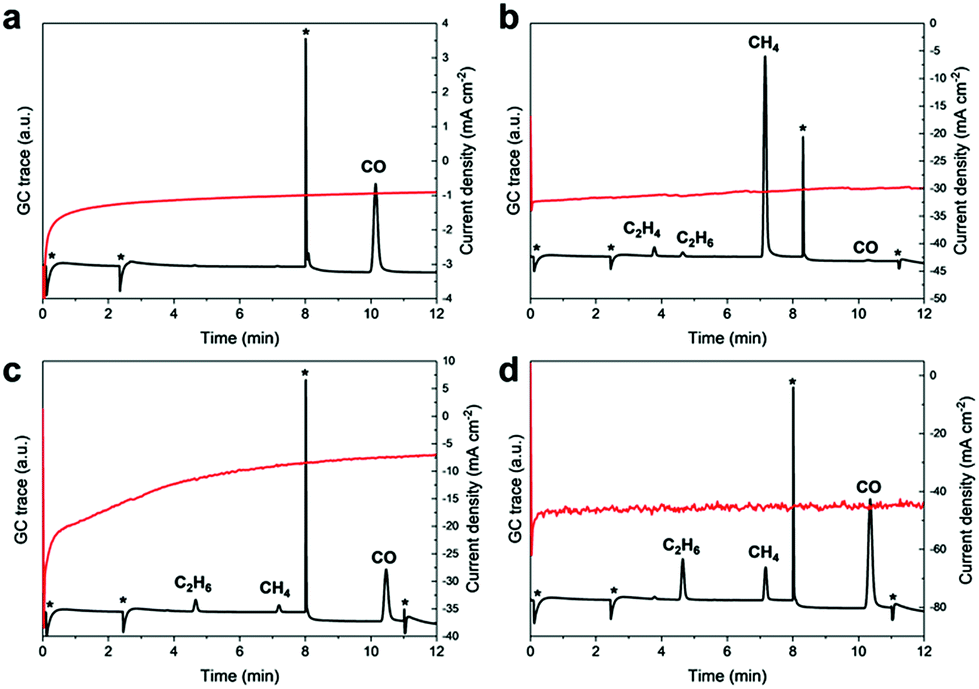 | ||
| Fig. 6 GC traces and recorded current densities starting from different carbon sources like Cs2CO3 (a), formaldehyde (b), oxalic acid (c) and formic acid (d). | ||
Conclusion
In conclusion we have shown, that Fe4.5Ni4.5S8, much like its natural congeners, [FeNi]-hydrogenase and CODHNi, can catalyze HER as well as CO2RR depending on the chemical environment and with seemingly comparable catalytic intermediates. In this regard, protic electrolytes such as aqueous KHCO3, methanol as well as aprotic pyridine promote HER with high faradaic efficiency. In addition, utilizing the same electrodes in aprotic solvents such as acetonitrile, dimethyl formamide, dimethyl sulfoxide and propylene carbonate with well-defined water contents, CO2RR is favored over HER. Under such conditions, CO and methane are the main products with FE of up to 87% and 13%, respectively, at moderate current densities (ca. 3 mA cm−2). Our experiments clearly show that controlling the availability of protons in the chemical environment of the electrocatalyst is key towards successful CO2RR. While we are aware that water is the preferred electrolyte to perform such reduction reactions, our study shows that investigating potential CO2RR electrocatalysts in non-aqueous, can lead to the discovery of novel catalysts that operate under alternative reactions conditions (e.g. gas diffusion electrodes or in a pure gas stream). In addition, we herein show that the materials can facilitate the hydrogenation of nitriles with simple iron and nickel-based electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr M. Reback for valuable hints during manuscript preparation. The authors thank for the financial support of the Fonds of the Chemical Industry (Liebig grant to U.-P. A.), the Deutsche Forschungsgemeinschaft (Emmy Noether grant to U.-P. A., AP242/2-1 and AP242/6-1) as well as the Fraunhofer Internal Programs under Grant No. Attract 097-602175. IS and BRC also acknowledge the financial support of the German Federal Ministry of Education and Research (Magisterium für Bildung und Forschung, BMBF) under grant #03SF0523C-“CO2EKAT.References
- T. R. Anderson, E. Hawkins and P. D. Jones, Endeavour, 2016, 40, 178–187 CrossRef PubMed.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- V. Pradeep Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 RSC.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- C. Liu, T. R. Cundari and A. K. Wilson, J. Phys. Chem. C, 2012, 116, 5681–5688 CrossRef CAS.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS PubMed.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- L. Dai, Q. Qin, P. Wang, X. Zhao, C. Hu, P. Liu, R. Qin, M. Chen, D. Ou, C. Xu, S. Mo, B. Wu, G. Fu, P. Zhang and N. Zheng, Sci. Adv., 2017, 3, e1701069 CrossRef PubMed.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531–535 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- J. Qiao, Y. Liu and J. Zhang, Electrochemical Reduction of Carbon Dioxide: Fundamentals and Technologies, CRC Press, Taylor & Francis group, 2016 Search PubMed.
- M. G. Vladimirov, Y. F. Ryzhkov, V. A. Alekseev, V. A. Bogdanovskaya, V. A. Otroshchenko and M. S. Kritsky, Origins Life Evol. Biospheres, 2004, 34, 347–360 CrossRef CAS.
- S. Zhao, S. Guo, C. Zhu, J. Gao, H. Li, H. Huang, Y. Liu and Z. Kang, RSC Adv., 2017, 7, 1376–1381 RSC.
- M.-R. Gao, Y.-R. Zheng, J. Jiang and S.-H. Yu, Acc. Chem. Res., 2017, 50, 2194–2204 CrossRef CAS PubMed.
- Y. Zhang, Y. Ma, Y.-Y. Chen, L. Zhao, L.-B. Huang, H. Luo, W.-J. Jiang, X. Zhang, S. Niu, D. Gao, J. Bi, G. Fan and J.-S. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 36857–36864 CrossRef CAS PubMed.
- J. Du, J. Wang, L. Ji, X. Xu and Z. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 30205–30211 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- G. Zhao, X. Huang, X. Wang and X. Wang, J. Mater. Chem. A, 2017, 5, 21625–21649 RSC.
- H. Dobbek, Science, 2001, 293, 1281–1285 CrossRef CAS PubMed.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci., 2001, 98, 11973–11978 CrossRef CAS PubMed.
- C. Darnault, A. Volbeda, E. J. Kim, P. Legrand, X. Vernède, P. A. Lindahl and J. C. Fontecilla-Camps, Nat. Struct. Mol. Biol., 2003, 10, 271–279 CrossRef CAS PubMed.
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem.–Eur. J., 2018, 24, 1471–1493 CrossRef PubMed.
- B. Konkena, K. Junge Puring, I. Sinev, S. Piontek, O. Khavryuchenko, J. P. Dürholt, R. Schmid, H. Tüysüz, M. Muhler, W. Schuhmann and U.-P. Apfel, Nat. Commun., 2016, 7, 12269 CrossRef CAS PubMed.
- S. Piontek, C. Andronescu, A. Zaichenko, B. Konkena, K. Junge Puring, B. Marler, H. Antoni, I. Sinev, M. Muhler, D. Mollenhauer, B. Roldan Cuenya, W. Schuhmann and U.-P. Apfel, ACS Catal., 2018, 8, 987–996 CrossRef CAS.
- J. Shen, R. Kortlever, R. Kas, Y. Y. Birdja, O. Diaz-Morales, Y. Kwon, I. Ledezma-Yanez, K. J. P. Schouten, G. Mul and M. T. M. Koper, Nat. Commun., 2015, 6, 8177 CrossRef PubMed.
- V. Artero, Nat. Energy, 2017, 2, 17131 CrossRef CAS.
- Y. Tang, H. Yang, J. Sun, M. Xia, W. Guo, L. Yu, J. Yan, J. Zheng, L. Chang and F. Gao, Nanoscale, 2018, 10, 10459–10466 RSC.
- S. Dey, M. E. Ahmed and A. Dey, Inorg. Chem., 2018, 57, 5939–5947 CrossRef CAS PubMed.
- D. Hong, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2017, 139, 6538–6541 CrossRef CAS PubMed.
- C. L. Bentley, C. Andronescu, M. Smialkowski, M. Kang, T. Tarnev, B. Marler, P. R. Unwin, U.-P. Apfel and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 4093–4097 CrossRef CAS PubMed.
- I. Zegkinoglou, A. Zendegani, I. Sinev, S. Kunze, H. Mistry, H. S. Jeon, J. Zhao, M. Y. Hu, E. E. Alp, S. Piontek, M. Smialkowski, U.-P. Apfel, F. Körmann, J. Neugebauer, T. Hickel and B. Roldan Cuenya, J. Am. Chem. Soc., 2017, 139, 14360–14363 CrossRef CAS PubMed.
- Q. Fu, D. I. Foustoukos and W. E. Seyfried, Geophys. Res. Lett., 2008, 35, 1–5 CrossRef.
- A. Roldan, N. Hollingsworth, A. Roffey, H.-U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. de Leeuw, Chem. Commun., 2015, 51, 7501–7504 RSC.
- A. Roldan and N. H. de Leeuw, Faraday Discuss., 2017, 197, 325–336 RSC.
- A. Roldan and N. H. de Leeuw, Faraday Discuss., 2016, 188, 161–180 RSC.
- K. Junge Puring, S. Piontek, M. Smialkowski, J. Burfeind, S. Kaluza, C. Doetsch and U.-P. Apfel, J. Visualized Exp., 2017, e56087 Search PubMed.
- J. Albo and A. Irabien, J. Catal., 2016, 343, 232–239 CrossRef CAS.
- W. Lv, R. Zhang, P. Gao and L. Lei, J. Power Sources, 2014, 253, 276–281 CrossRef CAS.
- K. Malik, S. Singh, S. Basu and A. Verma, Wiley Interdiscip. Rev.: Energy Environ., 2017, 6, e244 Search PubMed.
- Y. Tomita, S. Teruya, O. Koga and Y. Hori, J. Electrochem. Soc., 2000, 147, 4164 CrossRef CAS.
- P. G. T. Fogg, Carbon Dioxide in Non-aqueous Solvents at Pressures Less Than 200 kPa, Pergamon Press, 1992, vol. 50 Search PubMed.
- G. Kyriacou and A. Anagnostopoulos, J. Electroanal. Chem., 1992, 322, 233–246 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- L. Benoist, D. Gonbeau, G. Pfister-Guillouzo, E. Schmidt, G. Meunier and A. Levasseur, Thin Solid Films, 1995, 258, 110–114 CrossRef CAS.
- G. Wittstock, I. Kartio, D. Hirsch, S. Kunze and R. Szargan, Langmuir, 1996, 12, 5709–5721 CrossRef CAS.
- C. D. Wagner and J. A. Taylor, J. Electron Spectrosc. Relat. Phenom., 1982, 28, 211–217 CrossRef CAS.
- R. V. Siriwardane and J. M. Cook, J. Colloid Interface Sci., 1985, 104, 250–257 CrossRef CAS.
- P. A. Lindahl, Biochemistry, 2002, 41, 2097–2105 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03555e |
| This journal is © The Royal Society of Chemistry 2019 |