DOI:
10.1039/C8SC03006E
(Edge Article)
Chem. Sci., 2019,
10, 1082-1088
Selective utilization of methoxy groups in lignin for N-methylation reaction of anilines†
Received
6th July 2018
, Accepted 31st October 2018
First published on 1st November 2018
Abstract
The utilization of lignin as a feedstock to produce valuable chemicals is of great importance. However, it is a great challenge to produce pure chemicals because of the complex structure of lignin. The selective utilization of specific groups on lignin molecules offers the possibility of preparing chemicals with high selectivity, but this strategy has not attracted attention. In this work, we propose a protocol to produce methyl-substituted amines by the selective reaction of the methoxy groups of lignin and aniline compounds. It was found that LiI in the ionic liquid 1-hexyl-3-methylimidazolium tetrafluoroborate could catalyze the reaction efficiently and the selectivity to the N-methylation product could be as high as 98%. Moreover, the lignin was not depolymerized in the reaction. As it was rich in hydroxyl groups, the residual material left over after the reaction was used as an efficient co-catalyst for the cycloaddition of epoxy propane with CO2, using KI as the catalyst.
Introduction
Biomass is considered to be a renewable and sustainable alternative to fossil resources.1,2 The utilization of biomass as a raw material to produce useful chemical compounds is of great importance for the sustainable development of our society.3–8 As the second most abundant form of biomass,9 lignin is drawing increasing attention.10,11 Lignin is a byproduct in the pulp and paper industry, and it is usually burnt as low-grade fuel, which is recognized as a waste of its potential.12,13 In recent years, much effort has been devoted to converting lignin into alternative fuels, platform compounds, value-added chemicals, and functional materials.14–16 General approaches to transform lignin into valuable chemicals involve the depolymerization of lignin17–20 and further conversion into valuable products.21–25 Such approaches are very promising, although the depolymerized products are almost always mixtures, leading to serious separation and purification problems. Thus, it is of significant importance to improve the selectivity and obtain only a limited number of defined products instead of complex mixtures.16
Although great progress has been achieved for the selective transformation of model compounds,26–29 it is challenging to achieve high selectivity to a desired product, and much effort is yet to be made for lignin as a feedstock.30 The lignin-first approach is a very promising route to improving the selectivity and yield of aromatic monomers. This approach transforms the native lignin released from lignocellulose in situ and leaves other components as a solid residue.31,32 As one of the few notable examples, Abu-Omar and co-workers developed a bimetallic Zn/Pd/C catalyst for the selective conversion of intact woody biomass from a diverse selection of species.33,34 The diethylether extract liquor of the product contained mainly 4-propylguaiacol and 4-propylsyringol, with small amounts of unidentified impurities. A genetically-modified poplar enhanced in syringyl monomer content yielded 4-propylguaiacol as the single product, determined by gas chromatography, with a yield of 19 wt%. In contrast to yet unmodified native lignin, which contains a higher fraction of cleavable β-O-4 linkages, the case for technical lignin is more challenging. The structures of Kraft, Sulfite, Alkaline and Klason lignins that are readily available in the pulping industry are heavily modified under relatively harsh processing conditions.16 The easily cleavable β-O-4 and α-O-4 linkages are cleaved to varying degrees, and the random repolymerization forms highly recalcitrant C–C bonds.11 As a result, it is extremely difficult to selectively convert technical lignin into a limited number of desired products. On the other hand, the –OCH3 groups remain mostly untouched during the technical fraction process.16 It is known that the methoxy group is the most abundant group in lignin.2 Thus, lignin, whether native or technical, can be regarded as an abundant renewable methoxyl source. Selective utilization of methoxy groups in lignin, while leaving the other groups as a solid residue, provides a new approach to the selective transformation of lignin. We believe that using the methoxyl-first strategy to selectively produce valuable compounds is a very interesting protocol for the valorization of lignin.35
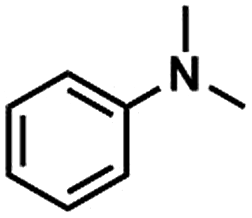
Methyl-substituted amines are important intermediates for the synthesis of dyes and can also be used as solvents, stabilizers and analytical reagents. Traditional methods to produce methyl-substituted amines are through the N-methylation reaction using methyl iodide, methanol or dimethyl sulfate as the methylating agent. These are highly toxic chemicals. Recently, CO2 combined with a reductant (hydrogen or hydrosilane) has been developed as a greener starting material for the N-methylation reaction.36–39 However, lignin, which is abundant in methyl groups, has never been recognized as a cheap and renewable methyl source.
The cycloaddition of epoxides with CO2 to produce cyclic carbonates is one of the most promising and extensively studied approaches for CO2 utilization.40 Numerous metal complexes, metal–organic hybrid materials41 and metal-free organic systems42 have been developed as efficient catalysts for this reaction. Hydrogen-bond donor groups (–OH, –NH2, –COOH, etc.) have been found to induce synergistic effects to facilitate CO2/epoxide coupling.43 It has been reported that substances with hydroxyl groups could act as an efficient co-catalyst for promoting the cycloaddition of epoxides with CO2 when using KI as the catalyst.44
Herein, we made the first attempt to utilize lignin as a methyl source to produce methyl-substituted amines via an N-methylation reaction. It was demonstrated that lignin could be used as a methylation agent for efficiently producing N,N-dimethylanilines from anilines. We found that a catalytic system comprising LiI and an ionic liquid (IL, e.g. 1-hexyl-3-methylimidazolium tetrafluoroborate, HMimBF4) could selectively cleave the C–O bond of methoxy groups in lignin and catalyze the N-methylation reaction of N-methylanilines using lignin as a methylating agent to produce N,N-dimethylanilines. When Kraft lignin was employed, the selectivity of N,N-dimethylanilines reached as high as 98%. Furthermore, the leftover material after the N-methylation reaction can be used as an efficient co-catalyst for the CO2/epoxide coupling reaction.
Results
Screen of catalytic systems
We first screened the catalytic system by testing the methylation of N-methylaniline, using anisole as the methylation reagent. The results are given in Table 1. In conventional organic solvents, such as CH3CN and toluene, the reaction did not occur (Table 1, entries 1 and 2). The C–O bond of anisole remained untouched under these reaction conditions. However, ionic liquids (ILs) containing BF4− were efficient solvents for this reaction when LiI was used as the catalyst (Table 1, entries 3–4). In particular, in HMimBF4, the yield of N,N-dimethylaniline could reach 72% (Table 1, entry 3). Other alkali halides, such as NaI, KI, CsI, LiBr, and LiCl were inactive for this reaction (Table 1, entries 5–9). We also tested other ionic liquids with different anions as the solvent, and the yields of N,N-dimethylaniline were very low (Table 1, entries 10–14). The results indicate that the BF4− anion and iodates with strong Lewis acidity were required, and LiI/HMimBF4 was the most active among the catalytic systems studied.
Table 1 Screening of the catalyst system of the reaction of anisole and N-methylanilinea
|
|
| Entry |
Solvent |
Catalyst |
Conversion of N-methylaniline (%) |
Yield of N,N-dimethylaniline (%) |
Yield of phenol (%) |
|
Reaction conditions: N-methylaniline 1 mmol, anisole 1 mmol, metal halide 2 mmol, reaction temperature 120 °C, reaction time 12 h, solvent 1 mL. The yield was calculated based on the NMR spectra using 1,3,5-trioxane as an internal standard.
|
| 1 |
CH3CN |
LiI |
0 |
0 |
0 |
| 2 |
Toluene |
LiI |
0 |
0 |
0 |
| 3 |
HMimBF4 |
LiI |
77 |
72 |
95 |
| 4 |
BMimBF4 |
LiI |
74 |
68 |
90 |
| 5 |
HMimBF4 |
NaI |
0 |
Trace |
0 |
| 6 |
HMimBF4 |
KI |
0 |
Trace |
0 |
| 7 |
HMimBF4 |
CsI |
0 |
Trace |
0 |
| 8 |
HMimBF4 |
LiBr |
0 |
Trace |
0 |
| 9 |
HMimBF4 |
LiCl |
0 |
Trace |
0 |
| 10 |
BMimAc |
LiI |
0 |
0 |
1 |
| 11 |
BMimN(CN)2 |
LiI |
1 |
1 |
2 |
| 12 |
BMimClO4 |
LiI |
20 |
10 |
9 |
| 13 |
BMimNO3 |
LiI |
6 |
2 |
3 |
| 14 |
BMimOTf |
LiI |
33 |
25 |
25 |
Optimization of reaction conditions
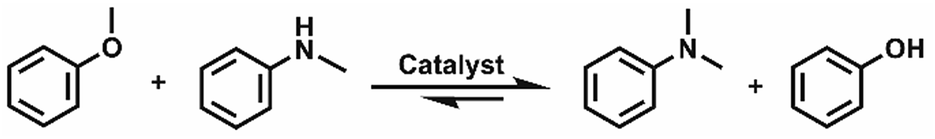
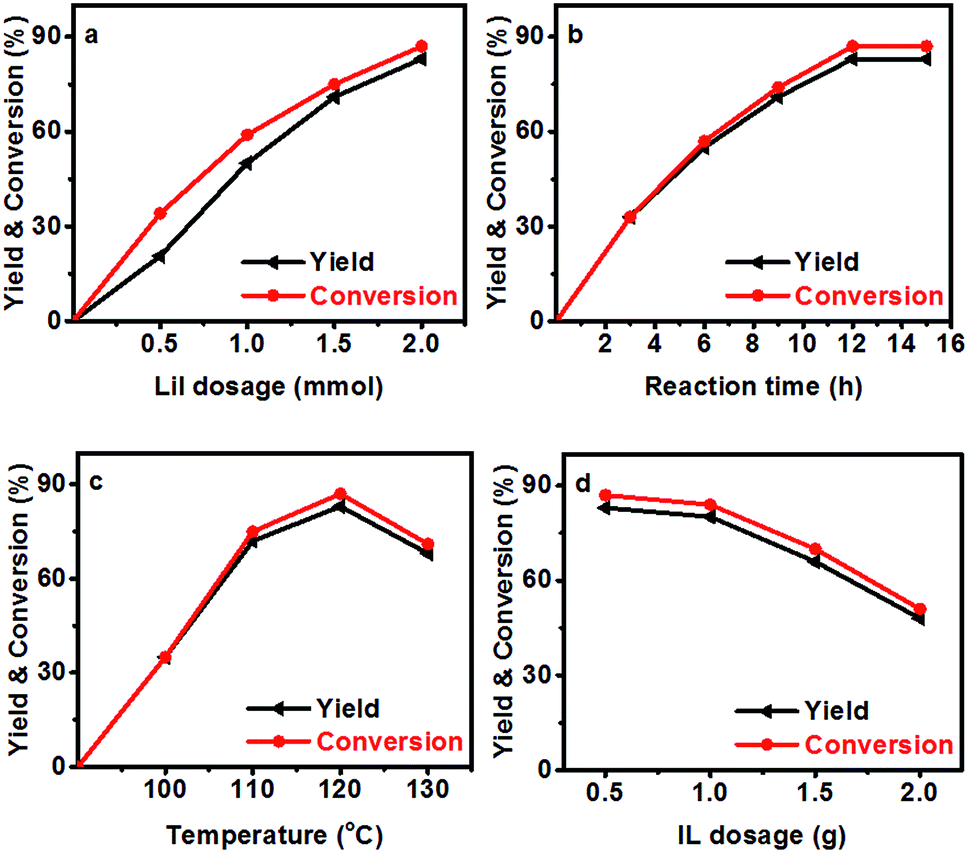
The effect of reaction parameters on the reaction was further studied using the LiI/HMimBF4 catalytic system, and the results are given in Fig. 1. As expected, the conversion of N-methylaniline and the yield of N,N-dimethylaniline increased with increasing amount of LiI, as shown in Fig. 1a. This may have resulted from the increased acidity of the catalytic system. Fig. 1b demonstrates the effect of reaction time on the reaction. The conversion and yield increased with time within 12 h, and then became independent of time. The standard Gibbs free energy change of the reaction is −15.7 kJ mol−1 at 120 °C, as calculated using the CBS-APNO method45 with the Gaussian 09 package.46 The corresponding equilibrium constant of the reaction is 122.5 if the activity coefficients of the reactants and products are supposed to be unity, and the equilibrium conversion of anisole should be 91.7%. This can explain the fact that the conversion and yield could not approach 100%. The discrepancy between the experimental and theoretical equilibrium conversion values stems mainly from the fact that the activity coefficients may not be unity. Moreover, the disproportionation reaction of N-methylaniline (Scheme 1) may also take place under the reaction conditions because the calculated standard Gibbs free energy change of this process is −1.9 kJ mol−1. The reactant, N-methylaniline, could be transformed into aniline and N,N-dimethylaniline in the absence of a methylation agent, and we detected the formation of a small amount of aniline in the experiment. This is another reason that the conversion of N-methylaniline and yield of N,N-dimethylaniline could not approach 100%. The conversion of N-methylaniline and the yield of N,N-dimethylaniline increased from 100 °C to 120 °C, and then decreased from 120 °C to 130 °C (Fig. 1c). The standard enthalpy of the reaction was calculated to be −14.8 kJ mol−1, i.e., the reaction is exothermic. At lower temperature, the reaction could not reach equilibrium within a reasonable reaction time. The reaction equilibrium was reached at 120 °C, and further increase of temperature resulted in the decrease of equilibrium conversion. Thus, a maximum conversion was observed in the conversion vs. temperature curve (Fig. 1c). The amount of HMimBF4 (IL) also affected the conversion of N-methylaniline and the yield of N,N-dimethylaniline. The reaction did not occur without the IL (Fig. 1d). However, the conversion and yield decreased with increasing amount of IL. This may be ascribed to the dilution of the reaction system and especially the decrease of the acidity, which resulted in slower reaction kinetics. Indeed, extending the reaction time slowly increased the conversion and yield.
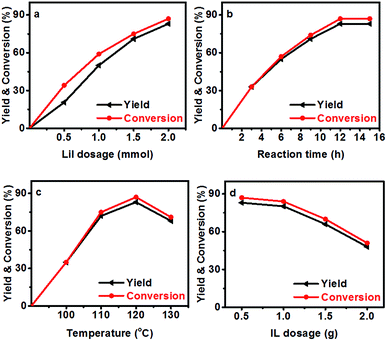 |
| | Fig. 1 Conversion of N-methylaniline and the yield of N,N-dimethylaniline (1 mmol N-methylaniline, 1 mmol anisole). (a) Effect of LiI amount with reaction time of 12 h at 120 °C in 0.5 g HMimBF4 solvent; (b) effect of reaction time at 120 °C using 2 mmol LiI in 0.5 g HMimBF4; (c) effect of temperature with reaction time of 12 h at 120 °C using 2 mmol LiI in 0.5 g HMimBF4; (d) effect of HMimBF4 amount with reaction time of 12 h at 120 °C using 2 mmol LiI. The yield is based on aniline compounds. | |
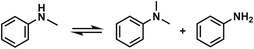 |
| | Scheme 1 The disproportionation reaction of N-methylaniline in the absence of lignin or anisole. | |
Lignin as the methylation agent
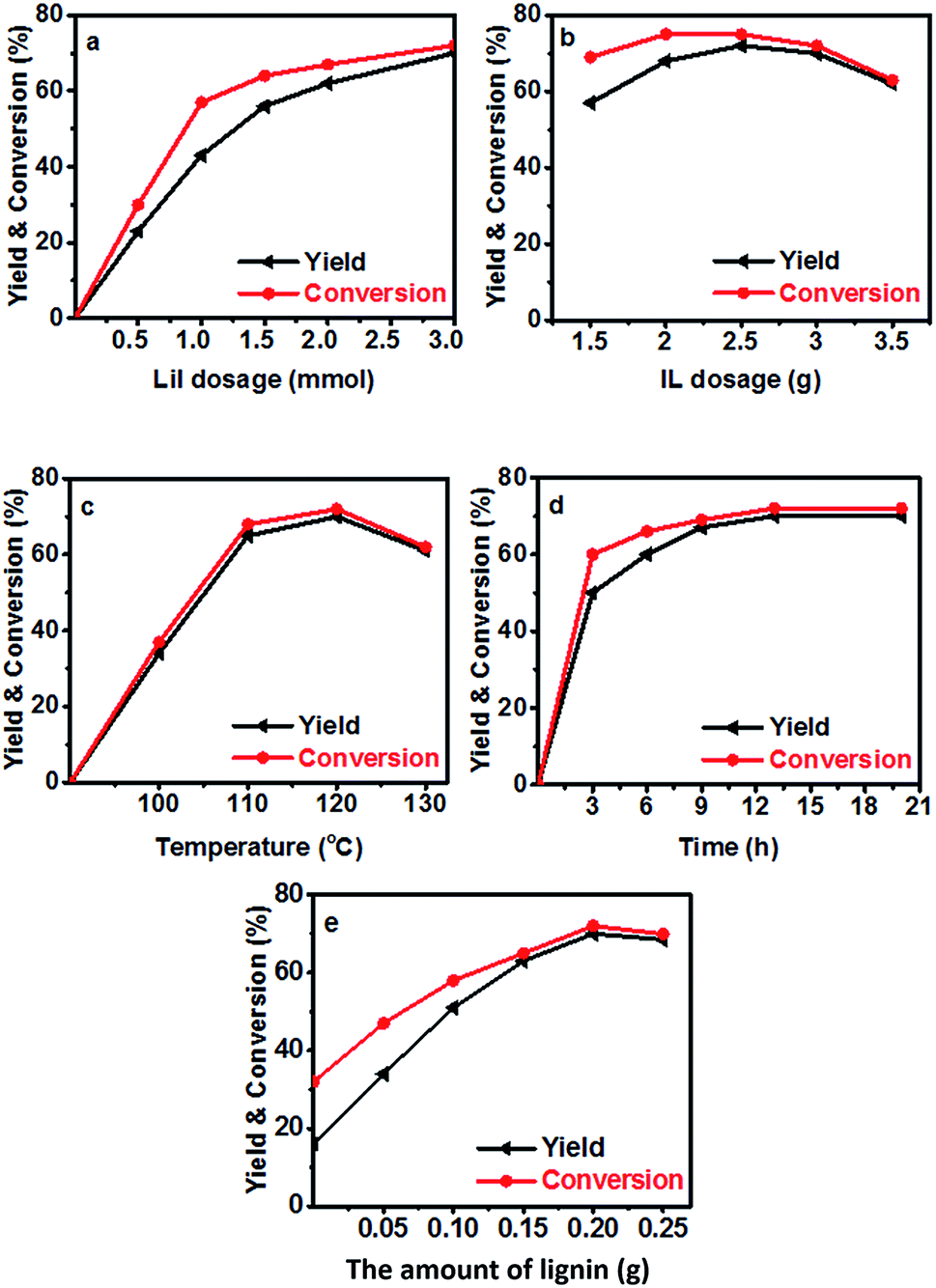
Then the performance of the LiI/HMimBF4 catalytic system using Kraft lignin as the methylation agent was investigated, and the results are shown in Fig. 2. The methoxyl content of the used lignin was 3.5 mmol g−1, as determined using the iodine stoichiometry titration method.35 The reaction could not proceed without LiI. In the conversion of N-methylaniline, the yield and selectivity of N,N-dimethylaniline increased with increasing amount of LiI. When the amount of LiI was 3 mmol, the selectivity of the desired product was 97% (Fig. 2a), and a trace amount of quaternary ammonium salt was detected. The conversion and yield increased with increasing amount of IL at the beginning, and then decreased (Fig. 2b) due to the dilution effect. The dependence of the conversion and yield on temperature was similar to that of the reaction of anisole and N-methylaniline, and the conversion of N-methylaniline and the yield of the product could reach 70% at 120 °C (Fig. 2c). Fig. 2d illustrates that the conversion and yield increased with time and reached equilibrium after about 13 h. The effect of the amount of lignin on the conversion of N-methylaniline and the yield of the desired product was also checked (Fig. 2e). The conversion and yield increased with increasing amount of lignin, while the gap between the conversion and yield diminished, indicating that the selectivity to the desired product N,N-dimethylaniline increased. When the amount of Kraft lignin was 0.2 g, the selectivity could reach 97% under the reaction conditions. This suggested that an adequate amount of lignin could inhibit the disproportionation reaction of N-methylaniline. The by-product was a small amount of quaternary ammonium salt.
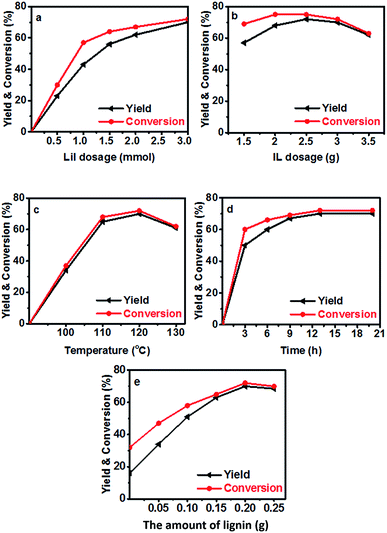 |
| | Fig. 2 The conversion of N-methylaniline and the yield of N,N-dimethylaniline (N-methylaniline: 0.5 mmol). (a) Effect of the amount of LiI in 3 g IL HMimBF4 at 120 °C with reaction time of 13 h and 0.2 g Kraft lignin; (b) effect of HMimBF4 amount at 120 °C with reaction time of 13 h using 3 mmol LiI and 0.2 g Kraft lignin; (c) effect of reaction temperature using 3 mmol LiI in 3 g HMimBF4 with reaction time of 13 h and 0.2 g Kraft lignin; (d) effect of reaction time at 120 °C using 3 mmol LiI in 3 g HMimBF4; (e) effect of the amount of Kraft lignin at 120 °C with reaction time of 13 h using 3 mmol LiI in 3 g HMimBF4. The yield is based on aniline compounds. | |
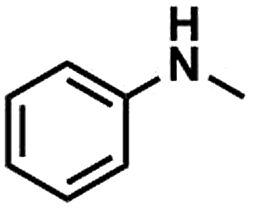
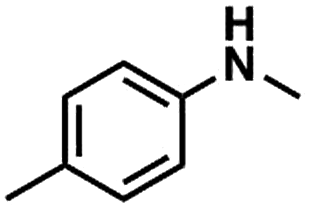
Substrate scope
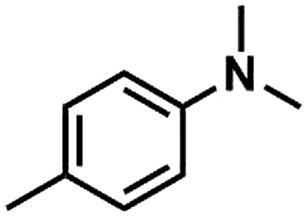
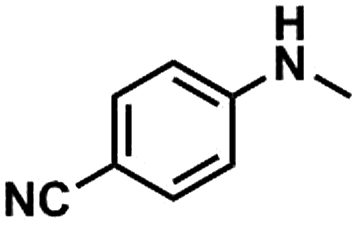
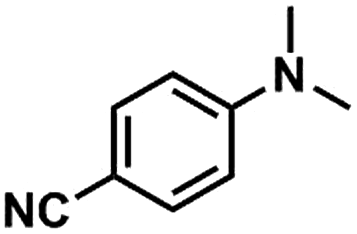
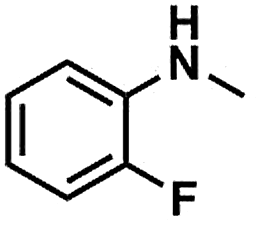
We also studied the N-methylation reaction of different aniline compounds to produce the corresponding N,N-dimethylanilines using Kraft lignin as the methylation agent, and the results are given in Table 2. Obviously, lignin could also be used as efficient methylation reagent for producing other N,N-dimethylanilines. The selectivity to the desired products is more than 96%. No other byproducts were detected except for a small amount of quaternary ammonium salt.
Table 2 The N-methylation reactions of aniline compounds using lignin as a methylating agenta
Discussions
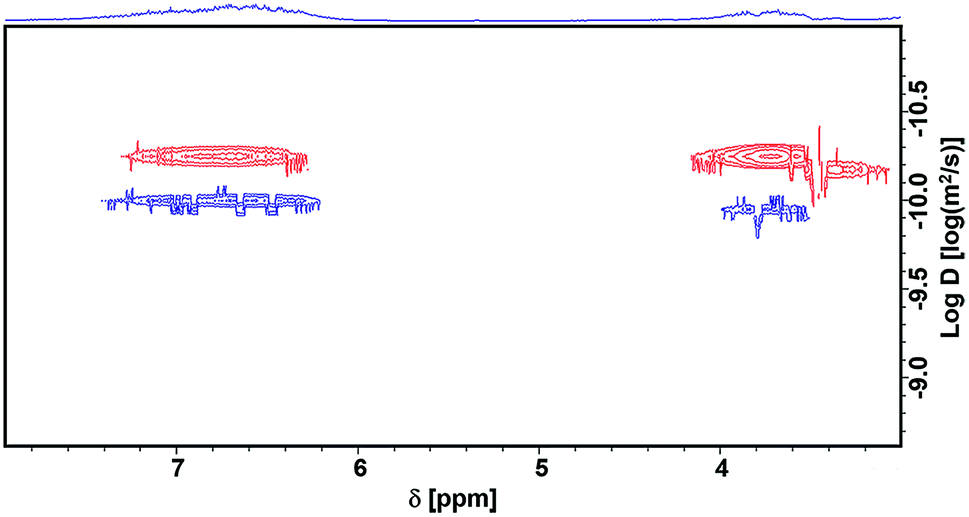
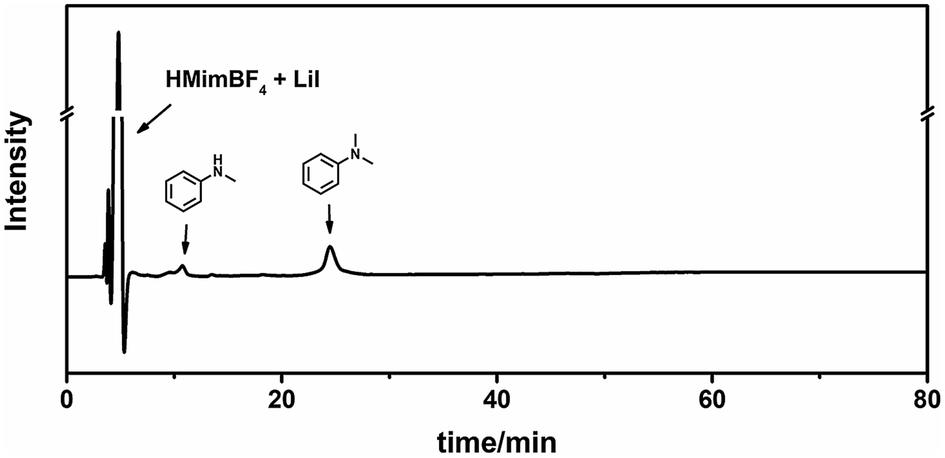
We characterized the lignin before and after the reaction using Diffusion-Ordered NMR Spectroscopy (DOSY)47 and two-dimensional heteronuclear single quantum coherence (2D-HSQC) NMR Spectroscopy.48 DOSY is a powerful method for size and molecular weight measurements for different classes of macromolecules and complex mixtures.49 It has been demonstrated to be applicable for constructive determination of lignin structure and molar mass.50,51 The measured diffusion coefficient (D) can be correlated to the molecular weight (MW) of the analyzed molecule using the Mark–Houwink equation, i.e., D = K MWα, where the scaling parameters (K and α, K < 0) depend on the molecular structure and the experimental conditions.51Fig. 3 shows that, after the reaction, the diffusion coefficient increased, indicating that the molecular weight decreased after the reaction. However, in the low molecular weight region (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D ≈ −9), there is no aromatic ring signal (δ 6–8), demonstrating that the lignin is not depolymerized into small molecules or low molecular weight fragments. The signal of –OCH3 is at 3.7 ppm, and it becomes much weaker after the reaction. Meanwhile, new signals appear at lower field (δ 7.2–7.4), which suggests the formation of catechol groups. These can also be figured out from 2D-HSQC NMR spectra, as shown in Fig. S1 and S2.† The methoxy group signal (56.4/3.70 ppm, colored yellow) becomes dramatically weaker after the reaction. Meanwhile, a new signal for catechyl units (colored brown) appears in the aromatic region. We also characterized the liquid phase after the reaction using HPLC (Fig. 4). The trace clearly shows that N,N-dimethylaniline is almost the sole product, and no signal for depolymerized fragments of lignin was observed. This further demonstrates the selective utilization of lignin.
D ≈ −9), there is no aromatic ring signal (δ 6–8), demonstrating that the lignin is not depolymerized into small molecules or low molecular weight fragments. The signal of –OCH3 is at 3.7 ppm, and it becomes much weaker after the reaction. Meanwhile, new signals appear at lower field (δ 7.2–7.4), which suggests the formation of catechol groups. These can also be figured out from 2D-HSQC NMR spectra, as shown in Fig. S1 and S2.† The methoxy group signal (56.4/3.70 ppm, colored yellow) becomes dramatically weaker after the reaction. Meanwhile, a new signal for catechyl units (colored brown) appears in the aromatic region. We also characterized the liquid phase after the reaction using HPLC (Fig. 4). The trace clearly shows that N,N-dimethylaniline is almost the sole product, and no signal for depolymerized fragments of lignin was observed. This further demonstrates the selective utilization of lignin.
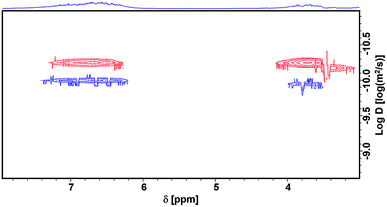 |
| | Fig. 3 The DOSY spectra of lignin before (red) and after (blue) reaction. | |
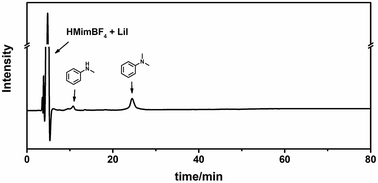 |
| | Fig. 4 HPLC trace of the liquid mixture after the reaction. | |
Reaction mechanism
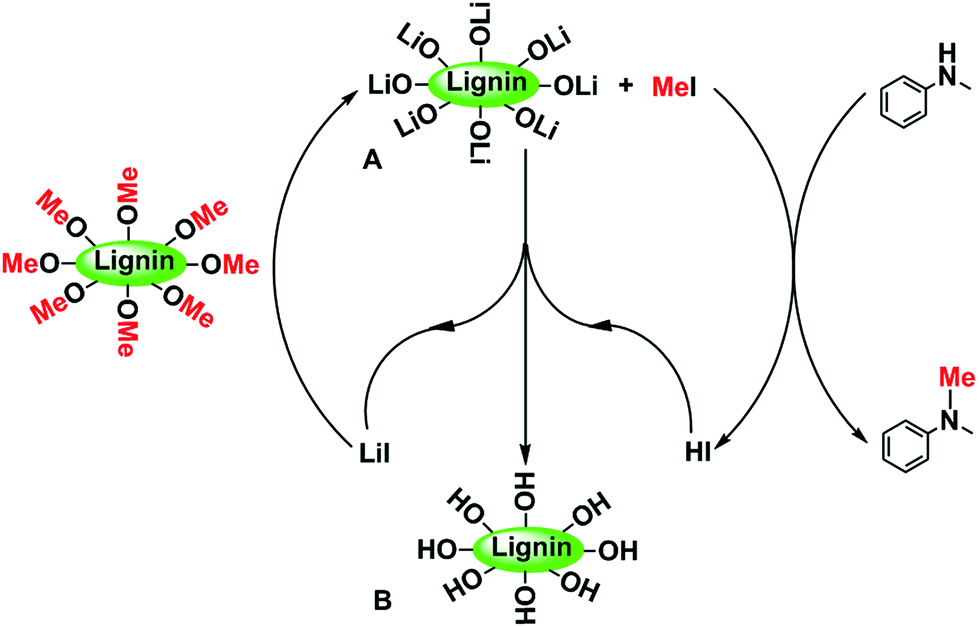
The possible reaction pathway is schematically shown in Scheme 2. In the reaction, LiI first cleaves the ether bond of the methoxy groups of lignin to form CH3I and intermediate A with the synergy of tetrafluoroborate. The mechanism of the BF4−-aided cleavage of the Ar-OCH3 ether bond is reported in our previous work.52 Then CH3I reacts with N-methylaniline to form N,N-dimethylaniline and HI, which immediately react with intermediate A to produce the leftover material B and regenerated LiI.
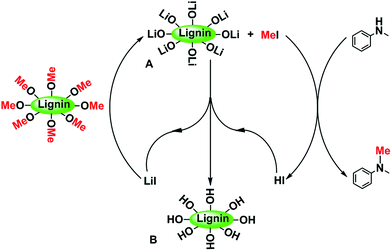 |
| | Scheme 2 The possible reaction pathway of the N-methylation reaction using lignin as a methylating agent. | |
Application of the leftover material
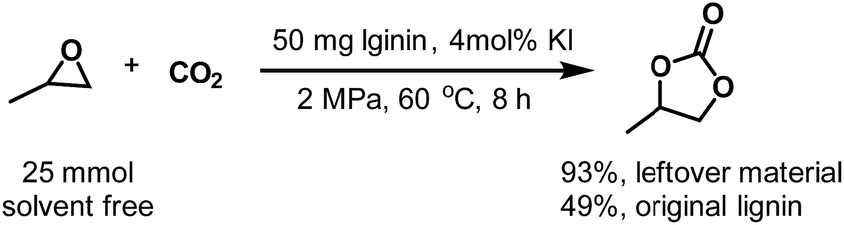
Because only the methoxy groups are selectively converted, the bulk structure is left as a solid residue. As suggested by the NMR analysis, the leftover material is rich in hydroxyl groups, which endows it with more applications than original lignin, such as in catalysis and the production of phenolic resin. Herein, we conducted a cycloaddition reaction of epoxy propane with CO2 using the residual lignin as the co-catalyst under solvent-free conditions (Scheme 3). At 60 °C under 2 MPa CO2 atmosphere, the yield of 1,2-propylene cyclic carbonate reached 93% at 8 h, while the original lignin only gave a yield of 49% under the same reaction conditions. Without lignin, the reaction did not take place. This result is consistent with the results reported in the literature.44
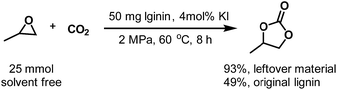 |
| | Scheme 3 Cycloaddition of epoxy propane with CO2 using the leftover material and original lignin as a catalyst under solvent-free conditions. | |
Conclusions
In summary, a route to produce N,N-dimethylanilines using the methoxy groups in lignin as the methylation reagent was developed. N,N-dimethylanilines can be produced with a selectivity of more than 96% using Kraft lignin and aniline compounds as the starting materials. This work reveals the possibility of using lignin as a methylation agent, which may benefit the production processes of some bulk chemicals that involve a methylation step. There are some obvious advantages to using lignin as methylation agent, such as its low cost, abundance and ease of separation. Moreover, the residual lignin, which is abundant in hydroxyl groups, is more favorable for use in certain applications than original lignin, such as the synthesis of functional materials. We believe that this strategy, which selectively transforms a class of functional groups in lignin into a product with high purity, will trigger much more research in the future.
Experimental
Chemicals
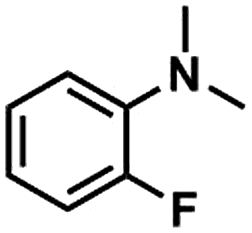
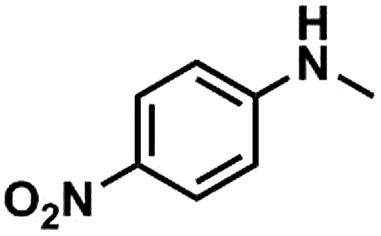
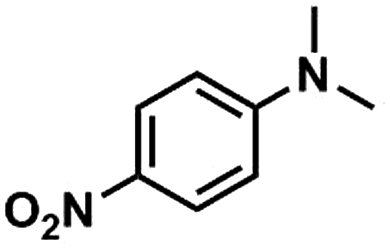
Kraft lignin was provided by Beijing InnoChem Science & Technology Co., Ltd. Anisole (99%), toluene (99%), acetonitrile (99%), N-methylaniline (99%), 4-(N-methylamino)benzonitrile (98%), N-methyl-4-nitroaniline (97%), N-methyl-p-toluidine (98%), 2-fluoro-N-methylaniline (98%), 1,3,5-trioxane (99%), epoxy propane (98%), LiI (99%), LiBr (99%), LiCl (99%), NaI (99%), KI (99%) and CsI (99%) were purchased from J&K scientific Ltd. HMimBF4 (1-hexyl-3-methylimidazolium tetrafluoroborate, >98%), HMimCl (1-hexyl-3-methylimidazolium chloride, >98%), HMimOAc (1-hexyl-3-methylimidazoliumacetate, >98%), BMimCl (1-butyl-3-methylimidazolium chloride, >98%) and BMimBF4 (1-butyl-3-methylimidazoliumacetate, >98%) were provided by Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. All chemicals were used as received.
Reaction procedures for N-methylation reaction
The reaction was carried out in a Teflon-lined stainless-steel reactor of 15 mL in capacity with an inner diameter of 16 mm, using a magnetic stirrer. In the experiment, desired amounts of catalyst, amines, Kraft lignin or anisole and solvent were loaded into the reactor. The reactor was purged with N2 to remove the air. The reaction mixture was stirred at the required temperature for the required time. The temperature was controlled by a PID temperature controller (model SX/A-1, Beijing Tianchen Electronic Company). After the reaction, the reactor was placed in ice water to cool down. About 0.02 g 1,3,5-trioxane as the internal standard was added to the liquid mixture and stirred for 5 min. The mixture was centrifuged, resulting in a liquid phase and a solid phase. The liquid phase (0.1 mL) was dissolved in MeCN-d3 (0.5 mL). The mixture was centrifuged and the supernatant liquid was collected for quantitative analysis using 1H NMR (Bruker Avance III 400 HD). The residue solid phase was washed with MeCN (5 × 3 mL), dilute HCl (pH = 2, 2 × 3 mL) and deionized water (2 × 3 mL) in sequence. Finally, it is dried under vacuum at 60 °C for 48 h over a P2O5 desiccant. The obtained solid was characterized by 2D-HSQC and DOSY analysis as well as used as the co-catalyst for the cycloaddition reaction of epoxy propane with CO2.
Reaction procedures for cycloaddition of epoxides with CO2
The reaction of CO2 and epoxide was also conducted in the same reactor. In a typical experiment, the desired amount of lignin or leftover material, KI and PO were added into the reactor. The reaction mixture was cooled down using liquid N2, and then the reactor was sealed and purged with CO2 to remove the air. After the temperature restored to room temperature, CO2 was charged into the reactor until a pressure of 2.0 MPa was reached. The pressure was determined by a pressure transducer (FOXBORO/ICT, Model 93), which could be accurate to ±0.025 MPa. Then the reactor was placed in an air bath at 60 °C for 8 h. After the reaction, the reactor was placed in ice water and the gas was released. The yield of the product was determined by 1H NMR (Bruker Avance III 400 HD) using 1,3,5-trioxane as the internal standard (Fig. S3†).
NMR and HPLC analysis
The samples for HSQC and DOSY analysis were prepared by dissolving 30 mg lignin or the leftover material in to 0.6 mL DMSO-d6. The samples were sonicated at room temperature for 1 h to ensure dissolution. The HSQC experiments were conducted on a Bruker Avance III 600 MHz spectrometer at room temperature. The DOSY experiments were conducted on a Bruker Avance III 500 MHz spectrometer, and the temperature was set and maintained at 348 K. The data analysis was performed using the Bruker Dynamics Center 3.7 according to the procedure reported in the literature. The HPLC analysis was conducted on an Agilent LC-20AT liquid chromatograph with a C18 chromatographic column and a differential refraction detector (RID-20C). After the reaction, the reaction mixture was filtered through a 0.22 μm PTFE syringe filter, and the filtrate was used for HPLC analysis. The mobile phase was methanol aqueous solution (MeOH:H2O volume ratio of 55:45) with a flow rate of 0.8 mL min−1, and the temperature was kept at 50 °C. The trace was collected for 80 min.
Computational details
The free energy of each compound was calculated using the CBS-APNO method.45 This approach is a compound method for calculating highly accurate values for thermochemical parameters, which involves a series of calculations on a particular geometry and a complete basis set model chemistry. All the Gibbs free energies (Gθ) calculated in this paper are for a standard state of 393.15 K and 1 atm. The equilibrium constant (K) is based on the equation, 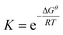 , where the ΔGθ, R and T are the standard free energy change of the reaction, the ideal gas constant and the temperature, respectively.
, where the ΔGθ, R and T are the standard free energy change of the reaction, the ideal gas constant and the temperature, respectively.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by National Key Research and Development Program of China (2017YFA0403103), the National Natural Science Foundation of China (21603235), and Chinese Academy of Sciences (QYZDY-SSW-SLH013), the Recruitment Program of Global Youth Experts of China.
Notes and references
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer, K. Huang, C. J. Houtman, N. Labbé, D. P. Harper, C. T. Maravelias, T. Runge and J. A. Dumesic, Sci. Adv., 2017, 3, e1603301 CrossRef PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS PubMed.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef.
- T. E. Amidon and S. Liu, Biotechnol. Adv., 2009, 27, 542–550 CrossRef CAS PubMed.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv., 2018, 4, eaau2968 CrossRef PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Chem., 2014, 6, 1035 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef.
- D. Forberg, T. Schwob, M. Zaheer, M. Friedrich, N. Miyajima and R. Kempe, Nat. Commun., 2016, 7, 13201 CrossRef CAS PubMed.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 1474–1478 CrossRef CAS PubMed.
- X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- Q. Mei, X. Shen, H. Liu and B. Han, Chin. Chem. Lett., 2018 DOI:10.1016/j.cclet.2018.04.032.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- I. Klein, C. Marcum, H. Kenttämaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- A. Tlili, X. Frogneux, E. Blondiaux and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 2543–2545 CrossRef CAS PubMed.
- S. Das, F. D. Bobbink, G. Laurenczy and P. J. Dyson, Angew. Chem., Int. Ed., 2014, 53, 12876–12879 CrossRef CAS PubMed.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery Jr, J. Chem. Phys., 1996, 104, 2598–2619 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09 Revision D.01, 2013, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579 CrossRef CAS PubMed.
- A. J. Simpson, Magn. Reson. Chem., 2002, 40, S72–S82 CrossRef CAS.
- J. Rönnols, A. Jacobs and F. Aldaeus, Holzforschung, 2017, 71, 563–570 Search PubMed.
- J. R. D. Montgomery, C. S. Lancefield, D. M. Miles-Barrett, K. Ackermann, B. E. Bode, N. J. Westwood and T. Lebl, ACS Omega, 2017, 2, 8466–8474 CrossRef CAS.
- Q. Mei, Y. Yang, H. Liu, S. Li, H. Liu and B. Han, Sci. Adv., 2018, 4, eaaq0266 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03006e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiaojun
Shen
a,
Xiaojun
Shen

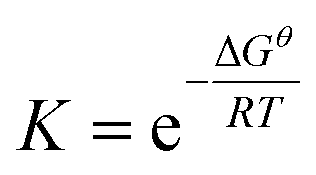 , where the ΔGθ, R and T are the standard free energy change of the reaction, the ideal gas constant and the temperature, respectively.
, where the ΔGθ, R and T are the standard free energy change of the reaction, the ideal gas constant and the temperature, respectively.