
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow to make nanobiosensors: surface modification and characterisation of nanomaterials for biosensing applications
Meral Yüce *a and
Hasan Kurtb
*a and
Hasan Kurtb
aSabanci University, Nanotechnology Research and Application Centre, Istanbul, 34956, Turkey
bIstanbul Medipol University, School of Engineering and Natural Sciences, Istanbul, 34810, Turkey. E-mail: meralyuce@sabanciuniv.edu; Tel: +90 5363826105
First published on 23rd October 2017
Abstract
This report aims to provide the audience with a guideline for construction and characterisation of nanobiosensors that are based on widely used affinity probes including antibodies and aptamers and nanomaterials such as carbon-based nanomaterials, plasmonic nanomaterials and luminescent nanomaterials. The affinity probes and major methodologies that have been extensively used to make nanobiosensors, such as thiol–metal interactions, avidin–biotin interaction, π-interactions and EDC–NHS chemistry, were described with the most recent examples from the literature. Characterisation techniques that have been practised to validate nanoparticle surface modification with antibodies and aptamers, including gel electrophoresis, ultraviolet-visible spectrophotometry, dynamic light scattering and circular dichroism were described with examples. This report mainly covers the reports published between 2014 and 2017.
1 Introduction
Interaction of nanomaterials with biological entities, such as proteins, enzymes, DNA and RNA oligonucleotides has emerged as an interdisciplinary field known as “nanobiotechnology”, which refers to the methodological approaches that nanoparticles or nanomaterials are combined to create tools for investigation of biological systems.1 Integration of nanomaterials into the sensing systems as the active elements (transducer or detector) has paved the way for a significant breakthrough in the field, resulting in stable sensing probes,2,3 enhanced detection signals in small sample volumes,4 miniaturised tools5 and systems for multiplex detection.6 The discovery of nanoparticles and following development of new instruments for nanoparticle characterisation between 1990 and 2000 has facilitated the fabrication of smart biosensing tools in the early 2000s. The number of publications that contained the keyword “nanoparticles” reached from a few hundred to hundreds of thousands within nearly two decades. Antibodies, the primary affinity reagents of the bio-analytical techniques over a century, have started accompanying the nanoparticles in the 1990s with a few thousand reports in the literature. The next generation synthetic affinity reagents called aptamers has followed the antibody-nanoparticle reports with hundreds of reports within the same period — since the aptamer discovery was only in 1990.7,8 The number of publications with affinity probe-conjugated nanoparticles has considerably increased over the years, representing around 8% of the total number of the publications made with the nanoparticles, according to the Google Scholar online database search (conducted in July 2017). The total number of reports that included the keywords “antibody and nanoparticle” appeared to be around 129![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 890 while it was 68740 for the keywords “aptamer and nanoparticle”. The nanomaterials that were functionalized with either affinity probe in these papers have varied from carbon-based nanomaterials (i.e., carbon nanotubes, graphene, fullerene) to plasmonic (i.e., gold nanoparticles) and photoluminescence nanoparticles (i.e., quantum dots and upconverting nanoparticles). Among those, carbon-based nanomaterials have seemed to be dominating the reports for both antibody and aptamer groups (around 45%), which has been immediately followed by plasmonic gold nanoparticles and luminescent nanoparticles such as quantum dots and upconverting nanoparticles. On the other hand, overall percentage of the reports with quantum dots in both probe groups was considerably high (20–25%) as compared to the percentage of other individual nanoparticles, such as upconverting nanoparticles (4–5%), graphene (13–17%), fullerene (5–12%) or carbon nanotubes (14–23%).
890 while it was 68740 for the keywords “aptamer and nanoparticle”. The nanomaterials that were functionalized with either affinity probe in these papers have varied from carbon-based nanomaterials (i.e., carbon nanotubes, graphene, fullerene) to plasmonic (i.e., gold nanoparticles) and photoluminescence nanoparticles (i.e., quantum dots and upconverting nanoparticles). Among those, carbon-based nanomaterials have seemed to be dominating the reports for both antibody and aptamer groups (around 45%), which has been immediately followed by plasmonic gold nanoparticles and luminescent nanoparticles such as quantum dots and upconverting nanoparticles. On the other hand, overall percentage of the reports with quantum dots in both probe groups was considerably high (20–25%) as compared to the percentage of other individual nanoparticles, such as upconverting nanoparticles (4–5%), graphene (13–17%), fullerene (5–12%) or carbon nanotubes (14–23%).
Carbon-based nanomaterials are the most commonly used nanomaterials in biological studies due to their versatile surface characteristics, electrical and optical merits.9,10 Combined with structural variations, the production method significantly affects nanomaterial's optical properties. For carbon nanotubes, similar size characteristics can show metal-like, semiconductor-like properties or chiral properties depending on their tubular axis indices and folding shapes. In case of graphene, the electrical and optical properties of the material can significantly change depending on the production method that embraces liquid phase exfoliation, Hummers method and chemical vapour deposition method. Each of these methods introduces a variety of defects in the two-dimensional structure of the material as well as new surface properties that are useful for surface biomodification. Incorporation of carbon nanomaterials, such as single wall carbon nanotubes,11,12 multiwall carbon nanotubes,13 chiral nanotubes, graphene, graphene oxide,14 reduced graphene oxide,15 carbon or graphene quantum dots16,17 into the biosensing platforms as signal transducers is a field growing rapidly. The biosensors fabricated with carbon-based nanomaterials could be classified as electrochemical biosensors, optical biosensors and piezoelectric-based biosensors. Carbon nanomaterial-based biosensor types, structural and physical properties of the sensing nanomaterials, key detection mechanisms and the recent advancements in the field can be found in the study reported by Tran and Mulchandani18 and Pasinszki et al.19
Noble metals such as gold and silver offer unique and robust optical properties for biosensing field.3,20 They are widely utilised in a plethora of biosensing platforms as signalling or signal enhancing elements. The unique optical properties of the noble metals arise from their ability to maintain surface-bound collective oscillation of electrons, called surface plasmon, on their dielectric–metal interfaces at visible to near infrared spectrum. Under certain size regimes, the surface-bound plasmon can be confined locally and can be excited resonantly at particular wavelengths of the incoming electromagnetic radiation. Localised surface plasmon resonance (LSPR) is highly susceptible to the changes of refractive index in the dielectric medium, and the resonance wavelength changes are highly confined in the vicinity of the nanoparticle surface. LSPR response can be easily tuned by using various geometries of nanoparticle substrates21,22 or directly in solution23 depending on the application of interest. Merging the unique optical properties of plasmonic nanomaterials with target-specific nature of affinity probes has constituted the base for plasmonic nano-biosensing.24 General working mechanisms of the plasmonic nanoparticles, including Surface Plasmon Resonance (SPR), Localized Surface Plasmon Resonance (LSPR), Surface Enhanced Raman Scattering (SERS) and the recent applications of plasmonic nanoparticles in biosensing, cancer diagnosis, drug delivery, photodynamic and photothermal therapy have been reviewed recently by Lim and Gao25 and Daraee et al.26
Luminescent nanoparticles, for example, upconverting nanoparticles are lanthanide or actinide-doped nanoscale ceramic crystals that have been utilized in biosensing applications since the last decade.27 Upconverting nanoparticles absorb and convert two or more low energy incident photons into a single higher energy photon emission. Usually, absorption energy of photons is realised using a higher concentration of dopant ions in the infrared region in order to avoid possible auto-fluorescence originating from biological entities in the ultraviolet-visible (UV-Vis) region. The fluorescent emission of higher energy photons occurs at specific visible spectrum wavelengths following the excitation with an infrared light source. Full-Width Half-Maximum values (FWHM) of the fluorescent emission is significantly narrower than the ones generated by quantum dots.28 Semiconductor quantum dots, on the other hand, are only several nanometers in size that can be engineered easily to produce a variety of different fluorescent emission signals in the visible spectrum. Size, surface properties and optical behaviours of quantum dots have enabled their use in several fields including energy,29 biosensing30 and bioimaging.31 Inorganic fluorescent nanoparticles have been immensely used in different sensing mechanisms including direct fluorescence, fluorescence resonance energy transfer, bioluminescence resonance energy transfer, chemiluminescence energy transfer, photon-induced electron transfer and electrochemiluminescence. General structures of the fluorescent inorganic nanomaterials, fundamentals of the molecular sensing strategies and a useful sensing optimisation guideline can be found in the recent review paper by Ng et al.32
The key methods used to functionalize these nanomaterials with antibodies and aptamers have been classified as thiol–noble metal interaction, streptavidin–biotin interaction, π–π interaction and NHS–EDC carbodiimide chemistry. Meanwhile, validation of surface bio-functionalization is performed by techniques such as UV-Vis spectrophotometry, Circular Dichroism (CD), Dynamic Light Scattering (DLS) and Gel Electrophoresis (GE). In this report, we reviewed the key methods used for surface biomodification of nanomaterials with antibodies and aptamers as well as the most common characterisation techniques that have been implemented for validation of the nanoparticle biomodification. Structures and properties of the affinity probes including antibodies and aptamers were briefly introduced, and core principles of the characterisation techniques were presented to provide the audience with a complete guideline for a successful surface modification and validation experiment.
2 Affinity probes for nanoparticle surface biomodification
Two major group of affinity probes that have been vastly used for surface functionalization of nanomaterials are protein-based antibodies and oligonucleotide-based aptamers. The antibodies were used to be the core affinity reagents of the sensing and imaging assays until the discovery of aptamers at the beginning of the 1990s. Both affinity molecules differ in their nature, structure and production methods and yet have their advantages and disadvantages, which are described below.2.1 Antibodies
The discovery of antibodies dates back to the pioneering research published by Emil Behring, and Shibasabura Kitasato from Robert Koch's Hygiene Institute in 1890.33 Behring and Kitasato reported that a lethal dose of diphtheria toxin could be neutralised in one animal by injecting a serum obtained from another animal actively immunised against diphtheria toxin. The production of the diphtheria antiserum to a large scale was achieved by Paul Ehrlich who moved to Koch's Institute in 1889. Ehrlich suggested that the toxicity was due to the protein side chains, that are what we know as “antibodies or Immunoglobulins” today.34The prototype antibody Immunoglobulin G (IgG) is a 150000 dalton “Y” shaped glycoprotein, comprising of two light (L) and two heavy (H) chains of amino acids. Each heavy chain and light chain contains one variable region (V) that are connected to the constant regions (C). The variable regions of the antibody are situated at the tips of “v” part, and they are responsible for the target-specific binding or antigen-specific binding (Fab, antigen-binding fragment). Each antibody has a unique amino acid sequence or “paratope” residue at the variable region as a result of the recombination of genes, to which the antibody binds to a specific site or “epitope” on the antigen molecule by complementarity in shape — supported with hydrogen bonds, electrostatic interactions, van der Waals forces and hydrophobic interactions.35 Thus, the variable regions make the antibodies suitable molecules for target-specific sensing and imaging applications. Possible variations of amino acids in different lengths in the variable Fab region has paved the way for the production of a wide variety of antibodies against several antigens or specific targets. Today, around 300 companies sell over two million antibodies for research purposes.36 According to the report by Ecker et al.,37 around 70 monoclonal antibody products developed only for therapeutic uses are expected to be on the market by 2020, and the combined worldwide sales are predicted to be around USD 125 billion. The global market for antibody production was reported as USD 7.4 billion in 2016, and it is expected to reach USD 22.6 billion by 2025, according to the report by Grand View Research, Inc.38
Although the antibody market is proliferating, standardisation of the production methods is necessary as there are serious concerns in the literature regarding the batch-to-batch variations during large-scale productions, causing conflicting and non-reproducible results in research.36 Structural instability of the antibodies due their organic amino acid assembly is another limiting factor in research conducted on-site or under varying experimental conditions. Antibodies need to be handled with caution during the assays to conserve their 3D structures, and so the target-specific binding properties. Antibodies require specific thermal conditions as well as buffer conditions, which ultimately confines the efficient utilisation of antibodies for on-site sensing applications. At this point, the conjugation of antibodies with nanoparticles could be useful in both ways; first, increasing the physiochemical stability and serum half-life of the antibodies and the second, giving a target-specific probe character to the nanoparticles.39
2.2 Aptamers
Aptamers are short, synthetic RNA, single-stranded DNA (ssDNA) or peptide chains that are selected from combinatorial libraries to target a molecule of interest through an in vitro iteration process, known as “Systematic Evolution of Ligands by Exponential Enrichment, SELEX”. The aptamers were first discovered in 1990 by two independent research groups, Ellington & Szostak7 and Tuerk & Gold.8 As we reviewed previously,40 target-specific aptamer selection can be made in several in vitro ways, unlike to conventional antibody production methods, only requiring an initial randomised library (with around 1013−14 unique sequences) flanked by primer binding sites and the purified target molecules. The randomised oligonucleotide library is enriched by iterative cycles of incubation with the target, elution of the binding sequences and amplification of the binding sequences for the next iteration, during which stringency of the steps can be tuned to find the best target-binding aptamer candidates. Aptamers can be selected in vitro from random oligonucleotide libraries for a variety of target molecules ranging from ions like Hg2+ and Cu2+,41 small chemical moieties or molecules like clenbuterol hydrochloride,42 bacterial surface proteins,43 virus proteins,44 enzymes,45 cell surface markers,46 and entire bacterial cells.47 Aptamers shows unique secondary structures like helixes and loops based on their nucleotide sequences, and a combination of these structures constitute a tertiary structure that allows aptamers to bind to their targets in a lock and key structure through van der Waals forces, hydrogen bonding and electrostatic interaction48–50 — similar to the interactions between antibodies and antigens (shape complementarity).In contrast to the antibodies, aptamers can be chemically synthesized in large scale with negligible batch-to-batch variation, and they can be modified or labelled at any desired nucleotide point for further applications, such as sensing,46 imaging,51 targeted drug delivery52 and therapeutics.53 The size of aptamers is determined by the number of nucleotides in the chain. Therefore, they could be significantly lighter in weight than the antibodies. For instance, an aptamer of 50 nucleotides is around 15 kDa whereas a typical antibody such as IgG is around 150 kDa, revealing the advantages of the aptamers over the antibodies for targeting hidden epitopes or small target molecules. Additionally, the shelf-life of aptamers is greater than the shelf-life of antibodies as a result of their synthetic structure, and aptamers can easily survive at different environmental conditions, allowing long-term storage at ambient conditions and on-site applications. According to the market analysis performed by Markets and Markets in 2015,54 the key companies in the global aptamer market are AM Biotechnologies, LLC (U.S.), Aptagen, LLC (U.S.), Aptamer Sciences, Inc. (South Korea), Aptamer Solutions (U.K.), Ltd., Aptus Biotech S.L. (Spain), Base Pair Biotechnologies, Inc. (U.S.), NeoVentures Biotechnology, Inc. (Canada), SomaLogic, Inc. (U.S.), TriLink BioTechnologies, Inc. (U.S.), and Vivonics, Inc. (U.S.). The global market value is expected to reach USD 244.93 million by 2020.
Despite their ease of use and low cost, aptamers have some disadvantages such as relatively lower sensitivity for the targets compared to the antibodies, overall negative charge due to the phosphate backbone that makes the structure hydrophilic, and the small size that makes the aptamer susceptible to renal filtration.55,56 Attachment of aptamers to nanoparticle surfaces may translate this disadvantage into an advantage, also imparts probe properties to the nanoparticles. A detailed comparison of antibodies and aptamers was previously reported by Zhou and Rossi.55 In Table 1, we prepared a similar comparison list for informative purposes.
| Properties | Aptamers | Antibodies |
|---|---|---|
| Chemical composition | ssDNA, RNA, peptide | Protein |
| Size | 5–30 kDa approximately ∼2 nm | 100–180 kDa, ∼10–15 nm |
| Structure | Rich secondary structures such as stem, loop, bulge, hairpins and G-quadruplex | α-Helix, β-sheet |
| Affinity mechanisms | 3D interactions via hydrogen bonding, electrostatic interactions and van der Walls forces (shape complementarity) | 3D epitope-based binding sites also shape complementarity |
| Targets | Ions, nucleic acids, proteins, whole cells, toxins, and chemicals | Limited to immune response |
| Applicable in all size regimes | Minimum target size ∼600 Da | |
| Selection | 1–3 months development cycle | 4–6 months development cycle |
| Automated SELEX systems can achieve ∼1 week | ||
| Cost | In vitro, solid-phase chemical synthesis | In vivo production |
| Low cost for short aptamers | High cost | |
| Physical stability | High-temperature resistance | Sensitive to temperature |
| Can be lyophilised for extended shelf life | Requires strict refrigeration | |
| Limited shelf life | ||
| Chemical modifications | Wide range of chemical modifications; sugar ring, base, and 3′ & 5′ ends | Limited chemical modifications |
| Chemical stability | High chemical resistance against enzymes through chemical modifications | Low chemical resistance |
| Low pH resistance |
3 Nanoparticle surface biomodification techniques
Key surface modification techniques that have been extensively practised to functionalize nanomaterial surfaces with biological entities such as antibodies and aptamers have appeared to be falling into four broad classes as: thiol–noble metal surface interaction (41%), streptavidin–biotin interaction (33.7%), π–π stacking interaction (23.8%) and NHS–EDC carbodiimide reaction (1.4%), according to Google Scholar publication database for 1990–2017 time period (search was conducted in July 2017). The number of publications in the field increased in a logarithmic trend by the time, and the thiol–gold nanoparticle surface interactions have dominated the field since gold is one of those ancient nanomaterials that has been used for centuries for medicinal purposes.57 Modification of colloidal gold particles with an antibody for the first time was done in 1971 by British researchers Faulk and Taylor who published the method for direct electron microscopy visualisation of Salmonellae surface antigens.58 On the other hand, the discovery of the strong interaction between avidin and biotin dated back to 1975 (ref. 59) whereas π-stacking interactions and EDC coupling technique became popular in the late 1990s60,61 during which nanotechnology was an emerging concept in the field of science and technology. Each of these major surface biomodification techniques is described below with examples from the current literature.3.1 Modification with thiol compounds
The inherently strong interaction between thiol compounds (–SH, mercaptans or sulfhydryl groups) and noble metal surfaces has allowed scientists to engineer self-assembled monolayers (SAM) on gold and silver nanoparticle surfaces for various biosensing applications. SAM of thiol molecules is simply prepared by submerging a clean noble metal substrate into a diluted solution of the desired thiol chemical. The non-reactive (inert) nature of gold has enabled the use of alkane chains with various terminal groups and formation of a vast range of functional groups on gold surfaces.62,63 The simple alkane chains create hydrophobic surfaces of gold, whereas the others with, for example, hydroxyl (OH) and carboxylic acid (COOH) terminal groups can form very hydrophilic surfaces. The strength of a single thiol–gold bond is dependent on several factors like pH of the solution, incubation time, quality of the thiol compound as well as the surface properties of the gold material itself.64 Assembly of a monolayer on gold requires a clean surface which can be achieved with sequential washes of the material in acetone, methanol, ethanol or piranha solution.65 Ultraviolet-ozone66 or oxygen plasma treatments are also used to dispose of any organic residue on the gold surface. Ultraviolet ozone treatment was found more successful than the oxygen plasma treatment method for cleaning nanostructured gold surfaces, which is in contrast to the bulk films of gold.67As presented in Table 2, a wide range of thiol compounds is available to prepare gold surfaces for biomodification. For example, 1,6-hexanedithiol being a dithiol, bears two –SH groups that are essential to attach nanoparticles or metallic ions to SAM surface.68 11-Mercaptoundecanoic acid has one –SH and one –COOH group that can be activated further with NHS–EDC reaction to covalently bind biological entities like antibodies onto the desired gold surface.69 4-Aminothiophenol bears one –SH group and one –NH2 group linked to a benzene ring that was employed as RAMAN signal reporter along with a thiolated aptamer probe.70 On the other hand, DNA and RNA oligonucleotides (aptamers) can be chemically modified with preferred thiol groups to functionalize the gold surfaces directly with affinity probes.64,70 In such cases, a spacer of carbon atoms or thymine/adenine nucleotide chain is added to the oligonucleotide chain (usually before the –SH residues) in order to prevent steric hindrance effects at the nanoscale. It should be noted that the spacer groups may interfere with the affinity of the oligonucleotide aptamers in either way. For instance, Waybrant et al.71 investigated the change in the affinity of fractalkine binding aptamer (FKN-S2) in different tail and spacer structures that included the aptamer samples with no spacer, different polyethylene glycol spacers (PEG4, PEG8, PEG24), alkyl spacers (C12 and C24), and oligonucleotide spacers (T10 and T5: and A10). Based on the radioactive competition binding assay results, the A and T oligonucleotide spacers gave the highest affinity to the aptamer while the hydrophobic alkyl spacers significantly decreased the binding affinity. The tail structure of hydrophobic dialkyl C16 also reduced the binding affinity over 7-fold as compared to the free FKN-S2 aptamer.
| Gold type | Thiol type | Target | Affinity probe | Ref. |
|---|---|---|---|---|
| Gold electrode surface | 1,6-Hexanedithiol, 1 mM, 3 h | Thrombin | Aptamer, 5′-SH-(CH2)6 GGT TGG TGT GGT TGG-3′ | 64 |
| 6-Mercaptohexanol 1 mM, 1 h | ||||
| Benzoquinone, 50 μM, 2 h | ||||
| Thiolated aptamer, 2 μM, 2 h | ||||
| Gold-silver nanorods (15–55 nm) | 4-Aminothiophenol, 1 mM, 3 h | Human protein tyrosine kinase-7 expressed on Hela (cervical cancer) cells | Aptamer, 5′-fluorescein-ATC TAA CTG CTG CGC CGC CGG GAA AAT ACT GTA CGG TTA GA (T)10-SH-3′ | 70 |
| Thiolated aptamer, 2.5 μM, 12 h | ||||
| Gold nanoparticles (5 nm) | Thiolated aptamer, 100 nM, 1.5 h | Lysozyme | Aptamer, 5′-SH-(T)10-ATC TAC GAA TTC ATC AGG GCT AAA GAG TGC AGA GTT ACT TAG-3′ | 72 |
| Thiolated PEG, 200 nM | ||||
| Gold nanoparticles | 1-Pentanethiol | Circulating tumour cells | Antibody, human EpCAM biotinylated goat antibody and anti-cadherin 11 (CDH11) | 69 |
| 11-Mercaptoundecanoic acid | ||||
| 12-Mercaptododecanoic acid | ||||
| N-Hydroxysuccinimide ester (to bind neutravidin) | ||||
| Gold nanorods (15–52 nm) | Thiol-terminated carboxypolyethylene glycol | The activated leukocyte cell adhesion molecule (ALCAM), CD166 | Antibody, Activated Leukocyte Cell Adhesion Molecule (ALCAM) antibody | 73 |
| Thiol-terminated methoxypolyethylene glycol (2.5:1 and 2:1 M ratio of COOH-PEG-SH and mPEG-SH was incubated in water with NPs) |
||||
| Colloidal gold nanoparticles (40 nm) | Direct incubation with NPs in buffer with a pH around 9 | Campylobacter jejuni subsp. jejuni ATCC® 33291 | Antibody, rabbit polyclonal and mouse monoclonal antibody against C. jejuni | 74 |
| Gold NPs deposited on a chip array surface | The thiol-modified aptamers were incubated with the chip surface for 12 h in water | Total and glycated haemoglobin (HbA1c) in whole human blood | Thiol-modified aptamers | 75 |
Thiol compounds with a hydrophilic –OH groups at one end are usually employed in reactions at excess amounts as competitors when forming SAMs on gold surfaces with other functional thiol compounds. For example, 6-mercaptohexanol or 11-mercaptoundecanol can be used with 11-mercaptoundecanoic acid or 4-aminothiophenol to form a monolayer of –COOH or –NH2 groups on gold surfaces, respectively, to anchor biomolecules or other nanoparticles. In such reactions, thiol compounds with free –OH groups naturally arrange in between the other thiol compounds covering the entire metal surface to avoid non-specific interactions.76,77 In Fig. 1, structures of some of the thiol compounds that are used to modify gold nanoparticle surfaces with protein-based and oligonucleotide-based affinity probes were illustrated. A complementary list of thiol compounds and the methods for modification of colloidal inorganic nanoparticles can be found in an earlier report by Sperling and Parak.78 The fundamentals of thiol and dithiol self-assembly process on planar surfaces, irregular surfaces and on the nanomaterials were previously reviewed by Vericat et al.68 who additionally elaborated the chemical reactivity and thermal stability of the SAMs in aqueous and ambient conditions. There are also examples of some gold nanoparticles that were functionalized with aptamers without the need for thiol molecules. In one report, for instance, aptamers were incubated with gold nanoparticles to construct different types of nanoparticle branches that produced different colours based on the number of aptamers attached.79 Finally, the aptamer-decorated gold nanoparticles functioned as colourimetric probes for detection of ochratoxin A, cocaine and 17β-estradiol with nanomolar sensitivity. Ochratoxin A was also detected with high sensitivity in grape juice and wine samples by using silver nanoparticles incorporated into a polyoxometalate-functionalized reduced graphene oxide electrode, without an immobilised affinity probe.3
 | ||
| Fig. 1 Surface biomodification of gold NPs with thiol derivatives. Chemical structures were not drawn to their original scales. (1) 11-Mercapto-1-undecanol, (2) 11-mercaptoundecanoic acid, (3) protein bound to 11-mercaptoundecanoic acid with amide bond, (4) DNA bound to 11-mercaptoundecanoic acid with amide bond, (5) DNA directly bound to gold surface through a carbon spacer and thiol group at one end, (6) DNA directly bound to the surface through a poly A tail and thiol group, (7) DNA bound to the gold surface through a partially complementary DNA strand with thiol modification, (8) protein bound to gold surface through an amide bond with 4-aminothiophenol, (9) dithiol molecule, (10) thiolated PEG, (11) mercaptopropionic acid. | ||
3.2 Modification with avidin–biotin interaction
The non-covalent interaction between avidin and biotin is known as one of the strongest bonds in nature with a dissociation constant (Kd) around 10−14 mol L−1.59 Streptavidin (also known as vitamin H or vitamin B7) is a tetrameric protein with four identical biotin binding sites. Neutravidin, on the other hand, is a de-glycosylated form of avidin with a neutral isoelectric point and it has the same specific binding affinity towards biotin molecules.Interaction of biotin with avidin/streptavidin/neutravidin has been immensely used in biotechnology for separation of biomolecules on streptavidin-coated solid supports,80 as recently reviewed by other groups.81 In nanotechnology, this robust interaction has become a routine to functionalize various nanoparticle surfaces with affinity probes, including gold nanoparticles,82 carbon nanotubes,83 quantum dots and upconversion nanoparticles.84
Construction of nanobiosensors based on streptavidin–biotin interaction usually begins with modification of the nanoparticle surface with streptavidin molecules, followed by incubation with the affinity probe that carries at least one biotin residue to bind with the streptavidin coated nanoparticle. Modification of nanoparticle surface with streptavidin, avidin or neutravidin proteins can be achieved in several ways. Based on the surface properties of nanoparticles, proteins can be deposited directly onto the surface without the need for a specific reagent85 or ideally, they are covalently attached to the surface with NHS–EDC carbodiimide chemistry by forming an amide bond.84 Glutaraldehyde method has been found effective for surface coating.86,87 In another example,88 amine-terminated biotin molecules were initially decorated onto carboxylated nanotube surfaces through NHS–EDC reaction. Streptavidin molecules were attached to the nanotube surface through the decorated biotin residues, which eventually aided to capture the molecules of interest with free biotin groups (i.e., biotinylated DNA, biotinylated fluorophore or biotinylated gold nanoparticles on the captured streptavidin).
In the first method, where no chemical reagent is necessary for streptavidin coupling, the nanoparticles are simply incubated with the streptavidin protein in a buffer solution that is followed by elimination of the unbound proteins from reaction media by centrifuge or filtration. In this case, adsorption of streptavidin molecules onto the nanoparticle surface may be explained by electrostatic interactions, van der Walls forces, hydrogen bonding or π–π stacking interactions.89 In the case of NHS–EDC carbodiimide procedure, streptavidin molecules are covalently attached to the nanoparticle surface through the amide bond formation between EDC-activated –COOH groups and primary –NH2 groups. Eventually, the nanoparticle surface covered with streptavidin molecules becomes available for modification with desired biotin-labelled affinity probes, establishing a sandwich type sensing platform. In Table 3, some examples of streptavidin–biotin interactions that were used to modify nanoparticle surfaces for biosensing applications were presented. As shown in the table, a variety of nanoparticles with streptavidin or neutravidin modifications are commercially available and can be directly used to make sandwich assays with biotinylated affinity probes. The avidin–biotin technique can be employed with almost any type of nanoparticle regardless of the material properties, which is in contrast to thiol–noble metal interaction technique and π-stacking interaction method, both require specific type of nanomaterials to bind with the affinity probes.
| NP type | Preparation of NP surface | Affinity probe | Target | Ref. |
|---|---|---|---|---|
| Upconverting NPs with streptavidin coat on the surface | Incubation of NPs directly with streptavidin solution | Aptamers with 3′ biotin modification | Enrofloxacin, a high-potency antibacterial agent | 90 |
| QDs with streptavidin modification | QDs with covalently attached streptavidin was acquired commercially from Invitrogen Life Technologies | Aptamers attached with a biotinylated, short complementary sequence for anchoring purposes | Ricin toxin chain A and light chain of botulinum toxin A | 91 |
| Cellulose paper and flexible plastic chips with printed graphene-modified silver electrodes | Paper chips were incubated directly with a solution of streptavidin | Antibody for HIV gp120 | Viruses and HIV-1 nucleic acids | 92 |
| Gold NPs (citrate coated) | Streptavidin-coated magnetic beads were commercially obtained and incubated with the affinity probes | Polyclonal anti-human EGFR antibody used as the signal probe and biotinylated EGFR aptamer as the capture probe | Epidermal growth factor receptor (EGFR), a cancer biomarker | 93 |
| CNT carpet (vertically aligned CNT carpet with neutravidin modification) | Amine groups of neutravidin and COOH groups of the CNTs were coupled with NHS–EDC chemistry in MES buffer | Aptamer-biotinylated | Lysozyme | 94 |
| Gold NPs | Gold NPs was incubated with a solution of streptavidin for 30 minutes at 37 °C; unbound molecules were removed by centrifugation | Biotinylated capture p53-antibody | p53 antigen as a cancer biomarker | 95 |
| Gold NPs | Streptavidin and gold NPs were coupled with NHS–EDC chemistry in MES buffer | Biotinylated polyclonal HIV-1 p24 antibody | HIV-1 p24 antigens | 96 |
| Upconversion NPs | Upconversion NPs were functionalized with biotin and used to cover the streptavidin-coated surface uniformly for the detection | Biotinylated oligonucleotides | Oligonucleotide sensor | 97 |
3.3 Modification through π-stacking interactions
Π-Effects are non-covalent interactions involving π electrons which can strongly interact with other π systems or aromatic molecules. This type of interaction was observed between the stacking nucleobases of DNA,98 cations like some amino acid side chains99 and aromatic compounds stacked onto the nanoparticles.100 The interactions containing π systems are essential for biological processes such as enzyme–ligand binding,101 protein–DNA binding102 and protein–RNA binding.103 Existence of π electrons–anion interactions was also reported in the literature,104 which holds a great potential for construction of novel materials for sensing (i.e., ion sensing).Presence of π electrons both in nanoparticles and biological molecules has simplified the exploitation of this interaction to develop smart nano-bio hybrid or conjugate systems for cost-effective and robust biosensing applications. As presented in Table 4, the nanobiosensors built with π systems were mainly based on the carbon nanomaterials such as graphene and carbon nanotube derivatives. Graphene is a carbon nanomaterial composing of hexagonally arranged carbon atoms with single-atom thickness. Carbon has six atoms which make sp2-hybridised crystal structure in graphene and many related carbon nanomaterials like graphene oxide, reduced graphene oxide, fullerene and carbon nanotubes. In the sp2-hybridised crystal structure, one s-orbital hybridises with two p-suborbital, namely px and py which forms a planar assembly with a particular angle of 120 degrees between the hybridised orbitals, forming a σ-bond. The remaining pz-orbital stays perpendicular to the sp2-hybrid orbitals which form a π-bond105 that lays the foundation for engineering nano-bio conjugates with π–π interaction.106 For example, nucleobases with aromatic rings can bind to the graphene surface107 with electrostatic interactions, hydrophobic forces and π–π stacking interactions. Because the nucleobases in double-stranded DNA is protected within the helix structure, the single-stranded DNA can interact better with the carbon nanomaterials. Equally, proteins with aromatic amino acid residues (i.e., histidine and tryptophan) can make strong complexes with carbon nanomaterials, unless those aromatic residues are obscured in the hydrophobic regions of proteins.108
| NP type | Preparation of NP surface | Affinity probe | Target | Ref. |
|---|---|---|---|---|
| SWCNT (oxidised and filtered) | Aptamers were incubated with SWNTs in Dulbecco's phosphate buffered saline with CaCl2 and MgCl2 at a relatively high concentration | Aptamer Sgc8c, fluorophore-labelled | Cell membrane protein tyrosine kinase-7 | 110 |
| SWCNT (near-infrared emissive nanotubes) | The aromatic character of the Cy3 dye caused dye stacking to the SWNT surface. Aptamers also assembled onto the surface through stacking | Aptamers with alternating AT nucleotide repeats at 5′ to bind with SWCNTs and with a 3′ terminal Cy3 dye | RAP1 GTPase and HIV integrase proteins | 111 |
| CNTs | The detection was based on the non-covalent assembly of the FAM dye-labelled aptamers on CNTs in a buffer solution for 15 min that was induced by π stacking of DNA bases on CNTs | Aptamers with fluorescent FAM dye at 5′ | Kanamycin | 112 |
| Graphene oxide nanosheets | 3 μL of 100 μg μL−1 GO-solution and 10 μL of 100 nM ATP or GTP aptamer stock solution were mixed in PBS-based binding buffer for around 5 min | Aptamers with fluorescent Cy5 and FAM modifications | ATP and GTP in MCF-7 breast cancer cells | 113 |
| Graphene oxide used as the fluorescence quenching agent | 10 nM FAM-ssDNA solution was incubated for 15 min at room temperature, and 20 μL of GO (0.1 mg mL−1) was then added to this mixture for another 5 min | Aptamer with 5′ fluorescent FAM label | Bisphenol-A | 114 |
| Graphene oxide | FAM-labelled theophylline binding aptamer was mixed with GO suspension to a final concentration of 100 nM and incubated at room temperature for an hour | Aptamer with 5′ fluorescent FAM label | Theophylline in serum | 115 |
| Graphene oxide | 0.04 mg mL−1 of graphene oxide was mixed with different concentrations of β-lactamase aptamer in Tris–EDTA buffer for around 10 minutes | Aptamer with 5′ fluorescent FAM label | β-Lactamase in milk | 116 |
For example, single wall carbon nanocorns and 3′ FAM (carboxyfluorescein) dye-labelled aptamers were effectively utilised as fluorescent sensing probes for ochratoxin A detection.109 The essential point was that the ochratoxin A-specific DNA aptamers could go into a G-quadruplex structure upon binding to the target. In the absence of the target, DNA aptamers and aromatic FAM labels interacted with negatively-charged single wall carbon nanocorn surface through π stacking, leaving the fluorescent dye-quenched as a result of the fluorescence resonance energy transfer (FRET) from dye to the carbon surface.
In a similar example, nanographene was used as the fluorescent quencher platform for detection of dopamine using the DNA aptamers that were chemically conjugated with fluorescent carbon nanodots.117 Aptamers were conjugated with the fluorescent carbon dots by EDC–NHS reaction that was later incubated with the nanographene to form nano-bio conjugates through π–π stacking and hydrophobic interactions. The resulting conjugates did not produce fluorescent signals in the absence of target because of the surface energy transfer from carbon dots to the nanographene whereas the fluorescent signal was recovered in the presence of target dopamine. In another example, Xia et al.14 reported a method using graphene oxide as the fluorescent quenching platform and N-terminal fluorescein isothiocyanate (FITC)-labelled peptide aptamers as the affinity probes for detection of human chorionic gonadotropin (a biomarker for pregnancy and some cancer types). It was demonstrated that some amino acids including lysine, histidine, glutamic acid and aspartic acid had negligible effects on fluorescent quenching efficiency while the other amino acids including arginine, phenylalanine, tryptophan, tyrosine and four proteins (bovine serum albumin, immune globulin G, recombinant human erythropoietin and thrombin) disturbed the interaction between the peptide aptamer and graphene oxide surface, thus recovered the signal. Additionally, isothermal titration calorimetry technique revealed that electrostatic interactions and π–π systems played a major role in the strong interaction between some amino acids and graphene oxide, in which the amino acids influenced the conjugation either individually (for lysine, arginine, histidine, phenylalanine, and tyrosine) or co-operatively (tryptophan).118
3.4 Modification through EDC–NHS chemistry
Carbodiimide chemistry is the most common method to covalently modify free carboxylic acids with primary amine groups for labelling and surface functionalization purposes. The most common carbodiimide is the water-soluble N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC). EDC is usually employed together with N-hydroxysuccinimide (NHS) to accelerate the reaction rate and the final coupling efficiency. EDC activates the free carboxyl groups on one molecule to bind with primary amine groups of the other molecule, constituting an amide bond. NHS or its water-soluble analogue (sulpho-NHS) reacts with the carboxyl groups, forming a stable amine-reactive NHS ester intermediate to be coupled with the primary amines at physiologic pH conditions. The intermediate produced by NHS is significantly more stable than the O-acylisourea that is produced in reactions only with EDC. Thus, NHS is frequently included in EDC reactions. Because none of these compounds leave residues on the final conjugate structure, carbodiimide molecules are considered as zero-length cross-linkers.119 Unlike the previous modification methods, EDC–NHS carbodiimide reaction produces a strong covalent bond between the reaction compounds reinforcing the lifetime of the conjugation. Because all proteins (for example, antibodies) are composed of amino acids that contain primary amine and carboxylic acid groups, NHS–EDC reaction can be used to bind biomolecules to any surface comprising a primary amine or carboxylic acid groups. Similarly, DNA or RNA molecules (for example, aptamers) can be synthesised chemically with free carboxylic acid or primary amine groups at the desired nucleotide points, facilitating their coupling onto the functionalized material surfaces through EDC–NHS chemistry.75,94,112,120As presented in Table 5, EDC–NHS chemistry has been widely used to functionalize nanoparticles with affinity probes to construct nanobiosensors. In some cases, other modification techniques like thiol–noble metal interaction and π-stacking systems were combined with EDC–NHS chemistry in order to fabricate sophisticated nano-bio conjugates. For example, in the study reported by Zhu and Lee,121 carbon nanotubes and silver nanoparticles were employed together to develop a nanobiosensor for the detection α-1 antitrypsin, a biomarker for Alzheimer's disease. The authors treated the nanotubes with perylene tetracarboxylic acid, which had a pyrenyl group that could stack on the nanotubes through π–π interaction and free –COOH groups that were further used to modify the nanotubes with 5′ NH2-aptamers through EDC–NHS chemistry. In another report, naive silver nanoparticles were treated with a mercaptopropionic acid solution that had thiol groups at one end to bind with the silver nanoparticles and free –COOH groups on the other end for conjugation with the biomolecules. Free –COOH groups of the silver nanoparticles were activated through EDC–NHS reaction to attach antibodies that eventually served as the signal probes for the detection of α-1 antitrypsin. In such reactions, the amount of the affinity probe (aptamers or antibodies) should be significantly high in molar concentration to ensure the uniform coating of the nanoparticle surface.
| NP type | Preparation of NP surface | Affinity probe | Target | Ref. |
|---|---|---|---|---|
| Carbon nanotubes and α-1 antitrypsin modified silver NPs as signal enhancers | Nanotubes were first functionalized with a pyrenyl group of perylene tetracarboxylic acid/carbon nanotubes through π–π stacking, which produced –COOH groups on the tubes to link with 5′ NH2-aptamers through EDC–NHS. Silver NPs were first treated with mercaptopropionic acid through the thiols which produced free –COOH on the surface to covalently link with antibodies using EDC–NHS chemistry | Antibody with an alkaline phosphatase label and aptamer with 5′ NH2 modification | α-1 antitrypsin (a recognised biomarker for Alzheimer's disease) | 121 |
| Graphene films transferred onto silicon surfaces to make field effect transistor devices | EDC and sulpho-NHS were used to couple PEG/ethanolamine (as the spacer), and PEG/aptamer to the carboxyl groups of the pyrene butyric acid treated graphene device surfaces | Aptamers with 5′ NH2 and C6 spacer modification | Prostate-specific antigen (PSA) | 124 |
| Carbon dots | EDC without NHS was used to link carboxylic carbon dots with 5′ NH2 modified aptamers in PBS buffer | Aptamers with 5′ NH2 modification | Dopamine | 117 |
| Gold NPs | Carboxylated gold NPs were activated with EDC and NHS to be modified with the side chains of the protein | Antibody of prostate-specific antigen | Prostate-specific antigen | 125 |
| Quantum dots with free carboxylate groups on the surface | Carboxyl-terminated quantum dots were activated by EDC/sulpho-NHS mixture in PBS buffer and ![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) ncubated for 2 h. Unbound aptamers and the quantum dots were removed using a centrifugal filter unit ncubated for 2 h. Unbound aptamers and the quantum dots were removed using a centrifugal filter unit |
Aptamer with 5′ NH2 and C6 modification | Thrombin | 126 |
| Quantum dots (ZnS:Mn) | Quantum dots were first treated with mercaptopropionic acid to form free –COOH groups on the surface, which was later treated with EDC and NHS to bind amino terminated aptamers | Aptamers with 5′ NH2 modification | Acetamiprid residues | 120 |
| Upconverting NPs with free carboxylate groups | Carboxyl group terminated upconverting nanoparticles were incubated with amine terminated aptamers in the presence of EDC and sulpho-NHS | Aptamers with 5′ NH2 modification | Foodborne pathogens | 28 |
In a recent study conducted by our group,28 the surfaces of carboxylic acid-coated upconverting nanoparticles and carboxylic acid-coated quantum dots were activated by EDC–NHS carbodiimide reaction to decorate the nanoparticle surfaces with foodborne pathogen-specific DNA aptamers that had primary amine groups at 5′ end. These fluorescent nano-bio conjugates served as the signal probes for simultaneous detection of two different pathogens. The luminescent nanobiosensors fabricated in the study were characterised by GE, DLS, CD and UV-Vis spectroscopy techniques to validate the presence of target-specific aptamers on the nanoparticle surfaces, which is essential for nanobiosensor studies. The advancements in the development of nanoprobes for detection of pathogenic bacteria,122 and several other food contaminants123 has been covered in the recent literature.
4 Characterisation of nanoparticle surface biomodification
Characterisation studies are performed to validate presence and activity of the affinity probes that are coupled with the nanomaterials. During the surface biomodification of nanomaterials, an excess amount of affinity probe is typically used to saturate nanomaterial surface which eventually prevents non-specific interactions of the probes. In the second step, unreacted affinity probes and unreacted nanoparticles in the reaction medium are separated from the probe-conjugated nanomaterials. Because the unreacted components would compete with nanosensors to bind with target molecules non-specifically and severely undermine the performance of the nanosensors, leading to a narrower detection window and higher limit of detection values. Therefore, several wash-out steps with centrifugation or commercial filters are recommended to discard the unbound reaction components. Purification of the conjugated probes from the reaction medium is followed by the characterisation of the sensor probes. Naïve nanomaterials and unconjugated affinity probe samples also participate in characterisation studies as the key control samples. Here we briefly described the basic principles of the most common nanobiosensor characterisation techniques with examples from the recent literature.4.1 Ultraviolet-visible absorption spectrophotometry
UV-Vis absorption spectroscopy is a standard tool for the characterisation of nanobiosensors. Many of the core nanomaterials used in nanobiosensor construction studies can be distinctively identified and quantified by the absorption spectroscopy. Affinity probes, oligonucleotide-based aptamers and protein-based antibodies, also show typical absorption peaks around 260 and 280 nm with UV-Vis technique and the absorption at these specific wavelengths are used to calculate molar concentrations and purities of the samples in buffer solutions. Although this method cannot be applied quantitatively in the presence of affinity probes and nanomaterials with strong UV absorption (for example quantum dots, plasmonic nanostructures and carbon nanomaterials), it can still be used to qualitatively identify the presence of aptamers or antibodies on nanomaterials based on the absorbance spectrum change upon successful surface biomodification. In such cases, absorption peaks of the affinity probes are often observed as slight shoulders in the absorption spectrum of the nanoprobe, which strongly depends on the extinction coefficient difference and the number of functional sites on the nanomaterial available for probe conjugation. Some examples of antibody and aptamer-functionalized gold nanoparticle probes that were characterised by UV-Vis absorption spectroscopy were represented in Fig. 2a and b,127,128 respectively. | ||
| Fig. 2 Characterisation of antibody and aptamer-functionalized gold nanoparticles (NPs) with UV-Vis spectrophotometry technique. (a) UV-Vis absorption spectra of naïve (blue), antibody-conjugated (green) and aggregated gold NPs (red). The inset shows the localised surface plasmon resonance shift of the gold NPs after conjugation with antibodies. Reprinted by permission from Macmillan Publishers Ltd: [Nature Protocols],127 Copyright 2008. (b) UV-Vis absorption spectra of gold NPs (black) and aptamer-functionalized gold NPs (red). The inset shows the shift in localised surface plasmon resonance signal of the gold NPs upon successful conjugation. Reprinted from ref. 128, Copyright (2017), with permission from Elsevier. (c) UV-Vis absorption spectra of (a) aptamer-quantum dot conjugates and (b) naïve quantum dots. The inset shows the absorption spectrum shift of the quantum dots after conjugation to the aptamers. Reproduced from ref. 129 with permission of The Royal Society of Chemistry. | ||
As seen in the figure, the functionalization of the gold nanoparticles with affinity probes resulted in a red shift in the localised surface plasmon signal of the nanoparticle since the refractive index of the affinity reagent was significantly larger than the refractive index of the surrounding media. Besides, the DNA shoulder in the absorbance spectrum around 260 nm in Fig. 2b confirmed the successful surface modification of the gold nanoparticles. The plasmonic nanostructures usually show exponentially increased refractive index sensitivity in the nanoscale vicinity of their surface. In fact, the functionalization of plasmonic nanostructures with aptamers can be further validated using the complementary oligonucleotide sequences since the refractive index (n) difference between ssDNA (nss ∼ 1.46) and dsDNA (nds ∼ 1.54)130 can be easily detected in nanomolar range.131
In the Fig. 2c,129 UV-Vis absorption spectrum of naïve quantum dots and aptamer-functionalized quantum dots were represented where a distinct oligonucleotide shoulder around 260 nm in the spectrum was detected. Unlike the absorbance behaviours of aptamer-conjugated plasmonic gold nanoparticles, aptamer-functionalized quantum dots showed a blue-shift that was indicated the disruption of the core nanoparticle structure upon aptamer modification. The slight expansion in FWHM of the quantum dots upon aptamer conjugation was another evidence for successful surface biomodification.28 The change in the core structure of the quantum dots upon surface biomodification could also affect their behaviours under an electric field that is discussed later in electrophoresis subsection.
4.2 Gel electrophoresis
GE remains to be one of the most fundamental characterisation technique for biochemistry and molecular biology. Depending on the size and surface charge, biomolecules such as DNA, RNA and proteins can be easily separated with high resolution in porous agarose or polyacrylamide matrix under an applied electric field. The velocity of the biomolecules within the matrix is governed by the pore size of the matrix, applied voltage and the specific electrophoretic mobility of the macromolecules. Careful optimisation of the gel electrophoresis parameters can, in fact, provide an extraordinarily high resolution that could differentiate between DNA samples with a few nucleotide difference in size. A wide range of gel staining options can be utilised for visualisation of numerous specimens, depending on the purpose of the application. On the other hand, these staining procedures can be skipped for some nanoparticles (i.e. quantum dots) that show intrinsic fluorescent signals upon exposure to the UV light.Nanoprobes can also be analysed effectively with gel electrophoresis technique due to their small sizes, physical and intrinsic optical properties.132 The fundamentals of gel electrophoresis for nanoparticles and nanoprobes were well-established in the last decade.133–135 Electrophoretic mobility of the nanoparticles depends not only on their size but also the zeta potential characteristics of the particles. In Fig. 3a,136 electrophoretic behaviours of the quantum dots conjugated with monovalent streptavidin molecules at various concentrations were represented. As the concentration of streptavidin protein was increased in the biomodification medium, the number of electrophoretic bands for quantum dot–streptavidin conjugates decreased, and finally became one sharp electrophoretic band that was carrying the nanoparticles-conjugated with the ideal amount of streptavidin molecules.
 | ||
| Fig. 3 Characterisation of nanobiosensors with gel electrophoresis technique. (a) 6-His tagged Cd/Se–Zn/Cd/S core–shell quantum dots incubated with a range of monovalent streptavidin molecules. The resulting nano-bio conjugates were directly visible under UV light without a staining step. Purified conjugates shown in the top right image appeared as sharp single electrophoretic bands without residues or aggregates. Reprinted with permission from Macmillan Publishers Ltd: [Nature Methods],136 Copyright (2008). (b) Modification of streptavidin-coated quantum dots with biotinylated A22 aptamer (against the hemagglutinin of influenza A virus) at increasing concentrations (lane (a) QD alone, lane (b) QD and A22 aptamer at 1:1, lane (c) QD and A22 at 1:2; lane (d) QD and A22 at 1:3; lane (e) QD and A22 at 1:4 ratios, respectively). Reproduced from ref. 137 with permission of The Royal Society of Chemistry. (c) Naïve quantum dots move faster than the aptamer-conjugated quantum dots in the electrophoretic environment. Reproduced from ref. 138, MDPI open access content. | ||
In fact, competition between size and surface charge (zeta potential) of nanoparticles could lead up to peculiar gel electrophoresis results that are often observed in quantum dot gel samples. As represented in Fig. 3b,137 the streptavidin-coated quantum dots formed several different conjugates when they were incubated with the biotinylated aptamers at various concentrations. One could expect to see a slower movement of the aptamer-conjugated quantum dots in the gel as compared to the naïve nanoparticles because the nanoparticles became heavier in size upon bio-conjugation. On the contrary, the aptamer-conjugated quantum dots moved faster than the naïve quantum dots, which might be explained by the increased negative charge on the surface due to the phosphate backbone of the coupled oligonucleotides. Another explanation could be the destruction of the core structure of quantum dots during the aptamer coupling where a layer of the atoms may have left the structure. That eventually may have caused a decrease in the overall nanoparticle size and an increase in the velocity of the conjugated particles in the gel environment. In Fig. 3c,138 a common behaviour was observed for the quantum dots-conjugated with aptamers. As theoretically expected, the conjugation resulted in an increase in the size and decrease in the mobility of the conjugated quantum dots. In such situations, DLS technique might be useful to control the hydrodynamic size of the particles before and after conjugation reaction.
In Fig. 4, the electrophoretic behaviour of carboxylic acid-coated upconverting nanoparticles that were functionalized with 5′-NH2 aptamers through EDC–NHS chemistry in our laboratory was presented. Aptamer-conjugated upconverting nanoparticles and the unreacted aptamer molecules were visualised on 1% agarose gel under UV light exposure following the standard ethidium bromide staining. In Fig. 4a, an excess amount of aptamers were employed in the reaction for complete saturation of the upconverting nanoparticle surfaces. The conjugated upconverting nanoparticles at various sizes and the unreacted aptamers appeared as discrete bands on an agarose gel. One the molar concentration of the aptamers in the reaction was decreased by half, a single and thick band of the conjugated upconverting nanoparticles was observed along with a faint band of the unreacted aptamers at the bottom of the gel (Fig. 4b). The purification of the conjugated nanoprobes from the unreacted aptamers by filtration resulted in a sharp single band, as can be seen in Fig. 4c.
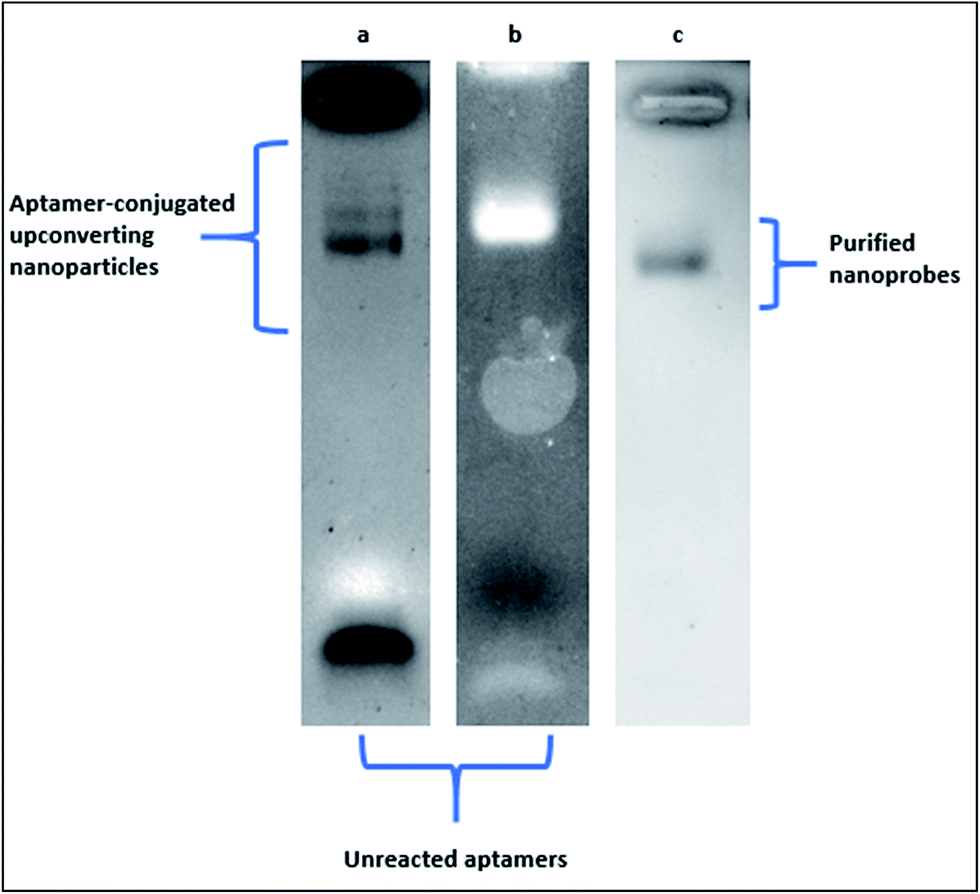 | ||
| Fig. 4 Electrophoretic behaviour of upconverting nanoparticles modified with aptamers. (a) Modification with an excess amount of aptamer in the reaction medium (nanoparticle aptamer ratio was 1:20). Aptamer-conjugated upconverting nanoparticles at different sizes accumulated at the upper part of the gel whereas the unreacted aptamers (lighter in size) accumulated at the bottom of the gel. (b) Modification with a standard amount of aptamer in the reaction medium (1:10 nanoparticle aptamer ratio was). Aptamer-conjugated upconverting nanoparticles at only one size accumulated at the upper part of the gel whereas the unreacted aptamers (lighter in size) accumulated at the bottom of the gel. (c) Purification of the conjugated upconverting nanoparticles from the reaction medium with filtration. Aptamer-conjugated upconverting nanoparticles at one size accumulated at the upper part of the gel. The band for unreacted aptamers disappeared after the purification step. | ||
4.3 Circular dichroism
CD spectroscopy is used to evaluate dichroic behaviour of materials over UV, visible and near-infrared spectrum. Dichroism concept originates from the chiral materials that interact with different states of circularly polarised light. CD spectroscopy is simply the polarisation-based UV-Vis absorption spectroscopy, utilising right or left-circularly polarised incident light instead of unpolarized light. Although UV-Vis absorption spectroscopy method remains to be one of the most robust techniques for characterisation of macromolecules, the high extinction coefficient of nanomaterials in UV spectrum renders the evaluation of nanobiosensors challenging. Because most of the nanoprobes utilised in biosensing applications do not show dichroic behaviour in the UV spectrum, CD spectroscopy can easily surpass overlapping absorption responses and deliver precise structural information. CD technique is often used to analyse the secondary structure of macromolecules (i.e., proteins, nucleic acids) and non-racemic mixtures of small chiral molecules (i.e., amino acids) under various experimental conditions. Biological molecules like DNA and proteins exhibit highly dichroic response due to their chiral monomers and constituents, unlike many nanoparticles. Therefore, CD technique can be performed to evaluate conformational changes of antibodies and aptamers upon binding to the nanomaterials. As presented in Fig. 5,28 CD spectra of quantum dots (a) and upconverting nanoparticles (b) resulted in zero ellipticity since they did not show any chiral properties (black lines). However, unconjugated ssDNA aptamer samples presented peaks around 220 and 280 nm due to their chiral properties (blue lines). The spectral difference between the naïve aptamers, nanoparticles and the aptamer-conjugated nanoparticles showed the changes in the helix structures of the DNA molecules, indicating the successful biomodification of the nanoparticle surfaces (red lines). | ||
| Fig. 5 Circular dichroism spectra of aptamer-conjugated luminescent nanoparticles. Reprinted from ref. 28 with permission from Elsevier. (a) Spectrum for naïve Cd/Te quantum dots (black), spectrum for ssDNA aptamer against Staphylococcus aureus (blue) and spectrum for aptamer-conjugated quantum dots (red). (b) Spectrum for naïve NaYF4:Yb,Er upconverting nanoparticles (black), spectrum for ssDNA aptamer against Salmonella typhimurium (blue) and spectrum for aptamer-conjugated upconverting nanoparticles (red). | ||
4.4 Dynamic light scattering
DLS technique combined with zeta potential measurements offers an extensive repertoire for nanobiosensor characterisation. DLS technique exploits the Brownian motion of the particles that are dispersed well in a solution. Temporal fluctuations of light and scattering events due to the movement of particles in solution are often analysed using photon autocorrelation function that can reveal hydrodynamic radii, size distribution and concentration of the samples. DLS technique is based on three fundamental requirements: (a) spherical shape of the scattering particles, (b) sufficiently dilute solution of the sample to refrain multiple scattering events and (c) the absence of interfering absorption events or purity of the sample solution. It is imperative to dilute the solution of nanoprobes (i.e., 1:1000) to obtain reproducible results with DLS technique. At high concentrations of the particles, multiple scattering events could result in artificially low mean size measurements. Additionally, viscosity dominates the size measurement and result in artificially high mean size. Apart from that, dispersion quality of the colloidal particle or bio-nano conjugate solution is necessary for the accurate evaluation of the final data. Nanoprobe absorption profile is often neglected in DLS measurements due to the integrated 633 nm red laser in standard DLS instruments. However, it could result in failed measurements for specific nanoparticle types. For example, quantum dots with deep-red fluorescent emissions absorb the incident laser light with high extinction coefficient, thus, prevent the data acquisition. Since DLS method exploits Rayleigh scattering, the scattering intensity is directly proportional to d.6 The presence of dust, artefacts or agglomerates severely jeopardises the accuracy of size distribution results. Therefore, disposable and sterile cut-off syringe filters can be used to remove unwanted artefacts. Additionally, DLS could only analyse the colloidal system in logarithmic size due to the incorporated data analysis method. In an ideal monodisperse colloidal nanoprobe solution, DLS should reveal slightly larger size distribution for bio-conjugated samples than the naïve nanoparticle sample. The difference in the determined size originates from the functionalization of the nanoprobes and the hydration shell of nanoprobes originating from the surrounding solvent molecules. As presented in Fig. 6, functionalization of nanoprobes with oligonucleotides and proteins increased the hydrodynamic radii of the nanoprobes that confirmed the successful functionalization of nanoparticle samples. In the case of aptamers, a sharp increase in the mean nanoprobe diameter can be seen as a result of the free aptamer tails that are in contact with the solvent molecules. Example of such interaction was shown in Fig. 6a139 in which the DLS data were further validated with gel electrophoresis technique. The nanoparticles with DNA and DNA + RNA conjugations became heavier in size. Thus, they moved rather slowly in the gel. In case of antibody samples, the increase in the hydrodynamic size distribution of the conjugates possibly originated from the bulk size of the antibodies as can be seen in Fig. 6b and c.140,141 DLS and zeta potential measurements are quite precise and easy to use for nanoprobe stability monitoring, as well. Periodic DLS measurements can reveal the colloidal stability of the functionalized nanoparticles over a defined period. Combined with the other methods, validation of the surface biomodification can be achieved for various nanoparticles.
 | ||
| Fig. 6 Characterisation of nanoparticle surface biomodification with dynamic light scattering technique. (a) Hydrodynamic size distribution of naïve gold NPs (pink), anchor DNA oligonucleotide-conjugated gold NPs (red) and hybridised RNA–anchor DNA oligonucleotide-conjugated gold nanoparticles (blue). Gel electrophoresis shows the electrophoretic behaviours of the conjugated gold nanoparticles.139 Copyright 2014, American Chemical Society, open access content. (b) Size distribution histogram of quantum dots (upper panel) and antibody-conjugated quantum dots (lower panel).140 Copyright 2011 Tiwari et al., publisher and licensee Dove Medical Press Ltd. (c) Hydrodynamic size distribution profile of upconverting nanoparticles (black) and antibody-conjugated upconverting nanoparticles (red). Reproduced from ref. 141 with permission of The Royal Society of Chemistry. | ||
A summary of these characterisation techniques is presented in Table 6.
| Technique | Type of NP | Advantages | Disadvantages | Expected results | Ref. |
|---|---|---|---|---|---|
| UV-Vis absorption spectroscopy | Nanomaterials with high extinction coefficients | Highly sensitive qualitative and quantitative measurements, easy to operate, low sample consumption | Overlapping absorption profiles with biomaterials can affect quantification quality | DNA and protein conjugation of nanoparticles results in an absorbance shoulder at 260 and 280 nm, respectively. In general, overall absorbance spectrum of the nanoparticle is expected to be different from those conjugated with the probes, conjugation of plasmonic nanoparticles additionally cause a shift in LSPR signal and may result in particle aggregation | 28 and 142–145 |
| Gel electrophoresis | Nanomaterials with sizes below the pore size of the gel, nanomaterials with a (−) or (+) surface charge | Electrophoretic separation of conjugated nanoprobes and non-conjugated nanomaterials, separation of probes with different sizes and surface charges, low sample consumption, easy to operate | Optimisation of gel parameters for precise separation could be cumbersome, for example, quantum dots may lose their fluorescent properties in gel buffers containing ethylenediaminetetraacetic acid (EDTA) | Change in electrophoretic mobility confirms successful conjugation, the level of functionalization can be tracked along with non-functionalized nanoprobes and excess unbound biomolecules, conjugation can be directly confirmed with specific DNA/protein staining methods, nanoparticles with intrinsic fluorescent properties can be directly visualized without staining | 28, 142, 143, 146 and 147 |
| Circular dichroism spectroscopy | Nanomaterials with chiral coatings (e.g. protein, nucleic acids) | Highly sensitive to the chiral moieties, no background noise from most of the nanomaterials, secondary structure changes can be tracked upon anchoring on nanomaterials, structural changes of the probes upon binding to the nanoparticles can be directly observed, easy to operate | Lower availability, requires samples at higher concentrations to reveal the structural properties | Most of the nanoparticles do not show chiral properties. Thus, CD spectrum of nanoparticle is collected around zero, affinity probes are chiral molecules with well-characterized CD peaks in negative and positive ellipticity regions, nanoparticle surface biomodification results in a distinct change in the CD spectrum of the samples | 28, 144, 145 and 147–149 |
| Dynamic light scattering | Nanomaterials preferably in spherical shapes and smaller than 1000 nm in size, nanomaterials that do not exhibit absorbance at the incident laser's wavelength | Sub-nanometer mean size detection, accurate size distribution determination, low sample consumption, easy to operate | Sample solutions are expected to be as diluted as possible and monodisperse, colloidal stability of the samples is also required to prevent false positives data | Change in hydrodynamic radius of the nanoparticle sample due to attachment of biomolecules, effects of functionalization on size distribution profile | 28, 142–144, and 148 |
5 Conclusion
There are several methods established in the literature for nanobiosensor construction, mainly differing in affinity probe type, nanoparticle type, sensing principle, selectivity, repeatability, assay time and the final cost. In this report, the most recent and versatile methods for nanoparticle surface biomodification and characterisation were reviewed. Prevalent affinity reagents for nanobiosensor construction including antibodies and aptamers were described, the key surface biomodification techniques and characterisation tools were explained in the context of the recent literature. Based on the reviewed reports, carbon-based nanomaterials along with plasmonic gold nanoparticles have been found to be the most utilised nanomaterials for nanobiosensor construction, which has been followed by the luminescent nanoparticles such as quantum dots and the upconverting nanoparticles. In line with these findings, π–π bonds and thiol–noble metal interactions have been classified as the leading methods for surface biomodification after avidin–biotin interaction and EDC–NHS carbodiimide methods. Finally, characterisation of the surface bio-modification by UV-Vis absorption spectrophotometry, DLS, CD and GE techniques were discussed, several examples from the literature were presented with some technical details. The development of reliable and affordable nanoparticle-based biosensors for on-site detection and clinical applications are expected to grow significantly by the discovery of aptamers for novel biomarkers and biocompatible nanoparticles. This report outlines the key methodologies for nanoparticle surface biomodification that could be a useful source for graduate students and academics in the field of nanotechnology and biotechnology.Conflicts of interest
There are no conflicts to declare.References
- C. M. Niemeyer and C. A. Mirkin, Nanobiotechnology, Wiley-VCH, 2004 Search PubMed.
- Y. Seok Kim, N. H. Ahmad Raston and M. Bock Gu, Biosens. Bioelectron., 2016, 76, 2–19 CrossRef CAS PubMed.
- M. L. Yola, V. K. Gupta and N. Atar, Mater. Sci. Eng., C, 2016, 61, 368–375 CrossRef CAS PubMed.
- C. Fenzl, T. Hirsch and A. J. Baeumner, TrAC, Trends Anal. Chem., 2016, 79, 306–316 CrossRef CAS.
- T. Lakshmipriya, U. Hashim, S. C. B. Gopinath and N. Azizah, Microsyst. Technol., 2016, 22, 2389–2395 CrossRef CAS.
- J. Li, Z. Zhu, B. Zhu, Y. Ma, B. Lin, R. Liu, Y. Song, H. Lin, S. Tu and C. Yang, Anal. Chem., 2016, 88, 7828–7836 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- P. Ramnani, N. M. Saucedo and A. Mulchandani, Chemosphere, 2016, 143, 85–98 CrossRef CAS PubMed.
- J. Wen, Y. Xu, H. Li, A. Lu and S. Sun, Chem. Commun., 2015, 51, 11346–11358 RSC.
- J. Bhardwaj, S. Devarakonda, S. Kumar and J. Jang, Sens. Actuators, B, 2017, 253, 115–123 CrossRef CAS.
- F. Khosravi, S. Loeian and B. Panchapakesan, Biosensors, 2017, 7, 17 CrossRef PubMed.
- W. Hou, Z. Shi, Y. Guo, X. Sun and X. Wang, Bioprocess Biosyst. Eng., 2017, 40, 1419–1425 CrossRef CAS PubMed.
- N. Xia, X. Wang and L. Liu, Sensors, 2016, 16, 1699 CrossRef PubMed.
- A. Tabasi, A. Noorbakhsh and E. Sharifi, Biosens. Bioelectron., 2017, 95, 117–123 CrossRef CAS PubMed.
- Z. S. Qian, X. Y. Shan, L. J. Chai, J. R. Chen and H. Feng, Biosens. Bioelectron., 2015, 68, 225–231 CrossRef CAS PubMed.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- T.-T. Tran and A. Mulchandani, TrAC, Trends Anal. Chem., 2016, 79, 222–232 CrossRef CAS.
- T. Pasinszki, M. Krebsz, T. T. Tung and D. Losic, Sensors, 2017, 17, 1919 CrossRef PubMed.
- T. Špringer, X. Chadtová Song, M. L. Ermini, J. Lamačová and J. Homola, Anal. Bioanal. Chem., 2017, 409, 4087–4097 CrossRef PubMed.
- H. Kurt and C. W. Ow-Yang, Phys. Status Solidi A, 2016, 213, 3165–3177 CrossRef CAS.
- S. Khabbaz Abkenar, A. Tufani, G. Ozaydin Ince, H. Kurt, A. Turak, C. W. Ow-Yang, J. K. Kruger, D. G. Castner, B. D. Ratner, J. P. Spatz, M. Möller, M. Ozawa, F. Banhart, M. G. Garnier, P. Oelhafen and C. A. Richter, Nanoscale, 2017, 9, 2969–2973 RSC.
- J. A. Webb, R. Bardhan, M. Carmen, M. C. I. la Cruz, F. Sanz-Rodríguez, Á. Juarranz, J. G. Solé, D. Jaque, Y.-X. J. Wang, K. C.-F. Leung, J. T. Jenkins, T. Wang, G. D. Clarke, R. Asmis, K. Sokolov, B. Chandrasekar, T. E. Milner and K. P. Johnston, Nanoscale, 2014, 6, 2502 RSC.
- H.-H. Jeong, A. G. Mark, M. Alarcón-Correa, I. Kim, P. Oswald, T.-C. Lee and P. Fischer, Nat. Commun., 2016, 7, 11331 CrossRef CAS PubMed.
- W. Q. Lim and Z. Gao, Nano Today, 2016, 11, 168–188 CrossRef CAS.
- H. Daraee, A. Eatemadi, E. Abbasi, S. Fekri Aval, M. Kouhi and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 410–422 CrossRef CAS PubMed.
- A. Lay, D. S. Wang, M. D. Wisser, R. D. Mehlenbacher, Y. Lin, M. B. Goodman, W. L. Mao and J. A. Dionne, Nano Lett., 2017, 17, 4172–4177 CrossRef CAS PubMed.
- H. Kurt, M. Yüce, B. Hussain and H. Budak, Biosens. Bioelectron., 2016, 81, 280–286 CrossRef CAS PubMed.
- A. Tolga Çolak, T. Eren, M. L. Yola, E. Beşli, O. Şahin and N. Atar, J. Electrochem. Soc., 2016, 163, F1237–F1244 CrossRef.
- T. Tang, J. Deng, M. Zhang, G. Shi and T. Zhou, Talanta, 2016, 146, 55–61 CrossRef CAS PubMed.
- J. Tang, N. Huang, X. Zhang, T. Zhou, Y. Tan, J. Pi, L. Pi, S. Cheng, H. Zheng and Y. Cheng, Int. J. Nanomed., 2017, 12, 3899–3911 CrossRef PubMed.
- S. M. Ng, M. Koneswaran and R. Narayanaswamy, RSC Adv., 2016, 6, 21624–21661 RSC.
- E. v. Behring and B. S. Kitasato, DMW, Dtsch. Med. Wochenschr., 1890, 16, 1113–1114 CrossRef.
- M. B. Llewelyn, R. E. Hawkins and S. J. Russell, Br. Med. J., 1992, 305, 1269–1272 CrossRef CAS PubMed.
- C. A. Janeway Jr, P. Travers, M. Walport, et al., The interaction of the antibody molecule with specific antigen, Immunobiology: The Immune System in Health and Disease, Garland Science, New York, 5th edn, 2001, available from: https://www.ncbi.nlm.nih.gov/books/NBK27160 Search PubMed.
- M. Baker, Nature, 2015, 521, 274–276 CrossRef CAS PubMed.
- D. M. Ecker, S. D. Jones and H. L. Levine, mAbs, 2015, 7, 9–14 CrossRef CAS PubMed.
- GVR, Gd. View Res. Inc., 2017.
- M. Arruebo, M. Valladares and A. Gonzalez-Fernandez, J. Nanomater., 2009, 2009, 1–24 CrossRef.
- M. Yüce, N. Ullah and H. Budak, Analyst, 2015, 140, 5379–5399 RSC.
- H. Qu, A. T. Csordas, J. Wang, S. S. Oh, M. S. Eisenstein and H. T. Soh, ACS Nano, 2016, 10, 7558–7565 CrossRef CAS PubMed.
- N. Duan, W. Gong, S. Wu and Z. Wang, J. Agric. Food Chem., 2017, 65, 1771–1777 CrossRef CAS PubMed.
- J. M. Escolano, B. Diaz-Duran, M. DeMiguel-Ramos, J. Olivares, M. A. Geday and E. Iborra, Sens. Actuators, B, 2017, 246, 591–596 CrossRef CAS.
- L. Zhou, P. Li, M. Yang, Y. Yu, Y. Huang, J. Wei, S. Wei and Q. Qin, Antiviral Res., 2016, 129, 104–114 CrossRef CAS PubMed.
- J. H. Lee and M. J. Lee, ChemBioChem, 2017, 18, 171–175 CrossRef CAS PubMed.
- P. Mallikaratchy, Molecules, 2017, 22, 215 CrossRef PubMed.
- P. S. R. Lavu, B. Mondal, S. Ramlal, H. S. Murali and H. V. Batra, ACS Comb. Sci., 2016, 18, 292–301 CrossRef CAS PubMed.
- M. Davlieva, J. Donarski, J. Wang, Y. Shamoo and E. P. Nikonowicz, Nucleic Acids Res., 2014, 42, 10795–10808 CrossRef CAS PubMed.
- R. Torabi, K. Bagherzadeh, H. Ghourchian and M. Amanlou, Org. Biomol. Chem., 2016, 14, 8141–8153 CAS.
- R. Amano, K. Takada, Y. Tanaka, Y. Nakamura, G. Kawai, T. Kozu and T. Sakamoto, Biochemistry, 2016, 55, 6221–6229 CrossRef CAS PubMed.
- F. Santos do Carmo, E. Ricci-Junior, C. Cerqueira-Coutinho, M. d. S. Albernaz, E. S. Bernardes, S. Missailidis and R. Santos-Oliveira, Int. J. Nanomed., 2016, 12, 53–60 CrossRef PubMed.
- J. Liu, T. Wei, J. Zhao, Y. Huang, H. Deng, A. Kumar, C. Wang, Z. Liang, X. Ma and X.-J. Liang, Biomaterials, 2016, 91, 44–56 CrossRef CAS PubMed.
- S. M. Nimjee, R. R. White, R. C. Becker and B. A. Sullenger, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 61–79 CrossRef CAS PubMed.
- MarketsandMarkets, MarketsandMarkets, 2015 Search PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2016, 16, 181–202 CrossRef PubMed.
- A. Ruscito and M. C. DeRosa, Front. Chem., 2016, 4, 14 Search PubMed.
- L. A. Dykman and N. G. Khlebtsov, Acta Naturae, 2011, 3, 34–55 CAS.
- W. P. Faulk and G. M. Taylor, Immunochemistry, 1971, 8, 1081–1083 CrossRef CAS PubMed.
- N. M. Green, in Advances in Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall and F. M. Richards, Academic Press, 1975, vol. 29, pp. 85–133 Search PubMed.
- N. Nakajima and Y. Ikada, Bioconjugate Chem., 1995, 6, 123–130 CrossRef CAS PubMed.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- E. B. Troughton, C. D. Bain, G. M. Whitesides, R. G. Nuzzo, D. L. Allara and M. D. Porter, Langmuir, 1988, 4, 365–385 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- Z. Su, H. Xu, X. Xu, Y. Zhang, Y. Ma, C. Li and Q. Xie, Talanta, 2017, 164, 244–248 CrossRef CAS PubMed.
- N. Viet Long, M. Ohtaki, M. Yuasa, S. Yoshida, T. Kuragaki, C. Minh Thi and M. Nogami, J. Nanomater., 2013, 2013, 1–8 CrossRef.
- A. B. Serrano-Montes, D. Jimenez de Aberasturi, J. Langer, J. J. Giner-Casares, L. Scarabelli, A. Herrero and L. M. Liz-Marzán, Langmuir, 2015, 31, 9205–9213 CrossRef CAS PubMed.
- C. A. Lancaster and J. S. Shumaker-Parry, Thin Solid Films, 2016, 612, 141–146 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805 RSC.
- M.-H. Park, E. Reategui, W. Li, S. N. Tessier, K. H. K. Wong, A. E. Jensen, V. Thapar, D. Ting, M. Toner, S. L. Stott and P. T. Hammond, J. Am. Chem. Soc., 2017, 139, 2741–2749 CrossRef CAS PubMed.
- S. Bamrungsap, A. Treetong, C. Apiwat, T. Wuttikhun and T. Dharakul, Microchim. Acta, 2016, 183, 249–256 CrossRef CAS.
- B. Waybrant, T. R. Pearce and E. Kokkoli, Langmuir, 2014, 30, 7465–7474 CrossRef CAS PubMed.
- X. Lin, A. P. Ivanov and J. B. Edel, Chem. Sci., 2017, 8, 3905–3912 RSC.
- J.-H. Pai, C.-T. Yang, H.-Y. Hsu, A. B. Wedding and B. Thierry, Anal. Chim. Acta, 2017, 974, 87–92 CrossRef CAS PubMed.
- N. A. Masdor, Z. Altintas and I. E. Tothill, Biosens. Bioelectron., 2016, 78, 328–336 CrossRef CAS PubMed.
- S. Eissa and M. Zourob, Sci. Rep., 2017, 7, 1016 CrossRef PubMed.
- C. Fenzl, C. Genslein, C. Domonkos, K. A. Edwards, T. Hirsch and A. J. Baeumner, Analyst, 2016, 141, 5265–5273 RSC.
- Y.-T. Tseng, R. Cherng, Z. Yuan, C.-W. Wu, H.-T. Chang and C.-C. Huang, Nanoscale, 2016, 8, 5162–5169 RSC.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- J. H. Soh, Y. Lin, S. Rana, J. Y. Ying and M. M. Stevens, Anal. Chem., 2015, 87, 7644–7652 CrossRef CAS PubMed.
- C. M. Dundas, D. Demonte and S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS PubMed.
- A. Jain and K. Cheng, J. Controlled Release, 2017, 245, 27–40 CrossRef CAS PubMed.
- R. D'Agata, P. Palladino and G. Spoto, Beilstein J. Nanotechnol., 2017, 8, 1–11 CrossRef PubMed.
- E. Villemin, E. Gravel, N. Izard, A. Filoramo, L. Vivien and E. Doris, Chem.–Eur. J., 2015, 21, 18649–18653 CrossRef CAS PubMed.
- L. Mattsson, K. D. Wegner, N. Hildebrandt and T. Soukka, RSC Adv., 2015, 5, 13270–13277 RSC.
- Z. Yang, J. Shen, J. Li, J. Zhu and X. Hu, Anal. Chim. Acta, 2013, 774, 85–91 CrossRef CAS PubMed.
- Z. Wu, E. Xu, M. F. J. Chughtai, Z. Jin and J. Irudayaraj, Food Chem., 2017, 230, 673–680 CrossRef CAS PubMed.
- S. Wu, H. Zhang, Z. Shi, N. Duan, C. Fang, S. Dai and Z. Wang, Food Control, 2015, 50, 597–604 CrossRef CAS.
- Z. Liu, F. Galli, K. G. H. Janssen, L. Jiang, H. J. van der Linden, D. C. de Geus, P. Voskamp, M. E. Kuil, R. C. L. Olsthoorn, T. H. Oosterkamp, T. Hankemeier and J. P. Abrahams, J. Phys. Chem. C, 2010, 114, 4345–4352 CAS.
- S.-T. Yang, Y. Liu, Y.-W. Wang and A. Cao, Small, 2013, 9, 1635–1653 CrossRef CAS PubMed.
- X. Liu, J. Ren, L. Su, X. Gao, Y. Tang, T. Ma, L. Zhu and J. Li, Biosens. Bioelectron., 2017, 87, 203–208 CrossRef CAS PubMed.
- A. Bogomolova and M. Aldissi, Biosens. Bioelectron., 2015, 66, 290–296 CrossRef CAS PubMed.
- M. Safavieh, V. Kaul, S. Khetani, A. Singh, K. Dhingra, M. K. Kanakasabapathy, M. S. Draz, A. Memic, D. R. Kuritzkes and H. Shafiee, Nanoscale, 2017, 9, 1852–1861 RSC.
- H. Ilkhani, M. Sarparast, A. Noori, S. Zahra Bathaie and M. F. Mousavi, Biosens. Bioelectron., 2015, 74, 491–497 CrossRef CAS PubMed.
- Q. Wang, P. Subramanian, A. Schechter, E. Teblum, R. Yemini, G. D. Nessim, A. Vasilescu, M. Li, R. Boukherroub and S. Szunerits, ACS Appl. Mater. Interfaces, 2016, 8, 9600–9609 CAS.
- H. Afsharan, B. Khalilzadeh, H. Tajalli, M. Mollabashi, F. Navaeipour and M.-R. Rashidi, Electrochim. Acta, 2016, 188, 153–164 CrossRef CAS.
- T. N. Ly, S. Park and S. J. Park, Sens. Actuators, B, 2016, 237, 452–458 CrossRef CAS.
- D. Mendez-Gonzalez, M. Laurenti, A. Latorre, A. Somoza, A. Vazquez, A. I. Negredo, E. López-Cabarcos, O. G. Calderón, S. Melle and J. Rubio-Retama, ACS Appl. Mater. Interfaces, 2017, 9, 12272–12281 CAS.
- L. R. Rutledge, L. S. Campbell-Verduyn and S. D. Wetmore, Chem. Phys. Lett., 2007, 444, 167–175 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- J. Björk, F. Hanke, C.-A. Palma, P. Samori, M. Cecchini and M. Persson, J. Phys. Chem. Lett., 2010, 1, 3407–3412 CrossRef.
- M. Stornaiuolo, G. E. De Kloe, P. Rucktooa, A. Fish, R. van Elk, E. S. Edink, D. Bertrand, A. B. Smit, I. J. P. de Esch and T. K. Sixma, Nat. Commun., 2013, 4, 1875 CrossRef PubMed.
- K. A. Wilson, J. L. Kellie and S. D. Wetmore, Nucleic Acids Res., 2014, 42, 6726–6741 CrossRef CAS PubMed.
- V. Sivasakthi, A. Anbarasu and S. Ramaiah, Cell Biochem. Biophys., 2013, 67, 853–863 CrossRef CAS PubMed.
- Y. Chen, Y. Xianyu and X. Jiang, Acc. Chem. Res., 2017, 50, 310–319 CrossRef CAS PubMed.
- G. Peschel, Freie Univ. Berlin website, 2005 Search PubMed.
- V. C. Sanchez, A. Jachak, R. H. Hurt and A. B. Kane, Chem. Res. Toxicol., 2012, 25, 15–34 CrossRef CAS PubMed.
- F. Li, J. Chao, Z. Li, S. Xing, S. Su, X. Li, S. Song, X. Zuo, C. Fan, B. Liu, W. Huang, L. Wang and L. Wang, Anal. Chem., 2015, 87, 3877–3883 CrossRef CAS PubMed.
- S. Marchesan and M. Prato, Chem. Commun., 2015, 51, 4347–4359 RSC.
- L. Lv, C. Cui, C. Liang, W. Quan, S. Wang and Z. Guo, Food Control, 2016, 60, 296–301 CrossRef CAS.
- L. Yan, H. Shi, X. He, K. Wang, J. Tang, M. Chen, X. Ye, F. Xu and Y. Lei, Anal. Chem., 2014, 86, 9271–9277 CrossRef CAS PubMed.
- M. P. Landry, H. Ando, A. Y. Chen, J. Cao, V. I. Kottadiel, L. Chio, D. Yang, J. Dong, T. K. Lu and M. S. Strano, Nat. Nanotechnol., 2017, 12, 368–377 CrossRef CAS PubMed.
- Q. G. Liao, B. H. Wei and L. G. Luo, Microchim. Acta, 2017, 184, 627–632 CrossRef CAS.
- Y. Wang, L. Tang, Z. Li, Y. Lin and J. Li, Nat. Protoc., 2014, 9, 1944–1955 CrossRef CAS PubMed.
- Y. Zhu, Y. Cai, L. Xu, L. Zheng, L. Wang, B. Qi and C. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 7492–7496 CAS.
- K. Ling, H. Jiang, Y. Li, X. Tao, C. Qiu and F.-R. Li, Biosens. Bioelectron., 2016, 86, 8–13 CrossRef CAS PubMed.
- J. Qin, X. Cui, P. Wu, Z. Jiang, Y. Chen, R. Yang, Q. Hu, Y. Sun and S. Zhao, Food Control, 2017, 73, 726–733 CrossRef CAS.
- L. Zhu, G. Xu, Q. Song, T. Tang, X. Wang, F. Wei and Q. Hu, Sens. Actuators, B, 2016, 231, 506–512 CrossRef CAS.
- S. Pandit and M. De, J. Phys. Chem. C, 2017, 121, 600–608 CAS.
- Z. Grabarek and J. Gergely, Anal. Biochem., 1990, 185, 131–135 CrossRef CAS PubMed.
- B. Lin, Y. Yu, R. Li, Y. Cao and M. Guo, Sens. Actuators, B, 2016, 229, 100–109 CrossRef CAS.
- G. Zhu and H. J. Lee, Biosens. Bioelectron., 2017, 89, 959–963 CrossRef CAS PubMed.
- J. Chen, S. M. Andler, J. M. Goddard, S. R. Nugen and V. M. Rotello, Chem. Soc. Rev., 2017, 46, 1272–1283 RSC.
- B. Hussain, M. Yüce, N. Ullah and H. Budak, in Nanobiosensors, ed. A. M. Grumezescu, Academic Press, 2017, pp. 93–127 Search PubMed.
- N. Gao, T. Gao, X. Yang, X. Dai, W. Zhou, A. Zhang and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14633–14638 CrossRef CAS PubMed.
- M. H. Jazayeri, H. Amani, A. A. Pourfatollah, A. Avan, G. A. Ferns and H. Pazoki-Toroudi, Cancer Gene Ther., 2016, 23, 365–369 CrossRef CAS PubMed.
- Y.-H. Lao, C.-W. Chi, S. M. Friedrich, K. Peck, T.-H. Wang, K. W. Leong and L.-C. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 12048–12055 CAS.
- S. Kumar, J. Aaron and K. Sokolov, Nat. Protoc., 2008, 3, 314–320 CrossRef CAS PubMed.
- B. Jin, S. Wang, M. Lin, Y. Jin, S. Zhang, X. Cui, Y. Gong, A. Li, F. Xu and T. J. Lu, Biosens. Bioelectron., 2017, 90, 525–533 CrossRef CAS PubMed.
- C. Liu, G. Mao, C. Su, X. Ji, Z. Chen and Z. He, Anal. Methods, 2015, 7, 7748–7752 RSC.
- S. Elhadj, G. Singh and R. F. Saraf, Langmuir, 2004, 20, 5539–5543 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- R. J. Hill, Soft Matter, 2016, 12, 8030–8048 RSC.
- R. W. O'Brien and L. R. White, J. Chem. Soc., Faraday Trans. 2, 1978, 74, 1607 RSC.
- S. Park, K. A. Brown and K. Hamad-Schifferli, Nano Lett., 2004, 4, 1925–1929 CrossRef CAS.
- T. Pons, H. T. Uyeda, I. L. Medintz and H. Mattoussi, J. Phys. Chem. B, 2006, 110, 20308–20316 CrossRef CAS PubMed.
- M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397–399 CrossRef CAS PubMed.
- Z.-Q. Cui, Q. Ren, H.-P. Wei, Z. Chen, J.-Y. Deng, Z.-P. Zhang and X.-E. Zhang, Nanoscale, 2011, 3, 2454 RSC.
- J. Lee, H. Kang, H. Jang, Y. Lee, Y. Lee, B. Ali, A. Al-Khedhairy and S. Kim, Sensors, 2015, 15, 8595–8604 CrossRef CAS PubMed.
- J. L. Rouge, L. Hao, X. A. Wu, W. E. Briley and C. A. Mirkin, ACS Nano, 2014, 8, 8837–8843 CrossRef CAS PubMed.
- D. K. Tiwari, T. Jin and J. Behari, Int. J. Nanomed., 2011, 6, 463–475 CAS.
- C. Liu, W. Ma, Z. Gao, J. Huang, Y. Hou, C. Xu, W. Yang and M. Gao, J. Mater. Chem. C, 2014, 2, 9637–9642 RSC.
- V. Nasirian, A. Chabok, A. Barati, M. Rafienia, M. S. Arabi and M. Shamsipur, Microchim. Acta, 2017, 1–8 Search PubMed.
- L. García-Fernández, J. Garcia-Pardo, O. Tort, I. Prior, M. Brust, E. Casals, J. Lorenzo and V. F. Puntes, Nanoscale, 2017, 9, 6111–6121 RSC.
- Y. Li, J. Xu, L. Wang, Y. Huang, J. Guo, X. Cao, F. Shen, Y. Luo and C. Sun, Sens. Actuators, B, 2016, 222, 815–822 CrossRef CAS.
- B. H. Lee, V. T. Nguyen and M. B. Gu, Biosens. Bioelectron., 2017, 88, 174–180 CrossRef CAS PubMed.
- M. Azhdarzadeh, F. Atyabi, A. A. Saei, B. S. Varnamkhasti, Y. Omidi, M. Fateh, M. Ghavami, S. Shanehsazzadeh and R. Dinarvand, Colloids Surf., B, 2016, 143, 224–232 CrossRef CAS PubMed.
- J.-H. Yeom, B. Lee, D. Kim, J. Lee, S. Kim, J. Bae, Y. Park and K. Lee, Biomaterials, 2016, 104, 43–51 CrossRef CAS PubMed.
- T. Hu, S. Lu, C. Chen, J. Sun and X. Yang, Sens. Actuators, B, 2017, 243, 792–799 CrossRef CAS.
- V.-T. Nguyen, H. Bin Seo, B. C. Kim, S. K. Kim, C.-S. Song and M. B. Gu, Biosens. Bioelectron., 2016, 86, 293–300 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |