DOI:
10.1039/C7RA10283F
(Paper)
RSC Adv., 2017,
7, 49404-49422
Synthesis and crystal structure of new monometallic Ni(II) and Co(II) complexes with an asymmetrical aroylhydrazone: effects of the complexes on DNA/protein binding property, molecular docking, and in vitro anticancer activity†
Received
16th September 2017
, Accepted 13th October 2017
First published on 23rd October 2017
Abstract
Two novel complexes, NiL2phen·CH3CN (1) and CoL2phen·CH3CN (2) (HL = 2-acetonaphthone salicylylhydrazone, phen = 1,10-phenanthroline) were synthesized and characterized by spectroscopy (IR, ESI-MS) and elemental analysis. The structures for the complexes were determined by X-ray crystallography proving the distorted octahedral coordination environment around the metal center with an MN4O2 chromophore [M = Ni(II) and Co(II)], with the hydrazone ligand acting as a monoanionic bidentate N,O-donor. Interaction of the ligand HL along with the corresponding copper complexes 1 and 2 with herring sperm DNA (HS–DNA) has been estimated by absorption and emission titration methods which revealed that the compounds interacted with HS–DNA through intercalation. Binding of the compounds with bovine serum albumin (BSA) protein investigated using UV-visible, fluorescence and synchronous fluorescence spectroscopic methods indicated that there occurred strong binding of nickel and cobalt complexes to BSA over the ligand HL. The alterations in the secondary structure of the protein by the complexes were confirmed by synchronous and three dimensional fluorescence spectroscopic studies. The interaction of the Ni(II) complex with DNA/BSA also has been supported by molecular docking studies. Further, the ligand HL and corresponding Ni(II) and Co(II) have been tested for their scavenging effect towards DPPH, NO, OH and O2− radicals. An in vitro cytotoxicity study of the complexes found significant activity against human breast (HeLa) and lung (A549) cancer cell lines, with the best results for Ni(II) complex, where the IC50 values are of 34.93 ± 2.05 and 29.19 ± 1.10 μM respectively after 24 h of incubation.
Introduction
In spite of the rapid development of novel anticancer drugs, cancer remains the major leading cause of death in the world due to drug resistance or undesirable side effects. Metal-based anticancer drug discovery remains one of the most advanced areas of pharmaceutical research. Metal complexes which can efficiently bind and cleave DNA under physiological conditions are considered as potential candidates for use as therapeutic agents in medicinal applications and for genomic research.1–3 It is a well known fact that cisplatin is arguably the most successful anticancer drug in the world. But it exhibits high toxicity to normal cells leading to undesirable side-effects, although minimized by careful administration protocols, and also it is inactive against many cancer cell lines and metastasis (secondary) cancers.4–6 Therefore, attempts are being made to replace cisplatin with suitable alternatives and hence numerous transition metal complexes have been synthesized and tested for their anticancer activities. Among the transition metals, cobalt is a biologically relevant element, and many enzymes that depend on cobalt for their activity have been identified, while nickel is an essential component in enzymes such as urease, carbon monoxide dehydrogenase, and hydrogenase.7–10 It should be noted that reviews devoted to the general use of nickel and cobalt complexes in medicine have been published.11,12 A large body of evidence indicates that nickel and cobalt complexes are an effective method to inhibit tumor growth, and they have become promising agents in the treatment of cancer.13–16
The pharmacological properties of metal complexes depend on the properties of both the metal and the ligand. In many cases, addition of metal ions to an organic ligand can lead to the increase of its biological activity. Hydrazones are the important class of ligands with interesting ligation properties due to the presence of NNO coordination sites. Various important properties of hydrazones along with their applications in medicine and analytical chemistry have led to increased interest in their complexation characteristics with nickel and cobalt ions. It is well established that the formation of nickel and cobalt complexes plays a very important role to enhance the biological activity of free hydrazones.17,18 In addition, the use of heterocyclic diimine as the co-ligand in ternary complexes is of considerable interest because some of the diimine containing transition metal complexes exhibit interesting biological as well as pharmacological properties.19,20 DNA is the primary intracellular target of anti-cancer drugs. Small molecules can interact with DNA through the following three non-covalent modes: intercalation, groove binding and external static electronic effects.21 Both the planarity of ligand and the coordination geometry of the metal ion play important roles in deciding the intercalating ability of complexes to DNA.22,23 It has been proved that increasing the size of the substituent and planarity of ligands can enhance the DNA interaction and protein binding of nickel hydrazones complexes.18,24 Therefore, planar aromatic groups, such as benzyl and napthyl group were introduced as substituent to enhance planarity of the ligand. On the other hand, like DNA, proteins are also considered to be one of the main molecular targets in the action of anticancer agents.25 Since proteins play an important role in the transport and deposition of a variety of endogenous and exogenous substances in blood the interactions of drugs with them result in the formation of a stable drug–protein complex, which can exert an important effect on the distribution, free concentration and metabolism of the drug in the blood stream.26 Such an interactions of drug compounds have aromatic rings are very important for protein sterilization and different regulation processes.27 Therefore, the activity of metal complexes towards DNA and protein BSA is useful in the design and synthesis of metal-based anticancer therapeutics.
Enlightened by the above-mentioned facts and as a continuation of our ongoing program in the field of design and synthesis transition-metal complexes with new asymmetrical aroylhydrazone and biological activities,28,29 in this paper, two new mononuclear nickel and cobalt complexes bearing asymmetrical aroylhydrazone and 1,10-phenanthroline were synthesized and structurally characterized by X-ray single-crystal diffraction. The interactions of the two complexes with herring sperm DNA (HS–DNA) are investigated by using UV absorption and fluorescence spectra and viscometry. The protein binding ability has been monitored by using UV-vis absorption and fluorescence quenching experiment in the presence of the complexes using bovine serum albumin (BSA) as model protein. We have also tried to give experimental evidence for the influence of the centre metal ion and the coordination geometry of metal complexes on antioxidant and cytotoxicity potential.
Experimental
Materials and methods
Ni(OAc)2·4H2O, Co(NO3)2·6H2O, 2-acetonaphthone, salicylhydrazine, 1,10-phenanthroline monohydrate, ethidium bromide (EB), herring sperm DNA (HS–DNA), tris(hydroxymethyl)aminomethane hydrochloride (Tris–HCl), bovine serum albumin (BSA) were purchased from Sigma-Aldrich Chemicals and used as received. Human hepatocellular carcinoma cell HeLa, human lung adenocarcinoma cell A549 and the normal mouse embryonic fibroblast cell line NIH-3T3 were purchased from Shanghai Cell Bank, Chinese Academy of Sciences. Fetal bovine serum and all other cell-culture reagents were obtained from Solarbio Science and Technology Co., Ltd. Beijing, China. All the other chemicals and reagents were of high quality and available from reputed suppliers.
Elemental analyses for C, H and N were performed on a Perkin Elmer 240 C elemental analyzer. Electron spray ionization mass spectrum (ESI-MS) was recorded on LTQ Orbitrap XL spectrometer in a positive ion mode. The FT-IR measurements (KBr pellets) were recorded in the range of 4000–400 cm−1 on the instrument of Thermo Nicolet Nexus 670 infrared spectrometer. UV-vis spectrum was obtained on a Varian Cary 300 UV-vis spectrophotometer. Fluorescence spectroscopy was measured with Perkin Elmer LS-55 spectrometer at room temperature.
Synthetic procedures
Preparation of 2-acetonaphthone salicylylhydrazone ligand (HL). To a solution of 2-acetonaphthone (1.360 g, 8 mmol) in anhydrous ethanol (40 mL) was added two drops of glacial acid. A solution of salicylhydrazine (1.217 g, 8 mmol) in anhydrous ethanol (20 mL) was added dropwise over a period of 1 h with stirring. The mixture was cooled after refluxing for 24 h. White needle-like solid was filtered and washed with ethanol several times. Yield 1.48 g (60.7%). Melting point: 244.0–244.4 °C. Anal. calc. (%) for C19H16N2O2 (Mol. wt = 304.34): C, 74.98; H, 5.30; N, 9.20; found: C, 74.94; H, 5.31; N, 9.23. 1H NMR (400 MHz, DMSO-d6, δ in ppm): 11.46 (s, 1 H, N–H), 8.35 (s, 1 H, 2-naphthalene ring), 8.17 (d, 1 H, J = 8.0 Hz, 2-naphthalene ring), 8.04 (d, 2 H, J = 4.0 Hz, 2-naphthalene ring), 7.95 (t, 2 H, J = 8.0 Hz, 2-naphthalene ring), 7.55–7.58 (m, 2 H, 2-naphthalene ring, Ar–H), 7.45 (t, 1 H, J = 8.0 Hz, Ar–H), 7.00–7.07 (m, 2 H, Ar–H), 2.47 (s, 3 H, –CH3); 13C NMR (100 MHz, DMSO-d6, δ in ppm): 162.6, 157.0, 152.4, 135.8, 133.9, 133.8, 133.2, 131.1, 128.2, 128.0, 127.4, 127.0, 124.1, 120.2, 118.4, 117.4, 14.1. Selected IR bands (υ in cm−1): 3277 (–NH–), 1630 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1555 (>CN–), 1161 (phenolic C–OH), 1095 (N–N<).
O), 1555 (>CN–), 1161 (phenolic C–OH), 1095 (N–N<).
Preparation of NiL2phen·CH3CN (1). HL (0.576 g, 2 mmol) was dissolved in 10 mL actonitrile, to which 2 mmol of triethylamine (0.201 g) was added. A solution of nickel acetate tetrahydrate (0.176 g, 1 mmol) in MeOH (10.0 mL) was added dropwise to the ligand solution, which was stirred at room temperature. After 1 h, a methanolic solution of 1,10-phenanthroline monohydrate (0.180 g, 1 mmol) was slowly added to the reaction mixture, resulting in a yellow green colour solution. The reaction mixture was allowed to stir for another 2 h, after which the precipitate that formed was filtered, washed successively with 2 mL of MeOH and 10 mL diethyl ether, and then dried at room temperature. X-ray quality single crystals, having a green colour and prime shape appeared on slow evaporation of the filtrate at room temperature. Yield 0.309 g (34%). Melting point: 268.4–269.6 °C. Anal. calc. (%) for C52H41N7O4Ni (Mol. wt = 886.63): C, 70.44; H, 4.66; N, 11.05; found: C, 70.70; H, 4.50; N, 11.16. UV-visible (solvent: 5% DMSO and 95% Tirs–HCl buffer): λmax, nm (ε, M−1 cm−1): 281(35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 784), 300(35364) and 349(34104). Selected IR bands (υ in cm−1): 1589 and 1494 (>CN–NC<), 1373 (–C–O), 1160 (phenolic C–OH), 1037 (N–N). ESI-MS (MeOH): m/z = 844 (NiL2phen+), (calculated m/z = 844 for (NiL2phen+)).
784), 300(35364) and 349(34104). Selected IR bands (υ in cm−1): 1589 and 1494 (>CN–NC<), 1373 (–C–O), 1160 (phenolic C–OH), 1037 (N–N). ESI-MS (MeOH): m/z = 844 (NiL2phen+), (calculated m/z = 844 for (NiL2phen+)).
Preparation of CoL2phen·CH3CN (2). HL (0.576 g, 2 mmol) was dissolved in 10 mL acetonitrile solution, to which 2 mmol of triethylamine (0.201 g) was added. A solution of cobalt nitrate hexahydrate (0.129 g, 1 mmol) in MeOH (10.0 mL) was added dropwise to the ligand solution, which was stirred at room temperature. After one hour a methanolic solution of 1,10-phenanthroline monohydrate (0.180 g, 1 mmol) was slowly added to the reaction mixture, resulting in a deep brown colour solution. The reaction mixture was allowed to stir for another 2 h, after which the precipitate that formed was filtered, washed successively with 2 mL of MeOH and 10 mL diethyl ether, and then dried at room temperature. X-ray quality single crystals, having a orange colour and square shape appeared on slow evaporation of the filtrate at room temperature. Yield 0.529 g (58%). Melting point: 260.3–261.5 °C. Anal. calc. (%) for C52H41N7O4Co (Mol. wt = 886.85): C, 70.42; H, 4.65; N, 11.05; found: C, 70.90; H, 4.57; N, 11.11. UV-visible (solvent: 5% DMSO and 95% Tris–HCl buffer): λmax, nm (ε, M−1 cm−1): 271(32524), 300(28728), 341(24096). Selected IR bands (υ in cm−1): 1600 and 1493 (>CN–NC<), 1372 (–C–O), 1160 (phenolic C–OH), 1038 (N–N). ESI-MS (MeOH): m/z = 845 (CoL2phen+), (calculated m/z = 845 for (CoL2phen+)).
Crystallography
The X-ray single-crystal data for complex 1 and 2 were recorded on a Brucker SMART Apex CCD detector diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The collected data were reduced using the SAINT program,30 and multi-scan absorption corrections were performed using the SADABS program.31 The structures were solved by direct methods and refined by full-matrix least squares on F2 using SHELXL-97.32 All non-hydrogen atoms were refined anisotropically and the hydrogen atoms in these structures were located via the difference Fourier map and constrained to ideal positions in the refinement procedure. Experimental details for X-ray data collection complexes are presented in Table 1.
Table 1 Crystallographic data and structure refinement parameter of complexes
| Parameter |
Complex 1 |
Complex 2 |
| Formula |
C52H41N7O4Ni |
C52H41N7O4Co |
| Formula weight |
886.63 |
886.85 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
| a (Å) |
10.252(5) |
10.2454(14) |
| b (Å) |
23.344(11) |
23.167(3) |
| c (Å) |
19.134(9) |
19.249(2) |
| α (°) |
90 |
90 |
| β (°) |
103.243(6) |
103.024(2) |
| γ (°) |
90 |
90 |
| V (Å3) |
4457(4) |
4451.3(10) |
| Z |
4 |
4 |
| ρcalcd (mg cm−3) |
1.321 |
1.323 |
| μ (mm−1) |
0.49 |
0.44 |
| Limiting indices |
−12 ≤ h ≤ 12 |
−12 ≤ h ≤ 13 |
| |
−27 ≤ k ≤ 27 |
−29 ≤ k ≤ 30 |
| |
−22 ≤ l ≤ 22 |
−22 ≤ l ≤ 24 |
| Crystal size (mm3) |
0.25 × 0.18 × 0.15 |
0.25 × 0.2 × 0.15 |
| F (000) |
1848 |
1844 |
| T (K) |
296 |
296 |
| Goodness of fit on F2 |
0.99 |
1.01 |
| R1, wR2 [I > 2σ(I)] |
0.1038, 0.2644 |
0.0517, 0.1157 |
| R1, wR2 [all data] |
0.1882, 0.3059 |
0.1123, 0.1489 |
| ρmax, ρmin (e·Å−3) |
1.07, −0.98 |
0.33, −0.41 |
DNA binding studies
Experiments involving the binding of compounds to HS–DNA were performed in double-distilled water with 5 mM Tris and 50 mM NaCl solution and the pH was adjusted to 7.2 with hydrochloric acid. DNA solution was prepared with 50 mM NaCl solution. The UV absorbance ratio at 260 and 280 nm (A260/A280) of 1.85 indicated that the DNA was sufficiently free of protein contamination.33 The concentrations of DNA were determined spectrophotometrically by assuming ε260 = 6600 M−1 cm−1,34 which was found to be 3.72 × 10−4 mol L−1 when the mass concentration was 200 μg mL−1. The ligand and complexes were dissolved in a combined solvent of 5% DMSO and 95% Tris–HCl buffer for all experiments. A stock solution of DNA was stored at 4 °C and used after no more than 4 days.
UV-vis titration experiments were performed by keeping a fixed concentration of the metal complex constant (25 μM) and varying the nucleotide concentration (0–125 μM). However, when obtaining the absorption spectra, equal amounts of DNA were added to both complex and reference before the absorption spectra were recorded. Further support for the binding of complexes to DNA via intercalation was obtained using fluorescence spectral techniques in order to point out whether a complex can displace EB from a DNA–EB complex. EB displacement experiments were carried out by adding solutions of the complexes to a Tris–HCl buffer solution (pH 7.2) of a DNA/EB mixture. DNA was pretreated with EB at a [DNA]/[EB] ratio of 1 for 30 min at room temperature, then a test solution was added to this mixture of EB–DNA and the change in fluorescence intensity was measured. The excitation wavelength was fixed at 545 nm for EB bound to DNA. Emissions were recorded with increasing concentrations of compounds and the emission range was adjusted before measurements. The ligand and complexes (0–100 μM) were then added to the mixture and their effect on the emission intensity was measured.
Viscosity experiments were carried on an Ubbelohde viscometer of 10 mL capacity, immersed in a thermostated water bath maintained at 37.0 ± 0.1 °C. The stock solutions of DNA (50 μM) and complexes (0–50 μM) were prepared in Tris–HCl/NaCl buffer. Mixing of the solutions was achieved by purging the nitrogen gas through viscometer. The flow time was measured three times for each sample with a digital stopwatch, and the mean flow time was calculated. Data were presented as (η/η0)1/3 versus binding ratio [complex]/[DNA], where η and η0 are the specific viscosity of DNA in the presence and absence of complex, respectively. The values of η and η0 were calculated from the relation: η0 = (t − tb)/tb, where tb is the flow time of buffer alone, and t is the observed flow time for DNA in the presence of complex. Relative viscosities for DNA were obtained from the relation, η/η0.
Cyclic voltammogram (CV) was recorded on a CHI 660C electrochemical work station using a three-electrode cell in which a platinum plate, a saturated Ag/AgCl and a platinum foil are used as the working, reference and auxiliary electrodes, respectively. A ferrocene/ferrocenium (Fc/Fc+) couple was used as an internal standard. Cyclic voltammograms of the samples were recorded in DMSO/Tris–HCl/NaCl buffer mixed solution at a scan rate of 50 mV s−1. Prior to every electrochemical assay, the solution was deoxygenated by purging with nitrogen. All electrochemical measurements were performed at room temperature.
Protein binding studies
The protein–binding interactions of the ligand and complexes with bovine serum albumin (BSA) were investigated using both of UV-vis absorption and fluorescence spectra. For UV absorption experiment, a 3 mL solution of BSA (1 μM) was titrated with various concentrations of the complexes. Equal solutions of complexes were added to the reference solutions to eliminate the absorbance of the complexes themselves. The UV-vis absorption spectra were measured from 200 to 400 nm. Fluorescence spectra were obtained with an excitation wavelength of 280 nm and an emission wavelength of 343 nm corresponding to those of free BSA. The excitation and emission slit widths and scan rates were maintained at a constant for all experiments. Samples were thoroughly degassed using pure nitrogen gas for 15 minutes using quartz cells (4 × 1 × 1 cm) with high-vacuum Teflon stopcocks. Stock solutions of BSA were prepared in 50 mM phosphate buffer (pH 7.2) and stored in the dark at 4 °C for additional use. Concentrated stock solutions of metal complexes were prepared by dissolving them in DMSO:Tris–HCl buffer (5:95) and diluted suitably with phosphate buffer to obtain appropriate concentrations. 2.5 mL of BSA solution (1 μM) was titrated by successive additions of a 5 μL stock solution of complexes (10−4 M) using a micropipette. Synchronous fluorescence spectra were also obtained using the same concentrations of BSA and complexes as mentioned above with two different values of Δλ (difference between the excitation and emission wavelengths of BSA) such as 15 and 60 nm. The three-dimensional fluorescence spectrums were performed under the following conditions: the emission wavelength scan range was recorded between 240 nm and 440 nm, the excitation wavelength scan range was recorded from 200 to 360 nm at 10 nm increments. The number of scanning curves was 17, and other scanning parameters were just the same as the fluorescence quenching spectra.
Antioxidant activity
The DPPH (2,2-diphenyl-1-picrylhydrazyl) radical-scavenging activity of the compounds was measured according to the method described by Blois.35 The DPPH radical is a stable free radical having a λmax of 517 nm. Various concentrations (10–60 mM) of the test compounds were added to a solution of DPPH (125 mM, 2 mL) in methanol and the final volume was made up to 4 mL with double-distilled water. The solution was incubated at 37 °C for 30 min in the dark. The decrease in the absorbance of DPPH was measured at 517 nm. The same experiment carried out without the test compounds served as a control.
Nitric oxide (NO˙) radical-scavenging activity was determined based on the reported method, in which sodium nitroprusside in an aqueous solution at physiological pH spontaneously generates nitric oxide, which interacts with oxygen to produce nitrate ions that can be estimated using the Griess reagent.36 For the experiment, sodium nitroprusside (10 mM) in phosphate buffered saline was mixed with a fixed concentration of the compound and incubated at room temperature for 150 min. After the incubation period, 0.5 mL of the Griess reagent containing 1% sulfanilamide, 2% H3PO4 and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride was added. The reaction mixture without the sample but with equivalent amount of solvent served as a control. The absorbance of the chromophore formed was measured at 546 nm.
The superoxide radical (O2˙) was produced by the system of MET/VitB2/NBT and determined spectrophotometrically by NBT photoreduction method with a little modification in the method adopted elsewhere.37,38 The amount of O2˙ could be calculated by measuring the absorbance at 560 nm, because NBT can be reduced quantitatively to blue formazan by O2˙. The solution of MET, VitB2 and NBT were prepared with 0.067 M phosphate buffer (pH = 7.8) under the condition of avoiding light. The tested compounds were dissolved in DMF. The assay mixture contained MET (10 mM), NBT (46 mM), VitB2 (3.3 mM), phosphate buffer (67 mM, pH = 7.8) and the tested compound (10–50 μM). After illuminating with a fluorescent lamp at 30 °C for 10 min, the absorbance of the samples was measured at 560 nm. The sample without the tested compound was used as control.
The hydroxyl radical (OH˙) in aqueous media was generated by the Fenton system.39 The solution of the tested complex was prepared with DMF. The 5 mL assay mixture contained following reagents: safranine T (20 μM), EDTA–Fe2+ (200 μM), H2O2 (196 mM), the tested compounds and a phosphate buffer (40 μM, pH = 7.4). The assay mixtures were incubated at 30 °C for 10 min in a water bath. The sample without the tested compound was used as the control. After that, the absorbance was measured at 520 nm.
In the case of the above three assays, all of the tests were run in triplicate. All data are expressed as mean ± standard deviation (SD). Various concentrations of the compounds were used to fix a concentration at which the compounds showed in and around 50% of activity. The percentage of activity was calculated using the formula, % of activity = [(A0 − AC)/A0] × 100. A0 and AC are the absorbance in the absence and presence of the tested compounds respectively. The 50% activity (IC50) can be calculated using the percentage of activity results.
Molecular docking
AutoDock 4.2 using Lamarckian genetic algorithm together with the AutoDock Tools was employed to set up and perform blind docking calculations of complex 1 binding to DNA and BSA.40 According to the literature method,41 the structure of DNA (PDB ID: 423D) with sequenced (ACCGACGTCGGT)2 and BSA (PDB ID: 4F5S) taken from the Protein Data Bank (http://www.rcsb.org/pdb) with the resolution of 1.60 and 2.47 Å, and the r-value of 0.206 and 0.259 for DNA and BSA, respectively, were employed to establish the docking mode, in which polar hydrogen atoms as well as Kollman charges were added to receptor molecules. The coordinate of the complex 1 was taken from the crystal structures as CIF files and converted to the PDB format using Mercury software (http://www.ccdc.cam.ac.uk/) and missing hydrogen atoms and Gasteiger charges were added. The receptor and “the ligand” (complex 1) files were prepared using AutoDock Tools. In the docking analysis, the binding site was assigned to include the entire biomacromolecule, which was enclosed in a box with number of grid points in x × y × z directions, 96 × 96 × 96 and a grid spacing of 0.375 Å. Initially, AutoGrid was run to generate the grid map of various atoms of “the ligands” and the receptor. After the generation of the grid map, AutoDock was run using parameters as follows: GA population size, 150; maximum number of energy evaluations, 2500000; numbers of generations, 27000. A total of 25 runs were carried out. A maximum of 50 conformers was considered for each molecule, and the root-mean-square cluster tolerance was set to 2.0 Å in each run. All calculations were performed on an Intel Core i7 based machine running GNU/Linux as operating system. For each of the docking cases, the lowest energy docked conformation, according to the AutoDock scoring function, was selected as the binding mode. Visualization of the docked pose has been done by using PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC) molecular graphics program.
In vitro cytotoxic activity evaluation by CCK8 assays
The in vitro cytotoxicity assays (IC50) were performed on the human cervical cancer cell lines HeLa, the human lung adenocarcinoma cell lines A549, and the normal mouse embryonic fibroblast cell lines NIH-3T3. The HeLa tumor cell lines and the NIH-3T3 normal cell lines used in this work were grown in Dulbeccos Modified Eagles Medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and the A549 were grown in Eagle Minimum Essential Medium (MEM) containing 10% FBS and 1% penicillin/streptomycin. For the screening experiments, the cells were seeded into 96-well plates in 100 μL of the respective medium containing 10% FBS, at a plating density of 10000 cellsper well. The cells were incubated at 37 °C in 5% CO2 and 95% air at a relative humidity of 100% for 24 h prior to the addition of the test compounds. The test compounds were solubilized in dimethylsulfoxide and diluted in the respective serum free medium. After 24 h, 100 μL of the medium containing the test compounds with various concentrations (e.g. 5, 10, 25, 50, 75, 100, 200 μM for tumor cell lines assays and 50, 100, 200, 400, 600, 800, 1000 μM for normal cell lines assays) was added and incubated at 37 °C in an atmosphere of 5% CO2 and 95% air with 100% relative humidity for 24 h, and the cell viability was assessed by the CCK8 method. Briefly, 10 mL of CCK8 stock solution (Dojindo China Co. Shanghai, China) was added to the treatment media per well, and the UV absorbance was recorded at 450 nm after 30 min with a 318C-microplate reader (Sanco Instrument Ltd. Shanghai, China). All measurements were made in triplicate and the medium containing no test compounds served as the control. The percentage cell inhibition was calculated as (ODcontrol − ODdrug)/ODcontrol × 100. The half maximal inhibitory concentration (IC50) values of each compound were obtained from the dose-dependent curve fit.
Results and discussion
Characterization of hydrazone ligand HL and complexes
Benzhydrazone ligand derivative, as a pale yellow crystalline solid, were prepared in 60.7% yield by the condensation of 2-acetonaphthone with salicylhydrazine in an equimolar ratio. It was then reacted with Ni(OAc)2·4H2O and Co(NO3)2·6H2O in a 1:2 molar ratio, respectively, followed by 1,10-phen in the presence of triethylamine in a 2:1:1:2 molar proportion in methanol/acetonitrile at room temperature (Scheme 1). After a few days, single crystals were obtained from the reaction mixture on slow evaporation at room temperature. 1H and 13C NMR spectra of the free hydrazone ligand HL were assigned on the basis of the observed chemical shift (assignment shown in Fig. S1†). The 1H NMR spectrum displayed a weak singlet at 11.46 ppm due to the NH proton. The appearance of peaks at 6.99–8.35 ppm suggests the presence of aromatic group. The signal corresponding to the protons of methyl group was observed at 2.47 ppm. The 13C NMR spectrum of the ligand revealed the resonance for the aromatic carbon atoms at 117.4–135.8 and 157.0 ppm, the resonance for the carbonyl carbon at 162.6 ppm, and the resonance for the CN carbon atom of the azomethine group at 152.4 ppm, respectively. These spectroscopic data result in affair agreement with the expected structures. And furthermore, the elemental analyses data, ESI-MS mass spectra and melting point of the complexes are consistent with their formulation as bis–hydrazone complexes of Ni(II) and Co(II) containing phen an ancillary ligand. The synthesized complexes were sparingly soluble in solvents such as ethanol, methanol or acetonitrile and readily soluble only in solvents such as DMF and DMSO, producing pale orange-coloured solutions.
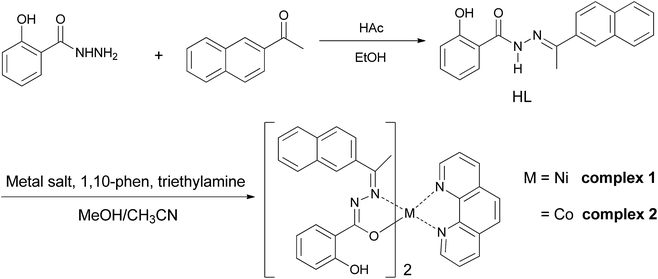 |
| | Scheme 1 Synthesis of the ligand HL and complexes 1 and 2. | |
Infrared spectroscopy. The infrared spectra of the ligand, 1 and 2 are shown in Fig. S2.† The IR spectra of the metal hydrazone complexes were compared with those of free ligand in the region 4000–400 cm−1. The free ligand displayed characteristic absorption bands at 3415, 3277, 1630, 1555, 1231 and 1095 cm−1 due to υ(O–H), υ(N–H), υ(CO), υ(CN), υ(C–OH) and υ(N–N) vibrations, respectively. The bands due to υ(N–H) and υ(CO) vibrations of the free ligands were absent for complexes 1 and 2, thus indicating that enolisation and deprotonation had taken place prior to coordination. This fact was further confirmed by the appearance of two new bands in the spectra of complexes around 1600–1534 cm−1 and 1372–1494 cm−1 that corresponds to υ(CN–NC) and υ(C–O) stretching vibrations, respectively. The bands attributed to υ(CN) stretching were shifted to higher frequencies while a positive shift of about 58 cm−1 was observed for the υ(N–N) stretching vibration in comparison with that of their respective free ligand, thus implying that the nitrogen atom of the azomethine group is coordinated to the metal in these complexes. All these facts suggested that the hydrazone ligand HL behaves as a monobasic bidentate (NO) chelating ligands in the two complexes.
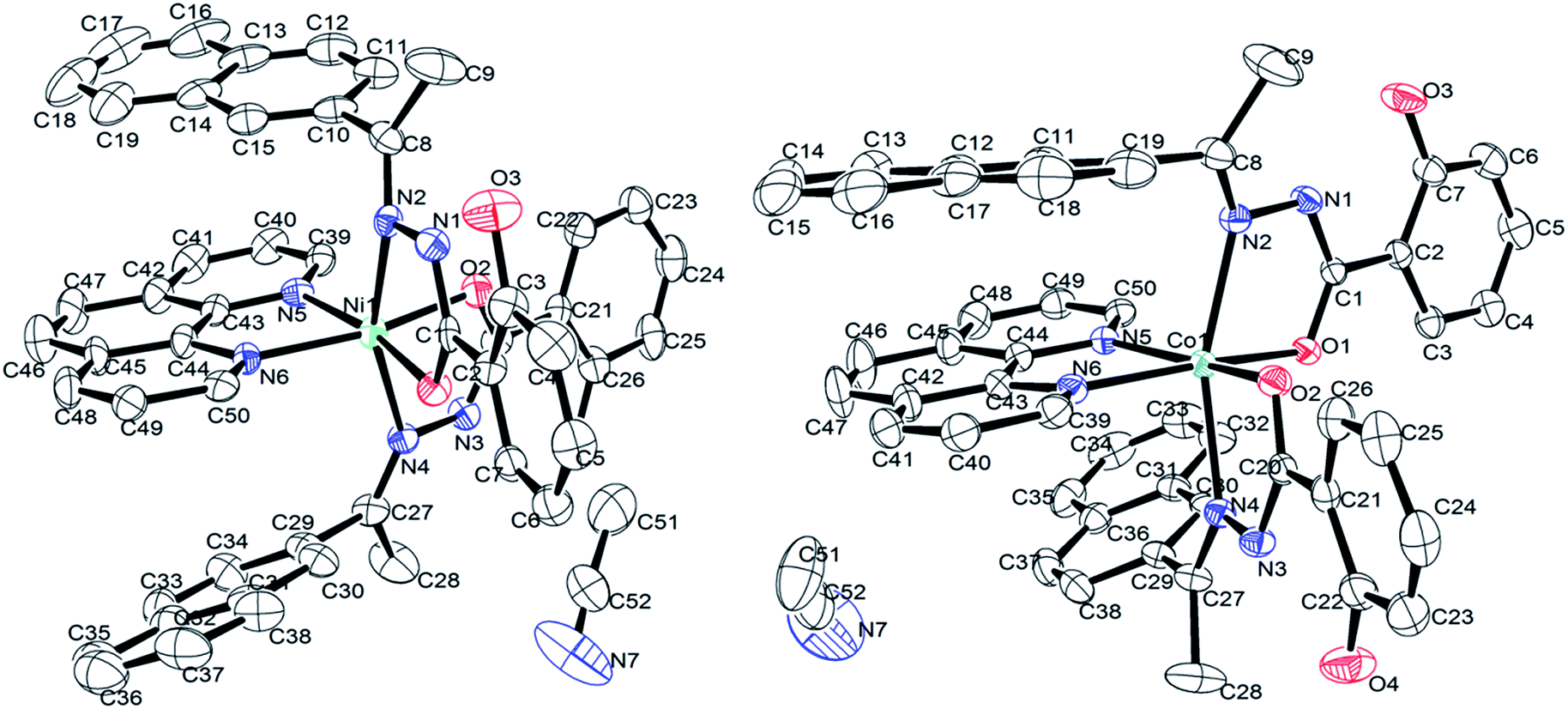
Description of solid state structures. The solid-state structure of complexes was analyzed with single crystal X-ray diffraction study. Details of the data collection conditions and the parameters of refinement process are given in Table 1. The crystal structures of nickel and cobalt complexes with atom numbering scheme are depicted in Fig. 1 and selected bond distances (Å) and bond angles (°) are given in Table 2. Both complexes are crystallized in the monoclinic space group P21/c with four molecules in the unit cell. In both the complexes [NiL2phen·CH3CN] and [CoL2phen·CH3CN], the coordination geometry around the metal ion is a distorted octahedral geometry with NiN4O2 and CoN4O2 chromophores, forming three five-membered metallocycle involving the metal ion. As expected, the metal atom is bonded to the hydrazone ligand via enolate oxygen atom and the imine nitrogen atom, along with two N atoms of phen. For [NiL2phen·CH3CN], Ni(II) center exhibits a distorted octahedral geometry comprising of two equivalent monoanionic hydrazone ligands coordinated in a meridional fashion using cis phen nitrogen, trans azomethine nitrogen and cis enolate oxygen atoms positioned very nearly perpendicular to each other. The hydrazone ligand has undergone keto–enol tautomerism and it is well known that the imino tautomers can exist as two geometrical isomers, syn (Z) and anti (E), but in this crystal, only the E isomer has been observed. The torsion angle C1–N1–N2–C8, −166.4(6)°, N1–N2–C8–C10, −171.9(7)°, C20–N3–N4–C27, −179.2(7)° and N3–N4–C27–C29, 177.9(7)° observed supports the E conformation of the ligand on coordination.42 The coordination around nickel is distorted octahedral with two oxygen atoms and four nitrogen atoms occupy the coordination sites with the ligand-metal-ligand bite angles varying among 107.7(2)° [N5–Ni1–N2], 87.0(2)° [O2–Ni1–N2], 91.5(2)° [O1–Ni1–N4] and 104.5(2)° [N4–Ni1–N6]. The [N2–Ni1–N4] trans angle was found as 160.5(2)° but the other trans angles [O2–Ni1–N5] 90.3(2)° and [O1–Ni1–N6] 93.9(2)° are constrained within the meridional ligands.42 These observations suggested that the coordination geometry around the nickel ion had undergone much distortion from a perfect octahedron. Further, the bond distances around the metal [Ni1–N2, 2.127(6); Ni–N4, 2.150(6); Ni1–N5, 2.132(6); Ni1–N6, 2.082(6); Ni1–O1, 2.033(5); Ni1–O2, 2.003(5) Å] are in good agreement with that of other Ni(II) complex in which the aroylhydrazone ligand takes enolic form.43
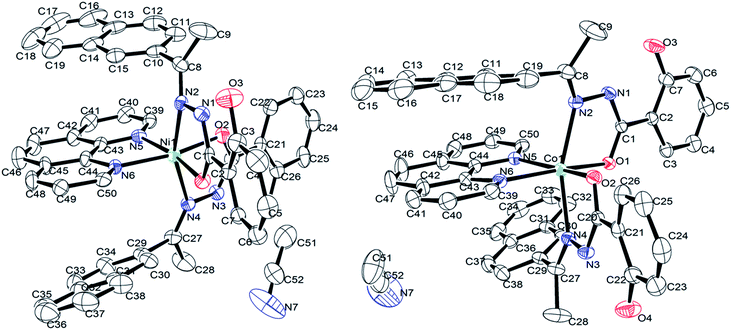 |
| | Fig. 1 ORTEP view of the structure of Ni(II) and Co(II)complexes. Thermal ellipsoids are scaled to the 25% probability level and hydrogen atoms omitted for clarity. | |
Table 2 Selected bond lengths (Å) and bond angles (°)
| C52H41N7O4Ni |
|
C52H41N7O4Co |
|
| Ni1–O1 2.033(5) |
O1–Ni1–N2 77.9(2) |
Co1–O1 2.0426(18) |
O1–Co1–N2 76.34(8) |
| Ni1–O2 2.003(5) |
O1–Ni1–N4 91.5(2) |
Co1–O2 2.0175(19) |
O1–Co1–N4 93.10(8) |
| Ni1–N2 2.127(6) |
O1–Ni1–N5 170.5(2) |
Co1–N2 2.174(2) |
O1–Co1–N5 94.04(8) |
| Ni1–N4 2.150(6) |
O1–Ni1–N6 93.9(2) |
Co1–N4 2.183(2) |
O1–Co1–N6 169.10(8) |
| Ni1–N5 2.132(6) |
O2–Ni1–O1 97.8(2) |
Co1–N5 2.124(2) |
O2–Co1–O1 101.03(8) |
| Ni1–N6 2.082(6) |
O2–Ni1–N2 87.0(2) |
Co1–N6 2.187(2) |
O2–Co1–N2 88.53(8) |
| O1–C1 1.274(8) |
O2–Ni1–N4 78.3(2) |
O1–C1 1.276(3) |
O2–Co1–N4 93.10(8) |
| O2–C20 1.300(8) |
O2–Ni1–N5 90.3(2) |
O2–C20 1.275(3) |
O2–Co1–N5 164.29(8) |
| O3–C3 1.352(10) |
O2–Ni1–N6 167.9(2) |
O3–C7 1.343(4) |
O2–Co1–N6 88.82(8) |
| O4–C26 1.339(10) |
N2–Ni1–N4 160.5(2) |
O4–C22 1.339(4) |
N2–Co1–N4 160.09(9) |
| N1–N2 1.406(8) |
N2–Ni1–N5 107.7(2) |
N1–N2 1.395(3) |
N2–Co1–N5 90.75(8) |
| N1–C1 1.326(9) |
N5–Ni1–N4 85.3(2) |
N1–C1 1.317(3) |
N5–Co1–N4 107.00(8) |
| N2–C8 1.282(9) |
N6–Ni1–N2 92.6(2) |
N2–C8 1.273(3) |
N6–Co1–N2 108.90(8) |
| N3–N4 1.402(8) |
N6–Ni1–N4 104.5(2) |
N3–N4 1.400(3) |
N6–Co1–N4 84.53(8) |
| N3–C20 1.314(9) |
N6–Ni1–N5 78.3(2) |
N3–C20 1.314(3) |
N6–Co1–N5 76.58(9) |
| N4–C27 1.273(9) |
C1–O1–Ni1 113.5(4) |
N4–C27 1.286(3) |
C1–O1–Co1 114.37(16) |
| |
C20–O2–Ni1 112.4(5) |
|
C20–O2–Co1 113.64(17) |
| C1–N1–N2–C8 −166.4(6) |
C1–N1–N2–C8 167.2(3) |
| N1–N2–C8–C10 −171.9(7) |
N1–N2–C8–C10 171.4(3) |
| C20–N3–N4–C27 −179.2(7) |
C20–N3–N4–C27 −177.6(3) |
| N3–N4–C27–C29 177.9(7) |
N3–N4–C27–C29 −178.5(3) |
Akin to complex 1, complex 2 features the coordination of two (HL) ligands and phen to the metal ion thus forming three fused five-membered chelate rings. Collectively, the bond angles N2–Co1–N5 [90.75(8)°], N2–Co1–O1 [76.34(8)°], N6–Co1–N4 [84.53(8)°] and O2–Co1–N4 [93.10(8)°] confirm that the coordination geometry is distorted from that of a perfect octahedron.43 The [N2–Co1–N4] trans bond angle is 160.09(9)°. However, the remaining trans bond angles of 90.04(8)° and 88.82(8)° for [N5–Co1–O1] and [N6–Co1–O2], respectively, also imply constraints within the meridional ligands. When compared the corresponding bond lengths of the Ni(II) complex with that of the Co(II) complex, it was found that the distances of Ni–N and Ni–O [Ni1–N2, 2.127(6); Ni–N4, 2.150(6); Ni1–N5, 2.132(6); Ni1–N6, 2.082(6); Ni1–O1, 2.033(5); Ni1–O2, 2.003(5) Å] are both shorter than the corresponding Co–N and Co–O [Co1–N2, 2.174(2); Co1–N4, 2.183(2); Co1–N5, 2.124(2); Co1–N6, 2.187(2); Co1–O1, 2.0426(18); Co1–O2; 2.0175(19) Å] bond distances, indicating the smaller size of Ni(II) complex.20 In addition, and the bite angles of the Co(II) complex were more deviate from the ideal bond angles of 90° and 180° than that of the Ni(II) complex, which indicate that Co(II) complex has a more distorted octahedral geometry.43 X-ray determination confirms the structure that was proposed on the basis of spectroscopic data, which is consistent with the bivalency of the metal and the monoionic nature of the ligand in the complexes.
Thermal analysis (TGA). The thermogravimetric analysis curves (Fig. S5†) for the complexes show that the two complexes undergo there similar decomposition processes because of similar geometries of the complexes. The first weight loss from room temperature to 200 °C for 1 and 100 to 200 °C for 2 corresponds to the removal of the uncoordinated acetonitrile molecule in both complexes (calculated value 4.6% for 1 and 2, while experimental value 4.0% for 1 and 2.3% for 2, respectively). The second weight loss from 200 °C to about 320 °C corresponds to the loss of one coordinated HL molecule in both complexes (experimental value about 35.0% and calculated value 34.2%). The further exothermic decomposition of the complex 1 began at around 500 °C and finished at about 900 °C, which is attributed to the complete removal of organic part of the compound. The main product was attributed to NiO and CoO with a residual value of 10.6% for both complexes (theoretical residual value 8.4% for both 1 and 2). This thermal-stable property makes the complexes suitable for long-term storage at normal ambient temperatures, with easier potential medical and biological usage.
DNA binding studies
UV-vis absorption studies. The electronic absorption spectra of hydrazone ligand and respective nickel and cobalt chelates in the absence and presence of HS–DNA are shown in Fig. 2. The absorption bands in the range of 270–400 nm for the hydrazone ligand have been assigned to charge transfer transitions of the type π → π* and n → π*. In the case of the complexes, the high energy absorption bands appeared between 250–320 nm and another absorption around 320–400 nm were assigned to the intra-ligand charge transfer transitions of the type π → π* and n → π*,14,44 respectively. Further, the bands appeared around 350 nm for complexes have been assigned to ligand-to-metal charge transfer (LMCT) as well as metal-to-ligand charge transfer (MLCT) transitions,45 respectively. Upon the addition of a solution of DNA to the compounds, a decrease in the absorption intensity (hypochromism) of the absorption bands with red shift (2–3 nm) are observed. These spectral characteristics reveal that both the ligand and the complex interact with CT–DNA most likely through the intercalation mode that involves π-π stacking interaction between the hydrazone chromophore and the base pairs of DNA.46,47 The magnitude of the hypochromism and red shift depends on the strength of the interaction between the complex and the DNA helix. These observations are similar to those that were reported earlier for various metallointercalators.48
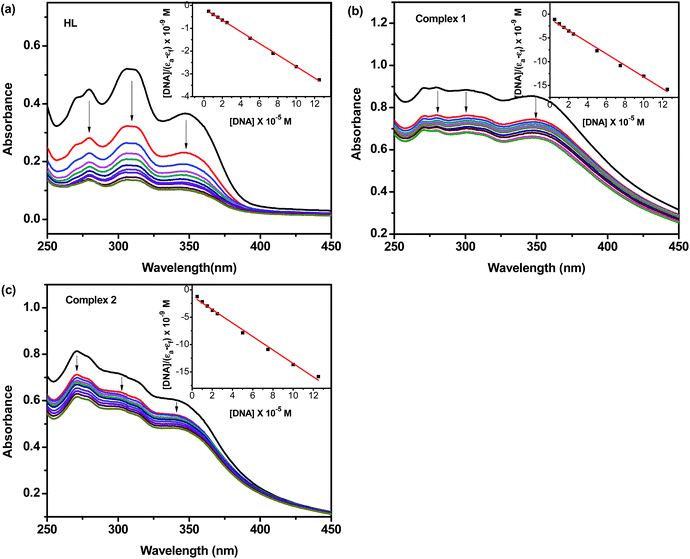 |
| | Fig. 2 Electronic absorption spectra of HL and complexes (25 μM) in the absence and presence of increasing amounts of HS–DNA (5, 10, 15, 20, 25, 50, 75, 100, 125 μM). Arrows show the changes in absorbance as a function of increasing DNA concentration (inset: plot of [DNA] vs. [DNA]/(εa−εf)). | |
In order to determine quantitatively the binding strength of the ligands and its complexes with HS–DNA, intrinsic binding constants were obtained by monitoring the changes in both the wavelength as well as their corresponding intensity of absorption of the high energy bands upon increasing concentration of added DNA. The following equation was applied to calculate the binding constant: [DNA]/(εa−εf) = [DNA]/(εb−εf) + 1/Kb(εb−εf), where [DNA] is the concentration of DNA in base pairs, the apparent extinction coefficient εa is obtained by calculating Aobs/[complex], εf corresponds to the extinction coefficient of the complex in its free form and εb refers to the extinction coefficient of the complex in the fully bound form. Each set of data, fitted to the above equation, gave a straight line with a slope of 1/(εb−εf) and y-intercept of 1/Kb(εb−εf). The Kb value was determined from the ratio of the slope to intercept,49 which were found to be 9.00 (±0.12) × 104 M−1, 1.20 (±0.15) × 105 M−1 and 1.05 (±0.09) × 105 M−1, corresponding to HL, 1 and 2, respectively. The magnitude of the binding constant value clearly showed that all compounds interact with DNA through an intercalative mode, and complex 1 bound more strongly with DNA than HL and complex 2.
Competitive binding between EB and ligands/complexes for HS–DNA. Fluorescence quenching of the EB–DNA complex is used to monitor the binding of test compounds to DNA regardless of their binding modes and only measures their ability to influence the EB luminescence intensities in the EB–DNA complex.50 It has been previously reported that the fluorescence intensity of EB–DNA could be decreased by addition of the ligands/complexes as quenchers, indicating the competition between the ligands/complexes and EB in binding to DNA that proved the intercalation of ligands/metal complexes to the base pairs of DNA.51,52 The emission spectra of EB bound to HS–DNA in the absence and presence of each compound have been recorded for [EB] = 50 μM and [DNA] = 50 μM upon the addition of increasing amounts of respective compounds (0–100 μM), as shown in Fig. 3. In all the cases, it is clear that a significant decrease in the intensity of the emission band of the DNA–EB system at 602 nm of about 44.3%, 80.7% and 71.7%, respectively, indicates that there existed a competition between the metal hydrazone complexes and EB towards binding to DNA. The observed significant quenching of DNA–EB fluorescence after the addition of 1 and 2 proved that they displace EB from the DNA–EB complex and they probably interact with HS–DNA by the intercalative mode.53 The quenching constant (Kq), obtained from the slope of the plot [Q] versus I0/I (shown as insets in Fig. 3) is used to evaluate the quenching efficiency for each compound according to the equation: I0/I = Kq[Q] + 1. The Stern–Volmer plots of DNA–EB illustrate that the quenching of EB bound to DNA by the ligand and complexes is in good agreement with the linear Stern–Volmer equation. Further, the values of Kq corresponding to the three compounds are found as 6.64 (±0.08) × 103 M−1, 4.45 (±0.12) × 104 M−1 and 2.91 (±0.10) × 104 M−1, respectively. The values of the apparent DNA binding constant (Kapp) were calculated using the equation:54 KEB[EB] = Kapp[complex]. The test compound concentrations were obtained from the value at 50% reduction of the fluorescence intensity of EB and KEB = 1.0 × 107 M−1 and [EB] = 5 μM. The calculated Kapp values for HL, 1 and 2 are 3.32 × 105 M−1, 1.36 × 106 M−1 and 1.10 × 106 M−1, respectively. These values suggested that the hydrazone ligand did not exhibit much potential to leach out the EB molecules that were originally bound to DNA. Also, it was assessed that complex 1 showed a higher quenching efficiency than the complex 2 that reflected the strong binding of complex 1 with DNA to leach out more number of EB molecules originally bound to DNA. The binding affinity of the compounds towards DNA increased in the order HL < 2 < 1. The binding as well as quenching constants determined in our experiments clearly demonstrates that the titled complexes possess better DNA interaction than other similar complexes reported in the literature.43
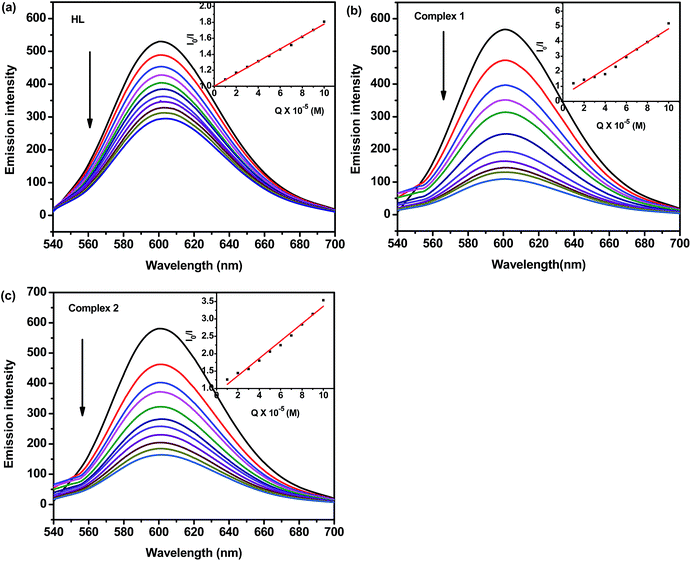 |
| | Fig. 3 Emission spectra of EB–DNA in the absence and presence of HL, 1 and 2 in Tris–HCl/NaCl buffer (pH = 7.2). [EB] = 50 μM, [DNA] = 50 μM, and [compound] = 10–100 μM. Arrow indicates the change in the emission intensity as a function of complex concentration (inset: Stern–Volmer plot of the fluorescence titration data corresponding to the compound). | |
Viscosities studies. Though optical photophysical methods are employed to monitor the binding mode of the metal complex with the DNA, hydrodynamic measurements are preferred as the confirmation tests. Viscosity measurements are sensitive to changes in the length of DNA molecule and regarded as least ambiguous and most critical test for the binding mode. EB as a classical intercalator shows a significant increase in relative viscosity of the DNA solution on intercalation due to an increase in overall DNA contour length on binding to DNA.55 In contrast, partial or non-classical intercalation of complex would bend or kink the DNA helix, shortening the effective length of the DNA, and reducing DNA viscosity accordingly, while the electrostatic and groove binding cause little or no effect on the relative viscosity of DNA solution.56 In order to clarify the binding mode and strength of complexes with DNA, the DNA viscosity variance at room temperature was studied by varying the concentration of complexes. The values of relative viscosity (η/η0)1/3 were determined and plotted against the values of [DNA]/[Complex]. The effect of HL, 1 and 2 on the viscosity of HS–DNA is shown in Fig. 4. The obtained plots show increase in the DNA viscosity after the increase in the concentration of metal complex due to its consecutive addition. The increase in viscosity of DNA by complexes suggests insertion of aromatic ring into the base pairs of DNA as discussed above. Complex 1 showed a much more significant effect on viscosity of DNA among all the synthetic compounds, the reason could be ascribed to the smaller size of Ni(II) complex fitting well into the interior of DNA grooves than the bulkier cation of Co(II) complex, therefore effectively increased the length of DNA biopolymer. The observed viscosity results were in accordance with optical photophysical titration results, suggesting that complexes can bind to DNA through intercalation.
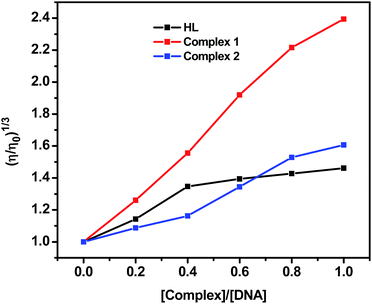 |
| | Fig. 4 Relative viscosity of DNA in Tris–HCl/NaCl buffer solution (pH 7.2) in the presence of the ligand and complexes at increasing amounts. ([DNA] = 50 μM, [compound] = 0–50 μM, 37 ± 0.1 °C). | |
Cyclic voltammetry. Cyclic voltammetry is one of the most important electrochemical techniques employed to examine the interface of a metal complex with biomolecules due to the similarity between many redox chemical and biological process.57 Also many biological processes can rely on chemical processes. The cyclic voltammetric study of complexes in the absence and in presence of DNA was performed in DMSO/Tris–HCl/NaCl buffer (pH = 7.2) medium at room temperature under dry argon atmosphere (Fig. S6†). Complex 1 showed quasi-reversible one-electron reduction responses with a reversible reduction at −0.981 V (vs. Ag/AgCl). This peak shifted to a more positive value (−0.969 V) when DNA (10 μM) was added to the complex solution. An oxidation peak was found at −0.602 V, which shifted to a more negative position (−0.633 V) upon the addition of DNA. On comparison with previously reported mono-nuclear Ni(II) systems the oxidation can be assigned to the redox of the metal center NiII–NiI.58 The shift of the reductive band to more positive values upon addition of DNA indicated electron deficiency due to interaction with the bases of the DNA double strands; the shift of the oxidative bands to more negative values confirmed this point again. So by monitoring the CV of 1 and a solution of 1 containing DNA, it could be suggested that some sort of interaction between 1 and the DNA bases took place through which the electron cloud of the complex could be “pulled” towards the DNA strands.59 Similar quasi-reversible one-electron reduction responses (CoII–CoI) were also obtained for the complex 2, as seen in Fig. S6.†
Protein binding studies
Fluorescence spectroscopy. Serum albumins are proteins that are amongst others involved in the transport of metal ions and metal complexes with drugs through the blood stream. Binding to these proteins may lead to loss or enhancement of the biological properties of the original drug, or provide paths for drug transportation. Hence, the binding experiments using the newly synthesized compounds with BSA were carried out. A generally known fact is that the fluorescence of a protein is caused by three intrinsic characteristics of the protein, namely tryptophan, tyrosine and phenylalanine residues.14,58 Fluorescence quenching refers to any process that decreases the fluorescence intensity of a fluorophores due to a variety of molecular interactions including excited state reactions, molecular rearrangements, energy transfer, ground state complex formation and collision quenching.14 Qualitative analysis of hydrazone ligand and its bivalent nickel and cobalt hydrazones bound to BSA has been undertaken by examining the respective fluorescence spectra. Fig. 5 showed the effect of the increase in the concentration of test compounds on the emission intensity of BSA. The intensity of the fluorescence band of BSA observed at 345 nm was quenched to an extent of about 47.9%, 63.3% and 65.1% from its initial intensity upon the addition of the corresponding HL, 1 and 2 together with a hypsochromic shift of 2, 4 and 4 nm due to the formation of a hydrazone–BSA complex/metal hydrazone–BSA complex. The observed blue shift is mainly due to the fact that the active site in the protein is buried in a hydrophobic environment. This result suggested a definite interaction of the compounds with the BSA protein. The fluorescence quenching is described by Stern–Volmer relation, I0/I = 1 + KSV[Q], where I0 and I are the fluorescence intensities of the fluorophore in the absence and presence of quencher, KSV is the Stern–Volmer quenching constant and [Q] is the quencher concentration. The KSV value obtained as a slope from the plot of [Q] vs. I0/I (as insets in Fig. 5a–c) in respect of HL, 1 and 2 are found to be 4.32(±0.20) × 105 M−1, 6.41(±0.15) × 105 M−1 and 5.91(±0.14) × 105 M−1, respectively.
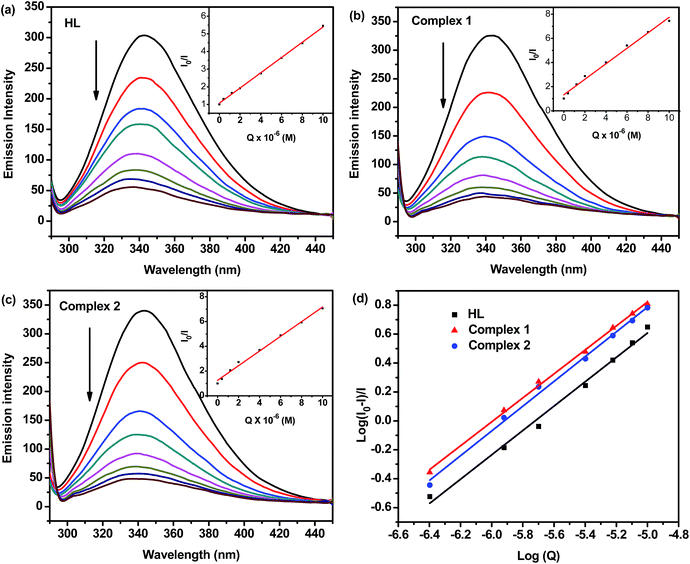 |
| | Fig. 5 (a), (b) and (c) Emission spectrum of BSA (1 μM; λex = 280 nm; λem = 343 nm) as a function of concentration of HL and complexes (0, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0, 10.0 μM). Arrow indicates the effect of metal complexes on the fluorescence emission of BSA. Inset: Stern–Volmer plot of the fluorescence titration data corresponding to the compounds. (d) Plot of log[Q] vs. log[(I0–I)/I]. | |
Binding constant and the number of binding sites. When small molecules bind independently to a set of equivalent sites on a macromolecule, the equilibrium between free and bound molecules is represented by the Scatchard equation:60 log[(I0–I)/I] = logK + nlog[Q], where K and n are the binding constant and the number of binding sites, respectively. Thus, a plot of log[Q] versus log[(I0–I)/I] (Fig. 5d) can be used to determine the values of both K and n and such values calculated for test compounds are listed in Table 3. From the values of n, it is inferred that there is only one independent class of binding sites for complexes on BSA and also a direct relation between the binding constant and number of binding sites. The results of binding experiments clearly explored that the complexes showed stronger interactions with both DNA and BSA than that of the free hydrazone ligand. Generally, these types of binding affinities of metal chelates towards DNA/BSA were due to the strong chelation phenomenon experienced by the hydrazone systems with the bivalent metal centers. However, the nickel complex showed stronger binding with BSA than the cobalt complex. The observed stronger binding efficiency of nickel complex could be attributed to the smaller size of Ni(II) complex to fit well into the DNA/BSA grooves than the bulkier cation of Co(II) complex. The binding ability is also in line with the natural order of stability. So, it is clear that the sizes of the metal complexes make a major contribution in determining the DNA/BSA binding affinities of 3d metal complexes.
Table 3 Comparison of interaction study results between compounds on DNA and BSA
| Compounds |
DNA binding |
Protein binding |
| Kb (M−1) |
Kq (M−1) |
Ksv (M−1) |
K (M−1) |
n |
| HL |
9.00(±0.12) × 104 |
6.64(±0.08) × 103 |
4.32(±0.20) × 105 |
8.34(±0.08) × 104 |
0.8426 |
| Complex 1 |
1.20(±0.15) × 105 |
4.45(±0.06) × 104 |
6.41(±0.15) × 105 |
1.17(±0.13) × 105 |
0.8566 |
| Complex 2 |
1.05(±0.09) × 105 |
2.91(±0.10) × 104 |
5.91(±0.14) × 105 |
9.12(±0.09) × 104 |
0.8281 |
Absorption spectral studies. Quenching can occur by different mechanisms, which are usually classified as dynamic quenching and static quenching. Dynamic quenching refers to a process in which the fluorophore and the quencher come into contact during the transient existence of the excited state. Static quenching refers to the fluorophore-quencher complex formation in the ground state.17 UV-visible absorption spectroscopy is a simple method to explore the type of quenching. BSA has two main absorption bands including: (i) one weak band at the wavelength range near 280 nm, where only the aromatic side chains of the protein absorb, (ii) one strong band at the wavelength range near 210–230 nm, where, in addition to the aromatic amino acids, histidine, cystine, and the peptide group all contribute to the absorption.61 Fig. 6, S7 and S8† show the absorption spectra of the solution of BSA upon addition of increasing amounts of 1, HL and 2, respectively at wavelength of around 200–320 nm. Upon addition of all compounds to the BSA solution, the intensity of the absorption peak of BSA at 230 nm obviously decreased, and the peak shifted toward longer wavelength (with 54.0% hypochromicity and 4 nm red shift for the complex 1). Meanwhile, the intensity of the peak of BSA at 278 nm increases obviously upon addition of complexes. These observations (hypochromic effect and red shift) indicate that there is the strong interaction between complex with aromatic amino acids of BSA that may cause the conformational changes in BSA and change the polarity of the microenvironment around amino acids of BSA.61,62 These results revealed that there exists a static interaction between BSA and the added compounds due to the formation of a ground state complex of the type BSA-compound as has been reported previously.14,53
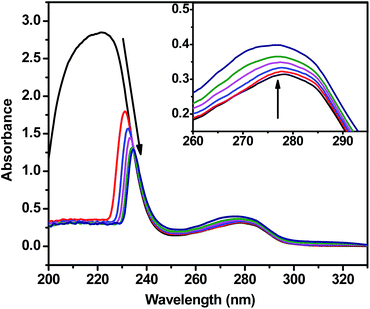 |
| | Fig. 6 UV-vis absorption spectra of BSA (10 μM) with increasing concentrations (2.0, 4.0, 6.0, 8.0 and 10.0 μM) of the complex 1 (PBS buffer, pH = 7.2). The arrows show the absorbance changes upon increasing amounts of the complex. | |
Synchronous fluorescence studies. Synchronous fluorescence spectra provide information on the molecular micro-environment, particularly in the vicinity of the fluorophore functional groups.63 The fluorescence of BSA is due to presence of tyrosine and tryptophan residues. Among them, tryptophan is the most dominant fluorophore, located at the substrate binding sites. Most of the drugs bind to the protein in the active binding sites. Hence, synchronous method is usually applied to find out the conformational changes around tryptophan and tyrosine region. In synchronous fluorescence spectroscopy, according to Miller,64 the difference between the excitation wavelength and emission wavelength (Δλ = λem−λex) indicates the type of chromophores. A higher Δλ value such as 60 nm is indicative of the characteristic of tryptophan residue while a lower Δλ value such as 15 nm is characteristic of tyrosine residue.65 The effect of the ligand HL and complexes on BSA synchronous fluorescence spectroscopy with Δλ = 15 nm and Δλ = 60 nm are shown in Fig. 7. In the synchronous fluorescence spectra of BSA at Δλ = 15 nm, the addition of the compounds to the solution of BSA results in small decrease in the fluorescence intensity of BSA at 287 nm, up to 23.9% (HL), 37.7% (1) and 36.6% (2) of the initial fluorescence intensity of BSA without any shift in emission wavelength. In the case of Δλ = 60 nm, the addition of the compounds to the solution of BSA results in a significant decrease in the fluorescence intensity of BSA at 280 nm, up to 44.5% (HL), 65.7% (1) and 62.5% (2) of the initial fluorescence intensity of BSA without any change in the position of the emission band. These experimental results indicate that the interaction of the complex or ligand with BSA protein affects the conformation of both the tryptophan and tyrosine micro-regions. Hence, during the binding process the polarity around the tyrosine and tryptophan residues were decreased and the hydrophobicity around the same residues was strengthened, however, the effect was more towards tryptophan than tyrosine. The hydrophobicity observed in fluorescence and synchronous measurements confirmed the effective binding of all the complexes with the BSA.
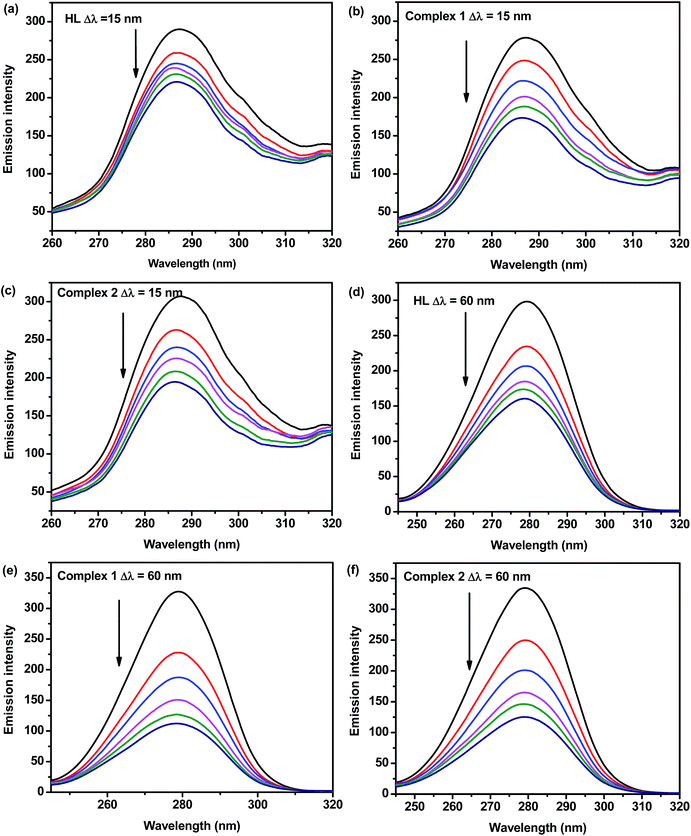 |
| | Fig. 7 Synchronous spectra of BSA (1 μM) in the absence and presence of increasing amounts of the ligand and complexes (0, 0.4, 0.8, 1.2, 1.6 and 2.0 μM) at a wavelength difference of Δλ = 15 nm and Δλ = 60 nm. The arrow shows the emission intensity changes upon an increase in the concentration of the compounds. | |
Three-dimensional fluorescence spectral studies. Excitation-emission matrix spectroscopy or three-dimensional emission spectroscopy can provide total information regarding the emission characteristics of fluorophores by changing the excitation and emission wavelength simultaneously. Accordingly, three-dimensional emission spectroscopy can be employed to yield information regarding the conformational change of protein bound to a probe. Application of three dimensional fluorescence techniques is seen in recent years for the investigation of conformational changes of proteins interacting with drugs. The excitation wavelength, emission wavelength and fluorescence intensity can be used as axes in the fluorescence emission spectra. The maximum emission wavelength and fluorescence intensity of the residues showed a close relation to the polarity of their microenvironment. The corresponding characteristic parameters from three-dimensional fluorescence spectra are presented in Table 4 and the corresponding spectra and contour diagrams for BSA, BSA + HL, BSA + 1, and BSA + 2 are shown in Fig. S9.† The Rayleigh scattering peak (λem = λex) and two typical fluorescence peaks 1 and 2 were observed. As referred to peak 1 (λex = 280 nm, λem = 340 nm), we think that it mainly reveals the spectral characteristic of tryptophan and tyrosine residues. The reason is that when protein is excited at 280 nm, it mainly reveals intrinsic fluorescence of tryptophan and tyrosine residues. Comparing with the UV-vis absorption spectra of BSA (Fig. 6), there is an absorption peak around 278 nm and this peak is mainly caused by the transition of π → π* of aromatic amino acids in BSA. After addition of the complexes, the fluorescence intensity of BSA decreased from 256.7 to 120.4 for the complex 1 and 256.7 to 135.1 for complex 2. This suggests a less polar environment of both residues with almost all the hydrophobic amino acid residues of the BSA being buried in the hydrophobic pocket. Also, the addition of the complex changed the polarity of this hydrophobic microenvironment and the conformation of the BSA.66 Besides peak 1, there is another strong fluorescence peak 2. And the excitation wavelength of this peak is around 220 nm, which is related to the conformation of the peptide backbone associated with the helix-coil. Comparing with the UV-vis absorbance spectra of BSA (Fig. 6), there is a strong absorption peak around 225 nm and this peak was mainly attributed to the n → π* transition of protein's characteristic polypeptide backbone structure, CO.66 After the addition of these complexes, the fluorescence intensity of peak 2 decreased a lot from 517.1 to 100.6 for complex 1 and 517.1 to 110.9 for complex 2, and the excitation peak has an obvious red shift (from 220 to 230 nm), which demonstrated that the peptide strands structure of protein was changed. These phenomenon and analytical results reveal that the interaction between BSA and the complex trigger micro environmental and conformational alterations in BSA. The investigation of the effect of both complexes on the conformation of BSA is quite helpful to the understanding in biological role of serum albumin. Furthermore, our work could also be a useful guideline for further drug design.
Table 4 Three-dimensional fluorescence spectral characteristics of BSA and BSA-complex system
| System |
Parameters |
Peak 1 |
Peak 2 |
| BSA |
Peak position (λex/λem, nm) |
280/343 |
220/343 |
| Relative intensity |
256.7 |
517.1 |
| BSA + HL |
Peak position (λex/λem, nm) |
280/342 |
230/342 |
| Relative intensity |
180.9 |
158.80 |
| BSA + 1 |
Peak position (λex/λem, nm) |
280/342 |
230/342 |
| Relative intensity |
120.4 |
100.6 |
| BSA + 2 |
Peak position (λex/λem, nm) |
280/342 |
230/343 |
| Relative intensity |
135.1 |
110.9 |
Molecular docking
Molecular docking of the Ni(II) complex with DNA sequence d(ACCGACGTCGGT)2. Computational docking is extremely useful tool to gain an understanding of synthesized compounds and biological drug target interactions which is very important in drug discovery. In our study, the synthesized nickel(II) complex were subjected to molecular docking studies using the AutoDock Tools (ADT) and AutoDock version 4.2 docking programs, which are interactive molecular graphics programs, to understand the drug–DNA interactions to investigate the potential binding mode and energy. The obtained results from docking of the complexes to DNA sequence d(ACCGACGTCGGT)2 reveal that the binding energy value for the Ni(II) complex is −7.3 kcal mol−1, which indicate strong binding ability. This Ni(II) complex interacts with the ADE5, CTY18 and GUA4 bases in the major groove involving hydrophobic and hydrogen bonding contacts (Fig. 8). The phenolic hydroxyl group of the complex interacts with the nitrogen of GUA4 (chain A) and the oxygen of CYT18 (chain B) forming GUA4:N⋯H–Ocomplex and CYT18:P:O⋯H–Ocomplex hydrogen bond with the bond length of 3.2 and 2.9 Å, respectively. Further, the uncoordinated nitrogen of the complex interacts with the oxygen of CYT18:O (chain B) forming CYT18:O–H⋯Ncomplex with the bond length of 3.2 Å. They are significantly preference of complex that bind to the minor groove of DNA, which would affect the stability of DNA, auxiliary express the effect of the complex with DNA.
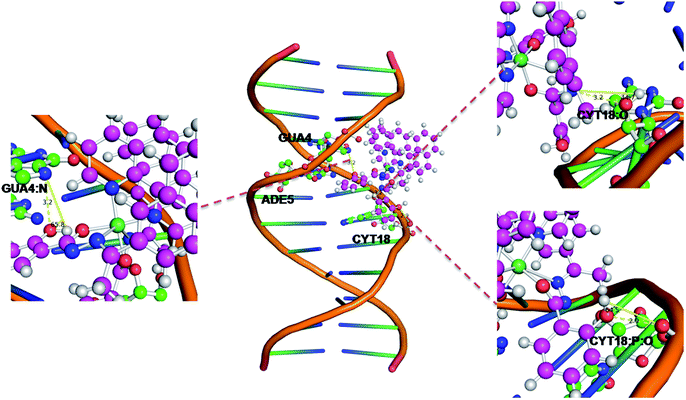 |
| | Fig. 8 Docking pose of Ni(II) complex with DNA (423D). | |
Molecular docking of the Ni(II) complex with BSA. Molecular docking was used to identify the preferential binding sites in BSA for the Ni(II) complex. Serum albumin as the most abundant carrier protein comprises three α-helical domains. Fig. 9 shows the best energy-ranked result of the interaction between the Ni(II) complex and BSA in all runs of docking procedure. The obtained results from molecular docking show that the complex is suited within the pocket with the binding energy of −9.0 kcal mol−1 involving van der Waals interaction, hydrophobic and hydrogen bonding contacts between the complex atoms and the amino acids of the binding site. First of all, the complex interacts with the Ser109, Thr526 and Lys523 residues via van der Waals interaction. Secondly, the complex is nearby to some hydrophobic residues in of BSA (Leu 112, Lys114, Val 423 and IIe 525). Some of the residues in the neighborhood of the complex 1 (Ser 109, Pro 110, Asp 111, Arg 427 and Arg 458) are polar. Further, there is a high possibility of an electrostatic interaction between the Ni(II) and the side-chain carboxyl group of Glu424 residue. Finally, the complex is able to form a π-π interaction with Pro420 and a hydrogen bonding interaction with Leu112 (lengths of the hydrogen bond: Leu112:O⋯H–Ocomplex = 2.7 Å and angles of the hydrogen bond: Leu112:O⋯H–Ocomplex = 135.4°). In conclusion, these interactions play significant role in stabilizing of the Ni(II) complex intercalative mode of binding. These results are in agreement with the fluorescence quenching of BSA emission in the presence of the complexes.
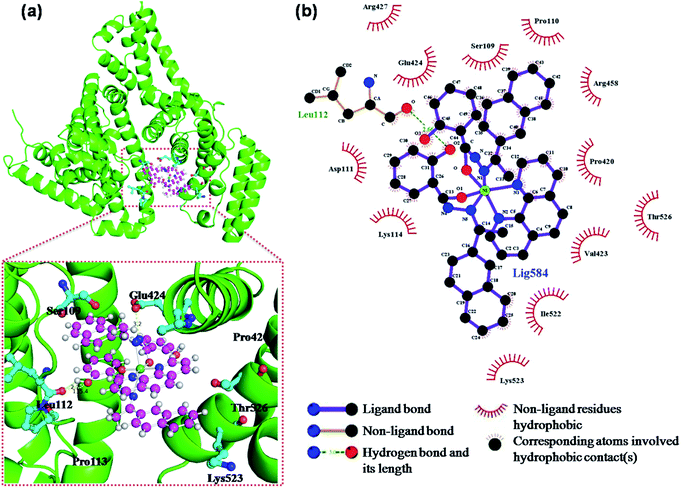 |
| | Fig. 9 (a) The Ni complex was docked in the binding pocket of BSA (4F5S) using PyMol, (b) two-dimensional interactions generated by LIGPLOT+. | |
Antioxidant activities
It is a well documented fact that hydrazones and their corresponding transition metal complexes displayed significant antioxidant activity.45,67,68 In order to elucidate the ability of the nickel and cobalt hydrazone complexes to act toward different reactive radical species, the radical scavenging activities of our compounds along with standard butylated hydroxytoluene (BHT) in a cell free system, have been examined with reference to DPPH radicals (DPPH˙), nitric oxide (NO˙) superoxide anion (O2˙) and hydroxyl radicals (OH˙), and their corresponding IC50 values have been presented in Fig. 10. The power of the ligand HL ability to scavenge various radicals was found to be slightly lower than that of the standard antioxidant BHT, except in case of DPPH˙ scavenging, which is almost fifty times higher. Even though the DPPH˙ scavenging ability of the Ni(II) complex was only slightly higher than the standard, the ability was excellent for the other three radicals when compared to BHT, HL and Co(II) complex. The IC50 values indicated that the various radical scavenging activities of the complex are in the order of DPPH˙ > O2˙ > OH˙ > NO˙. It is to be noted that no significant radical scavenging activities were observed in all the experiments carried out with Ni(OAc)2·4H2O and Co(NO3)2·6H2O, even up to 1.0 mM of concentration under the same experimental conditions. From the above results, it can be concluded that the scavenging effects of the free ligand is significantly less when compared to that of its corresponding Ni(II) and Co(II) complexes, which is mainly due to the chelation of the organic ligand with the metal ions. Although the IC50 values observed in NO and HO scavenging assays show much lower scavenging ability than those of the reported nickel(II) benzhydrazone complexes15,58 and cobalt(II) hydrazone complexes,15,69 the IC50 values observed in DPPH and superoxide scavenging assays do demonstrate that both complexes have a comparable effective in arresting the formation DPPH and superoxide radicals. Therefore, the complexes have significant potential to be applied as scavengers to eliminate the radicals.
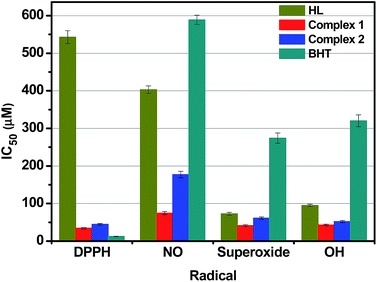 |
| | Fig. 10 The radical scavenging activity of the compounds. | |
In vitro cytotoxicity studies
Cytotoxicity is a common limitation in terms of the introduction of new compounds into the pharmaceutical industry. In general, macrocyclic complexes exhibit only slight cytotoxic effects and no dose–response effects are observed. In order to understand the in vitro cytotoxicities of the hydrazone and its metal complexes 1 and 2 experiments were carried out using HeLa and A549 human cancer cell lines, as well as the normal cell line NIH-3T3. HL and complexes 1 and 2 were dissolved in DMSO and blank samples containing same volume of DMSO were taken as controls to indentify the activity of solvent in this cytotoxicity experiment. Assays of compounds against cancerous cell lines HeLa and A549 were performed in the concentration range from 5 to 200 μM, and assays of compounds against NIH-3T3 cell line were performed in the concentration range from 50 to 800 μM. Cisplatin was used as a standard to assess the cytotoxicity of compounds (not shown in graph). Fig. 11a and b display the effect of the compounds on cell inhibition at different concentration after 24 h incubation on HeLa and A549 cancer cell lines, respectively. Upon increasing the concentration of compounds, an increase in the percentage of cell inhibition was observed. Table 5 shows the IC50 values obtained from CCK8 assay. From the results, it is inferred that both complexes exhibit significant activity against HeLa and A549 cell lines. However, they do not reach the potential of the well-known anticancer drug cisplatin (IC50 = 13.2 ± 0.6 μM for HeLa and IC50 = 17.2 ± 0.5 μM for A549).70,71 The in vitro cytotoxic activity test also shows that the IC50 value of the complex against NIH-3T3 (non-cancerous cells) was found to be above 450 μM, which confirms that the complexes are very specific for cancer cells compared with cisplatin. Among the ligand and complexes studies here, complex 1 exhibits greater cytotoxic effect than the other compounds for HeLa and A549 cancer cell lines. Further, complex 2 are more toxic towards A549 than HeLa. The cytotoxic activity of the complexes may be attributed to the extended planar structure induced by the π-π* conjugation resulting from the chelation of the metal ions with aroylhydrazone primary ligand and phen diimine co-ligand and the cationic nature of the complexes.18,27,72 It should be noted that significantly enhanced activities can be seen for the Ni(II) complex compared to free HL and the corresponding Co(II) complex, suggesting better synergistic effect between HL and nickel ion. The results for the in vitro cytotoxic activities further confirm the binding of the complexes to DNA, which consequently leads to cell death. The IC50 values of the Ni(II) complex are much lower than those previously reported for other nickel complex containing a coordinated hydrazone with planar napthyl group, which is responsible for the selectivity and the potential inhibition against the tumour cells.14 These findings suggest a strategy that increases the synergistic planarity and the cationic nature in mononuclear systems for the development of transition metal-based chemotherapeutic agents.
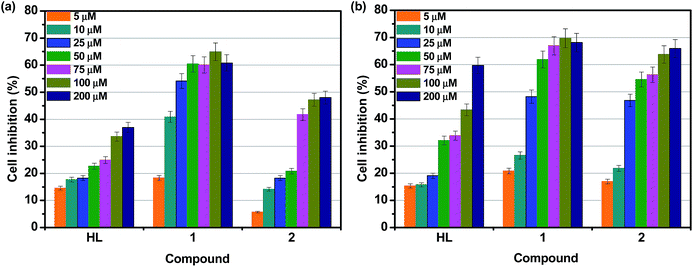 |
| | Fig. 11 Cytotoxicity of HL, 1 and 2 after 24 h incubation on (a) HeLa and (b) A549 cell lines. | |
Table 5 The IC50 values (μM) obtained for the hydrazone ligand and complexes against human cell lines
| Compounds |
IC50 (μM) |
| HeLa |
A549 |
NIH-3T3 |
| Data from ref. 58, 70 and 71. |
| HL |
170.28 ± 7.45 |
149.83 ± 4.79 |
453.21 ± 4.50 |
| Complex 1 |
34.93 ± 2.05 |
29.19 ± 1.10 |
664.32 ± 2.79 |
| Complex 2 |
149.91 ± 6.04 |
33.40 ± 2.45 |
560.45 ± 3.67 |
| Cisplatina |
13.2 ± 0.6 |
17.2 ± 0.5 |
240.5 ± 0.6 |
Conclusions
Two new divalent transition metal hydrazone complexes have been synthesized and characterized using various spectroscopic methods. Single-crystal X-ray diffraction study revealed that the hydrazone primary ligand and diimine co-ligand used in this study forms mononuclear Co(II) and Ni(II) complexes of distorted octahedral geometry with 1:2:1 stoichiometry among metal and ligands. The newly synthesized complexes were evaluated for DNA binding, protein binding and cytotoxicity studies. The DNA binding of the hydrazone ligand and metal complexes examined by absorption and fluorescence spectral techniques revealed an intercalative interaction between them and HS–DNA in the order of HL < Co(II) complex < Ni(II) complex. Binding of the metal complexes with BSA monitored by UV-vis spectroscopy revealed the presence of static quenching and the results of synchronous and three-dimensional fluorescence spectroscopic studies indicated that the complexes bound with BSA in both tyrosine and tryptophan residues and change the secondary structure of the BSA. From the above results we observed that Ni(II) complex exhibited higher DNA and BSA binding affinity that Co(II) complex, which could be ascribed to the smaller size of Ni(II) complex to fit well into the DNA/BSA grooves than the bulkier cation of Co(II) complex. Molecular docking technique has been used to evaluate and understand the interaction mode of the Ni(II) complex with DNA and BSA, revealed that hydrogen bonding between the phenolic hydroxyl group of Ni(II) complex and base pairs of DNA and amino acids of BSA, which play significant role in stabilizing of the complex interaction. Also, the antioxidant and cytotoxicity properties of the newly synthesized complexes showed that they can be investigated in detail as potential drugs. In vitro cytotoxicity of complexes showed that the Ni(II) hydrazone complex has moderate ability towards the inhibition of HeLa and A549 tumour cell growth without greatly affecting normal NIH-3T3 cells. The present study highlighted that synergistic planarity of aroylhydrazone primary ligand and diimine co-ligand, as well as the cationic nature of the complex may play an important role in biological activities, suggesting that the DNA/BSA-binding activities as well as the cytotoxicity can be tuned by changing the ligands and the geometry in these nickel and cobalt systems. This is a step toward enabling the rational design of novel metallodrugs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully thanked the financial support from the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the facilities support from Jiangsu Key Lab for the Chemistry & Utilization of Agricultural and Forest Biomass.
References
- C. R. Munteanu and K. Suntharalingam, Dalton Trans., 2015, 44, 13796–13808 RSC.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728 CrossRef CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- N. Raman and S. Sobha, Inorg. Chem. Commun., 2012, 17, 120–123 CrossRef CAS.
- S. Betanzos-Lara, C. Gómez-Ruiz, L. R. Barrón-Sosa, I. Gracia-Mora, M. Flores-Álamo and N. Barba-Behrens, J. Inorg. Biochem., 2012, 114, 82–93 CrossRef CAS PubMed.
- V. Oliveri and G. Vecchio, Eur. J. Med. Chem., 2016, 120, 252–274 CrossRef CAS PubMed.
- X. Totta, A. A. Papadopoulou, A. G. Hatzidimitriou, A. Papadopoulos and G. Psomas, J. Inorg. Biochem., 2015, 145, 79–93 CrossRef CAS PubMed.
- M. C. Heffern, N. Yamamoto, R. J. Holbrook, A. L. Eckermann and T. J. Meade, Curr. Opin. Chem. Biol., 2013, 17, 189–196 CrossRef CAS PubMed.
- G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. García, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef CAS.
- M. D. Hall, T. W. Failes, N. Yamamoto and T. W. Hambley, Dalton Trans., 2007, 3983–3990 RSC.
- P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2012, 41, 6842–6854 RSC.
- P. Krishnamoorthy, P. Sathyadevi, P. T. Muthiah and N. Dharmaraj, RSC Adv., 2012, 2, 12190–12203 RSC.
- J. Haribabu, K. Jeyalakshmi, Y. Arun, N. S. P. Bhuvanesh, P. T. Perumal and R. Karvembu, RSC Adv., 2015, 5, 46031–46049 RSC.
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Dalton Trans., 2012, 41, 4365–4377 RSC.
- P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2011, 40, 9690–9702 RSC.
- V. Rajendiran, M. Murali, E. Suresh, M. Palaniandavar, V. S. Periasamy and M. A. Akbarsha, Dalton Trans., 2008, 2157–2170 RSC.
- S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2011, 40, 3245–3256 RSC.
- A. Erdem and M. Ozsoz, Electroanalysis, 2002, 14, 965–974 CrossRef CAS.
- H. Xu, K.-C. Zheng, Y. Chen, Y.-Z. Li, L.-J. Lin, H. Li, P.-X. Zhang and L.-N. Ji, Dalton Trans., 2003, 2260–2268 RSC.
- M. Asadi, E. Safaei, B. Ranjbar and L. Hasani, New J. Chem., 2004, 28, 1227–1234 RSC.
- E. Ramachandran, D. S. Raja, J. L. Mike, T. R. Wagner, M. Zeller and K. Natarajan, RSC Adv., 2012, 2, 8515–8525 RSC.
- T. M. Sielecki, J. F. Boylan, P. A. Benfield and G. L. Trainor, J. Med. Chem., 2000, 43, 1–18 CrossRef CAS PubMed.
- T. Kosa, T. Maruyama and M. Otagiri, Pharm. Res., 1997, 14, 1607–1612 CrossRef CAS.
- G. Ayyannan, M. Mohanraj, G. Raja, N. Bhuvanesh, R. Nandhakumar and C. Jayabalakrishnan, Inorg. Chim. Acta, 2016, 453, 562–573 CrossRef CAS.
- Y. Li, Z. Yang, B. Song, H. Xia and Z. Wang, Inorg. Nano-Met. Chem., 2017, 47, 966–972 CrossRef CAS.
- Y. Li, Z. Yang, M. Zhou, J. He, X. Wang, Y. Wu and Z. Wang, J. Mol. Struct., 2017, 1130, 818–828 CrossRef CAS.
- Bruker SMART and SAINT, Syntheses, crystal structures and DNA-binding studies of Cu(II) and Zn(II) complexes bearing asymmetrical aroylhydrazone ligand, Bruker AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- G. M. Sheldrick, SADABS. Program for Empirical Absorption Correction of Area Detector, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- G. Felsenfeld and S. Z. Hirschman, J. Mol. Biol., 1965, 13, 407–427 CrossRef CAS PubMed.
- M. S. Blois, Nature, 1958, 181, 1199–1200 CrossRef CAS.
- L. C. Green, D. A. Wagner, J. Glogowski, P. L. Skipper, J. S. Wishnok and S. R. Tannenbaum, Anal. Biochem., 1982, 126, 131–138 CrossRef CAS PubMed.
- C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276–287 CrossRef CAS PubMed.
- T. Ak and İ. Gülçin, Chem.–Biol. Interact., 2008, 174, 27–37 CrossRef CAS PubMed.
- C. C. Winterbourn, Biochem. J., 1981, 198, 125–131 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- M. Anjomshoa, M. Torkzadeh-Mahani, J. Janczak, C. Rizzoli, M. Sahihi, F. Ataei and M. Dehkhodaei, Polyhedron, 2016, 119, 23–38 CrossRef CAS.
- P. Sathyadevi, P. Krishnamoorthy, M. Alagesan, K. Thanigaimani, P. Thomas Muthiah and N. Dharmaraj, Polyhedron, 2012, 31, 294–306 CrossRef CAS.
- S. Mondal, B. Pakhira, A. J. Blake, M. G. B. Drew and S. K. Chattopadhyay, Polyhedron, 2016, 117, 327–337 CrossRef CAS.
- K. Chichak, U. Jacquemard and N. R. Branda, Eur. J. Inorg. Chem., 2002, 2002, 357–368 CrossRef.
- P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Metallomics, 2012, 4, 498–511 RSC.
- J. K. Barton, A. Danishefsky and J. Goldberg, J. Am. Chem. Soc., 1984, 106, 2172–2176 CrossRef CAS.
- Z.-F. Chen, Y.-F. Shi, Y.-C. Liu, X. Hong, B. Geng, Y. Peng and H. Liang, Inorg. Chem., 2012, 51, 1998–2009 CrossRef CAS PubMed.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- J. Liu, T. Zhang, T. Lu, L. Qu, H. Zhou, Q. Zhang and L. Ji, J. Inorg. Biochem., 2002, 91, 269–276 CrossRef CAS PubMed.
- G. Han and P. Yang, J. Inorg. Biochem., 2002, 91, 230–236 CrossRef CAS PubMed.
- G.-Y. Bai, K.-Z. Wang, Z.-M. Duan and L.-H. Gao, J. Inorg. Biochem., 2004, 98, 1017–1022 CrossRef CAS PubMed.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376–3387 CrossRef CAS PubMed.
- P. Krishnamoorthy, P. Sathyadevi, K. Senthilkumar, P. T. Muthiah, R. Ramesh and N. Dharmaraj, Inorg. Chem. Commun., 2011, 14, 1318–1322 CrossRef CAS.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS PubMed.
- J. Ravichandran, P. Gurumoorthy, M. A. Imran Musthafa and A. Kalilur Rahiman, Spectrochim. Acta, Part A, 2014, 133, 785–793 CrossRef CAS PubMed.
- I.-u.-H. Bhat and S. Tabassum, Spectrochim. Acta, Part A, 2009, 72, 1026–1033 CrossRef PubMed.
- R. Raj Kumar and R. Ramesh, RSC Adv., 2015, 5, 101932–101948 RSC.
- S. Banerjee, S. Mondal, S. Sen, S. Das, D. L. Hughes, C. Rizzoli, C. Desplanches, C. Mandal and S. Mitra, Dalton Trans., 2009, 6849–6860 RSC.
- D. Senthil Raja, G. Paramaguru, N. S. P. Bhuvanesh, J. H. Reibenspies, R. Renganathan and K. Natarajan, Dalton Trans., 2011, 40, 4548–4559 RSC.
- H. Polet and J. Steinhardt, Biochemistry, 1968, 7, 1348–1356 CrossRef CAS PubMed.
- G. Zhang, Y. Ma, L. Wang, Y. Zhang and J. Zhou, Food Chem., 2012, 133, 264–270 CrossRef CAS PubMed.
- D. Senthil Raja, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2011, 46, 4584–4594 CrossRef PubMed.
- E. U. Akusoba and J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 92–95 RSC.
- E. A. Burstein, N. S. Vedenkina and M. N. Ivkova, Photochem. Photobiol., 1973, 18, 263–279 CrossRef CAS PubMed.
- D. Li, M. Zhu, C. Xu and B. Ji, Eur. J. Med. Chem., 2011, 46, 588–599 CrossRef CAS PubMed.
- T.-R. Li, Z.-Y. Yang, B.-D. Wang and D.-D. Qin, Eur. J. Med. Chem., 2008, 43, 1688–1695 CrossRef CAS PubMed.
- P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2012, 41, 4423–4436 RSC.
- S. Ramakrishnan, V. Rajendiran, M. Palaniandavar, V. S. Periasamy, B. S. Srinag, H. Krishnamurthy and M. A. Akbarsha, Inorg. Chem., 2009, 48, 1309–1322 CrossRef CAS PubMed.
- D. Senthil Raja, E. Ramachandran, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2013, 64, 148–159 CrossRef CAS PubMed.
- K.-B. Huang, Z.-F. Chen, Y.-C. Liu, M. Wang, J.-H. Wei, X.-L. Xie, J.-L. Zhang, K. Hu and H. Liang, Eur. J. Med. Chem., 2013, 70, 640–648 CrossRef CAS PubMed.
- F. A. Beckford, J. M. Shaloski, G. Leblanc, J. Thessing, L. C. Lewis-Alleyne, A. A. Holder, L. Li and N. P. Seeram, Dalton Trans., 2009, 10757–10764 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of HL (Fig. S1), FTIR (Fig. S2), ESI-mass of complexes (Fig. S3 and S4), TG curves of complexes (Fig. S5), cyclic voltammograms of complexes (Fig. S6), absorption spectra of binding of HL and Co(II) complex with BSA (Fig. S7 and S8), three-dimensional fluorescence projects (Fig. S9). CCDC 1518027 and 1518022. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10283f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Zhiwei Yanga,
Minya Zhoua and
Yun Lia
*ab,
Zhiwei Yanga,
Minya Zhoua and
Yun Lia