
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot, two-step synthesis and photophysical properties of 2-(5-phenylindol-3-yl)benzimidazole derivatives†
Liu Lyua,
Liu Caia,
Yun Wang b,
Jinfeng Huanga,
Xiangchao Zeng*a and
Porun Liu*b
b,
Jinfeng Huanga,
Xiangchao Zeng*a and
Porun Liu*b
aCollege of Chemistry and Materials Science, Jinan University, Guangzhou, Guangdong 510632, China. E-mail: xczeng@126.com
bCentre for Clean Environment and Energy, Griffith University, Gold Coast Campus, Queensland 4222, Australia. E-mail: p.liu@griffith.edu.au
First published on 23rd October 2017
Abstract
An efficient one-pot, two-step procedure has been successfully applied for the synthesis of a series of 2-(5-phenylindol-3-yl)benzimidazoles. The first step was cyclocondensation–oxidation of 5-bromoindole-3-aldehydes with o-phenylenediamines in 1,4-dioxane, which was promoted by activated carbon and used atmospheric air as a “green” oxidant. The resulting 2-(5-bromoindol-3-yl)benzimidazoles in the pot were in situ coupled with postadded phenylboronic acids, and catalyzed with PdCl2(dppf) in 1,4-dioxane-H2O to afford the desired compounds with satisfactorily high yields. The relationship between the synthesized compounds' absorption, and fluorescence spectra with molecular structures has been investigated with experimental data and theoretical calculations.
Introduction
Nitrogen-containing heterocycles are fundamental targets in the pharmaceutical industry as well as in materials science.1 Benzimidazoles and their derivatives have attracted much interest due to their outstanding physiological and biological properties.2 They have been successfully explored as drug-like scaffolds which can be found in a variety of natural products as well as a number of biologically active compounds.3 As electron-transporting and emission functional moieties, benzimidazole derivatives also serve in numerous optical applications, such as liquid crystals,4 fluorimetric chemosensors5/biosensors,6 organic luminescent materials like molecular switches,7 electron-transporters and light-emitting layers for organic light-emitting diodes (OLEDs),8 and for solar cells.9 As another group of heterocycles, indole derivatives display a broad spectrum of biological activity,10 such as anti-tumor,11 anti-inflammation,12 and anti-bacteria.13 They have been also reported to exhibit excellent photophysical properties that leads to numerous optical applications, for instance, fluorimetric chemosensors,14 light-emitting materials for OLEDs,15 and light absorbing material for dye-sensitized solar cells (DSSCs).16In particular, benzimidazoles and indoles are well established as “privileged scaffolds” given their electron accepting and donating properties that give rise to enhanced molecular photoluminescence. Some extended π-conjugated compounds with benzimidazoles and/or indole moieties exhibit synergic properties, for instance, 2-(6-(1H-benzo[d]imidazol-2-yl)naphthalen-2-yl)-1-dodecyl-1H-benzo[d]imidazole, a benzimidazole derivative bearing naphthalen block, can be applied as a colorimetric and ratiometric fluorescent sensor with high sensitivity and fast response for the detection of F− from inorganic origins and in the solid state.5a The compound, 2-aryl-6-(aryleneethynylene)-1H-indole derivative, displays intense fluorescence with high fluorescence quantum yields.17 Considering the unique fluorescent properties of benzimidazoles, indoles and their derivatives, it is envisaged that introduction of the electron donating and accepting structural moiety to an expanded π-conjugated structure could largely enhance fluorescence.18 Nevertheless, this has yet been fully understood. Hence, in the present work, we focus on the synthesis and photophysical characterization of indole–benzimidazole compounds (Fig. 1) with additional structural groups that could expand the conjugated system and modify their properties, in order to gain detailed understanding of the relationship between structure and optical property.
 | ||
| Fig. 1 One-pot, two-step synthetic procedure for the 2-(5-phenylindol-3-yl)benzimidazole compounds. | ||
A typical approach to synthesize benzimidazole moieties includes the condensation–dehydration of o-phenylenediamine with carboxylic acids, or their derivatives (nitriles, amidates, or esters).19 However, the reaction is generally carried out under harsh reaction conditions with strong acid as catalyst for extended reaction time. In another method, benzimidazoles have been synthesized by cyclocondensation of o-phenylenediamine with the corresponding aldehydes followed by oxidation. Various oxidative and catalytic reagents, such as TiCl4,20 H2O2/CAN,21 In(OTf)3,22 Sc(OTf)3,23 Yb(OTf)3,24 I2 in aqueous THF,25 BF3–OEt2,26 polyaniline-sulfate salt,27 and NH4OAc28 have been employed. Nevertheless, practically a large majority of these reactions suffer from multiple disadvantages, e.g., prolonged reaction time, high reaction temperature, tedious workup procedure and toxic oxidizing agents/catalysts. Therefore there is an urgent need to modify the synthesis methods with improved operational simplicity, milder conditions and more environmental-friendliness.
Air is an abundant, green substance, it has been used as oxidant for the syntheses of benzimidazoles in simple, mild, and efficient procedures.29 Activated carbon is environmentally-friendly, and can be separated easily after the reaction completion. It was reported that activated carbon could promote the reaction of o-phenylenediamine with benzaldehyde in xylene at 120 °C under oxygen atmosphere or air to produce 2-phenylbenzimidazole in moderate yields.30 We have also synthesized 2-(indol-3-yl)benzimidazoles in excellent yields by reaction of indole-3-aldehydes with o-phenylenediamines promoted by activated carbon using air as oxidant at relatively low reaction temperature.31 Yet this reported method involves a separate two-step processes, namely Suzuki coupling reaction of 5-bromoindole-3-aldehydes with phenylboronic acids in step one, followed by the cyclocondensation–oxidation of the isolated 5-phenylindole-3-aldehydes with o-phenylenediamines catalysed with activated carbon under air in step two. To further improve the synthesis reaction efficiency, a cost and time effective one-pot three-component reaction strategy is proposed as it adopts step-economical transformations to introduce structural complexity by allowing multiple bond-forming events to occur in one pot.32 Herein, for the first time, we report a facile yet efficient one-pot two-step synthesis of 2-(5-phenylindol-3-yl)benzimidazole compounds (Fig. 1).
Results and discussion
Synthesis parameter optimization
Our initial attempt to synthesize the 2-(5-phenylindol-3-yl)benzimidazole compounds using all materials including phenylboronic acid (PhB(OH)2), 5-bromoindole-3-aldehyde and o-phenylenediamine (o-PD), catalyst (Pd/C, or PdCl2(dppf)-activated carbon), alkali, hexadecyl trimethyl ammonium bromide (CTAB, as phase transfer catalyst), and the solvent (Diox–H2O) in one pot resulted to a trace amount of desired product after reaction at 80 °C or 90 °C for 12 h. This strongly indicated that an unfavourable reaction pathway dominated using a simple one-pot synthesis strategy.33 Contrastingly, when a one-pot two-step reaction was employed, i.e., a cyclocondensation–oxidation reaction of 5-bromoindole-3-aldehyde with o-PD, catalysed with activated carbon in 1,4-dioxane (Diox) to obtain 2-substituted benzimidazole, and a subsequent Suzuki coupling reaction in situ with PhB(OH)2 catalysed with Pd catalyst in alkali, CTAB, Diox and H2O under N2 (product 3, Fig. 1), the desired product was gained in a proper yield. In comparison, the synthesis route of initial Suzuki coupling reaction of bromoindolaldehyde with PhB(OH)2 followed by cyclocondensation–oxidation reaction suffered from a poor yield.In order to establish the optimal conditions for the one-pot, two step reaction, a model reaction among 5-bromo-1-ethylindole-3-aldehyde, o-PD and PhB(OH)2 was examined under various conditions. The catalyst was initially investigated as it largely determines the reaction pathways and the product yield. As shown in Table 1, the reaction with Pd/C catalysts without activated C only resulted to trace amount of target product (Table 1, entry 1). We then improved the reaction recipe using activated carbon and different Pd complexes as the catalysts for the cyclocondensation–oxidation and Suzuki coupling reactions respectively. The PdCl2(dppf)-activated carbon group was found to be the most effective catalyst in terms of reaction rate, selectivity and product yield (Table 1, entry 4). The reactions with other catalyst combinations, e.g., Pd(PPh3)Cl2-activated carbon or Pd(PPh3)4-activated carbon, resulted in much lower yields of 51% or 65% (Table 1, entry 2, 3), respectively. An increase in the molar ratio of added PdCl2(dppf) catalyst (>6%) did not further promote the product yield (Table 1, entries 4–6). With this optimal amount of PdCl2(dppf) catalyst, the amount of activated carbon has a paramount effect on product yields: 62% using 0.05 g of activated carbon with respect to 1 mmol of 1a, while extremely low yield be resulted without activated carbon (Table 1, entry 7, 8). Our experiment also confirmed that optimal reaction temperature for the cyclocondensation–oxidation reaction and Suzuki coupling reaction are 80 (Table 1, entries 5, 9 and 10) and 90 °C (Table 1, entries 5, 11) respectively.
|
|||||
|---|---|---|---|---|---|
| Entry | Pd catalystb (mol%) | Activated C (g) | T1c (°C) | T2d (°C) | Yielde/% |
| a Reaction conditions: 1a (1 mmol), o-phenylenediamine (1 mmol), activated carbon (X g), Diox (15 mL), 8 h under air (for entry 9 and entry 10, the time are 12 h and 5 h respectively); then K2CO3 (2 mmol) and CTAB (0.06 mmol) dissolved in H2O (5 mL), Pd catalyst (X mmol), and phenylboronic acid (1.1 mmol) were added, definite temperature for 5 h under N2.b Molar equivalent of the Pd catalysts used with respect to 1a.c Temperature for cyclocondensation–oxidation reaction.d Temperature for Suzuki coupling reaction.e Isolated yields of product by recrystallization. | |||||
| 1 | Pd/C (5%) | None | 80 | 90 | Trace |
| 2 | Pd(PPh3)2Cl2 (5%) | 0.1 | 80 | 90 | 51 |
| 3 | Pd(PPh3)4(5%) | 0.1 | 80 | 90 | 65 |
| 4 | PdCl2(dppf) (5%) | 0.1 | 80 | 90 | 77 |
| 5 | PdCl2(dppf) (6%) | 0.1 | 80 | 90 | 83 |
| 6 | PdCl2(dppf) (7%) | 0.1 | 80 | 90 | 83 |
| 7 | PdCl2(dppf) (6%) | 0.05 | 80 | 90 | 62 |
| 8 | PdCl2(dppf) (6%) | None | 80 | 90 | 0 |
| 9 | PdCl2(dppf) (6%) | 0.1 | 70 | 90 | 57 |
| 10 | PdCl2(dppf) (6%) | 0.1 | 90 | 90 | 70 |
| 11 | PdCl2(dppf) (6%) | 0.1 | 80 | 80 | 44 |

It has been demonstrated that optimal level of alkalis species plays a crucial role in promoting the transmetalation step and also facilitating the reductive elimination step in Suzuki coupling reaction.34 Our comparative experiments using different alkalis and amounts revealed that more than 87% yield can be achieved using 2.5 mmol K2CO3 as alkali (Table 2, entries 1–6). In addition, the effect of the reagent ratios on the reaction yield was also examined. Stoichiometric molar amount of PhB(OH)2, o-PD and 5-bromoindole-3-aldehyde afforded the product in yield of 70% (Table 2, entry 7). When the molar ratio of the reagents (acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diamine:aldehyde) was 1.1:1:1 or 1.2:1.1:1, the yield was improved to 87% or 91%, respectively (Table 2, entries 5, 8). Further increase in either acid or diamine ratio could not lead to higher yield (Table 2, entries 9, 10).
:diamine:aldehyde) was 1.1:1:1 or 1.2:1.1:1, the yield was improved to 87% or 91%, respectively (Table 2, entries 5, 8). Further increase in either acid or diamine ratio could not lead to higher yield (Table 2, entries 9, 10).
| Entry | Alkali (mmol) | Acid:amine:1ab |
Solvent (V/V, mL) | Timec/h | Yieldd/% |
|---|---|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), o-phenylenediamine (X mmol), activated carbon (0.1 g), Diox (X mL), 80 °C for 8 h under air; then phenylboronic acids (X mmol), PdCl2(dppf) (0.06 mmol), and the solution of K2CO3 (X mmol) and CTAB (0.06 mmol) in H2O (X mL) were added, 90 °C for the time indicated (see table) under N2.b Equiv. of phenylboronic acid, diamine and 1a.c Reaction time for Suzuki coupling reaction.d Isolated yields of product by recrystallization. | |||||
| 1 | K3PO4 (2) | 1.1:1:1 |
Diox–H2O (3/1, 20) | 5 | 74 |
| 2 | KOH (2) | 1.1:1:1 |
Diox–H2O (3/1, 20) | 5 | 78 |
| 3 | Na2CO3 (2) | 1.1:1:1 |
Diox–H2O (3/1, 20) | 5 | 65 |
| 4 | K2CO3 (2) | 1.1:1:1 |
Diox–H2O (3/1, 20) | 5 | 83 |
| 5 | K2CO3 (2.5) | 1.1:1:1 |
Diox–H2O (3/1, 20) | 5 | 87 |
| 6 | K2CO3 (3) | 1.1:1:1 |
Diox–H2O (3/1, 20) | 5 | 81 |
| 7 | K2CO3 (2.5) | 1:1:1 |
Diox–H2O (3/1, 20) | 10 | 70 |
| 8 | K2CO3 (2.5) | 1.2:1.1:1 |
Diox–H2O (3/1, 20) | 4 | 91 |
| 9 | K2CO3 (2.5) | 1.3:1.1:1 |
Diox–H2O (3/1, 20) | 4 | 91 |
| 10 | K2CO3 (2.5) | 1.2:1.2:1 |
Diox–H2O (3/1, 20) | 4 | 90 |
| 11 | K2CO3 (2.5) | 1.2:1.1:1 |
Diox–H2O (4/1, 20) | 7 | 80 |
| 12 | K2CO3 (2.5) | 1.2:1.1:1 |
Diox (20) | 12 | 21 |
Solvent exerts a significant effect on determining the reaction rate, isolated yield, and selectivity, thus different solvents have been examined in this work. The reagents dissolve in dry Diox easily, but dry Diox has higher viscosity and shown poor dissolving capability towards K2CO3 alkali. As a result, the poor mass transfer of the reagent and insufficient dissolved alkali lead to only 21% of product (Table 2, entry 12). Better product yields could be obtained in Diox–H2O than dry Diox with the same reagent ratios (Table 2, entries 8, 11, 12). This is due to the fact that a proper amount of water could improve the solubility of phenylboronic acids as well as inorganic alkalis in reaction mediums and meliorate Suzuki coupling reaction.35 In present work, when 20 mL of Diox–H2O (V/V = 3/1) was used as reaction medium, high yield of 91% could be comfortably achieved (Table 2, entry 8). Although other solvents, such as DMF, DMF–H2O and ethanol, have been also examined, the overall yields were unfavourable. The optimized condition for the one-pot, two-step synthesis of 3a was thus ascertained as 1 mmol of 1a and 1.1 equiv. of o-phenylenediamine, in the presence of 0.1 g activated carbon, reacting in 15 mL Diox at 80 °C for 8 hours under air for the cyclocondensation–oxidation, followed by Suzuki coupling reaction after in situ addition of 1.2 mmol of phenylboronic acid, K2CO3 (2.5 mmol), CTAB (0.06 mmol) in 5 mL H2O, 0.06 mmol of PdCl2(dppf) to the reaction mixtures and kept at 90 °C for 4 hours under N2.
Synthesis of 2-(5-phenylindol-3-yl)benzimidazole derivatives
To test the general scope and versatility of this procedure in the synthesis of a variety of 2-(5-bromoindol-3-yl)benzimidazoles, we examined a number of differently substituted o-phenylenediamines, 5-bromoindole-3-aldehydes and phenylboronic acids. It has been confirmed that moderate to high yields were obtained for the reactions of virtually all phenylboronic acids and phenylenediamines examined. As shown in Table 3, phenylboronic acids bearing either electron donating (Table 3, compounds 3b–d) or electron-withdrawing (Table 3, compounds 3e and 3f) substituents gave desired products, 2-(5-phenylindol-3-yl)benzimidazoles with good yields. These two-step reactions in one-pot were generally completed in ca. 12 hours, except for the cases of 4-chlorophenylboronic acid or 4-fluorophenylboronic acid used, where over 14 hours were required (Table 3, compounds 3e, 3f, 3s). All the reactions were completed as indicated by TLC. The title 2-(5-bromoindol-3-yl)benzimidazoles products were conveniently obtained by recrystallization.
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Time 1b/h | Time 2c/h | Yieldd/% |
| a Reaction conditions: 1 (1 mmol), o-phenylenediamine (1.1 mmol), activated carbon (0.1 g), 15 mL of Diox at 80 °C for the time indicated (see table, Time 1) under air; then phenylboronic acids (1.2 mmol), PdCl2(dppf) (0.06 mmol), and the solution of K2CO3 (2.5 mmol) and CTAB (0.06 mmol) in 5 mL of H2O were added, 90 °C for the time indicated (see table, Time 2) under N2.b Reaction time for cyclocondensation–oxidation.c Reaction time for Suzuki coupling reaction.d Isolated yields of product by recrystallization. | ||||||
| 3a | C2H5 | H | H | 8 | 4 | 91 |
| 3b | C2H5 | H | CH3 | 8 | 4 | 90 |
| 3c | C2H5 | H | OCH3 | 8 | 4 | 92 |
| 3d | C2H5 | H | N(Ph)2 | 8 | 4 | 86 |
| 3e | C2H5 | H | Cl | 8 | 7 | 80 |
| 3f | C2H5 | H | F | 8 | 7 | 82 |
| 3g | C2H5 | CH3 | H | 7 | 4 | 87 |
| 3h | C2H5 | CH3 | OCH3 | 7 | 4 | 92 |
| 3i | n-Bu | H | H | 8 | 4 | 85 |
| 3j | n-Bu | H | CH3 | 8 | 4 | 86 |
| 3k | n-Bu | H | OCH3 | 8 | 4 | 90 |
| 3m | n-Bu | CH3 | H | 7 | 4 | 83 |
| 3n | n-Bu | CH3 | OCH3 | 7 | 4 | 88 |
| 3o | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2 CHCH2 |
H | H | 8 | 4 | 81 |
| 3p | CH2CHCH2 |
H | CH3 | 8 | 4 | 83 |
| 3q | CH2CHCH2 |
H | OCH3 | 8 | 4 | 85 |
| 3r | CH2CHCH2 |
CH3 | H | 7 | 4 | 81 |
| 3s | CH2CHCH2 |
CH3 | Cl | 7 | 7 | 76 |
| 3t | PhCH2 | H | H | 8 | 5 | 85 |
| 3u | PhCH2 | H | CH3 | 8 | 4 | 84 |
| 3v | PhCH2 | H | OCH3 | 8 | 4 | 87 |
| 3w | PhCH2 | CH3 | H | 7.5 | 5 | 80 |
| 3x | PhCH2 | CH3 | CH3 | 7.5 | 5 | 83 |
| 3y | PhCH2 | CH3 | OCH3 | 7.5 | 5 | 85 |

The procedure that we employed to synthesize the target 2-(5-phenylindol-3-yl)benzimidazole compounds 3a–y is envisaged as a straightforward sequence of condensing of 5-bromoindole-3-aldehydes (compounds 1) with o-phenylenediamines, followed by cyclization–oxidation and Suzuki coupling reaction, the generation of intermediate 2 is one of the determinant factors (Fig. 1). The electron donating and withdrawing nature of substituents R2 on benzene ring of o-phenylenediamines is important to the cyclocondensation–oxidation reaction. Electron donating action of electron-rich indole fragment enhances the electron density of the electrophilic aldehyde carbon atom center, which is disadvantageous to nucleophilic addition of amine nitrogen in the initial stage of cyclocondensation of compounds 1 with o-phenylenediamines. Thus, compounds 1 tend to react with more nucleophilic aromatic diamines with electron donating substituents, e.g. 4-methyl-o-phenylenediamine, rather than those with electron withdrawing substituents or low electron density. Indeed, reaction using 4-nitro-o-phenylenediamine or pyridine-2,3-diamine or pyridine-3,4-diamine as diamine failed to produce intermediate benzimidazole compounds despite of extended duration (12 hours) or elevated reaction temperature (100 °C).
Photophysical properties
The organic compounds 3 synthesized in this work possess a π-conjugated backbone with indole as electron donor moieties and benzimidazole as electron acceptor moieties and therefore render typical photophysical properties. Their photophysical properties in ethanol solvent (1.0 × 10−5 mol L−1) are analyzed with UV-vis absorption and fluorescence spectrometers at room temperature (data summarized in Table 4).| Compound | UV-vis absorption | Emission λem/nm | Stoke's shift/cm−1 | ||
|---|---|---|---|---|---|
| λmax/nm | εa (M−1 cm−1) | ||||
| a Coefficients of the absorption band around 262 nm except 3d that at 311 nm. | |||||
| 3a | 260 | 327 | 68618 |
375 | 3914 |
| 3b | 262 | 325 | 66480 |
375 | 4103 |
| 3c | 264 | 325 | 65749 |
378 | 4314 |
| 3d | 311 | 347 | 49184 |
398 | 3693 |
| 3e | 265 | 325 | 43362 |
379 | 4384 |
| 3f | 258 | 325 | 55537 |
374 | 4031 |
| 3g | 261 | 328 | 48613 |
378 | 4033 |
| 3h | 262 | 324 | 43828 |
380 | 4548 |
| 3i | 260 | 328 | 51330 |
375 | 3821 |
| 3j | 261 | 328 | 55798 |
375 | 3821 |
| 3k | 264 | 328 | 46804 |
378 | 4033 |
| 3m | 260 | 329 | 51084 |
378 | 3940 |
| 3n | 265 | 329 | 47117 |
380 | 4079 |
| 3o | 260 | 324 | 79833 |
374 | 4126 |
| 3p | 261 | 325 | 80011 |
374 | 4031 |
| 3q | 266 | 326 | 53301 |
380 | 4359 |
| 3r | 264 | 328 | 50891 |
377 | 3963 |
| 3s | 265 | 329 | 70885 |
376 | 3799 |
| 3t | 260 | 325 | 56953 |
375 | 4102 |
| 3u | 262 | 324 | 51684 |
374 | 4126 |
| 3v | 264 | 325 | 36488 |
377 | 4244 |
| 3w | 261 | 328 | 43374 |
377 | 3963 |
| 3x | 261 | 326 | 39793 |
377 | 4150 |
| 3y | 264 | 329 | 36134 |
379 | 4010 |
The absorption spectra of most compounds 3 (except 3d) in ethanol solution (Table 4) evidently show two bands, the more intensive one at about 262 nm can be attributed to π–π* transition, and another less intensive one at about 327 nm is assigned to be π–π* transition.36 Moreover, absorption spectra of the compounds (e.g. 3b) using other solvents (dichrloroethane and n-hexane) did not show noticeable shift in band absorption maxima (Fig. 2a, b and Table S1†), indicating small dipole moments and also comparable ground-state electronic structures. Generally, electron donating moieties will increase the molar absorption coefficient whereas moieties that capable of enhancing π-conjugation will shift the band towards low energy direction. However, despite of the various moieties investigated in this work, it is observed that their substitution on the mainly three location of the molecular structure has minimal effect on the absorption maxima of most of the compounds 3 (Table 4). This strongly indicates the moieties are not able to significantly affect the π–π* transition or to extend the conjugation length to alter the electronic properties of the compounds. The only exception is compound 3d, with triphenylamine moiety connects to the position 5 of indole ring, demonstrating pronounced red shifts of 50 nm for larger-energy band and 18 nm for lowest-energy band (see Fig. S1† for fitted curve), compared to its counterpart compound 3a. It is well recognized that the triphenylamine is a strong electron donating moiety, as such, the intensive red shift of lowest-energy absorption band for 3d is rationally attributed to the intramolecular charge transfer (ICT) from triphenylamine donor towards indole–benzimidazole acceptor, while the extended conjugation is expected to alter the molecular electronic band structure and originate the red shift of higher-energy absorption band (see theoretical calculation below). The ICT state is generated upon π–π* transition and the charge-transfer characteristics can be confirmed by the sensitivity of the emission spectrum towards the solvent polarity (bathochromic effect), as shown in Fig. 2c, d and Table S1.† Notably, emission spectrum in ethanol solvent show blue shift compared to dichloroethane. This is probably due to the hypsochromic effect arise from hydrogen bonding at the terminal amino nitrogen atoms.37
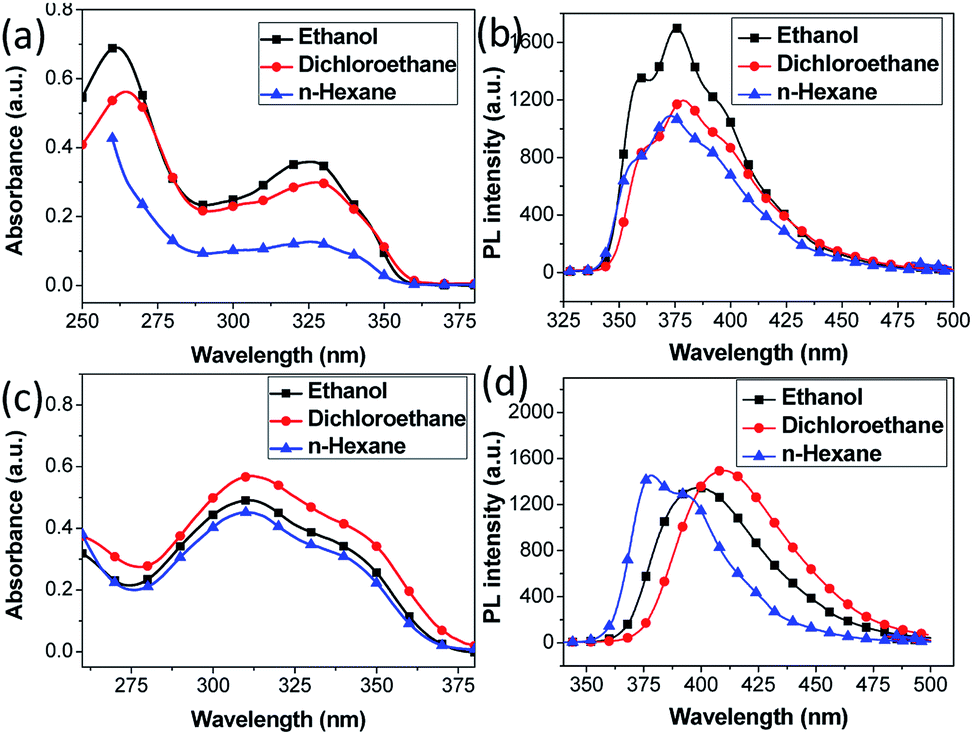 | ||
| Fig. 2 UV-visible and fluorescence spectra of (a, b) 3b and (c, d) 3d in ethanol, dichloroethane and n-hexane solutions. | ||
Fig. 3b and S2† depict the fluorescence emission spectra of selective samples of compounds 3 in ethanol solution at room temperature with an excitation at ∼330 nm (3d at 350 nm). The Stoke shifts measured are within 4000–5000 cm−1 range except 3d (Tables 4 and S2†). Evidently, the electron withdrawing and donating natures of the moieties exert an impact on the fluorescence intensities, although their influence on the emission wave length maxima is confirmed to be minimal. Also, the capability of the moiety to extend the π-conjugation in the molecule plays an important role in the fluorescence intensity. For example, for compounds with same R2 and R3 (e.g., compounds 3p vs. 3j, 3p vs. 3u, Fig. S2†), fluorescence intensity is larger when an allyl group (3p) connects to the nitrogen on indole ring as substituent. The fluorescence intensity of 3u is weaker compared to that of 3p. This may be resulted from the fact that the benzyl group and indole ring in 3u are not perfectly co-planar, giving rise to a disruptive effect on the σ–π hyper-conjugation of the indole ring–CH2–phenyl ring. Moreover, electron donating moiety connected to electron-acceptor benzimidazole, for example, methyl substituent group, is unfavorable to the charge-transfer capability for indole–benzimidazole derivatives, leading to weaker fluorescence intensities (compounds 3a vs. 3g, 3c vs. 3h, 3u vs. 3x, Fig. 3 and S2†).
 | ||
| Fig. 3 UV-visible and fluorescence spectra of compounds 3a–h in ethanol solution. | ||
As for the groups connected to phenyl of position 5 of the indole, enhanced fluorescence can be achieved by employing electron donating substituents such as methyl and methoxy groups (e.g., compounds 3a vs. 3b; 3c vs. 3f, Fig. 3). Furthermore, the more intensive photoluminescence of 3u compared with 3v can be rationalized by the σ–π hyper-conjugation of the indole ring–CH3 in 3u (enhancing the photoluminescence) and the negligible p–π conjugation of the partly protonated methoxy moiety in 3v in ethanol solution (Fig. S2†). Likewise, substitution of H with F or Cl atom on this location will expectedly decrease the fluorescence intensity given the strong electron-withdrawing effect of F and Cl atoms (3a vs. 3f, 3a vs. 3e, 3r vs. 3s, Fig. 3 and S2†). Consistent with the absorption spectra data set, 3d demonstrates distinctive emission peak (red shift of about 20 nm) compared to the rest of the compounds (Table 4 and Fig. 3). The photoluminescence quantum yield of selective sample compounds have been measured to be about 35% at excitation wavelength of 330 nm (Table S2†).
To gain insight into the molecular structure and its relationship with the spectroscopic properties, the geometric structure and electron distribution of some of the samples, i.e., 3a, 3b, 3d and 3e, have been optimized using the density functional theory (DFT) with the B3LYP/6-31G* level (see Experimental section for details). As the optimized geometric structures of the four compounds shown in Fig. 4, distorted conformations are adapted in all cases: atoms in the core rings of benzimidazole and indole moieties are mostly located in the same plane with a dihedral angle with respect to the plane formed by the benzyl group connected on 5 position of indole moiety. The dihedral angles for 3a, 3b and 3e (36.9°, 36.7°, 36.8°) are higher than that for 3d (34.9°), indicating a more disruptive effect on π-conjugation for these three compounds. Also, consistent with other studies, three benzene rings in the triphenylamine moiety of 3d are nonplanar,38 showing dihedral angles of about 41°. It is expected that the molecular geometries and moiety electron donating and accepting properties will exert significant impact on the electronic structure. Thus, the TD-DFT calculation of the singlet–singlet ground states with the optimized molecular structures was performed. As the calculated Frontier molecular orbital shown in Fig. 4, for compounds 3a, 3b and 3e the density of the highest occupied molecular orbital (HOMO) is mainly centered on the indole–benzimidazole segment. In comparison to 3a, the benzene ring on 5 position of indole moiety and the connected group on its para position (i.e., methyl in 3b or Cl in 3e) have insignificant influence on the density of molecular orbital. In strong contrast, the HOMO of 3d is localized in the triphenylamine moiety, similar to the reported results of triphenylamine–benzimidazole compounds.38a,38b,39 As for the lowest unoccupized molecular orbital (LUMO), for compounds 3a, 3b and 3e the density is spread over the whole molecule with more moderate dependence on the electron withdrawing/donating ability of substituent on para position of benzene ring: LUMO of 3e shows more distribution on electron withdrawing Cl end and that of 3b is dispersed more over the benzimidazole moiety. For 3d, the density of LUMO is mainly located in the indole–benzimidazole segment. In comparison of the HOMO and LUMO for all four compounds, it is evident that a more noticeable ICT feature is shown for 3d: a more density relocation of orbitals shifted from electron donating triphenylamine moiety to electron-accepting indole–benzimidazole moiety. In good agreement with the measured photophysical properties, the calculated results further confirm much obvious ICT character for transition of 3d molecules compared to others.
 | ||
| Fig. 4 Optimized ground state geometric conformation, molecular Frontier orbital electron distribution for the HOMOs and LUMOs of 3a, 3b, 3d and 3e. Atoms in grey, light blue, white and green represent carbon, nitrogen, hydrogen and chlorine respectively. | ||
The singlet–singlet excited state calculations of optimized molecular structures have also been performed to investigate the absorption bands measured in the experiments. As TD-DFT absorption spectra shown in Fig. 5, the simulated results (the red one) generally demonstrate excellent agreement with the experimental data (the black one). For 3a, 3b and 3e, the observed energy absorption bands (around 325 nm and 262 nm) are associated to the predicted π–π* transition from HOMO orbitals to LUMO and LUMO + 1 orbitals respectively (Fig. 5). Distinctively, band in 3d molecule with electron donating triphenylamine moiety centred at 347 nm can be assigned to be the π–π* transition with significant ICT character from HOMO orbital to LUMO orbital.40 The other higher energy absorption bands that involve transitions from HOMO-2 and HOMO-1 to LUMO orbitals can be ascribed to the locally excited π–π* transitions.40
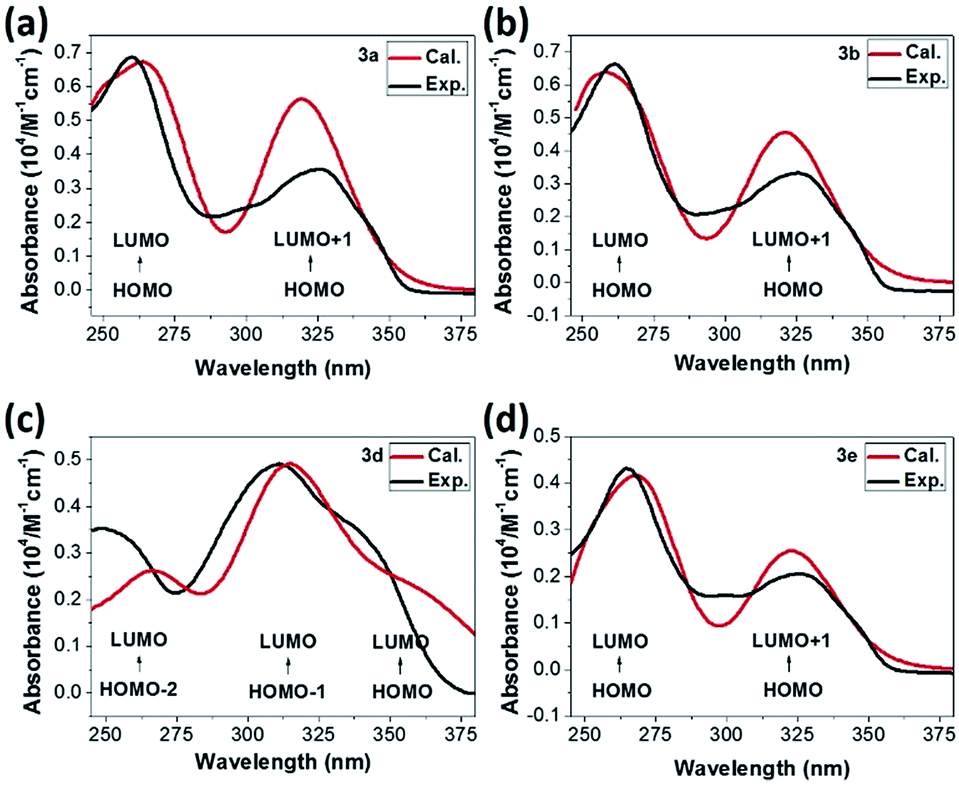 | ||
| Fig. 5 UV-vis spectrum of (a) 3a, (b) 3b, (c) 3d and (d) 3e. The black lines present absorption bands in ethanol while red ones shows calculated excited energies under vacuum, respectively. | ||
π-Conjugated donor–acceptor organic molecules with unique electronic and photonic properties have demonstrated various potentials in numerous applications, such as organic light emitting diodes (OLEDs), organic field effect transistors (OFETs), organic photovoltaics (OPVs). The deep-blue fluorescence of 3d sample demonstrates that the nondoped molecule may be used as a single emitting material in OLEDs without using complex host-dopant codeposition process. Also, the strong fluorescent molecules can be used as colorimetric and ratiometric fluorescent sensor for direct visual detection of cations, anions or gas in solution.
Conclusions
In summary, we have demonstrated a simple yet efficient one-pot two-step synthesis of 2-(5-phenylindol-3-yl)benzimidazoles with high yield under mild conditions. The procedure comprised an activated carbon catalysed cyclocondensation–oxidation of 5-bromoindole-3-aldehydes with o-phenylenediamines using air oxidant to yield 2-(5-bromoindol-3-yl)benzimoidazoles, followed by an in situ Suzuki coupling reaction after addition of phenylboronic acids without isolating the intermediates. The conjugated 2-(5-phenylindol-3-yl)benzimidazole compounds exhibit distinctive fluorescence. The photophysical properties of individual compounds show dependence on the electron withdrawing/donating capability of the substituents and also stabilization effect of π-conjugation. This is further confirmed with density functional theory computations. The demonstrated synthesis method can be extended to synthesize novel conjugated D–A type fluorescent molecules for various applications.Experimental section
Synthesis
All the chemicals used in the work were purchased from Shanghai D&R Finechem Co. or Sigma Aldrich. The mixture of 5-bromoindole-3-formaldehyde (1.0 mmol), o-phenylenediamine (1.1 mmol), 0.1 g activated carbon and 15 mL of 1,4-dioxane in a three-neck flask was stirred at 80 °C in air atmosphere for the appropriate time (see Table 3, Time 1) until the aldehyde was consumed (monitored by TLC). Subsequently, nitrogen gas was bubbled into the resulting mixture. Fifteen minutes later, henylboronic acids (1.2 mmol), PdCl2(dppf) (0.06 mmol), and the solution of K2CO3 (2.5 mmol) and CTAB (0.06 mmol) in 5 mL of H2O were added to the above mixture through a constant-pressure dropping funnel, and stirred at 90 °C in nitrogen atmosphere for the time indicated (see Table 3, Time 2) until completion of the reaction (monitored by TLC). The reacted mixture was cooled to room temperature and filtered; then 25 mL of EtOAc/H2O (2:1, V/V) was added to the filtrate and separated, the water layer was extracted with EtOAc (3 × 15 mL). The joined organic layer was dried with anhydrous MgSO4 overnight, and filtered, the filtrate was evaporated under reduced pressure; the residue was purified by recrystallization from EtOAc to obtain the desired benzimidazoles.
CH2), 5.00 (d, J = 5.1 Hz, 2H, CH2); 13C NMR (75 MHz, DMSO) δ: 149.31, 142.09, 136.50, 134.36, 133.93, 130.35, 129.34, 127.52, 127.08, 126.52, 122.33, 121.83, 120.11, 118.04, 111.60, 106.93, 49.02; IR (KBr) v: 3054, 2955, 1622, 1574, 1505, 1448, 1080, 928, 742 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C24H20N3 350.1652, found 350.1652.CH2), 4.98 (s, 2H, CH2), 2.38 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 149.46, 144.70, 139.21, 136.37, 136.19, 134.67, 134.39, 133.80, 130.15, 129.94, 127.31, 126.60, 122.15, 121.97, 121.45, 119.85, 118.50, 117.98, 111.49, 110.87, 107.10, 48.99, 21.15; IR (KBr) v: 3050, 2918, 1622, 1573, 1509, 1447, 1085, 924, 740 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C25H22N3 364.1808, found 364.1806.CH2), 4.97 (d, J = 5.3 Hz, 2H, CH2), 3.82 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6) δ: 158.80, 149.46, 136.16, 134.53, 134.38, 133.64, 130.13, 128.50, 126.58, 122.06, 121.73, 119.55, 117.97, 114.79, 111.46, 106.97, 55.63, 48.99; IR (KBr) v: 3052, 2954, 1619, 1573, 1514, 1447, 1111, 929, 742 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C25H22N3O 380.1757, found 380.1756.CH2), 4.99 (d, J = 4.8 Hz, 2H, CH2), 2.44 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 149.04, 142.08, 136.47, 134.36, 133.81, 130.85, 130.06, 129.34, 127.48, 127.06, 126.52, 123.12, 122.26, 120.14, 117.98, 111.54, 107.16, 48.99, 21.80; IR (KBr) v: 3033, 2974, 1623, 1576, 1508, 1446, 1082, 931, 756 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C25H22N3 364.1808, found 364.1811.CH2), 4.98 (d, J = 4.9 Hz, 2H, CH2), 2.44 (s, 3H, CH3); 13C NMR (75 MHz, DMSO) δ: 148.98, 140.88, 136.62, 134.33, 132.38, 131.90, 130.86, 130.21, 129.28, 129.13, 126.54, 123.14, 122.09, 120.13, 117.99, 111.67, 107.27, 48.98, 21.80; IR (KBr) v: 3039, 2919, 1626, 1579, 1508, 1445, 1091, 931, 798 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C25H21ClN3 398.1419, found 398.1415.Characterization
Melting point measurements were performed on an X-6 digital melting point apparatus without calibration. 1H and 13C NMR spectra were recorded on Varian Inova 300 at 300 and 75 MHz instruments respectively. For NMR spectra, d6-DMSO was used as solvents using TMS as an internal standard, chemical shifts are expressed in δ units and 1H–1H coupling constants in Hz. Infrared (IR) samples were prepared as KBr pellets, and spectra were obtained in the 400–4000 cm−1 range using a Bruker Eqinox55 FTIR spectrometer. HRMS (ESI) were recorded on a Bruker micro TOF-QII mass spectrometer or Agilent 6210 LC/MSD TOF mass spectrometer. UV-vis spectra were recorded by using a PRERSEE TU-1810 spectrometer in double-beam mode at room temperature. Quartz cells of 1 cm path length were used throughout and the absorbance were recorded with a spectral resolution of 0.1 nm. Fluorescence spectra were recorded with a Hitachi F-4500 fluorescence spectrometer at room temperature in ethanol or other solvents; and the slit width was 5 nm for both excitation and emission. Excitation maxima were determined from excitation spectra covering the range of 200–500 nm. Emission spectra were recorded from 300 to 600 nm. The quantum yield of photoluminescence (ΦS) was measured using quinine sulfate (QS) as standard (in 0.1 mol L−1 H2SO4, ΦQS = 54% at 330 nm) using the equation:
IS and IQS refer to the integrated luminescence intensity of the samples and QS using excitation wavelength of 330 nm. A and η are the UV absorbance at 330 nm (adjusted to be <0.1 by changing the concentration) and refractive index of the ethanol solvent, respectively.
Theoretical calculation
DFT calculations were performed by using the B3LYP exchange correlation functional in Gaussian 16 program package,41 version A01. A general basis set 6-31G* was used. Frequency analysis was employed to ensure the optimized structure has located at the minimum point. Ten excited states were calculated in TD-DFT computation. Three Frontier orbitals were drawn by VMD and POV-Ray software.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We are grateful to the National University Student Innovation Program (Grant No. 201610559039) for supporting this work. Research supports from JNU High Performance Computation Platform for providing quantum calculation resource is also acknowledged.Notes and references
- L. Q. Tran, J. H. Li and L. Neuville, J. Org. Chem., 2015, 80, 6102–6108 CrossRef CAS PubMed.
- (a) R. Kardanpour, S. Tangestaninejad, V. Mirkhani, M. Moghadam, I. Mohammadpoor-Baltork and F. Zadehahmadi, J. Solid State Chem., 2015, 226, 262–272 CrossRef CAS; (b) D. Cressier, C. Prouillac, P. Hernandez, C. Amourette, M. Diserbo, C. Lion and G. Rima, Bioorg. Med. Chem., 2009, 17, 5275–5284 CrossRef CAS PubMed.
- (a) P. Ghosh and R. Subba, Tetrahedron Lett., 2015, 56, 2691–2694 CrossRef CAS; (b) M. Q. Zhou, Y. J. Eun, I. A. Guzei and D. B. Weibel, ACS Med. Chem. Lett., 2013, 4, 880–885 CrossRef CAS PubMed; (c) T. M. A. Eldebss, A. M. Farag, M. M. Abdulla and R. K. Arafa, Mini-Rev. Med. Chem., 2016, 16, 67–83 CrossRef CAS PubMed; (d) A. Ravindernath, M. S. Reddy and V. Sunil, Med. Chem. Res., 2014, 23, 759–764 CrossRef CAS; (e) H. Z. Zhang, J. M. Lin, S. Rasheed and C. H. Zhou, Sci. China: Chem., 2014, 57, 807–822 CrossRef CAS; (f) M. Bouchouit, M. E. Said, M. K. Ali, S. Bouacida, H. Merazig, N. K. Chaouche, A. Chibani, B. Zouchoune, A. Belfaitah and A. Bouraiou, Polyhedron, 2016, 119, 248–259 CrossRef CAS; (g) F. C. Torres, M. E. Garcia-Rubino, C. Lozano-Lopez, D. F. Kawano, V. L. Eifler-Lima, G. L. von Poser and J. M. Campos, Curr. Med. Chem., 2015, 22, 1312–1323 CrossRef CAS PubMed; (h) Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208–6236 CrossRef CAS PubMed; (i) P. Singla, V. Luxami and K. Paul, Bioorg. Med. Chem. Lett., 2016, 26, 518–523 CrossRef CAS PubMed.
- J. F. Xiong, S. H. Luo, J. P. Huo, J. Y. Liu, S. X. Chen and Z. Y. Wang, J. Org. Chem., 2014, 79, 8366–8373 CrossRef CAS PubMed.
- (a) Y.-C. Wu, J.-Y. You, K. Jiang, J.-C. Xie, S.-L. Li, D. Cao and Z.-Y. Wang, Dyes Pigm., 2017, 140, 47–55 CrossRef CAS; (b) H. Goh, Y. G. Ko, T. K. Nam, A. Singh, N. Singh and D. O. Jang, Tetrahedron Lett., 2016, 57, 4435–4439 CrossRef CAS; (c) K. Aich, S. Das, S. Goswami, C. K. Quah, D. Sarkar, T. K. Mondal and H.-K. Fun, New J. Chem., 2016, 40, 6907–6915 RSC.
- (a) R. M. F. Batista, R. C. M. Ferreira, M. M. M. Raposo and S. P. G. Costa, Tetrahedron, 2012, 68, 7322–7330 CrossRef CAS; (b) J. F. Xiong, J. X. Li, G. Z. Mo, J. P. Huo, J. Y. Liu, X. Y. Chen and Z. Y. Wang, J. Org. Chem., 2014, 79, 11619–11630 CrossRef CAS PubMed; (c) B. Chetia, P. J. Goutam, F. A. S. Chipem and P. K. Iyer, J. Mol. Struct., 2013, 1042, 32–36 CrossRef CAS.
- (a) Y. He, G. Wang and M. X. Wang, J. Mol. Struct., 2016, 1116, 109–115 CrossRef CAS; (b) L. Z. Chen, G. Wang and X. C. Zhao, J. Lumin., 2011, 131, 1617–1620 CrossRef CAS.
- (a) S. L. Gong, Y. H. Chen, C. L. Yang, C. Zhong, J. G. Qin and D. G. Ma, Adv. Mater., 2010, 22, 5370–5373 CrossRef CAS PubMed; (b) J. Pina and J. S. S. de Melo, J. Org. Chem., 2013, 78, 11389–11395 CrossRef CAS PubMed.
- C. W. Lee, O. Y. Kim and J. Y. Lee, J. Ind. Eng. Chem., 2014, 20, 1198–1208 CrossRef CAS.
- T. Guo, Y. Liu, Y. H. Zhao, P. K. Zhang, S. L. Han and H. M. Liu, Tetrahedron Lett., 2016, 57, 4629–4632 CrossRef CAS.
- (a) J. Yan, J. H. Hu, B. J. An, L. Huang and X. S. Li, Eur. J. Med. Chem., 2017, 125, 663–675 CrossRef CAS PubMed; (b) M. P. Fortes, P. B. N. da Silva, T. G. da Silva, T. S. Kaufman, G. C. G. Militao and C. C. Silveira, Eur. J. Med. Chem., 2016, 118, 21–26 CrossRef CAS PubMed; (c) K. Mahal, B. Biersack, S. Schruefer, M. Resch, R. Ficner, R. Schobert and T. Mueller, Eur. J. Med. Chem., 2016, 118, 9–20 CrossRef CAS PubMed.
- P. F. Lamie, W. A. M. Ali, V. Bazgier and L. Rarova, Eur. J. Med. Chem., 2016, 123, 803–813 CrossRef CAS PubMed.
- C. Zhang, Y. Qu and B. X. Niu, Bioorg. Med. Chem., 2016, 24, 5781–5786 CrossRef CAS PubMed.
- (a) X. M. Liu, Y. P. Li, Y. H. Zhang, Q. Zhao, W. C. Song, J. Xu and X. H. Bu, Talanta, 2015, 131, 597–602 CrossRef CAS PubMed; (b) D. Jeyanthi, M. Iniya, K. Krishnaveni and D. Chellappa, Spectrochim. Acta, Part A, 2015, 136, 1269–1274 CrossRef CAS PubMed; (c) S. H. Mashraqui, S. S. Ghorpade, S. Tripathi and S. Britto, Tetrahedron Lett., 2012, 53, 765–768 CrossRef CAS; (d) S. O. Tumay, E. Okutan, I. F. Sengul, E. Ozcan, H. Kandemir, T. Doruk, M. Cetin and B. Cosut, Polyhedron, 2016, 117, 161–171 CrossRef CAS.
- (a) Q. C. Dong, H. Lian, Z. X. Gao, Z. L. Guo, N. Xiang, Z. Zhong, H. G. Guo, J. H. Huang and W. Y. Wong, Dyes Pigm., 2017, 137, 84–90 CrossRef CAS; (b) N. Xiang, Z. X. Gao, G. J. Tian, Y. Chen, W. Q. Lang, J. H. Huang, Q. C. Dong, W. Y. Wong and J. H. Su, Dyes Pigm., 2017, 137, 36–42 CrossRef CAS; (c) Y. Y. Shan, Q. Q. Wu, N. Sun, Y. H. Sun, D. X. Cao, Z. Q. Liu, R. F. Guan, Y. X. Xu and X. Y. Yu, Mater. Chem. Phys., 2017, 186, 295–300 CrossRef CAS.
- D. D. Babu, R. Su, A. El-Shafei and A. V. Adhikari, Electrochim. Acta, 2016, 198, 10–21 CrossRef CAS.
- M. Flores-Jarillo, A. Alvarez-Hernandez, R. A. Vazquez-Garcia, E. Arias, I. Moggio and J. R. Torres, Dyes Pigm., 2016, 133, 41–50 CrossRef CAS.
- R. Selvam and K. Subramanian, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 997–1007 CrossRef CAS.
- (a) J. Azizian, P. Torabi and J. Noei, Tetrahedron Lett., 2016, 57, 185–188 CrossRef CAS; (b) M. S. Mayo, X. Q. Yu, X. Y. Zhou, X. J. Feng, Y. Yamamoto and M. Bao, Org. Lett., 2014, 16, 764–767 CrossRef CAS PubMed; (c) Y. Y. Li, Y. H. Zhou, Y. F. Guo, W. G. Sun, Z. Y. Li and D. Q. Shi, Chin. J. Org. Chem., 2006, 26, 1097–1099 CAS.
- L. Zamani, B. B. F. Mirjalili, K. Zomorodian, M. Namazian, S. Khabnadideh and E. F. Mirzaei, Farmacia, 2014, 62, 459–474 CAS.
- H. Naeimi and N. Alishahi, J. Chin. Chem. Soc., 2012, 59, 1001–1005 CrossRef CAS.
- D. Kumar, A. Kumar, M. M. Qadri, M. I. Ansari, A. Gautam and A. K. Chakraborti, RSC Adv., 2015, 5, 2920–2927 RSC.
- L. Y. Fan, L. L. Kong and W. Chen, Heterocycles, 2015, 91, 2306–2314 CrossRef CAS.
- M. Curini, F. Epifano, F. Montanari, O. Rosati and S. Taccone, Synlett, 2004, 10, 1832–1834 CrossRef.
- K. Srinivas and P. K. Dubey, Synth. Commun., 2011, 41, 1584–1592 CrossRef CAS.
- X. R. Song, Y. F. Qiu, B. Song, X. H. Hao, Y. P. Han, P. Gao, X. Y. Liu and Y. M. Liang, J. Org. Chem., 2015, 80, 2263–2271 CrossRef CAS PubMed.
- U. Srinivas, C. Srinivas, P. Narender, V. J. Rao and S. Palaniappan, Catal. Commun., 2007, 8, 107–110 CrossRef CAS.
- A. A. Kalinin and V. A. Mamedov, Chem. Heterocycl. Comp., 2014, 50, 195–203 CrossRef CAS.
- S. N. Lin and L. Yang, Tetrahedron Lett., 2005, 46, 4315–4319 CrossRef CAS.
- (a) Y. Kawashita, N. Nakamichi, H. Kawabata and M. Hayashi, Org. Lett., 2003, 5, 3713–3715 CrossRef CAS PubMed; (b) Y. Tagawa, K. Yarnagata and K. Sumoto, Heterocycles, 2008, 75, 415–418 CrossRef CAS.
- Y. X. Meng, Y. Y. Gui, Q. Ji, Y. L. Pan, Z. Q. Lin, L. Lu and X. C. Zeng, Chin. J. Org. Chem., 2016, 36, 384–392 CrossRef CAS.
- Y.-S. Hsiao, B. D. Narhe, Y.-S. Chang and C.-M. Sun, ACS Comb. Sci., 2013, 15, 551–555 CrossRef CAS PubMed.
- T. Öztürk, M. N. Atalar, M. Göktaş and B. Hazer, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2651–2659 CrossRef.
- C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2011, 17, 2492–2503 CrossRef CAS PubMed.
- N. A. Bumagin and D. N. Korolev, Tetrahedron Lett., 1999, 40, 3057–3060 CrossRef CAS.
- T.-S. Yeh, T. J. Chow, S.-H. Tsai, C.-W. Chiu and C.-X. Zhao, Chem. Mater., 2006, 18, 832–839 CrossRef CAS.
- J. Figueras, J. Am. Chem. Soc., 1971, 93, 3255–3263 CrossRef CAS.
- (a) S. Jeong, M.-K. Kim, S. H. Kim and J.-I. Hong, Org. Electron., 2013, 14, 2497–2504 CrossRef CAS; (b) J. Pina, J. S. Seixas de Melo, R. M. F. Batista, S. P. G. Costa and M. M. M. Raposo, J. Org. Chem., 2013, 78, 11389–11395 CrossRef CAS PubMed; (c) S. Gong, Y. Zhao, M. Wang, C. Yang, C. Zhong, J. Qin and D. Ma, Chem.–Asian J., 2010, 5, 2093–2099 CrossRef CAS PubMed.
- S. Manoharan and S. Anandan, Dyes Pigm., 2014, 105, 223–231 CrossRef CAS.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M.-a. Kakimoto, Chem. Mater., 2008, 20, 2532–2537 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09864b |
| This journal is © The Royal Society of Chemistry 2017 |