
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion-driven tetrel bond-induced engineering of lead(II) architectures with N′-(1-(2-pyridyl)ethylidene)nicotinohydrazide: experimental and theoretical findings†
Ghodrat
Mahmoudi
*a,
Damir A.
Safin
*b,
Mariusz P.
Mitoraj
*c,
Mojtaba
Amini
*a,
Maciej
Kubicki
d,
Thomas
Doert
e,
Franziska
Locherer
e and
Michel
Fleck
f
aDepartment of Chemistry, Faculty of Science, University of Maragheh, P.O. Box 55181-83111, Maragheh, Iran. E-mail: mahmoudi_ghodrat@yahoo.co.uk; mamini@maragheh.ac.ir
bInstitute of Condensed Matter and Nanosciences, Molecules, Solids and Reactivity (IMCN/MOST), Université catholique de Louvain, Place L. Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: damir.a.safin@gmail.com
cDepartment of Theoretical Chemistry, Faculty of Chemistry, Jagiellonian University, R. Ingardena 3, 30-060 Cracow, Poland. E-mail: mitoraj@chemia.uj.edu.pl
dFaculty of Chemistry, Adam Mickiewicz University in Poznan, Umultowska 89b, 61-614 Poznan, Poland
eDepartment of Chemistry and Food Chemistry, Dresden University of Technology, Helmholtzstrasse 10, 01069 Dresden, Germany
fInstitute for Mineralogy and Crystallography, University of Vienna, Althanstrasse 14, 1090 Vienna, Austria
First published on 25th November 2016
Abstract
The evaluation of N′-(1-(2-pyridyl)ethylidene)nicotinohydrazide (HL) as a linker for the PbII tagged extended structures is described. The reaction of Pb(ClO4)2 or Pb(OAc)2 with HL in MeOH at 60 °C and room temperature, respectively, leads to heteroleptic complexes {[PbL]ClO4}n·nH2O and [PbL(OAc)]2, while the same reaction of Pb(ClO4)2 with HL at 60 °C in the presence of two equivalents of NaOAc or NaNO2 leads to heteroleptic complexes {[Pb(HL)(OAc)]ClO4}n and [PbL(NO2)]n, respectively. Using Pb(NO3)2 as a source of PbII in the same reaction with HL and two equivalents of NaN3 or NaNCS at room temperature yields [PbLN3]n and [Pb2(HL)2(NO3)2(NCS)2], respectively. The room temperature reaction of Pb(NO3)2 with HL in the presence of two equivalents of NaClO4 leads to the transformation of the parent ligand to its perchlorate salt [H2L]ClO4. In all the obtained PbII structures, HL or its deprotonated form L acts both as a chelating and a bridging ligand. The nature of the inorganic anion also influences the final structure. In all complexes the PbII center exhibits a hemidirected coordination geometry with all the covalent bonds being concentrated on one hemisphere of the coordination sphere with the closest approach of two atoms on the other side varying from 151° to 232°. The sterically available PbII ion participates in tetrel bonding as evidenced from the detailed structural analysis of the described complexes. As a result of tetrel bonding, the structures of all the six compounds can be extended to a higher dimensional framework, which is further stabilized by π⋯π stacking interactions between the aromatic rings. The DFT based charge and energy decomposition (ETS-NOCV) calculations are performed in order to shed light on the nature of non-covalent interactions that determine the stability of the obtained structures.
Introduction
In recent years metal–organic supramolecular systems have attracted great interest for their extremely versatile coordination motifs and potential applications as functional materials in numerous and diverse fields including separation, drug delivery, catalysis, molecular magnetism, gas adsorption and photoluminescence.1–21 The development of new synthetic strategies to produce metal–organic frameworks (MOFs) and coordination polymers (CPs) has become a great challenge because it depends on many factors such as the nature of the metal ion and its counter ion, structural features of the organic ligand and experimental conditions (reaction temperature, reagent ratio, pH, solvent(s)), and crystallization procedures.22–26 Furthermore, among the most powerful tools to design and control the structure of supramolecular systems, non-covalent inter- and intramolecular interactions are the most efficient. Non-covalent interactions play pivotal roles in many aspects of chemistry and biology, with van der Waals interactions and hydrogen bonding being among the best known examples. Nowadays the construction of any host to bind guest species is possible thanks to modern synthetic chemistry methods even in competitive media.27,28A more recently defined class of non-covalent interactions, that continues to garner a significant degree of attention, are classified as σ-hole bonds, i.e. interactions between a covalently-bonded atom of groups IV–VII acting as a Lewis acid and a Lewis base centre.29–32 σ-Hole bonding is a primarily electrostatic interaction between a localized region of positive electrostatic potential (ESP) on a molecular species (the σ-hole) and an electron-rich center nearby. Among σ-hole bonds, halogen,33–41 chalcogen42–50 and pnictogen51–53 interactions are the most studied bonds, where atoms of groups VII, VI and V, respectively, act as Lewis acid centres. More recently atoms of the group IV have also been considered as Lewis acid centres forming σ-hole bonds.32,54–62 The σ-hole bond formed by the group IV elements has been named a tetrel bond.55,59
Intrigued by this new class of σ-hole bonding, i.e. the tetrel bond, we have recently directed our attention towards PbII,63–66 as a perfect template for the formation of tetrel bonds. Particularly, lead is the heaviest group IV element and thus can be expected to form more positive σ-holes than other elements of this group. Furthermore, the experimental coordination geometry around lead, reveals two distinct structural features, namely holodirected, in which the bonds to ligand atoms are directed throughout the surface of the encompassing globe, and hemidirected, in which the bonds to ligand atoms are directed throughout only part of the globe, that is, there is an identifiable void (or gap) in the distribution of bonds to the ligands (Chart 1).67,68 While all PbIV compounds show a holodirected coordination geometry, for PbII both holodirected and hemidirected coordination geometries are found.67 A hemidirected coordination geometry around PbII, with a pronounced gap in the coordination sphere, would substantially facilitate closer approach of electron donors to the σ-hole of the PbII ion than to those of the other group IV atoms and thus enable the formation of a stronger tetrel bond with a more predictable geometry.
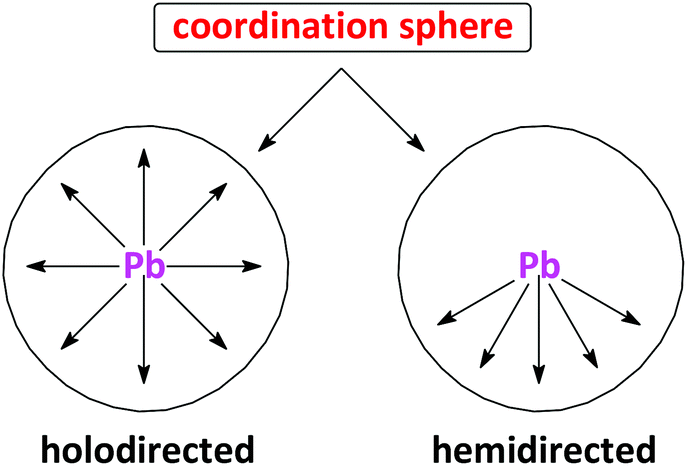 | ||
| Chart 1 The simplified diagram of the holodirected and hemidirected coordination spheres around lead. | ||
In this work we have been continuing our comprehensive investigations toward the synthesis of new PbII containing supramolecular assemblies driven by non-covalent interactions. Namely, we report for the first time that depending on the temperature a reaction of N′-(1-(2-pyridyl)ethylidene)nicotinohydrazide (HL)69 with PbII salts, comprising a wide set of anions that differ structurally (NO2−, NO3−, ClO4−, N3−, NCS− and OAc−), yields either a new perchlorate salt [H2L]ClO4 of a helice-like structure, or new PbII structures with intriguing architectures including, e.g., a very rare example of PbII⋯PbII interaction.
The structure of the ligand is designed with the potential to form tridentate chelation of one metal center, with a meridional N2O coordination motif analogous to the classical 2,2′:6′,2′′-terpyridine or bis(imino)pyridine ligands,70,71 while the remaining nitrogen atom of the 3-pyridyl fragment remains free to bind to another metal center, forming a bridge and leading to either di- or polymeric coordination compounds. The polydentate nature of both the ligand HL and its conjugate anion L facilitates the hemidirected coordination of PbII, while the nature of inorganic anions ensures the presence of electron donor atoms capable of forming tetrel bonds. Indeed, in the crystal structures of all the reported herein PbII complexes, tetrel bonds with hemidirected coordination spheres of the metal ions are mainly responsible for the formation of 3D supramolecular aggregates. The nature of non-covalent interactions in all the structures was studied for the first time by the DFT based charge and energy decomposition calculations (ETS-NOCV).
Results and discussion
An equimolar one-pot reaction of Pb(ClO4)2 or Pb(OAc)2 with HL in MeOH at 60 °C and room temperature, respectively, leads to heteroleptic complexes {[PbL]ClO4}n·nH2O and [PbL(OAc)]2, each containing the deprotonated form of the organic ligand L (Scheme 1). The same reaction of Pb(ClO4)2 with HL at 60 °C but in the presence of two equivalents of NaOAc or NaNO2 also leads to heteroleptic complexes {[Pb(HL)(OAc)]ClO4}n and [PbL(NO2)]n, respectively (Scheme 1). While the latter structure is also constructed from the deprotonated ligand L, the former compound, however, contains the parent ligand in its neutral form HL. Using Pb(NO3)2 as a source of PbII in the same reaction with HL and two equivalents of NaN3 or NaNCS at room temperature yields [PbLN3]n and [Pb2(HL)2(NO3)2(NCS)2], respectively (Scheme 1). Interestingly, the room temperature reaction of Pb(NO3)2 with HL in the presence of two equivalents of NaClO4 leads to the transformation of the parent ligand to its perchlorate salt [H2L]ClO4 (Scheme 1). | ||
| Scheme 1 Syntheses of [H2L]ClO4 and PbII complexes described in this work. Tetrel bonds are shown as dashed lines. | ||
All compounds were obtained with good yields, and were fully characterized by elemental analysis, FTIR spectroscopy, single crystal X-ray diffraction and theoretical calculations.
According to the single crystal X-ray diffraction data, {[PbL]ClO4}n·nH2O and [Pb2(HL)2(NO3)2(NCS)2] crystallize in the monoclinic space group P21/n, while [PbL(NO2)]n, [H2L]ClO4 and [PbL(OAc)]2 crystallize in the monoclinic space group P21/c, respectively. Compound {[Pb(HL)(OAc)]ClO4}n crystallizes in the orthorhombic space group Pbca, while the complex [PbLN3]n crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .
.
The ligand cation in the structure of [H2L]ClO4 is almost flat (Fig. 1) and the dihedral angle between the mean planes of rings is 9.9(4)°. The bond length and angles establish the double-bond character of the N–C(CH3) bond (1.266(10) Å). The protonation nitrogen site of the 2-pyridyl ring is confirmed by the localization of the hydrogen atom in the difference Fourier map and the successful refinement of this atom and the bond angles pattern around the nitrogen atom. In the crystal structure of [H2L]ClO4 the ligand cations are linked through intermolecular N–H⋯N hydrogen bonds (Table 1), formed between the pyridinium NH hydrogen atom of one cation and the nitrogen atom of the 3-pyridyl fragment of an adjacent cation. As a result of these interactions, left and right handed helices are formed (Fig. 1). Furthermore, the amide NH hydrogen atom of the ligand cation is involved in the intermolecular N–H⋯O hydrogen bond with one of the oxygen atoms of an adjacent ClO4− anion (Fig. 1 and Table 1). The structure of [H2L]ClO4 is further stabilized by π⋯π stacking interactions (Table 2), formed between the polytypic aromatic rings of neighbouring ligand cations.
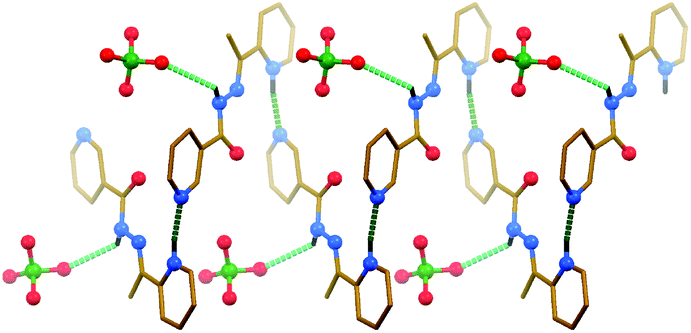 | ||
| Fig. 1 Left handed helix, constructed through hydrogen bonds, in the crystal structure of [H2L]ClO4 (CH hydrogen atoms are omitted for clarity). Hydrogen bonds are shown as dashed lines. Stronger non-covalent interactions are shown in a darker shade of green than the weaker non-covalent interactions. Color code: C = gold, H = black, N = blue, Cl = green, O = red. | ||
| Compound | D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(DHA) |
|---|---|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: #1 x, −1 + y, z; #2 1 − x, 1/2 + y, 1/2 − z. b Symmetry transformations used to generate equivalent atoms: #1 1 − x, 1 − y, 1 − z. c Symmetry transformations used to generate equivalent atoms: #1 1/2 − x, −1/2 + y, 1/2 − z. | |||||
[H2L]ClO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
N(8A)–H(8A)⋯O(3B)#1 | 0.92 | 2.47 | 3.085(11) | 125 |
| N(12A)–H(12A)⋯N(1A)#2 | 0.98 | 1.82 | 2.752(9) | 158 | |
|
{[Pb(HL)(OAc)]ClO4}nb |
N(8A)–H(8A)⋯O(11B)#1 | 0.88 | 2.15 | 2.923(7) | 146 |
| [Pb2(HL)2(NO3)2(NCS)2] | N(8A)–H(8A)⋯O(2C)#1 | 0.88 | 2.18 | 2.961(3) | 147 |
| Compound | Cg(I) | Cg(J) | d[Cg(I)–Cg(J)] | α | β | γ | Slippage |
|---|---|---|---|---|---|---|---|
| a Cg(I)–Cg(J): distance between ring centroids; α: dihedral angle between planes Cg(I) and Cg(J); β: angle Cg(I) → Cg(J) vector and normal to plane I; γ: angle Cg(I) → Cg(J) vector and normal to plane J; slippage: distance between Cg(I) and perpendicular projection of Cg(J) on ring I. b Symmetry transformations used to generate equivalent atoms: #1 1 + x, y, z; #2 −1 + x, y, z. Cg(1): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A), Cg(2): N(12A)–C(11A)–C(16A)–C(15A)–C(14A)–C(13A). c Symmetry transformations used to generate equivalent atoms: #1 1 − x, 1 − y, 1 − z. Cg(4): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A); Cg(4): N(12A)–C(11A)–C(16A)–C(15A)–C(14A)–C(13A). d Symmetry transformations used to generate equivalent atoms: #1 1 − x, 1 − y, 1 − z. Cg(1): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A); Cg(2): N(12A)–C(11A)–C(16A)–C(15A)–C(14A)–C(13A). e Symmetry transformations used to generate equivalent atoms: #1 1 − x, 2 − y, 1 − z. Cg(4): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A). f Symmetry transformations used to generate equivalent atoms: #1 x, −1 + y, z; #2 1 + x, −1 + y, z; #3 −1 + x, 1 + y, z; #4 x, 1 + y, z. Cg(4): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A), Cg(5): N(12A)–C(11A)–C(16A)–C(15A)–C(14A)–C(13A). g Symmetry transformations used to generate equivalent atoms: #1 1/2 − x, −1/2 + y, 1/2 − z; #2 1 − x, −y, 1 − z; #3 1/2 − x, 1/2 + y, 1/2 − z. Cg(1): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A), Cg(2): N(12A)–C(11A)–C(16A)–C(15A)–C(14A)–C(13A). h Symmetry transformations used to generate equivalent atoms: #1 −x, 1 − y, 1 − z. Cg(1): N(1A)–C(2A)–C(3A)–C(4A)–C(5A)–C(6A), Cg(2): N(12A)–C(11A)–C(16A)–C(15A)–C(14A)–C(13A). | |||||||
|
[H2L]ClO4b |
Cg(1) | Cg(2)#1 | 3.757(5) | 9.9(4) | 21.8 | 22.9 | 1.394 |
| Cg(2) | Cg(1)#2 | 3.756(5) | 9.9(4) | 22.9 | 21.8 | 1.463 | |
| {[PbL]ClO4}n·nH2O | Cg(4) | Cg(5)#1 | 3.675(3) | 11.6(3) | 25.0 | 19.4 | 1.555 |
| Cg(5) | Cg(4)#1 | 3.675(3) | 11.6(3) | 19.4 | 25.0 | 1.223 | |
|
{[Pb(HL)(OAc)]ClO4}nd |
Cg(1) | Cg(2)#1 | 3.694(5) | 8.7(4) | 25.6 | 18.6 | 1.595 |
| Cg(2) | Cg(1)#1 | 3.695(5) | 8.7(4) | 18.6 | 25.6 | 1.179 | |
|
[PbL(NO2)]ne |
Cg(4) | Cg(4)#1 | 3.615(3) | 0.0(3) | 26.0 | 26.0 | 1.586 |
|
[PbLN3]nf |
Cg(4) | Cg(5)#1 | 3.930(3) | 12.0(2) | 20.1 | 32.0 | 1.348 |
| Cg(4) | Cg(5)#2 | 3.809(3) | 12.0(2) | 20.4 | 22.8 | 1.325 | |
| Cg(5) | Cg(4)#3 | 3.809(3) | 12.0(2) | 22.8 | 20.4 | 1.478 | |
| Cg(5) | Cg(4)#4 | 3.930(3) | 12.0(2) | 32.0 | 20.1 | 2.085 | |
| [Pb2(HL)2(NO3)2(NCS)2] | Cg(1) | Cg(2)#1 | 3.7404(19) | 14.76(16) | 7.7 | 10.6 | 0.499 |
| Cg(1) | Cg(2)#2 | 3.7947(19) | 5.02(16) | 27.1 | 22.9 | 1.729 | |
| Cg(2) | Cg(1)#3 | 3.7404(19) | 14.76(16) | 10.6 | 7.7 | 0.689 | |
| Cg(2) | Cg(1)#2 | 3.7948(19) | 5.02(16) | 22.9 | 27.1 | 1.477 | |
|
[PbL(OAc)]2h |
Cg(1) | Cg(2)#1 | 3.839(3) | 10.7(2) | 20.0 | 30.7 | 1.313 |
| Cg(2) | Cg(1)#1 | 3.839(3) | 10.7(2) | 30.7 | 20.0 | 1.960 | |
Single-crystal X-ray diffraction studies showed that in all the six complexes the PbII ion exhibits a hemidirected coordination geometry, and the metal ion participates in tetrel bonding (Table 3).
| Complex | Bond | Ligand | Length | Bond type |
|---|---|---|---|---|
| {[PbL]ClO4}n·nH2O | Pb–N | L | 2.408(4) | Covalent |
| L | 2.507(4) | Covalent | ||
| L | 2.513(4) | Covalent | ||
| L | 2.934(4) | Tetrel | ||
| Pb–O | L | 2.382(4) | Covalent | |
| ClO4− | 3.201(5) | Tetrel | ||
| {[Pb(HL)(OAc)]ClO4}n | Pb–N | L | 2.612(6) | Covalent |
| L | 2.633(6) | Covalent | ||
| L | 2.755(7) | Covalent | ||
| Pb–O | L | 2.572(5) | Covalent | |
| OAc− | 2.372(5) | Covalent | ||
| OAc− | 2.515(6) | Covalent | ||
| ClO4− | 3.047(7) | Tetrel | ||
| ClO4− | 3.309(10) | Tetrel | ||
| [PbL(NO2)]n | Pb–N | L | 2.452(4) | Covalent |
| L | 2.535(5) | Covalent | ||
| L | 2.752(5) | Covalent | ||
| L | 3.258(4) | Tetrel | ||
| Pb–O | L | 2.384(5) | Covalent | |
| NO2− | 2.547(4) | Covalent | ||
| NO2− | 2.904(5) | Covalent | ||
| NO2− | 3.299(4) | Tetrel | ||
| [PbLN3]n | Pb–N | L | 2.571(3) | Covalent |
| L | 2.740(3) | Covalent | ||
| L | 2.837(3) | Covalent | ||
| L | 3.436(4) | Tetrel | ||
| N3− | 2.321(3) | Covalent | ||
| N3− | 2.883(3) | Covalent | ||
| Pb–O | L | 2.335(3) | Covalent | |
| [Pb2(HL)2(NO3)2(NCS)2] | Pb–N | L | 2.663(3) | Covalent |
| L | 2.677(3) | Covalent | ||
| L | 2.809(3) | Covalent | ||
| NCS− | 2.460(4) | Covalent | ||
| Pb–O | L | 2.555(2) | Covalent | |
| NO3− | 2.744(2) | Covalent | ||
| NO3− | 2.882(2) | Covalent | ||
| Pb–S | NCS− | 3.2246(11) | Tetrel | |
| [PbL(OAc)]2 | Pb–N | L | 2.490(3) | Covalent |
| L | 2.613(4) | Covalent | ||
| L | 3.030(4) | Tetrel | ||
| L | 3.409(4) | Tetrel | ||
| L | 3.489(3) | Tetrel | ||
| Pb–O | L | 2.383(3) | Covalent | |
| OAc− | 2.294(3) | Covalent | ||
| OAc− | 2.753(4) | Covalent |
The asymmetric unit of {[PbL]ClO4}n·nH2O contains one [PbL]+ cation, in which the deprotonated parent ligand L is κ3-coordinated, one ClO4− anion, with three oxygen atoms being disordered over two positions with a 58% and 42% ratio, and one molecule of water, disordered over three positions with a 40%, 30% and 30% ratio. The PbII atom is covalently linked (d[Pb–N/O] = 2.382(4)–2.513(4) Å) to two nitrogens and one oxygen atom of L and one 3-pyridyl nitrogen atom of another ligand L with the formation of a 1D zig-zag-like polymeric polycation (Fig. 2 and Table 3). All the covalent bonds are concentrated on one quarter of the globe of the coordination sphere with the closest approach of two atoms on the other side of about 230°. This leaves a large gap on the PbII ion, which enables a close approach of the non-disordered ClO4− oxygen atom (d[Pb⋯O] = 3.201(5) Å) and, remarkably, the amide nitrogen atom (d[Pb⋯N] = 2.934(4) Å) from the ligand L of an adjacent polymeric polycation (Fig. 2 and Table 3). The latter contacts interconnect the polymeric polycations into 2D sheets, which are further stabilized by π⋯π stacking interactions (Table 2), formed between the polytypic aromatic rings of neighbouring ligands L. These 2D sheets are separated by layers of ClO4− anions and H2O molecules. Taking all the covalent and tetrel bonds into account, the coordination environment around the PbII cation in the structure of {[PbL]ClO4}n·nH2O is best described as a distorted octahedron.
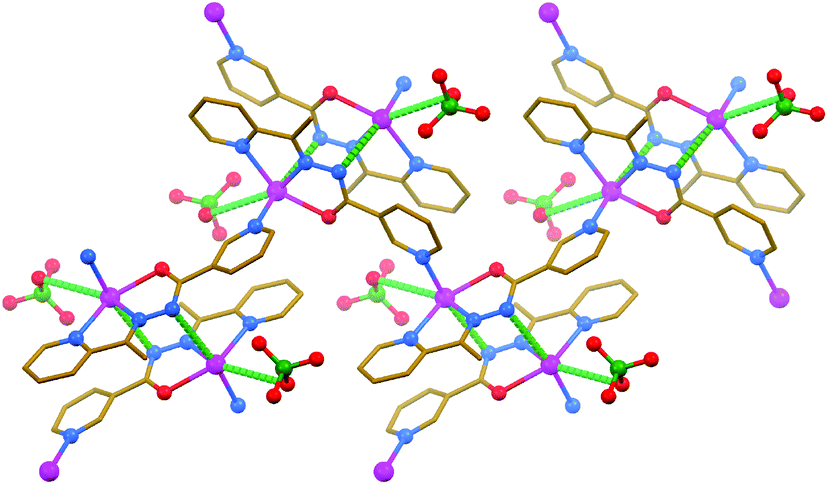 | ||
| Fig. 2 Crystal structure of {[PbL]ClO4}n·nH2O (hydrogen atoms and water molecules are omitted for clarity). Tetrel bonds Pb⋯N and Pb⋯O are shown as dashed lines. Stronger non-covalent interactions are shown in a darker shade of green than the weaker non-covalent interactions. Color code: C = gold, N = blue, Cl = green, O = red, Pb = magenta. | ||
The asymmetric unit of {[Pb(HL)(OAc)]ClO4}n contains one [Pb(HL)(OAc)]+ cation, in which the neutral form of the parent ligand HL is κ3-coordinated and the OAc− anion is κ2-coordinated to the same metal center, and one ClO4− anion. The PbII atom is covalently linked (d[Pb–N/O] = 2.372(5)–2.755(7) Å) to two nitrogens and one oxygen atom of HL, one 3-pyridyl nitrogen atom of another ligand HL, with the formation of a 1D zig-zag-like polymeric polycation (Fig. 3 and Table 3) similar to that in the structure of {[PbL]ClO4}n·nH2O (Fig. 2), and two OAc− oxygen atoms. All the covalent bonds are concentrated on one hemisphere of the coordination sphere with the closest approach of two atoms on the other side of about 205°. This also leaves a large gap on the PbII ion, which enables a close approach of the two ClO4− oxygen atoms (d[Pb⋯O] = 3.047(7) and 3.309(10) Å) (Fig. 3 and Table 3). The polymeric polycations are interconnected into 2D sheets through intermolecular N–H⋯O hydrogen bonds (Table 1), formed between the amide NH hydrogen atom of one cation and one of the OAc− oxygen atoms of an adjacent cation, which are further stabilized by π⋯π stacking interactions (Table 2), formed between the polytypic aromatic rings of the same species. These 2D sheets are also separated by layers of ClO4− anions.
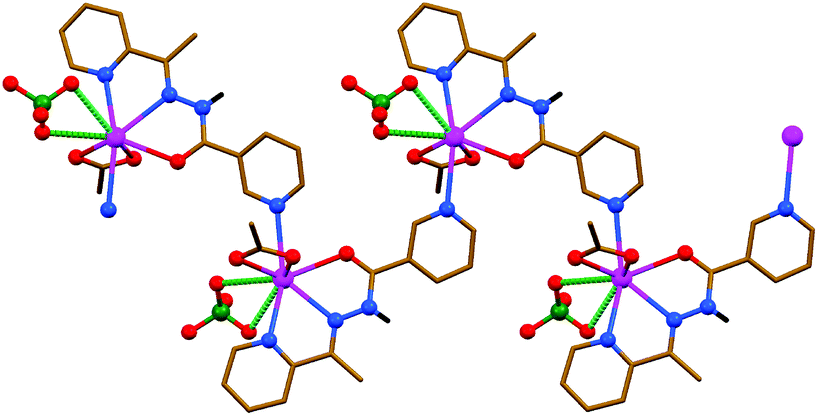 | ||
| Fig. 3 Crystal structure of {[Pb(HL)(OAc)]ClO4}n (CH hydrogen atoms are omitted for clarity). Tetrel bonds Pb⋯O are shown as dashed lines. Stronger non-covalent interactions are shown in a darker shade of green than the weaker non-covalent interactions. Color code: C = gold, H = black, N = blue, Cl = green, O = red, Pb = magenta. | ||
The structure of [PbL(NO2)]n is similar to that of {[PbL]ClO4}n·nH2O. The main difference, influencing the overall 1D zig-zag-like polymeric structure of the former complex (Fig. 4), however, arises from the NO2− anion, which is fully ordered and covalently bound to the metal center, yielding the [PbL(NO2)]-containing asymmetric unit. The deprotonated form of the κ3-coordinated parent ligand L and the κ2-coordinated NO2− anion as well as the 3-pyridyl nitrogen atom of an adjacent ligand L, all being linked to the same metal cation, provides six covalent bonds (d[Pb–N/O] = 2.384(5)–2.904(5) Å) (Table 3). These bonds are concentrated on one hemisphere of the coordination sphere with the closest approach of two atoms on the other side of about 154°. This gap on the PbII ion enables a close approach of the amide nitrogen atom (d[Pb⋯N] = 3.258(4) Å) and one of the NO2− oxygen atoms (d[Pb⋯O] = 3.299(4) Å), arising from the same [PbL(NO2)] fragment of an adjacent polymeric chain (Fig. 4). The 1D polymeric chains are further interlinked through π⋯π stacking interactions (Table 2), formed between the 3-pyridyl rings, yielding 2D sheets.
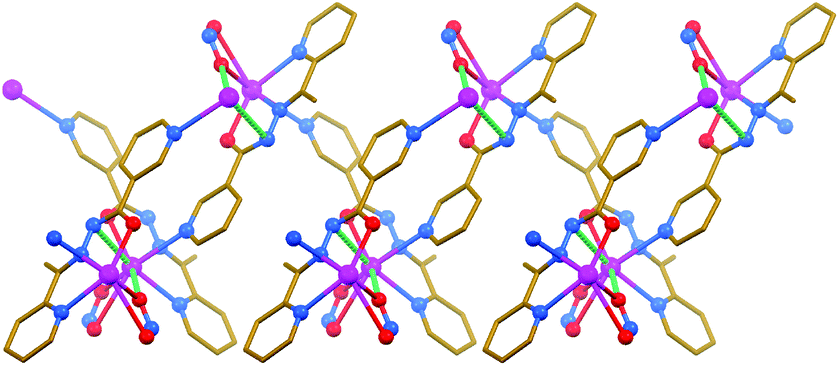 | ||
| Fig. 4 Crystal structure of [PbL(NO2)]n (hydrogen atoms are omitted for clarity). Tetrel bonds Pb⋯N and Pb⋯O are shown as dashed lines. Stronger non-covalent interactions are shown in a darker shade of green than the weaker non-covalent interactions. Color code: C = gold, N = blue, O = red, Pb = magenta. | ||
The structures of [PbLN3]n, [Pb2(HL)2(NO3)2(NCS)2] and [PbL(OAc)]2 are constructed from similar secondary building units (SBUs), each of which represent a dimeric aggregate with covalently κ3-coordinated organic ligands (Fig. 5–7 and Table 3). This dimer is, however, of the [Pb2L2]2+ structure in the N3−- and OAc−-containing complexes, and of the [Pb2(HL)2]4+ structure in the NCS−-containing compound. The asymmetric unit of [PbLN3]n and [PbL(OAc)]2 comprises [PbLX] (X = N3− or OAc−, respectively) species, while [Pb(HL)(NO3)(NCS)] was found in the asymmetric unit of [Pb2(HL)2(NO3)2(NCS)2]. Notably, these mononuclear species in the structures of [PbLN3]n and [Pb2(HL)2(NO3)2(NCS)2] are combined in SBUs through the Pb–N (2.837(3) and 2.809(3) Å, respectively) covalent bonds between the metal center of one monomeric species and the 3-pyridyl nitrogen atom of the other monomeric species (Fig. 5 and 6). The same aggregation in the structure of [PbL(OAc)]2 is, however, due to the Pb⋯N (3.030(4) Å) tetrel bonding, and is further stabilized by the C–H⋯O hydrogen bonds (Fig. 7).
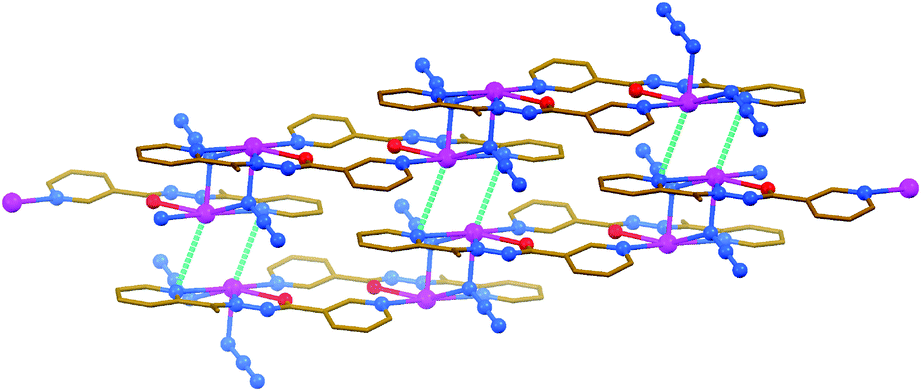 | ||
| Fig. 5 Crystal structure of [PbLN3]n (hydrogen atoms are omitted for clarity). Tetrel bonds Pb⋯N are shown as dashed lines. Color code: C = gold, N = blue, O = red, Pb = magenta. | ||
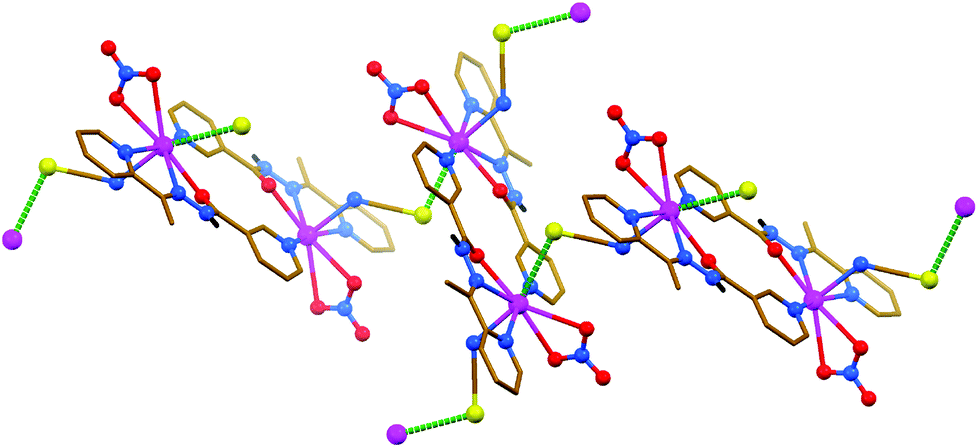 | ||
| Fig. 6 Crystal structure of [Pb2(HL)2(NO3)2(NCS)2] (CH hydrogen atoms are omitted for clarity). Tetrel bonds Pb⋯S are shown as dashed lines. Color code: C = gold, H = black, N = blue, O = red, Pb = magenta, S = yellow. | ||
 | ||
| Fig. 7 Crystal structure of [PbL(OAc)]2 (hydrogen atoms not-involved in H-bonding are omitted for clarity). Tetrel bonds Pb⋯N and hydrogen bonds are shown as dashed lines. Stronger non-covalent interactions are shown in a darker shade of green than the weaker non-covalent interactions. Color code: C = gold, H = black, N = blue, O = red, Pb = magenta. | ||
In the structure of [PbLN3]n, dimeric SBUs are linked in 1D ladder-like polymeric chains through the μ-1,1 (end-on) covalently coordinated N3− anions (Fig. 5 and Table 3). These 1D chains are interlinked through the Pb⋯N tetrel (3.436(4) Å) bonds, formed between the metal centers of the chain and the 2-pyridyl nitrogen atoms of an adjacent chain (Fig. 5). These interactions are also realized due to the concentration of the covalent bonds on one hemisphere of the coordination sphere with the closest approach of two atoms on the other side of about 197°. The 1D polymeric chains are further interlinked through π⋯π stacking interactions (Table 2), formed between the polytypic aromatic rings of neighbouring ligand cations.
In the structures of [Pb2(HL)2(NO3)2(NCS)2] and [PbL(OAc)]2, the inorganic anions NO3− and OAc−, respectively, are each covalently κ2-coordinated to the metal center through two oxygen atoms, and the coordination sphere of the PbII ion in the former complex is further filled by the covalently bound nitrogen atom of the terminally coordinated NCS− anion (Fig. 6 and 7 and Table 3). SBUs in the structure of [Pb2(HL)2(NO3)2(NCS)2] are linked into 2D sheets through the Pb⋯S tetrel (3.2246(11) Å) bonds, formed between the metal centers of the SBU and the NCS− sulfur atoms of adjacent SBUs (Fig. 6). Such kind of tetrel bonding is not only due to the free sulfur donor atom of the terminally coordinated NCS− anions but also due to a relatively small gap around the PbII ions (∼151°). Tetrel bonded 2D sheets in the structure of [Pb2(HL)2(NO3)2(NCS)2] are linked into a 3D framework through the intermolecular N–H⋯O hydrogen bonds (Table 1), formed between the amide NH hydrogen atom of one SBU and one of the coordinated NO3− oxygen atoms of an adjacent SBU. This 3D framework is further stabilized by π⋯π stacking interactions (Table 2), formed between the polytypic aromatic rings of different SBUs. A significantly larger gap around the PbII ion (∼232°) in the structure of [PbL(OAc)]2 is responsible for the formation of Pb⋯N tetrel (3.409(4) and 3.489(3) Å) bonds with both hydrazide nitrogen atoms of an adjacent SBU (Fig. 7 and Table 3). As a result of tetrel bonding between SBUs, 2D sheets are formed in the structure of [PbL(OAc)]2. These 2D sheets are additionally stabilized by π⋯π stacking interactions (Table 2), formed between the polytypic aromatic rings of different SBUs.
In order to rationalize the formation of various architectures of the synthesized compounds as well as to describe intermolecular interactions we have performed theoretical studies at the DFT/BLYP-D3/TZP level of theory as implemented in the Amsterdam Density Functional (ADF) package.72,73 The relativistic effects are accounted by the ZORA approximation as implemented in the ADF program. We have discussed predominantly the DFT/BLYP-D3/TZP results because it has been shown that it provides satisfactory results for noncovalent interactions as compared with the accurate CCSD(T) results.74–76 At the first stage we briefly analyze molecular electrostatic potentials (MEP) as they have been shown to provide very useful quantity to rationalize chemical structures and reactivity29–32 followed by in depth characterization of bonding patterns by means of the charge and energy decomposition scheme ETS-NOCV77–79 as implemented in the ADF program. It must be emphasized that qualitative and quantitative descriptions of various non-covalent interactions in the PbII complexes with the nicotinohydrazine, based on the ETS-NOCV energy decomposition scheme, will be provided for the first time.
It was established that, although the Pb–L bond is formed due to a charge donation L → PbII, a significant electrophilic region (blue color) around the metal center is noticed (Fig. 8), which, in turn, promotes binding of anions. Further binding of an anion (e.g. OAc−) decreases a positive charge at the PbII atom, resulting in less positive MEP values. However, still a large positive MEP area is noted around the metal (Fig. S1 in the ESI†). This explains that the entire PbII sphere participates in various binding modes in the synthesized compounds.
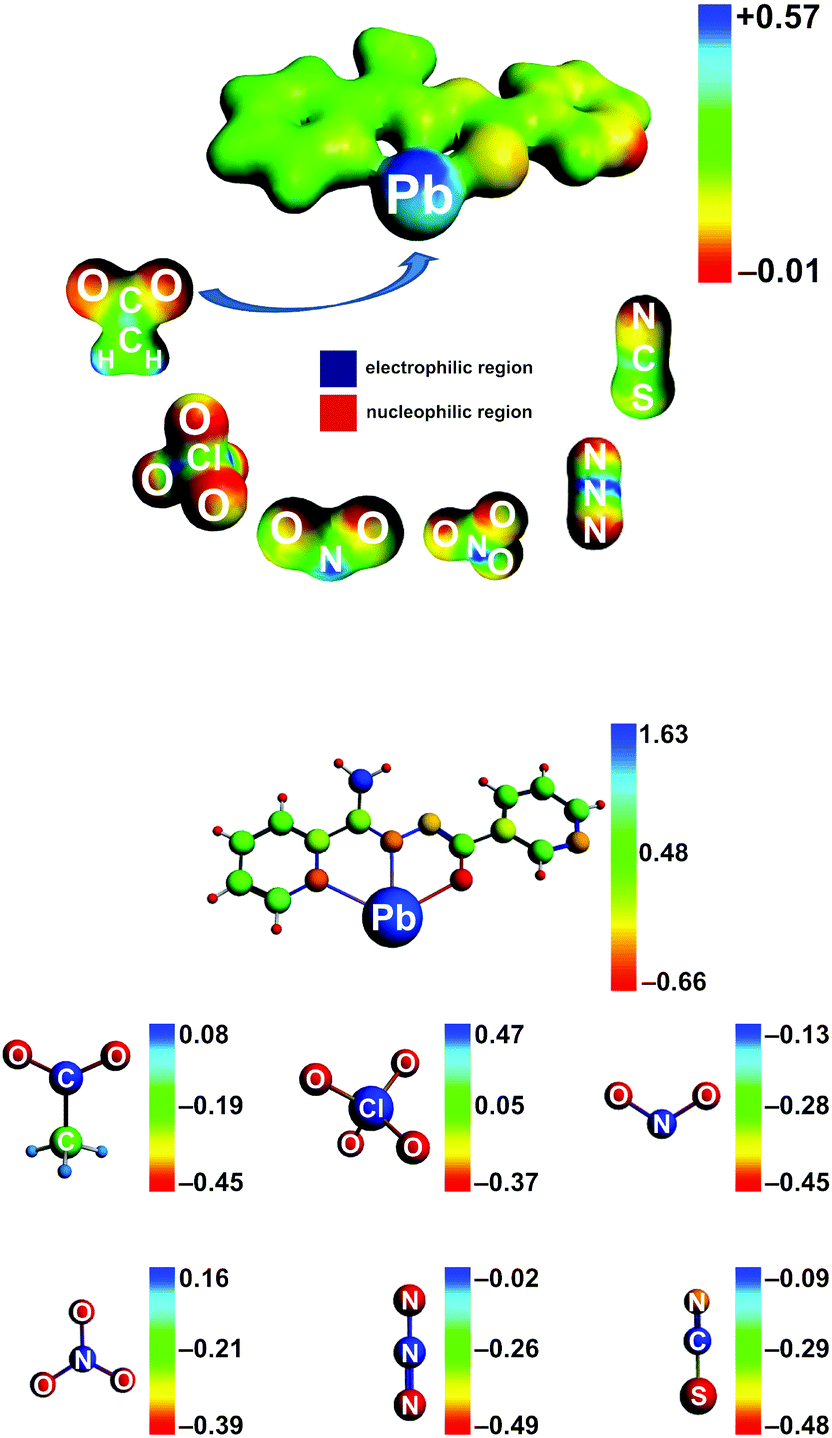 | ||
| Fig. 8 (Top) Molecular electrostatic potentials (DFT/BLYP-D3/ZORA/TZP), describing the [PbL]+ unit and anions (OAc−, ClO4−, NO2−, NO3−, N3− and NCS−). (Bottom) The Hirshfeld atomic charges of the [PbL]+ unit and anions (OAc−, ClO4−, NO2−, NO3−, N3− and NCS−). In order to differentiate the charge distribution at various regions of the selected ions, the optimal electron density contour value was chosen as 0.03 a. u. | ||
We have further performed an in depth bonding analysis on the example of [PbL(OAc)]2 by means of the ETS-NOCV method, using the cluster models (Fig. 9). This allowed us to shed light on various bonding modes, labeled as 1–3 in Fig. 9, that stabilize [PbL(OAc)]2. Polytypic rings of the two parallel oriented units [PbL(OAc)] give rise to the π⋯π interaction energy ΔEint = −29.14 kcal mol−1. The main stabilizing factor appeared to be the dispersion (ΔEdispersion = −34.59 kcal mol−1) followed by the electrostatic (ΔEelstat = −17.9 kcal mol−1) and charge transfer/polarization (ΔEorb = −7.49 kcal mol−1) contributions (Fig. 9). It is important to note that the presence of heteroatoms in the [PbL]+ species enforces additionally the formation of the Pb⋯N tetrel bonding, as evidenced from the contour of the overall deformation density Δρorb. In particular, both the charge donation from the PbII atom (6s electrons) to the empty π*(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) as well as back-donation from the occupied π(NN) to the metal center are noted (Fig. 9). Therefore, the PbII atom acts here as both the electron donor and acceptor. Hence, the red and blue colors of Δρorb are visible on PbII. A closer inspection of Δρorb reveals a charge transfer from the methyl σ(C–H) bonds of L onto the PbII, which demonstrates the formation of the agostic interaction Pb⋯CH. The π⋯π stacking, Pb⋯N and Pb⋯CH interactions are all together present in this system. It is to be referenced that such interactions have been also found in other PbII compounds.63–66,80–83 Considering the remaining bonds Pb–OAc− and Pb–L (2 and 3, respectively, in Fig. 9), far lower interaction energies (ΔEint = −139.47 and −180.96 kcal mol−1, respectively) are noticed. They are dominated by the electrostatic factor followed by the orbital and dispersion terms (Fig. 9). As far as changes in the electron density are concerned (Δρorb), the Pb–OAc− and Pb–L bonds are due to the donation (AcO−/L → PbII) and back-donation (PbII → AcO−/L) channels (Fig. 9). Notably, although the interaction energies change significantly when comparing 1–3, the overall picture, emerging from the deformation density (Δρorb), remains essentially very similar. In particular, all bonding types result from the donor–acceptor (dative) interactions giving rise additionally to the charge accumulation in the binding region (covalent contribution). Accordingly, these interactions, including the tetrel one, fall within the group of dative-covalent connections. It should be added that 2 and 3 are evidently more covalent as compared with 1 as it can be deduced from the contours of Δρorb (Fig. 9).
N) as well as back-donation from the occupied π(NN) to the metal center are noted (Fig. 9). Therefore, the PbII atom acts here as both the electron donor and acceptor. Hence, the red and blue colors of Δρorb are visible on PbII. A closer inspection of Δρorb reveals a charge transfer from the methyl σ(C–H) bonds of L onto the PbII, which demonstrates the formation of the agostic interaction Pb⋯CH. The π⋯π stacking, Pb⋯N and Pb⋯CH interactions are all together present in this system. It is to be referenced that such interactions have been also found in other PbII compounds.63–66,80–83 Considering the remaining bonds Pb–OAc− and Pb–L (2 and 3, respectively, in Fig. 9), far lower interaction energies (ΔEint = −139.47 and −180.96 kcal mol−1, respectively) are noticed. They are dominated by the electrostatic factor followed by the orbital and dispersion terms (Fig. 9). As far as changes in the electron density are concerned (Δρorb), the Pb–OAc− and Pb–L bonds are due to the donation (AcO−/L → PbII) and back-donation (PbII → AcO−/L) channels (Fig. 9). Notably, although the interaction energies change significantly when comparing 1–3, the overall picture, emerging from the deformation density (Δρorb), remains essentially very similar. In particular, all bonding types result from the donor–acceptor (dative) interactions giving rise additionally to the charge accumulation in the binding region (covalent contribution). Accordingly, these interactions, including the tetrel one, fall within the group of dative-covalent connections. It should be added that 2 and 3 are evidently more covalent as compared with 1 as it can be deduced from the contours of Δρorb (Fig. 9).
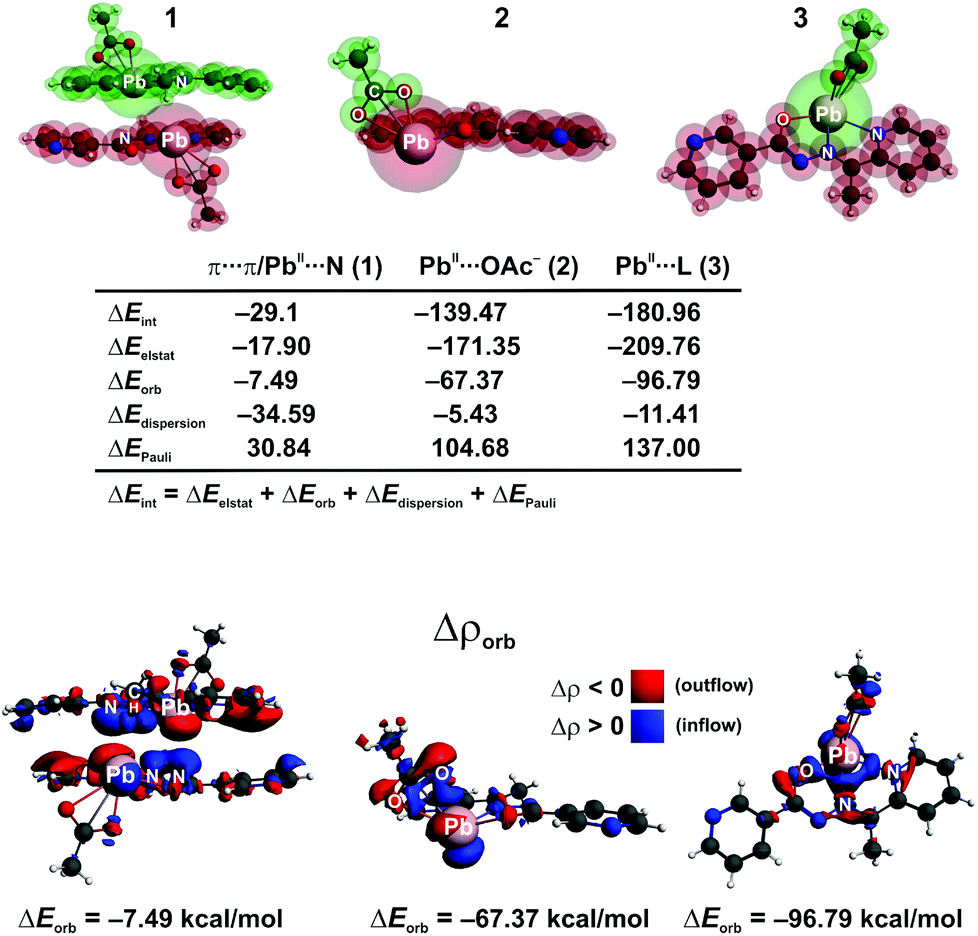 | ||
| Fig. 9 (Top) Cluster models of [PbL(OAc)]2 together with the fragmentation patterns 1–3 applied in the ETS-NOCV analysis. (Bottom) The overall deformation densities Δρorb of 1–3 with the corresponding energies ΔEorb. | ||
We have also performed the charge and energy decomposition calculations for various cluster models of the remaining compounds and the obtained results are shown in Fig. S2–S6 in the ESI.† For the sake of brevity, we will not discuss in detail each type of the bonding pattern and only the most relevant and interesting observations will be highlighted below.
The [PbL(NO)2] units in the structure of [PbL(NO)2]n are quasi-perpendicular (Fig. 4). This leads to the formation of “pure” tetrel bonding Pb⋯N (labelled as 1 in Fig. S2 in the ESI†). This strong interaction (ΔEint = −20.55 kcal mol−1) is from two equally important stabilizing factors, namely the dispersion (ΔEdispersion = −19.83 kcal mol−1) and electrostatic (ΔEelstat = −18.38 kcal mol−1) factors (Fig. S2 in the ESI†). The overall deformation density Δρorb demonstrates that the Pb⋯N interaction leads, similarly to [PbL(OAc)]2, to donor–acceptor electron density delocalizations PbII → π*(NN) and π(NN) → PbII (Fig. S2 in the ESI†). Similar to the Pb–OAc− and Pb–L bonds, the Pb–NO2− connection (labeled as 2 in Fig. S2 in the ESI†) is electrostatically dominated and it contains the dative-covalent contributions.
In the case of the [PbLN3]n structure, the [PbLN3] units are also aligned. However, the bonding pattern (labelled as 1 in Fig. S3 in the ESI†), emerging from the deformation density Δρorb, is quite distinctive as compared with the remaining structures (Fig. S3 in the ESI†). Surprisingly, not only the charge accumulation (blue color of Δρorb) in the Pb⋯N region is observed, but predominantly between the PbII centers (Fig. S3 in the ESI†). The latter is entirely non-intuitive because of the electrophilic nature of the lead centers. It is more clearly visible when considering the specific NOCV-based deformation density contributions Δρ1(1) + Δρ2(2), corresponding to ΔEorb(1) + ΔEorb(2) = −2.0 kcal mol−1, which clearly depicts the PbII⋯PbII interaction, and Δρ3(3), displaying the formation of the tetrel-bonding Pb⋯N as a part of the π⋯π stacking interaction (Fig. S3 in the ESI†). It could be anticipated, based on the above findings, that novel supramolecular architectures induced by non-covalent PbII⋯PbII interactions are expected to be prepared in the future.
It should be noted that the existence of weak and rare PbII⋯PbII bonding has also been suggested by Caruso and co-workers in the polymeric bis(pyrrolidinecarbodithioato)lead(II),84 and by Power et al. in Pb(C6H4-4-But)ArPr2.85 More recently, the AuI⋯PbII interaction has been determined in the structure of [Pb{HB(pz)3}Au(C6Cl5)2].86
The model 2 is suitable for the description of “pure” tetrel bonding Pb⋯N in [PbLN3]n (Fig. S3 in the ESI†). The results depicted as the second entry of the table demonstrate that the tetrel bonding in [PbLN3]n is predominantly electrostatic in nature (ΔEelstat = −35.57 kcal mol−1) followed by the orbital (ΔEorb = −14.66 kcal mol−1) and dispersion (ΔEdispersion = −10.76 kcal mol−1) contributions. It should be noted that the electrostatic and dispersion terms are equally important in the case of [PbL(NO2)]n (Fig. S2 in the ESI†). This clearly demonstrates that the same type of bonding may be constituted from quantitatively different forces in various crystal motifs.
As far as binding of anions is concerned, the results of ETS-NOCV calculations allowed us to observe that each bond Pb–X (X = NO3−, ClO4−, N3−, NCS− and OAc−) can generally be ascribed as dative-covalent (donor–acceptor) as it can be inferred from the change in the electron density (Δρorb) due to the formation of Pb–X bonds (Fig. 9 and Fig. S2–S6 in the ESI†). Furthermore, they are dominated by the electrostatic contribution followed by the orbital interaction and dispersion components. As far as the strength of the interaction is concerned one can summarize that the obtained interaction energies cover a wide range starting from very weak for X = L, ΔEint = −6.15 kcal mol−1 (Fig. S6 in the ESI†) in {[PbL]ClO4}n·nH2O through ΔEint = −74.53 kcal mol−1 for X = ClO4− in {[Pb(HL)(OAc)]ClO4}n (Fig. S4 in the ESI†) and ending up with the strongest one ΔEint = −180.96 kcal mol−1 for X = L in [PbL(OAc)]2 (Fig. 9). Similarly, the ClO4− anions in [H2L]ClO4 appeared to strongly interact with the cations [H2L]+ (ΔEint = −67.4 or −69.43 kcal mol−1 depending on the cluster model) predominantly through the electrostatic forces. These contributions constitute predominantly the ClO4−⋯π and NH⋯O interactions (Fig. S7 in the ESI†).
Conclusions
In summary, we have designed six new PbII complexes with the N′-(1-(2-pyridyl)ethylidene)nicotinohydrazide (HL) ligand. The nature of the inorganic anion is also important for the final structure. In all the complexes the PbII center exhibits a hemidirected coordination geometry with all the covalent bonds being concentrated on one hemisphere of the coordination sphere with the closest approach of two atoms on the other side varying from 151° to 232°. The sterically available PbII ion participates in tetrel bonding, as evidenced from the detailed structural analysis of the described complexes. These tetrel bonds play a key role in the supramolecular aggregation of building units of all the structures in the solid state. As a result of tetrel bonding, the structures of all the six compounds can be extended to a higher dimensional framework, which is further stabilized by π⋯π stacking interactions between the aromatic rings. The charge and energy decomposition calculations (ETS-NOCV) allowed us to identify and quantify the nature of non-covalent interactions that stabilize the obtained structures. Importantly, apart from the mentioned non-covalent interactions (π⋯π stacking and tetrel bonds), the ETS-NOCV method allowed us to identify and quantify for the first time a rare example of PbII⋯PbII interactions. It allows one to envisage that novel supramolecular architectures, driven by non-covalent PbII⋯PbII interactions, might be prepared in the close future. Finally, it has been determined that the anions bind to PbII predominantly through the electrostatic forces, however, the dative-covalent contributions also appeared to be important.Experimental
Materials
The Schiff base HL was prepared by following the reported method as described elsewhere69 and used without further purification. All other reagents and solvents used for synthesis and analysis were commercially available and used as received.Physical measurements
FTIR spectra were recorded on a Bruker Tensor 27 FTIR spectrometer. Microanalyses were performed with a Heraeus CHN-O-Rapid analyzer.DFT calculations
We have performed the DFT calculations based on the BLYP-D3/ZORA/TZP level as implemented in the ADF suit of programs.72,73 The charge and energy decomposition scheme ETS-NOCV77–79 has been applied in order to characterize the stability of the obtained structures.ETS-NOCV bonding analysis
The natural orbitals for chemical valence (NOCV)77–79 are eigenvectors that diagonalize the deformation density matrix:where Ci is a vector of coefficients, expanding Ψi in the basis of fragment orbitals λj; N is a total number of fragment λj orbitals. It was shown that the natural orbitals for chemical valence pairs (ψ−k,ψk) decompose the differential density Δρ into NOCV-contributions (Δρk):
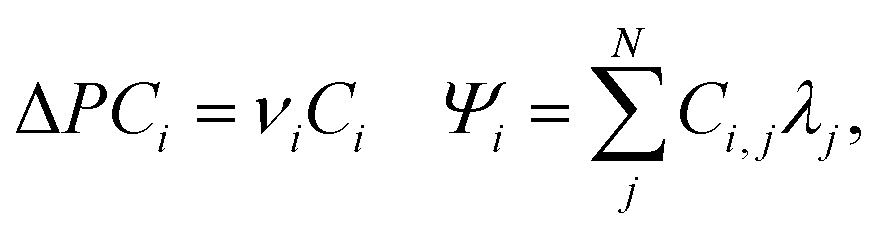
where νk and M stand for the NOCV eigenvalues and the number of basis functions, respectively. Visual inspection of deformation density plots (Δρk) helps to attribute symmetry and the direction of the charge flow. In addition, these pictures are enriched by providing the energetic estimations, ΔEorb(k), for each Δρk within the ETS-NOCV scheme.76–78 The exact formula, which links the ETS and NOCV methods, will be given in the next paragraph, after we briefly present the basic concept of the ETS scheme. In this method the total bonding energy, ΔEint, between interacting fragments, exhibiting the geometry as in the combined complex, is divided into three components: ΔEtotal = ΔEelstat + ΔEPauli + ΔEorb. The first term, ΔEelstat, corresponds to the classical electrostatic interaction between the promoted fragments as they are brought to their positions in the final complex. The second term, ΔEPauli, accounts for the repulsive Pauli interaction between occupied orbitals on the two fragments in the combined molecule. Finally, the last stabilizing term, ΔEorb, represents interactions between the occupied molecular orbitals of one fragment with the unoccupied molecular orbitals of the other fragment as well as mixing of occupied and virtual orbitals within the same fragment (inner-fragment polarization). This energy term may be linked to the electronic bonding effect coming from the formation of a chemical bond. In the combined ETS-NOCV scheme the orbital interaction term (ΔEorb) is expressed in terms of NOCV's eigenvalues (vk) as:
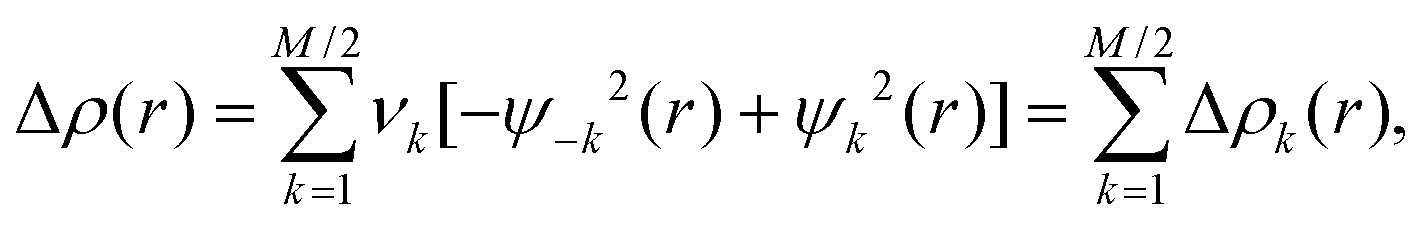
where Fi,iTS are diagonal Kohn–Sham matrix elements defined over NOCV with respect to the transition state (TS) density at the midpoint between density of the molecule and the sum of fragment densities. The above components ΔEorb(k) provide the energetic estimation of Δρk that may be related to the importance of a particular electron flow channel for the bonding between the considered molecular fragments. The ETS-NOCV analysis was done based on the Amsterdam Density Functional (ADF) package72,73 in which this scheme was implemented.
Synthesis of {[PbL]ClO4}n·nH2O
HL (0.024 g, 0.1 mmol) and Pb(ClO4)2 (0.041 g, 0.1 mmol) were placed in the main arm of a branched tube. MeOH (15 mL) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. X-ray suitable yellow prism-like crystals were formed during the next days in the cooler arm and were filtered off, washed with acetone and diethyl ether, and dried in air. Yield: 0.049 g (86.9%). FTIR, ν: 624, 910 and 1078 (ClO4−), 1638 and 3452 (H2O), 1590 (CO) cm−1. Anal. Calc. for C13H13ClN4O6Pb (563.92) (%): C 27.69, H 2.32 and N 9.94; found: C 28.1, H 2.2 and N 10.2.
Synthesis of {[Pb(HL)(OAc)]ClO4}n and [PbL(NO2)]n
HL (0.024 g, 0.1 mmol), Pb(ClO4)2 (0.041 g, 0.1 mmol) and NaOAc or NaNO2 (0.016 and 0.014 g, respectively; 0.2 mmol) were placed in the main arm of a branched tube. MeOH (15 mL) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. X-ray suitable crystals were formed during the next days in the cooler arm and were filtered off, washed with acetone and diethyl ether, and dried in air.O), 3469 (NH) cm−1. Anal. Calc. for C15H15ClN4O7Pb (605.96) (%): C 29.73, H 2.50 and N 9.25; found: C 30.1, H 2.6 and N 9.3.
O) cm−1. Anal. Calc. for C13H11N5O3Pb (492.46) (%): C 31.71, H 2.25 and N 14.22; found: C 31.5, H 2.4 and N 13.9.
Synthesis of [H2L]ClO4, [PbLN3]n and [Pb2(HL)2(NO3)2(NCS)2]
A solution of HL (0.024 g, 0.1 mmol) in MeOH (10 ml) was added dropwise to a solution of Pb(NO3)2 (0.033 g, 0.1 mmol) in the same solvent (10 ml). To this mixture, a solution of NaClO4 or NaN3 or NaNCS (0.024, 0.013 and 0.016 g, respectively; 0.2 mmol) in MeOH (5 ml) was added slowly under stirring. The resulting mixture was stirred at room temperature for 1 h and was left undisturbed for slow evaporation. X-ray suitable crystals were formed during the next days.O), 3448 and 3747 (NH) cm−1. Anal. Calc. for C13H13ClN4O5 (340.72) (%): C 45.83, H 3.85 and N 16.44; found: C 45.6, H 3.7 and N 16.2.
O), 1365 and 2035 (N3−) cm−1. Anal. Calc. for C13H11N7OPb (488.48) (%): C 31.97, H 2.27 and N 20.07; found: C 32.0, H 2.4 and N 19.4.
O), 2030 (NCS−), 3421 (NH) cm−1. Anal. Calc. for C28H24N12O8Pb2S2 (1135.09) (%): C 29.63, H 2.13 and N 14.81; found: C 29.8, H 2.0 and N 14.5.
Synthesis of [PbL(OAc)]2
A solution of HL (0.024 g, 0.1 mmol) in MeOH (10 ml) was added dropwise to a solution of Pb(OAc)2·3H2O (0.038 g, 0.1 mmol) in the same solvent (10 ml). The resulting mixture was stirred at room temperature for 1 h and left undisturbed for slow evaporation. X-ray suitable green plate-like crystals were formed during the next days. Yield: 0.040 g (79.1%). FTIR, ν: 1457 and 1507 (OAc−), 1574 (CO) cm−1. Anal. Calc. for C30H28N8O6Pb2 (1011.00) (%): C 35.64, H 2.79 and N 11.08; found: C 35.0, H 2.6 and N 11.4.
Single-crystal X-ray diffraction study
The X-ray data were collected on a Bruker four-circle single crystal diffractometer with a sealed graphite-monochromatised Mo-Kα (λ = 0.71073 Å) radiation tube and APEX-II CCD or SMART1000 CCD detectors. The structures were solved with SIR9287 and refined with the full-matrix least-squares procedure on |F2| by SHELXL-2013.88 Non-hydrogen atoms were anisotropically refined and the hydrogen atoms were placed at calculated positions in riding mode with temperature factors fixed at 1.2 times Ueq of the parent atoms. Figures were generated using the program Mercury.89
736 collected, 3288 unique, Rint = 0.069, R1(all) = 0.0604, wR2(all) = 0.0936.
487 collected, 2521 unique, Rint = 0.044, R1(all) = 0.0331, wR2(all) = 0.0829.
, a = 7.4765(3), b = 9.8748(5), c = 10.3866(9) Å, α = 85.982(6), β = 77.230(7), γ = 71.633(6)°, V = 709.77(8) Å3, Z = 2, ρ = 2.286 g cm−3, μ(Mo-Kα) = 11.899 mm−1, reflections: 14106 collected, 4097 unique, Rint = 0.038, R1(all) = 0.0263, wR2(all) = 0.0619.
335 collected, 4989 unique, Rint = 0.036, R1(all) = 0.0327, wR2(all) = 0.0535.
250 collected, 4587 unique, Rint = 0.044, R1(all) = 0.0413, wR2(all) = 0.0920.
Acknowledgements
We are grateful to the University of Maragheh for the financial support of this research. DFT calculations were partially performed using the PL-Grid Infrastructure and resources provided by the ACC Cyfronet AGH (Cracow, Poland).References
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376 CrossRef CAS PubMed.
- M. C. Das and P. K. Bharadwaj, J. Am. Chem. Soc., 2009, 131, 10942 CrossRef CAS PubMed.
- R. Wang, J. Zhang and L. Li, Inorg. Chem., 2009, 48, 7194 CrossRef CAS PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS PubMed.
- H. L. Jiang, Y. Tatsu, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586 CrossRef CAS PubMed.
- L. Duan, Z. H. Wu, J. P. Ma, X. W. Wu and Y. B. Dong, Inorg. Chem., 2010, 49, 11164 CrossRef CAS PubMed.
- P. Lama, A. Aijaz, E. C. Sañudo and P. K. Bharadwaj, Cryst. Growth Des., 2010, 10, 283 CAS.
- G. C. Xu, W. Zhang, X. M. Ma, Y. H. Chen, L. Zhang, H. L. Cai, Z. M. Wang, R. G. Xiong and S. Gao, J. Am. Chem. Soc., 2011, 133, 14948 CrossRef CAS PubMed.
- M. K. Sharma, I. Senkovska, S. Kaskel and P. K. Bharadwaj, Inorg. Chem., 2011, 50, 539 CrossRef CAS PubMed.
- P. Pachfule, R. Das, P. Poddar and R. Banerjee, Inorg. Chem., 2011, 50, 3855 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334 CrossRef CAS PubMed.
- N. B. Shustova, A. F. Cozzolino and M. Dincă, J. Am. Chem. Soc., 2012, 134, 19596 CrossRef CAS PubMed.
- P. Kanoo, S. K. Reddy, G. Kumari, R. Haldar, C. Narayana, S. Balasubramanian and T. K. Maji, Chem. Commun., 2012, 48, 8487 RSC.
- S. S. Nagarkar, A. K. Chaudhari and S. K. Ghosh, Inorg. Chem., 2012, 51, 572 CrossRef CAS PubMed.
- M. Ahmad, M. K. Sharma, R. Das, P. Poddar and P. K. Bharadwaj, Cryst. Growth Des., 2012, 12, 1571 CAS.
- B. A. Blight, R. G. Nicolas, F. Kleitz, R. Y. Wang and S. Wang, Inorg. Chem., 2013, 52, 1673 CrossRef CAS PubMed.
- Y. Z. Tang, M. Zhou, J. Huang, Y. H. Tan, J. S. Wu and H. R. Wen, Inorg. Chem., 2013, 52, 1679 CrossRef CAS PubMed.
- A. Santra, I. Senkovska, S. Kaskel and P. K. Bharadwaj, Inorg. Chem., 2013, 52, 7358 CrossRef CAS PubMed.
- Z.-W. Wang, C.-C. Ji, J. Li, Z.-J. Guo, Y.-Z. Li and H.-G. Zheng, Cryst. Growth Des., 2009, 9, 475 CAS.
- F. Luo, Y.-X. Che and J.-M. Zheng, Cryst. Growth Des., 2009, 9, 1066 CAS.
- R. Sarma, D. Kalita and J. B. Baruah, Dalton Trans., 2009, 7428 RSC.
- D. Sun, H.-R. Xu, C.-F. Yang, Z.-H. Wei, N. Zhang, R.-B. Huang and L.-S. Zheng, Cryst. Growth Des., 2010, 10, 4642 CAS.
- K.-L. Zhang, Y. Chang, C.-T. Hou, G.-W. Diao, R. Wu and S. W. Ng, CrystEngComm, 2010, 12, 1194 RSC.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366 CrossRef CAS PubMed.
- J. M. Zayed, N. Nouvel, U. Rauwald and O. A. Scherman, Chem. Soc. Rev., 2010, 39, 2806 RSC.
- J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 723 CrossRef CAS PubMed.
- J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef.
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725 CrossRef CAS.
- P. Metrangolo and G. Resnati, Chem. – Eur. J., 2001, 7, 2511 CrossRef CAS.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772 RSC.
- P. Metrangolo and G. Resnati, Halogen Bonding: Fundamentals and Applications (Stucture and Bonding), Springer, Heidelberg, 2010 Search PubMed.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2013, 15, 7249 RSC.
- P. Metrangolo and G. Resnati, Chem. Commun., 2013, 49, 1783 RSC.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072 CrossRef CAS PubMed.
- M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613 CrossRef CAS PubMed.
- C. Bleiholder, D. B. Werz, H. Köppel and R. Gleiter, J. Am. Chem. Soc., 2006, 128, 2666 CrossRef CAS PubMed.
- W. Z. Wang, B. M. Ji and Y. Zhang, J. Phys. Chem. A, 2009, 113, 8132 CrossRef CAS PubMed.
- P. Metrangolo and G. Resnati, Nat. Chem., 2012, 4, 437 CrossRef CAS PubMed.
- M. Bai, S. P. Thomas, R. Kottokkaran, S. K. Nayak, P. C. Ramamurthy and T. N. G. Row, Cryst. Growth Des., 2014, 14, 459 CAS.
- M. D. Esrafili, F. Mohammadian-Sabet and M. Solimannejad, Struct. Chem., 2014, 25, 1197 CrossRef CAS.
- U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2014, 118, 3183 CrossRef CAS PubMed.
- V. de P. N. Nziko and S. Scheiner, J. Phys. Chem. A, 2014, 118, 10849 CrossRef CAS PubMed.
- L. M. Azofra, I. Alkorta and S. Scheiner, J. Phys. Chem. A, 2015, 119, 535 CrossRef CAS PubMed.
- M. S. Balakrishna, D. J. Eisler and T. Chivers, Chem. Soc. Rev., 2007, 36, 650 RSC.
- S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem. – Eur. J., 2011, 17, 6034 CrossRef CAS.
- S. Scheiner, Acc. Chem. Res., 2013, 46, 280 CrossRef CAS PubMed.
- J. S. Murray, P. Lane and P. Politzer, J. Mol. Model., 2009, 15, 7239 Search PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef PubMed.
- A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model., 2013, 19, 2739 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Commun., 2014, 50, 12626 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. – Eur. J., 2014, 20, 10245 CrossRef PubMed.
- S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Phys. Chem. Chem. Phys., 2014, 16, 19192 RSC.
- E. C. Escudero-Adán, A. Bauzá, A. Frontera and P. Ballester, ChemPhysChem, 2015, 16, 2530 CrossRef PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Rec., 2016, 16, 473 CrossRef PubMed.
- M. Servati Gargari, V. Stilinović, A. Bauzá, A. Frontera, P. McArdle, D. Van Derveer, S. W. Ng and G. Mahmoudi, Chem. – Eur. J., 2015, 21, 17951 CrossRef CAS PubMed.
- G. Mahmoudi, A. Bauzá and A. Frontera, Dalton Trans., 2016, 45, 4965 RSC.
- G. Mahmoudi, A. Bauzá, M. Amini, E. Molins, J. T. Mague and A. Frontera, Dalton Trans., 2016, 45, 10708 RSC.
- G. Mahmoudi, V. Stilinović, A. Bauzá, A. Frontera, A. Bartyzel, C. Ruiz-Pérez and A. M. Kirillov, RSC Adv., 2016, 6, 60385 RSC.
- L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Inorg. Chem., 1998, 37, 1853 CrossRef CAS.
- R. L. Davidovich, V. Stavila, D. V. Marinin, E. I. Voit and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316 CrossRef CAS.
- F. Yi-Min and T. Xi-Shi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o65 Search PubMed.
- A. L. Gavrilova and B. Bosnich, Chem. Rev., 2004, 104, 349 CrossRef CAS PubMed.
- H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 379 RSC.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS and references therein.
- E. J. Baerends, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. Fonseca Guerra, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, G. van Kessel, F. Kootstra, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, J. I. Rodríguez, P. Ros, P. R. T. Schipper, G. Schreckenbach, M. Seth, J. G. Snijders, M. Solà, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo, A. L. Yakovlev and T. Ziegler, ADF2012.01, Theoretical Chemistry, Vrije Universiteit, Amsterdam Search PubMed.
- T. van der Wijst, C. Fonseca Guerra, M. Swart, F. M. Bickelhaupt and B. Lippert, Angew. Chem., Int. Ed., 2009, 48, 3285 CrossRef CAS PubMed.
- C. Fonseca Guerra, T. van der Wijst, J. Poater, M. Swart and F. Matthias Bickelhaupt, Theor. Chem. Acc., 2010, 125, 245 CrossRef.
- W. Gao, H. Feng, X. Xuan and L. Chen, J. Mol. Model., 2012, 18, 4577 CrossRef CAS PubMed.
- M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347 CrossRef CAS PubMed.
- A. Michalak, M. Mitoraj and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933 CrossRef CAS PubMed.
- M. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS PubMed.
- A. Morsali, A. R. Mahjoub, S. J. Darzi and M. J. Soltanian, Z. Anorg. Allg. Chem., 2003, 629, 2596 CrossRef CAS.
- F. Marandi, B. Mirtamizdoust, S. Chantrapromma and H. K. Fun, Z. Anorg. Allg. Chem., 2007, 633, 1329 CrossRef CAS.
- F. Marandi, M. Mottaghi, G. Meyer and I. Pantenburg, Z. Anorg. Allg. Chem., 2009, 635, 165 CrossRef CAS.
- V. D. Schwade, E. Faoro and E. Schulz Lang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2016, 72, 525 CAS.
- F. Caruso, M. L. Chan and M. Rossi, Inorg. Chem., 1997, 36, 3609 CrossRef CAS PubMed.
- S. Hino, M. Olmstead, A. D. Phillips, R. J. Wright and P. P. Power, Inorg. Chem., 2004, 43, 7346 CrossRef CAS PubMed.
- R. Echeverría, J. López-de-Luzuriaga, M. Monge and M. E. Olmos, Chem. Sci., 2015, 6, 2022 RSC.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7 and cif files. CCDC 1059556–1059562. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00477f |
| This journal is © the Partner Organisations 2017 |