
Synthesis, crystal structure and spectroscopic studies of a series of hexavanadate hybrids with multiple functional groups†
Bo
Huang‡
a,
Zicheng
Xiao‡
a,
Baolin
Wu
a,
Xiaokang
Hu
a,
Xunliang
Hu
a,
Pingfan
Wu
*a and
Yongge
Wei
*ab
aInstitute of POM-based Materials, Hubei University of Technology, Wuhan 430068, China. E-mail: pingfanwu-111@163.com; yonggewei@tsinghua.edu.cn
bDepartment of Chemistry, Tsinghua University, Beijing 100084, China
First published on 15th November 2016
Abstract
Three new derivatives of hexavanadate with remote methacryl, phthalyl and tetrahydrophthalyl groups, respectively, were synthesized by esterification of pentaerythritol-derivatized hexavanadate. These new compounds were characterized by FT-IR, 1H NMR, UV-vis, elemental analysis and single-crystal X-ray diffraction. Their thermal stabilities were also tested by TGA analyses. Through such syntheses, we have successfully introduced the carbon–carbon double bond and/or carboxylic groups into the polyoxovanadates which could be utilized to build more complex structures. We also discuss the influence of anhydrides with different functional groups on the esterification of the pentaerythritol derivatized hexavanadate.
Introduction
Polyoxometalates (POMs) are important inorganic materials which are widely favored by researchers on account of their diverse structures and versatile physical and chemical properties.1–4 In recent years, covalently linking the POMs with organic molecules to obtain more excellent hybrid materials has become an important research direction as it can be more beneficial to the organic–inorganic interaction and the stability of the hybrids.5–7As one important family of the POMs, polyoxovanadates (POVs) have attracted much attention in recent years because of their fascinating photochemical,8–10 magnetic,11,12 catalytic13,14 and biochemical properties.15,16 Although the organic modification of POMs has made great progress in the past few decades and large numbers of POM-based hybrid materials have been obtained, the synthesis of complicated POV hybrids with multiple functional groups is still a challenge.17–19 One synthetic approach for the preparation of POV hybrids is based on direct functionalization, where an alkoxo ligand reacted with the POV precursors.20–25 However, this approach often faces problems such as redox side reactions and low yield of the hybrid products because of the strong oxidative properties of the high valence-state vanadium. The second approach to construct POV hybrids relies on post functionalization where an organic substrate will react further with the preformed POV hybrid containing reactive groups in its organic segments.26,27 The trialkoxo-derived hexavanadates which were firstly reported by Zubieta are usually employed as POV building blocks for further functionalization.28,29 Regretfully, this approach is still limited by the lack of suitable POV platforms and convenient protocols. Thus it is meaningful to develop new ways for the functionalization of POVs.
Our group has devoted itself to research on the post functionalization of POVs. In the case of constructing complicated POM hybrids with multiple functional groups, one promising strategy is step-by-step functionalization, which firstly introduces a bridging ligand into the POM cluster to obtain a hybrid that can be further modified. By introducing different bridging ligands, more organic reactions can be utilized for the post functionalization of POMs. This strategy has already been successfully applied to the post functionalization of Anderson, Keggin and Dawson type POMs in the past few years.30–33 Recently, our group has developed a synthetic protocol which utilizes succinic acid as a bridging ligand to combine the pentaerythritol-derivatized hexavanadate (Bu4N)2[V6O13{(OCH2)3CCH2OH}2] (compound 1) with organic amines.34 However, on account of the lack of exploration of new ligands, the organic modification of POVs was limited to few reactions hitherto, such as esterification and amidation, which are easy for operation but difficult to link complex or inactive groups. Thus we hope to employ different bridging ligands to develop new synthetic approaches to construct POV-based materials, especially the bridging ligands with multiple functional groups which can be used to build complicated hybrids. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond is a highly reactive group which has already attracted much attention in POM chemistry and the variety of reactions it offers, such as Diels–Alder35,36 and free radical reactions,37,38 have been taken advantage of for further modification of POMs. Although a few suitable polymer-based POM hybrids have been reported to solve the poor processability of pure POMs as inorganic crystalline materials,39,40 this strategy was often limited by the steric hindrance and electrostatic effect of POMs. Towards the rational design and syntheses of POV-based hybrids, it is significant to introduce the CC double bond into the POV system. However, the interaction between the CC double bond and the POVs faces several problems as the strong oxidation property of POVs and the reactive unsaturated bond often cause unanticipated side reactions. Five years ago, our group developed a post modification protocol which employs DMAP catalyzed esterification between the pentaerythritol-derivatized hexavanadate and saturated fatty acid anhydrides.41 In this paper, we discover that this protocol can also be extended to unsaturated organic compounds containing the CC double bond group by the careful optimization of reaction conditions, and three new derivatives of hexavanadates with remote groups can be obtained (Bu4N)2[V6O13{(OCH2)3CCH2OOCC(CH3)CH2}2] (compound 2),§ (Bu4N)2[V6O13{(OCH2)3CCH2OOC(C6H4)COOH}2] (compound 3)¶ and (Bu4N)2[V6O13{(OCH2)3CCH2OOC(C6H8) COOH}2] (compound 4).|| To the best of our knowledge, this is the first example to combine phthalyl and tetrahydrophthalyl groups to the POM cluster. In particular, the tetrahydrophthalyl group was selected as an organic ligand with a bifunctional structural unit for further modification.
C double bond is a highly reactive group which has already attracted much attention in POM chemistry and the variety of reactions it offers, such as Diels–Alder35,36 and free radical reactions,37,38 have been taken advantage of for further modification of POMs. Although a few suitable polymer-based POM hybrids have been reported to solve the poor processability of pure POMs as inorganic crystalline materials,39,40 this strategy was often limited by the steric hindrance and electrostatic effect of POMs. Towards the rational design and syntheses of POV-based hybrids, it is significant to introduce the CC double bond into the POV system. However, the interaction between the CC double bond and the POVs faces several problems as the strong oxidation property of POVs and the reactive unsaturated bond often cause unanticipated side reactions. Five years ago, our group developed a post modification protocol which employs DMAP catalyzed esterification between the pentaerythritol-derivatized hexavanadate and saturated fatty acid anhydrides.41 In this paper, we discover that this protocol can also be extended to unsaturated organic compounds containing the CC double bond group by the careful optimization of reaction conditions, and three new derivatives of hexavanadates with remote groups can be obtained (Bu4N)2[V6O13{(OCH2)3CCH2OOCC(CH3)CH2}2] (compound 2),§ (Bu4N)2[V6O13{(OCH2)3CCH2OOC(C6H4)COOH}2] (compound 3)¶ and (Bu4N)2[V6O13{(OCH2)3CCH2OOC(C6H8) COOH}2] (compound 4).|| To the best of our knowledge, this is the first example to combine phthalyl and tetrahydrophthalyl groups to the POM cluster. In particular, the tetrahydrophthalyl group was selected as an organic ligand with a bifunctional structural unit for further modification.
Results and discussion
Synthesis
Compound 1 was prepared according to the literature method.42 Compounds 2–4 were synthesized by esterification of compound 1 with methacrylic anhydride, phthalic anhydride and tetrahydrophthalic anhydride (THPA) with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in acetonitrile at 50 °C, respectively (Scheme 1). DMAP (4-(N,N-dimethylamino)pyridine) was used as a catalyst to raise the yield of the target products, and was added in a catalytic amount, and triethylamine was used as an auxiliary base. The crude products were obtained on evaporation of the acetone/water mixture within 3 days and the X-ray quality single crystals were obtained by the slow diffusion of diethyl ether into an acetone solution of the crude product. The yield of compounds 2 and 3 is a bit lower than compound 4. This is due to the conjugative effect in methacrylic anhydride and phthalic anhydride which reduces the activity of the acyl cation, further making the nucleophilic attack more difficult.
:2 in acetonitrile at 50 °C, respectively (Scheme 1). DMAP (4-(N,N-dimethylamino)pyridine) was used as a catalyst to raise the yield of the target products, and was added in a catalytic amount, and triethylamine was used as an auxiliary base. The crude products were obtained on evaporation of the acetone/water mixture within 3 days and the X-ray quality single crystals were obtained by the slow diffusion of diethyl ether into an acetone solution of the crude product. The yield of compounds 2 and 3 is a bit lower than compound 4. This is due to the conjugative effect in methacrylic anhydride and phthalic anhydride which reduces the activity of the acyl cation, further making the nucleophilic attack more difficult.
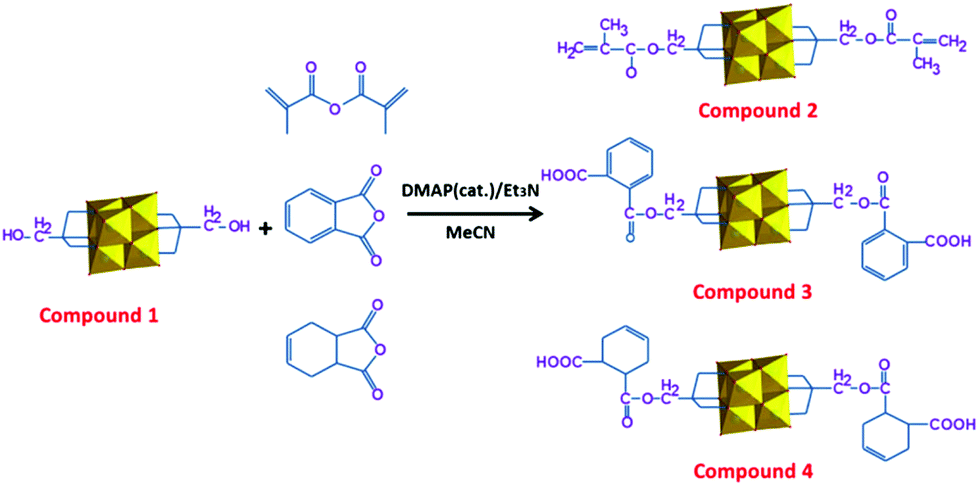 | ||
| Scheme 1 Synthetic approaches to compounds 2–4. | ||
Structure of the polyanions
The structures of the compounds 2–4 were determined by single-crystal X-ray diffraction. All the three compounds crystallize in a monoclinic system with the space group symmetry P21/c. Their structures are all composed of an organically-derivatized hexavanadate cluster anion and two tetrabutyl ammonium cations in an asymmetric unit, indicating that the valence of the vanadium atoms does not change after the reaction. As shown in Fig. 1, the cluster anion structures of compounds 2–4 are all derived from the Lindqvist-type polyanion {V6O19}, in which the six vanadium atoms form an octahedron and two trialkoxo ligands occupy the opposite sites of the octahedron. The acyl groups are linked to the pentaerythritol-derived hexavanadate through a C–O bond with a length of 1.423(6), 1.441(4) and 1.433(4) Å in compounds 2–4, respectively. In the anion structure of compound 3, there is a remarkable distortion between the benzene ring and the acyl group with the torsion angle (O12–C6–C7–C12) as 55.2° because of the steric hindrance. There is an interesting supramolecular assembly originating from the hydrogen bonding interactions between the terminal oxygen atoms of the V6 cluster and the carboxyl group in compound 3, in which each polyanion is linked to two adjacent polyanions by a couple of O–H⋯O hydrogen bonds, O14–H⋯O3, 1.987 Å, forming a 2D network (Fig. 2). The anion structure of compound 4 shows a C–C double bond (C9–C10, 1.30 Å) and a carboxylic group. Similar to compound 3, the anions of compound 4 also form a 2D network through hydrogen bonds between the carboxyl group and the terminal oxygen of the cluster (O14A–H⋯O6, 1.98 Å). Interestingly, compound 4 can also form intramolecular hydrogen bonding (O13B–H⋯O5, 2.82 Å), which results in a disorder in the crystal structure (Fig. 3). This is because the cyclohexenyl ring in compound 4 can rotate through the carbon–carbon single bond, which is different from compound 3, as the rotation of the benzene ring is limited by conjugative effects.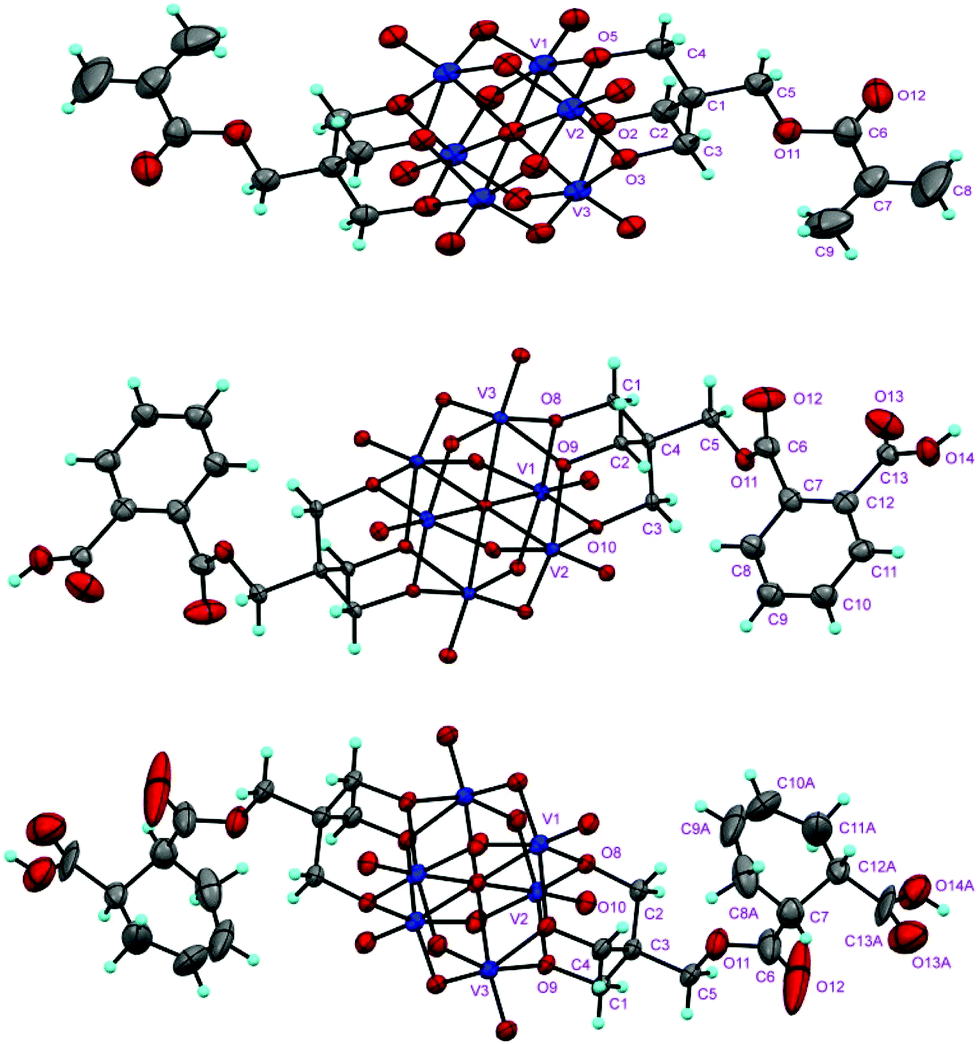 | ||
| Fig. 1 ORTEP drawings of the cluster anion structures of compounds 2 (upper), 3 (middle) and 4 (below). Blue sphere: V; red sphere: O; grey sphere: C; light blue sphere: H. Thermal ellipsoids are drawn at the 30% probability level. | ||
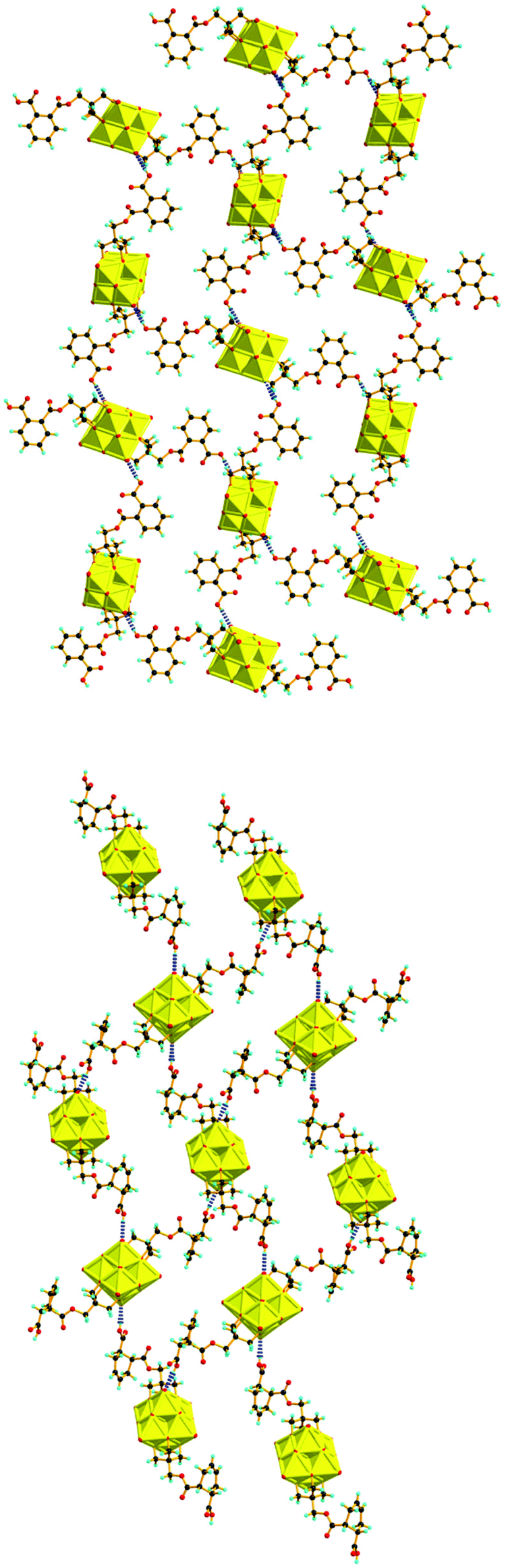 | ||
| Fig. 2 2D network formed through C–H⋯O hydrogen bonds in the crystals of compound 3 (upper) and compound 4 (lower). | ||
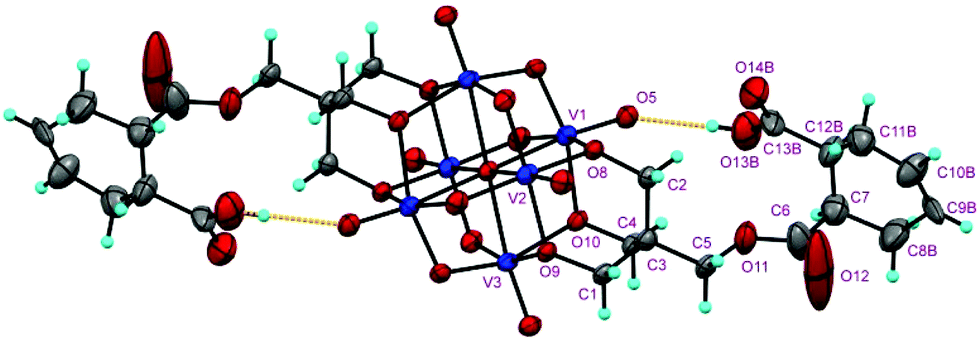 | ||
| Fig. 3 Intramolecular hydrogen bond of the anion of compound 4 in the crystal. | ||
IR spectroscopy
Fig. S1–S3† show the FT-IR spectra of compounds 2–4 in the range of 4000–500 cm−1. The characteristic features of the hexavanadate cluster can be found near 950 cm−1 (ν(V–Ot)), 810 cm−1 and near 715 cm−1 (ν(V–Ob–V)) in all spectra. The bands around 2960 cm−1 and 2800 cm−1 are assigned to νas(C–H) and νs(C–H) of the C–H bonds in these compounds, respectively, and the bands around 1470 cm−1 and 1380 cm−1 are assigned to δas(C–H) and δs(C–H) of the methylene and methyl groups in the ligand and (Bu4N)+ cations. The absorption peak near 3430 cm−1 in the spectra of compounds 3 and 4 was attributed to ν(O–H) of the carboxyl group. The strong absorption peak around 1720 cm−1 was attributed to the CO vibration, indicating that the carboxylic group has been bonded to the POV cluster by esterification and the esterification will not destroy the structure of hexavanadates. Obviously, the wavenumbers of the CO vibration for compounds 2 and 3 are found near 1722 and 1730 cm−1, respectively, which are lower than the wavenumber near 1735 cm−1 for compound 4 because of the conjugative effect between the ester group and carbon–carbon double bond in compound 2 and the benzene ring in compound 3, respectively. The bands around 1000–1100 cm−1 are assigned to ν(C–O) of the tris(hydroxymethyl) ligand; compared with compound 1, the signal of ν(C–O) for compounds 2–4 shifted from 1040 cm−1 to 1062, 1055, and 1095 cm−1, respectively. In the high-frequency region of compound 3, the aromatic ν(Ar–H) bands (ν > 3000 cm−1) are hardly visible due to their low intensity, but absorption peaks of benzene rings around 1500 cm−1 can be found.
NMR spectroscopy
The 1H NMR spectra of compounds 2–4 in d6-DMSO show clearly resolved signals, all of which can be unambiguously assigned and the integration matched well with the assumed structures (see Fig. S4–S6†). The carboxyl proton in compound 3 gives a singlet at 13.44 ppm, while the proton of the carboxyl group in compound 4 appears at 12.36 ppm. The chemical shift of the proton of the carboxyl group is usually seen near 12 ppm, and the carboxyl group is connected with a saturated carbon atom in compound 4, which has little effect on the chemical shift of the carboxyl proton. For compound 3, the carboxyl is in the unmasking area of the benzene ring, which results in the chemical shift of the carboxyl proton shifting downfield. The aryl protons show obviously resolved signals at 7.55–7.77 ppm in compound 3. The protons on the carbon–carbon double bond show two obviously resolved signals at 5.66 and 5.95 ppm in compound 2, and at 5.55 and 5.59 ppm in compound 4, which are due to the magnetically non-equivalent protons on the carbon–carbon double bond in compound 2, and the different chemical environment of the carbon–carbon double bond in compound 4. The protons of methyl on the carbon–carbon double bond in compound 3 shift downfield because of the negative inductive effect of the carbon–carbon double bond, which reduces the electron density and the protons show resolved signals at 1.84 ppm. As seen from the 1H NMR spectrum of each compound, the ratio of ligands to hexavanadate ion is approximately 2:1 by integrating the methylene in the ligands against the tetrabutylammonium ion, which is consistent with each compound being the post-functionalized product.
Thermal stability analysis
Fig. 4 shows the TGA curves of compounds 2–4. The weight loss is due to the decomposition of the organic ligands and the collapse of the whole framework. For compounds 2–4, their structure remains intact until it is heated to 248, 214 and 216 °C, respectively. Compared to compound 1 (see Fig. S10†), the stability of compounds 3 and 4 becomes low because of the increase of the organic area by the organic modification of polyoxovanadates. Interestingly, compound 2 is more stable than compounds 3 and 4, and even superior to compound 1, which gives better conditions to further modify the highly active carbon–carbon double bond. On the other hand, methacrylate compounds have a long research history, and are widely studied by researchers in view of their various applications. As a methacrylate type pentaerythritol derivatized hexavanadate, compound 2 shows broad prospects for further development. | ||
| Fig. 4 Thermogravimetric curves for compounds 2 (green), 3 (red), and 4 (blue). | ||
Conclusions
In conclusion, we have successfully synthesized a series of pentaerythritol-derivatized hexavanadates with multiple functional groups by DMAP-catalyzed esterification and the products have been characterized by FT-IR, 1H NMR, UV-vis, elemental analysis and single-crystal X-ray diffraction. Thermal stability has been characterized by TGA. The self-assembly of these crystals has also been explored. These compounds could be further modified through their functional groups step-by-step, such as the varied reactions of the CC double bond and the carboxyl groups. Compared to succinic acid, phthalic acid was rigid and has a π–π conjugative effect between the benzene ring and carbonyl group which might be employed to build photosensitive POV hybrids with large conjugated systems. The tetrahydrophthalyl group was a bridging ligand with a bifunctional structural unit which could be used to construct complicated POV-based hybrids. Furthermore, the crystal structures of compounds 3 and 4 are different from succinic acid derivatized hexavanadates, which show a special steric configuration and may open a new way to design and synthesize POV-based inorganic–organic hybrid materials.
Experimental section
Materials and general measurements
All chemicals were purchased from commercial sources and used without further purification except for CH3CN, which was dried by distillation in the presence of CaH2 prior to use. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Vector22 infrared spectrometer using KBr pellets; 1H NMR spectra were recorded on an AV600 NMR spectrometer with DMSO-d6 solvent; UV-vis absorption spectra were recorded on a TU-1901 double-beam ultraviolet-visible spectrophotometer; single-crystal X-ray diffraction was performed on an Agilent Xcalibur Gemini diffractometer; elemental analyses were carried out using a vario EL CHNS elemental analyser; TGA was performed at a rate of 10 K min−1 under an Ar stream using a METTLER TOLEDO apparatus.Synthesis of (Bu4N)2[V6O13{(OCH2)3CCH2OOCC(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH2}2] (compound 2)
CH2}2] (compound 2)
A mixture of compound 1 (2.0 g, 1.6 mmol), methacrylic anhydride (0.49 g, 3.2 mmol), 4-(N,N-dimethylamino)pyridine (0.008 g, 0.07 mmol), triethylamine (0.32 g, 3.2 mmol) and 50 ml acetonitrile was heated to 50 °C in an oil bath and stirred for 24 h. The resulting red solution was cooled to room temperature and filtered to remove the insoluble impurities. The filtrate was then evaporated to give a viscous oil. The oil was washed carefully with ethyl acetate several times and the residue was resolved in 50 ml acetonitrile. The crude products were obtained on evaporation of the acetone/water mixture within 3 days in about 48% yield based on compound 1. X-ray quality single crystals were obtained by the slow diffusion of diethyl ether into an acetone solution of the crude product.
NMR (600 MHz, d6-DMSO, ppm): 5.95 (m, CH2, 2H), 5.66 (m, CH2, 2H), 4.94 (s, CH2, 12H), 4.01 (s, CH2, 4H), 1.84 (m, CH3, 6H), 3.18 (t, CH2, 16H), 1.58 (m, CH2, 16H), 1.34 (m, CH2, 16H), 0.95 (t, CH3, 24H); IR spectrum (cm−1, KBr pullets): 2959 (m), 2932 (m), 2873 (m), 1722 (vs), 1637 (w), 1470 (m), 1384 (s), 1293 (w), 1174 (w), 1062 (w), 950 (vs), 810 (s), 720 (s) cm−1; UV-vis (MeCN): λmax = 354 nm. Elemental analysis: (Mw = 1400.96 g mol−1), % calculated: N 1.99, C 42.87, H 7.05; % found: N 2.12, C 42.79, H 6.08.
Synthesis of (Bu4N)2[V6O13{(OCH2)3CCH2OOC(C6H4)COOH}2] (compound 3)
The synthesis of compound 3 was similar to compound 2 except using phthalic anhydride instead of methacrylic anhydride in about 50% yield. X-ray quality single crystals were obtained by the slow diffusion of diethyl ether into an acetonitrile solution of the crude product.NMR (600 MHz, d6-DMSO, ppm): 13.44 (s, COOH, 2H), 7.77 (d, aromatic, 2H), 7.61 (m, aromatic, 4H), 7.55 (m, aromatic, 2H), 4.88 (s, CH2, 12H), 4.07 (s, CH2, 4H), 3.18 (t, CH2, 16H), 1.57 (m, CH2, 16H), 1.31 (m, CH2, 16H), 0.93 (t, CH3, 24H); IR spectrum (cm−1, KBr pullets): 3438 (m), 2960 (m), 2873 (m), 1731 (s), 1467 (m), 1379 (w), 1265 (w), 1127 (w), 1055 (m), 952 (vs), 807 (s), 719 (s); UV-vis (MeCN): λmax = 362 nm. Elemental analysis: (Mw = 1561.04 g mol−1), % calculated: N 1.79, C 44.63, H 6.33; % found: N 1.99, C 43.55, H 6.68.
Synthesis of (Bu4N)2[V6O13{(OCH2)3CCH2OOC(C6H8)COOH}2] (compound 4)
The synthesis of compound 4 was similar to compound 2 except using tetrahydrophthalic anhydride instead of methacrylic anhydride in about 65% yield. X-ray quality single crystals were obtained by the slow diffusion of diethyl ether into an acetonitrile solution of the crude product.NMR (600 MHz, d6-DMSO, ppm): 12.36 (s, COOH, 2H), 5.59 (m, CH, 2H), 5.55 (m, CH, 2H), 2.95 (m, CH, 2H), 2.89 (m, CH, 2H), 2.25 (m, CH2, 4H), 2.39 (m, CH2, 4H), 4.89 (s, CH2, 12H), 3.87 (s, CH2, 4H), 3.17 (t, CH2, 16H), 1.57 (m, CH2, 16H), 1.32 (m, CH2, 16H), 0.94 (t, CH3, 24H); IR spectrum (cm−1, KBr pullets): 3423 (m), 2960 (m), 2873 (m), 1735 (s), 1468 (m), 1382 (w), 1130 (w), 1095 (w), 952 (vs), 808 (s), 719 (s); UV-vis (MeCN): λmax = 353 nm. Elemental analysis: (Mw = 1569.10 g mol−1), % calculated: N 1.79, C 44.40, H 6.81; % found: N 2.36, C 41.88, H 6.27.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (no. 21271068, 21401050 and 21471087), the Natural Science Foundation of Hubei Province (no. 2015CFA131), and the Wuhan Applied Basic Research Program (no. 2014010101010020).Notes and references
- M. T. Pope, in Comprehensive Coordination Chemistry II, ed. A. G. Wedd, Elsevier, Oxford, 2004, p. 635 Search PubMed.
- Polyoxometalate Chemistry: From Topology via Self-Assembly to Applications, ed. M. T. Pope and A. Müller, Kluwer, Dordrecht, 2001 Search PubMed.
- C. L. Hill, in Comprehensive Coordination Chemistry II, ed. A. G. Wedd, Elsevier, Oxford, 2004, p. 679 Search PubMed.
- Polyoxometalate Chemistry for Nano-Composite Design, ed. T. Yamase and M. T. Pope, Kluwer Academic, New York, 2002 Search PubMed.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837 RSC.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC.
- J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217 CrossRef CAS.
- M. Santoni, G. La Ganga, V. M. Nardo, M. Natali, F. Puntoriero, F. Scandola and S. Campagna, J. Am. Chem. Soc., 2014, 136, 8189 CrossRef CAS PubMed.
- A. Seliverstov and C. Streb, Chem. – Eur. J., 2014, 20, 9733 CrossRef CAS PubMed.
- P. Kogerler, B. Tsukeblat and A. Müller, Dalton Trans., 2010, 39, 21 RSC.
- C. Daniel and H. Hartl, J. Am. Chem. Soc., 2009, 131, 5101 CrossRef CAS PubMed.
- P.-C. Yin, A. Bayaguud, P. Cheng, F. Haso, L. Hu, J. Wang, D. Vezenov, R. E. Winans, J. Hao, T. Li, Y.-G. Wei and T.-B. Liu, Chem. – Eur. J., 2014, 20, 9589 CrossRef CAS PubMed.
- P.-C. Yin, J. Wang, Z.-C. Xiao, P.-F. Wu, Y.-G. Wei and T.-B. Liu, Chem. – Eur. J., 2012, 18, 9174 CrossRef CAS PubMed.
- J. C. Pessoa, S. Etcheverry and M. Gambino, Coord. Chem. Rev., 2015, 301, 24 CrossRef.
- M. Aureliano and C. A. Ohlin, J. Inorg. Biochem., 2014, 137, 123 CrossRef CAS PubMed.
- K. Y. Monakhov, W. Bensch and P. Kogerler, Chem. Soc. Rev., 2015, 44, 8443 RSC.
- Y. Hayashi, Coord. Chem. Rev., 2011, 255, 2270 CrossRef CAS.
- Y.-Z. Gao, Y.-N. Chi and C.-W. Hu, Polyhedron, 2014, 83, 242 CrossRef CAS.
- J. W. Han, K. I. Hardcastle and C. L. Hill, Eur. J. Inorg. Chem., 2006, 2598 CrossRef CAS.
- L. Zhang and W. Schmitt, J. Am. Chem. Soc., 2011, 133, 11240 CrossRef CAS PubMed.
- C. Allain, S. Favette, L.-M. Chamoreau, J. Vaissermann, L. Ruhlmann and B. Hasenknopf, Eur. J. Inorg. Chem., 2008, 3433 CrossRef CAS.
- C. Aronica, G. Chastanet, E. Zueva, S. A. Borshch, J. M. Clemente-Juan and D. Luneau, J. Am. Chem. Soc., 2008, 130, 2365 CrossRef CAS PubMed.
- M. P. Santoni, A. K. Pal, G. S. Hanan, M. C. Tang, K. Venne, A. Furtos, P. Menard-Tremblay, C. Malveau and B. Hasenknopf, Chem. Commun., 2012, 48(2), 200–202 RSC.
- J. W. Han and C. L. Hill, J. Am. Chem. Soc., 2007, 129(49), 15094–15095 CrossRef CAS PubMed.
- D. Li, J. Song, P. C. Yin, S. Simotwo, A. J. Bassler, Y. Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. B. Liu, J. Am. Chem. Soc., 2011, 133, 14010 CrossRef CAS PubMed.
- P. C. Yin, P. F. Wu, Z. C. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. B. Liu and Y. G. Wei, Angew. Chem., Int. Ed., 2011, 50, 2521 CrossRef CAS PubMed.
- Q. Chen, D. P. Goshorn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667 CrossRef CAS.
- M. I. Khan, Q. Chen, H. Hope, S. Parkin, C. Oconnor and J. Zubieta, Inorg. Chem., 1993, 32, 2929 CrossRef CAS.
- C. Yvon, A. J. Surman, M. Hutin, J. Alex, B. O. Smith, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2014, 53, 3336 CrossRef CAS PubMed.
- C. Yvon, A. Macdonell, S. Buchwald, A. J. Surman, N. Follet, J. Alex, D.-L. Long and L. Cronin, Chem. Sci., 2013, 4, 3810 RSC.
- V. Duffort, R. Thouvenot, C. Afonso, G. Izzet and A. Proust, Chem. Commun., 2009, 6062 RSC.
- B. Matt, S. Renaudineau, L.-M. Chamoreau, C. Afonso, G. Izzet and A. Proust, J. Org. Chem., 2011, 76, 3107 CrossRef CAS PubMed.
- Z. C. Xiao, K. Chen, B. L. Wu, W. J. Li, P. F. Wu and Y. G. Wei, Eur. J. Inorg. Chem., 2016, 808 CrossRef CAS.
- H. K. Yang, M. M. Su, L. J. Ren, J. Tang, Y. K. Yan, W. K. Miao, P. Zheng and W. Wang, Eur. J. Inorg. Chem., 2013, 1381–1389 CrossRef CAS.
- J. F. W. Keana, M. D. Ogan, Y. Lu, M. Beer and J. Varkey, J. Am. Chem. Soc., 1986, 108, 7957–7963 CrossRef CAS.
- C. R. Mayer, V. Cabuil, T. Lalot and R. Thouvenot, Angew. Chem., Int. Ed., 1999, 38, 3672 CrossRef CAS.
- A. R. Moore, H. Kwen, A. M. Beatty and E. A. Maatta, Chem. Commun., 2000, 1793 RSC.
- W. F. Bu, H. L. Li, H. Sun, S. Y. Yin and L. X. Wu, J. Am. Chem. Soc., 2005, 127, 8016–8017 CrossRef CAS PubMed.
- H. L. Li, W. Qi, W. Li, H. Sun, W. F. Bu and L. X. Wu, Adv. Mater., 2005, 17, 2688–2692 CrossRef CAS.
- P. F. Wu, Z. C. Xiao, J. Zhang, J. Hao, J. K. Chen, P. C. Yin and Y. G. Wei, Chem. Commun., 2011, 47, 5557 RSC.
- A. Müller, J. Meyer, H. Bögge, A. Stammler and A. Botar, Z. Anorg. Allg. Chem., 1995, 621, 1818 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Analysis, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: IR, 1H NMR, UV-Vis spectra and crystallographic data for compounds 2–4. CCDC 1484788–1484790. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00302h |
| ‡ These two authors contributed equally to this work. |
| § Crystal data for compound 2: C50H98N2O23V6, Mr = 1400.94, crystal size 0.3 × 0.2 × 0.1 mm3, monoclinic, space group P21/c, a = 11.006(5), b = 15.153(6), c = 19.360(4) Å, β = 97.416(3)°, V = 3201.7(2) Å3, Z = 45, ρcalc = 1.351 mg cm−3, m = 7.673 mm−1, F(000) = 1277.5, a total of 20599 reflections were collected, of which 6679 were unique (Rint = 0.0301, 2Θ range for data collection: 7.44 to 153.44°, index (hkl) range measured: [−13, −19, −24] < [hkl] < [13, 13, 18]). The R1 value is 0.0988 for 6679 independent reflections with [I ≥ 2σ(I)] and the wR2 value is 0.2832 with [I > 2σ(I)]. The R1 value is 0.1064 for all data and the wR2 value is 0.2951 for all data. The goodness-of-fit on F2 is 1.002. The data were collected at 99.99 K on an Agilent Gemini E diffractometer with MoKα monochromated radiation (λ = 0.71073 Å). The structure was solved by direct methods using SHELXS-97 and refined using SHELXL-97.43 |
| ¶ Crystal data for compound 3: C58H98N2O27V6, Mr = 1561.02, crystal size 0.12 × 0.08 × 0.08 mm3, monoclinic, space group P21/c, a = 11.3754(2), b = 16.3585(3), c = 18.4016(4) Å, β = 99.0881(18)°, V = 3381.27(11) Å3, Z = 2, ρcalc = 1.533 mg cm−3, m = 0.878 mm−1, F(000) = 1628.0. A total of 15669 reflections were collected, of which 6633 were unique (Rint = 0.0364, 2Θ range for data collection: 5.86 to 52°, index (hkl) range measured: [−11, −17, −22] < [hkl] < [14, 20, 15]). The R1 value is 0.0489 for 6633 independent reflections with [I ≥ 2σ(I)] and the wR2 value is 0.1132 with [I > 2σ(I)]. The R1 value is 0.0663 for all data and the wR2 value is 0.1213 for all data. The goodness-of-fit on F2 is 1.045. The data were collected at 103.7 K on an Agilent Gemini E diffractometer with MoKα monochromated radiation (λ = 0.7107 Å). The structure was solved by direct methods using SHELXS-97 and refined using SHELXL-97. |
| || Crystal data for compound 4: C58H106N2O27V6, Mr = 1569.09, crystal size 0.15 × 0.15 × 0.12 mm3, monoclinic, space group P21/c, a = 11.5756(3), b = 15.3260(4), c = 20.1152(5) Å, β = 101.354(2)°, V = 3498.75(15) Å3, Z = 2, ρcalc = 1.489 mg cm−3, m = 0.849 mm−1, F(000) = 1644.0. A total of 17371 reflections were collected, of which 8064 were unique (Rint = 0.0301, 2Θ range for data collection: 5.98 to 58.64°, index (hkl) range measured: [−14, −17, −26] < [hkl] < [15, 20, 18]). The R1 value is 0.0554 for 8064 independent reflections with [I ≥ 2σ(I)] and the wR2 value is 0.1416 with [I > 2σ(I)]. The R1 value is 0.0759 for all data and the wR2 value is 0.1550 for all data. The goodness-of-fit on F2 is 1.022. The data were collected at 104.1 K on an Agilent Gemini E diffractometer with MoKα monochromated radiation (λ = 0.71073 Å). The structure was solved by direct methods using SHELXS-97 and refined using SHELXL-97. |
| This journal is © the Partner Organisations 2017 |