 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selection of boron reagents for Suzuki–Miyaura coupling
Alastair J. J.
Lennox
and
Guy C.
Lloyd-Jones
*
School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: guy.lloyd-jones@ed.ac.uk
First published on 3rd October 2013
Abstract
Suzuki–Miyaura (SM) cross-coupling is arguably the most widely-applied transition metal catalysed carbon–carbon bond forming reaction to date. Its success originates from a combination of exceptionally mild and functional group tolerant reaction conditions, with a relatively stable, readily prepared and generally environmentally benign organoboron reagent. A variety of such reagents have been developed for the process, with properties that have been tailored for application under specific SM coupling conditions. This review analyses the seven main classes of boron reagent that have been developed. The general physical and chemical properties of each class of reagent are evaluated with special emphasis on the currently understood mechanisms of transmetalation. The methods to prepare each reagent are outlined, followed by example applications in SM coupling.
 Alastair J. J. Lennox | Alastair Lennox is a graduate of Manchester University (2008, 1st class, MChem), where he conducted a final year research project with Dr Ian Watt and spent a year studying at the University of California, Los Angeles. He obtained his PhD in 2012 at the University of Bristol, where he worked under the supervision of Professor Guy Lloyd-Jones and studied the reactivity of potassium organotrifluoroborate salts in Suzuki–Miyaura couplings. He is currently co-authoring a book with Lloyd-Jones, on organic reaction mechanisms. |
 Guy C. Lloyd-Jones | Guy Lloyd-Jones studied at Huddersfield Polytechnic (BSc 1989) and Oxford University (DPhil with John M. Brown FRS, 1992) before tenure of a Royal Society postdoctoral fellowship, at the University of Basel with Andreas Pfaltz. He joined the University of Bristol in 1996 and was promoted to full Professor in 2003. In 2013 he was elected to the Royal Society (FRS) and moved to Edinburgh University as the Forbes Professor of Organic Chemistry. Recent research includes Au- and Pd-catalyzed arylation, the chemistry of organotrifluoroborate salts, Tsuji–Trost allylation, metathesis, Ar–S bond-formation, amide activation, diazomethane reactions, aryne chemistry and phosphine/amine borane complexes. |
1. Introduction
1.1. Suzuki–Miyaura coupling
The Suzuki–Miyaura (SM) coupling reaction conjoins chemically differentiated fragments that participate in electronically divergent processes with the metal catalyst. Oxidative addition occurs with formally electrophilic organic groups, whereby palladium becomes oxidized through its donation of electrons to form the new Pd–C bond. However, transmetalation occurs with formally nucleophilic organic groups, which are transferred from boron to palladium, Scheme 1. This complimentary reactivity sequence between oxidative addition and transmetalation allows two similar, but distinct, components to be cross-coupled, thereby forming the basis of this important methodology.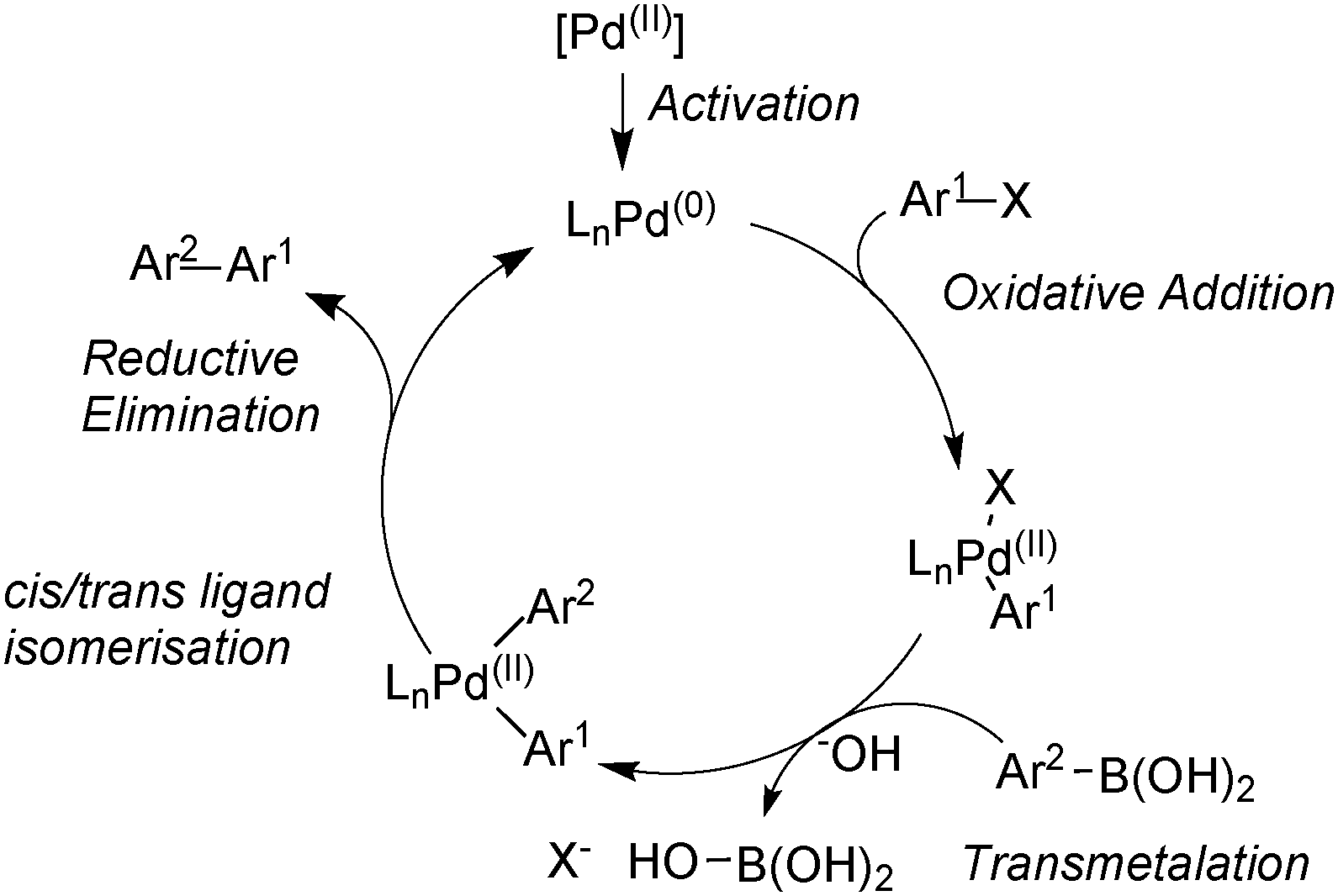 | ||
| Scheme 1 A generic mechanism for aryl–aryl SM coupling. | ||
The broad application of SM coupling arises from the exceptionally mild and functional group tolerant reaction conditions, the relatively stable, readily prepared and generally environmentally benign nature of the oroganoboron reagents, and their rapid transmetalation with palladium(II) complexes. These features contribute to the practical up-scaling of the reaction, and together with the low cost of the reagent, explain its lasting value to the fine chemical, pharmaceutical, agrochemical, and modern-materials industries. Indeed, SM coupling has become the “gold standard” for biaryl construction, arguably resulting in the ubiquity of this moiety in medicinal chemistry.1
Since its inception (1979) a series of major advancements in SM coupling technology have occurred; including expansion of the substrate scope,2,3 reaction at lower temperatures4,5 and reduction in the catalyst loading.6,7 Many of these aspects have been reviewed in detail elsewhere.8–11 However, although the boron reagent itself has also received significant development, reviews tend not to focus on this integral aspect of the reaction. Each reagent exhibits a unique range of physical, chemical and reactivity characteristics. This ability to tailor the reagent for the reaction in hand has allowed SM coupling to be employed in the synthesis of a number of natural products, pharmaceutical targets and lead compounds,12 as well as being applied in scale-up for clinical trials, process development, and even manufacture. This review considers the seven main classes of boron reagent employed in SM coupling, analysing their properties and mechanism of activation, the common methods for their preparation, and selected examples of their application.
1.2. Boron reagents
The outer shell bonding electrons (2s2, 3p1) in neutral boron can engage in three sp2 hybridised bonds, resulting in a trigonal planar geometry, with the resulting non-bonding vacant p-orbital orthogonal to the plane. This empty p-orbital dominates the reactivity patterns and physical characteristics of all neutral sp2 boron compounds and renders them susceptible towards electron donation from Lewis bases. Upon coordination, an anionic (or zwitterionic) tetrahedral ‘ate’ complex is formed with very different properties to the neutral trigonal precursor.The boron reagents initially employed for SM coupling were alkenylboranes and catechol boronic esters, both conveniently obtained through the hydroboration of terminal alkynes. However, by the 1990s boronic acids had become the reagents of choice, especially for aryl couplings, primarily due to their enhanced reactivity and high atom-economy. Pinacol boronic esters also became popular, particularly in the context of Miyaura-borylation. Over the last decade or so, a wide range of new reagents for SM coupling have been developed, with stabilities that allow distal manipulation and expansion of substrate scope. Organotrifluoroborate salts and MIDA boronates are two of the most developed systems, but several alternatives have also been well advanced, Fig. 1.
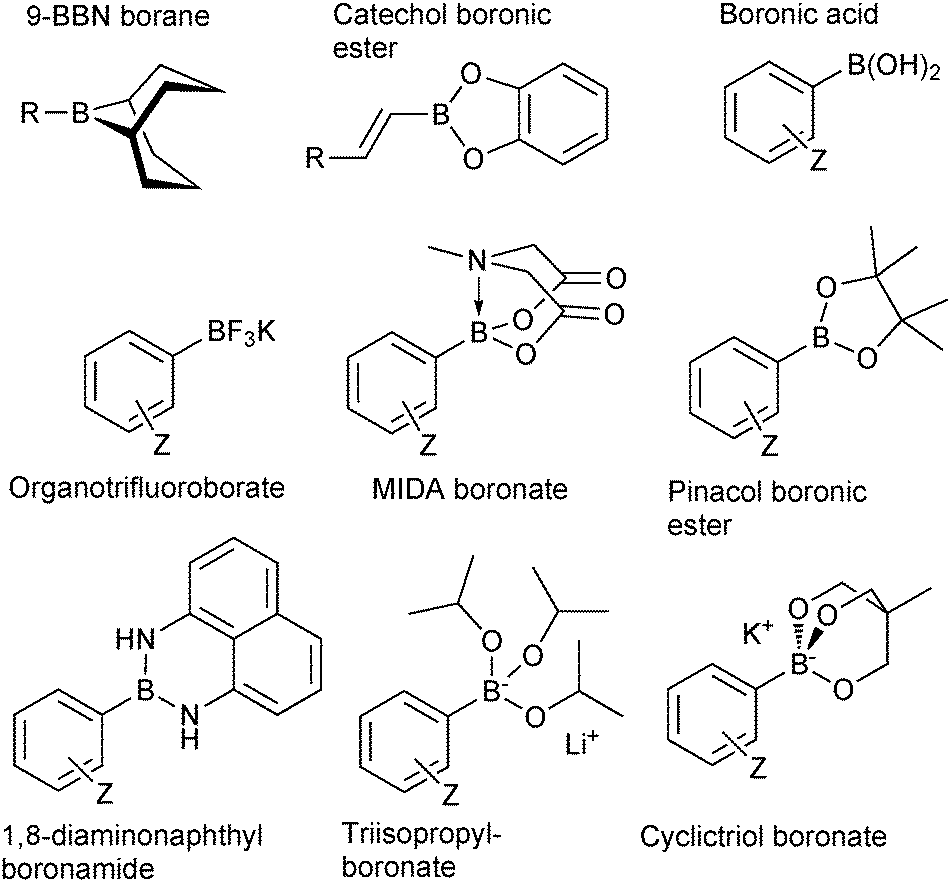 | ||
| Fig. 1 Examples of some of the most popular types boron reagents used directly or indirectly in SM coupling reactions. | ||
As the boron reagent tends to be the nucleophilic component in SM coupling, an insightful method for their comparison is to compare their nucleophilicity. Mayr has developed electrophilicity and nucleophilicity scales that allow one to directly compare reagents and thus predict the outcome of a huge range nucleophile–electrophile combinations.13 The addition rates of a range of 2-borylated furan moieties to an electrophilic benzhydrylium ion proved highly informative, Fig. 2.14 Sp2 pinacol boronic esters were found to be marginally less reactive towards carbocations than the parent non-borylated furan. The addition of an extra ligand hybridises boron to sp3, which increased the observed nucleophilicity, as would be expected from a formally anionic species. However, the most nucleophilic reagent was an intramolecular trialkoxy-ligated boronate salt that reacted with the standard electrophile some 10 orders of magnitude faster than pinacol boronic esters; with nucleophilicity comparable to that of ketene acetals and enamines. The high electronegativity of fluoride rendered organotrifluoroborates less nucleophilic than trialkoxyboronate salts. Interestingly, MIDA boronates proved to be least nucleophilic of all those measured. The electron withdrawing effect of the carbonyl groups evidently out-competes the quaternisation by nitrogen, which did increase the nucleophilicity of the N-methyl diethanolamine adduct.
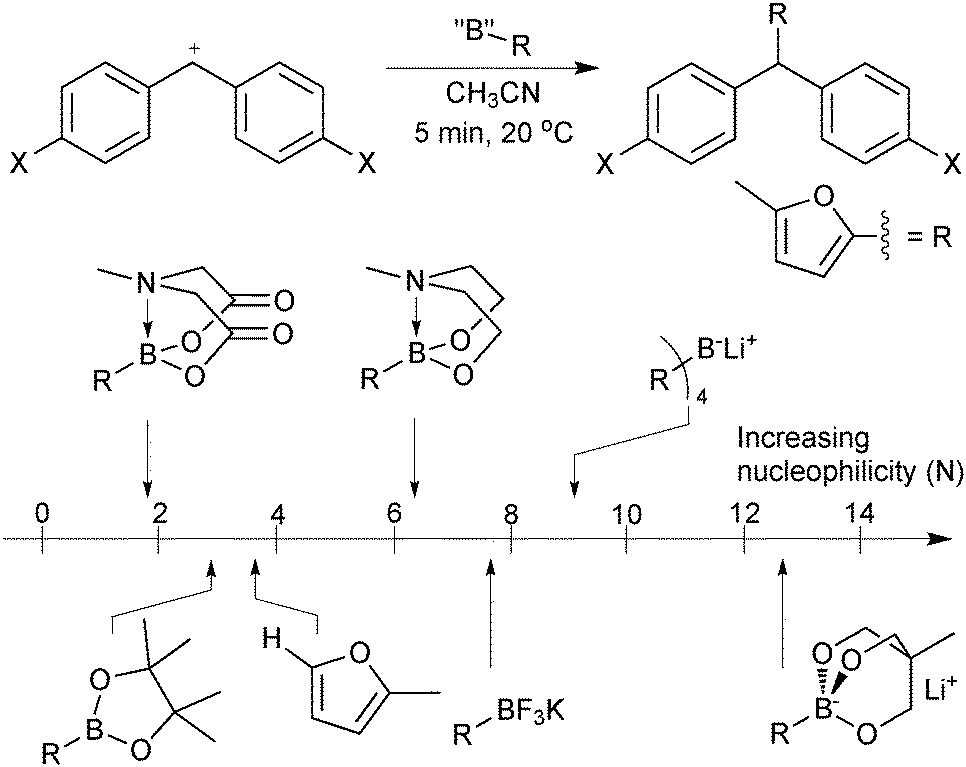 | ||
| Fig. 2 Comparison of boron reagents with furan on Mayr's nucleophilicity scale.14 | ||
1.3. Nomenclature
The nomenclature employed for boron reagents in the SM coupling literature is varied and exhibits little consistency. In addition, to the best of our knowledge, the IUPAC system does not appear to fully cover the range of species available.15,16 As a logical and uniform model to follow was not easily sourced, the nomenclature employed herein is outlined in Fig. 3. It is predominantly based on recent formats adopted in the literature, and is not intended to be definitive or to replace the IUPAC system.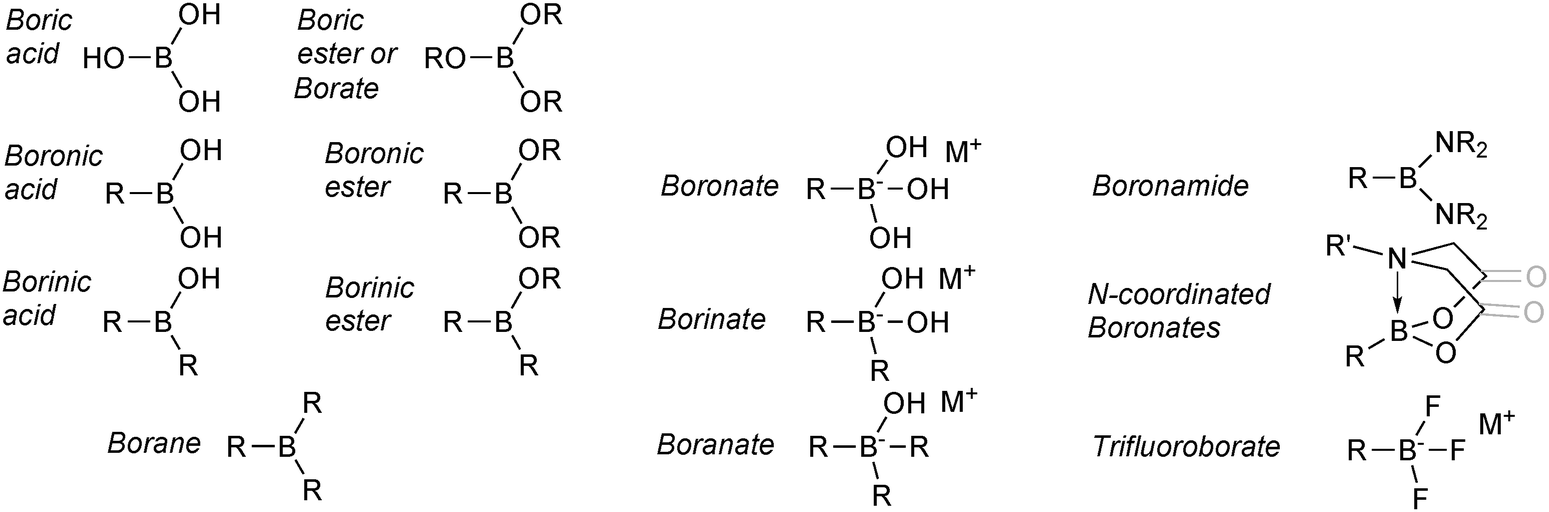 | ||
| Fig. 3 Overview of the nomenclature employed for the boron reagents and intermediates covered in this review. | ||
2. Organoboranes
2.1. Properties and mechanism
The organoboranes most commonly employed in SM coupling reactions are based on 9-borabicyclo[3.3.1]nonane (9-BBN), disiamylborane (sia) and dicyclohexylborane building blocks, Scheme 2.17 However, much of their use was during the initial stages of the reaction development,18,19 primarily due to their ease of preparation via alkene and alkyne hydroboration. Boranes with secondary alkyl ligands are best suited to the coupling reaction so that sufficient differentiation between the “R” groups in trialkyl boranes can be achieved during transmetalation. The difference in the rate of transmetalation between primary alkyl or alkenyl groups and secondary alkyl groups is large enough for the selective transfer of the desired group to palladium.20 However, the structural rigidity and bulk of the 9-BBN moiety allows greater selectivity compared to other trialkylboranes.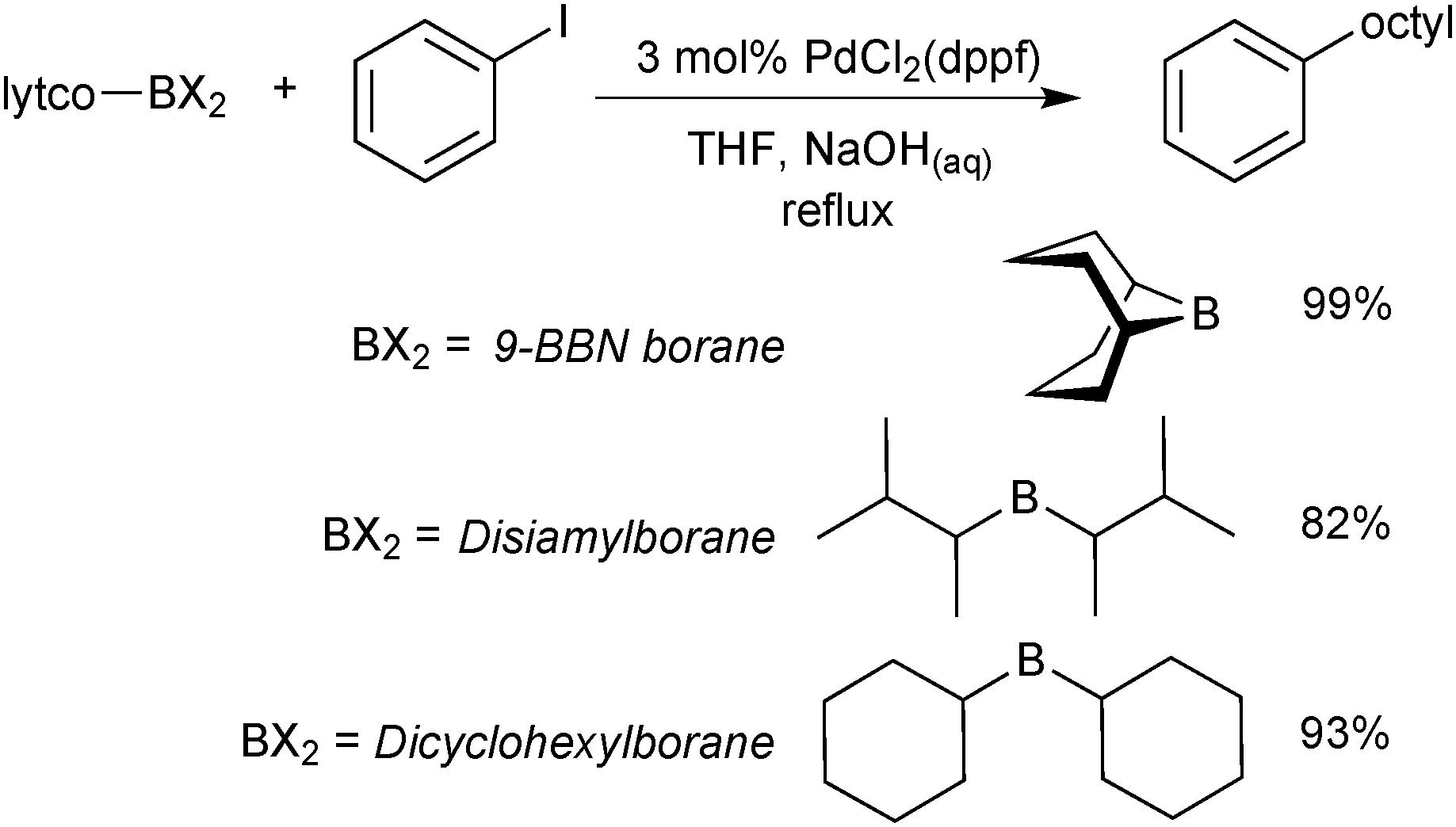 | ||
| Scheme 2 Example application of the three most commonly employed organoborane moieties, in this case to deliver an octyl group for SM coupling.20 | ||
A primary disadvantage to the use of organoboranes is their propensity towards aerobic oxidation, which not only limits their application, but also decreases yields during coupling reactions if the solvent is incompletely degassed, or the reaction head space not fully anaerobic. Dehydroboration can also be a problematic decomposition pathway for these motifs. In addition, protodeboronation of the alkenyl moiety in alkenyl dialkylboranes can readily occur in alcoholic solvents, with a substrate-dependent requirement for acid catalysis.21 Rates of organoborane protodeboronation were observed to decrease in the following order: 9-BBN > B(cyclohexyl)2 > B(sia)2 (≫B(OR)2). Therefore, as would be expected, lower yields have been reported in the cross-coupling of disiamyl and dicyclohexylborane compared to that of the diisopropylboronic ester.22
Suzuki and Miyaura conducted mechanistic studies on the coupling between alkenylboranes and bromoalkenes using alkoxide bases.23 They considered whether the role of the base was to initially react with the borane to form a more nucleophilic tetrahedral boronate, or whether it reacted with palladium to form a more reactive alkoxo–palladium species, Scheme 3.24 These two pathways are named the boronate pathway and oxo–palladium pathway respectively.
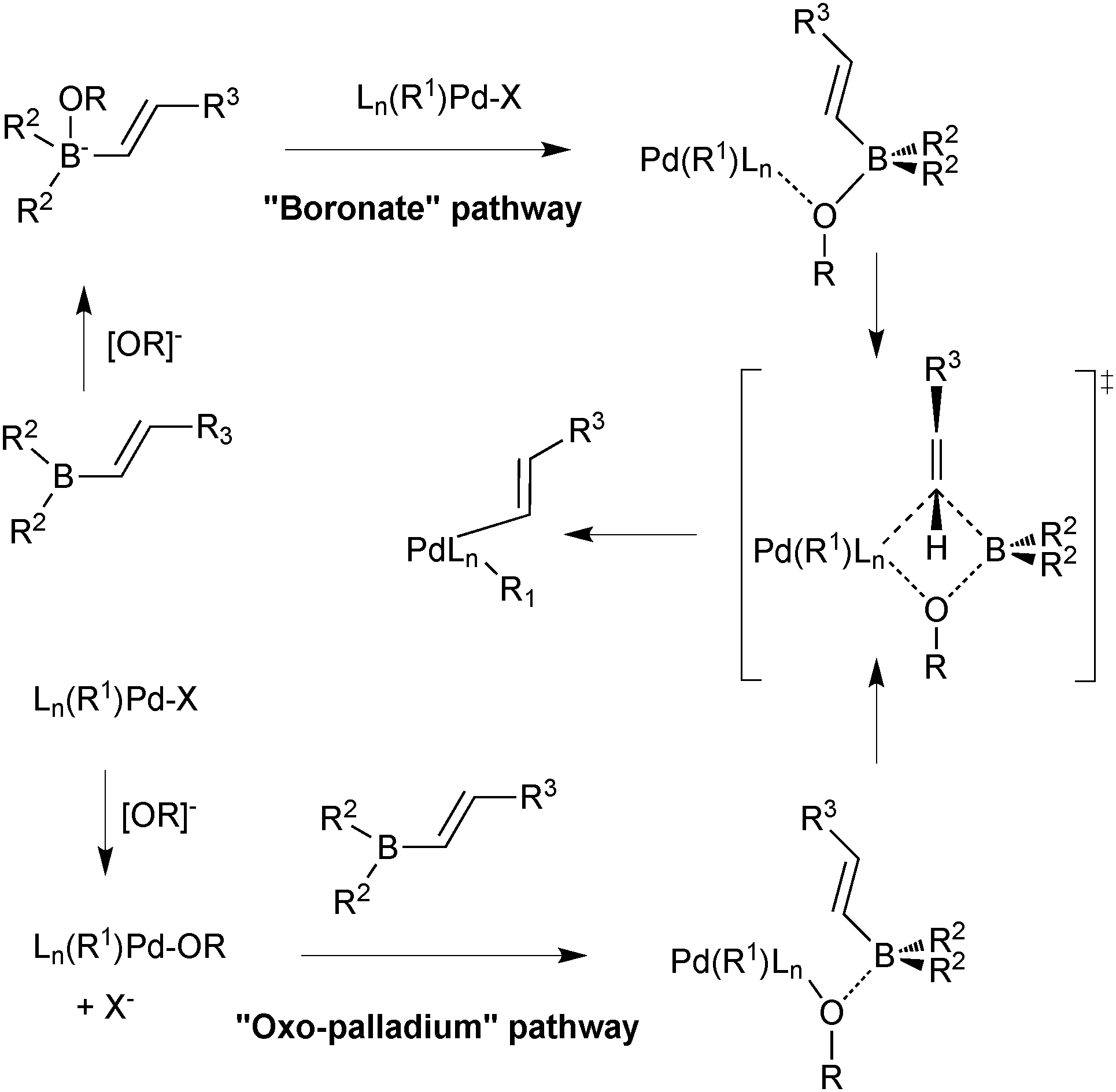 | ||
| Scheme 3 Two mechanisms considered for transmetalation of alkenylboranes with palladium complexes.23 | ||
Suzuki and Miyaura also tested the direct coupling between a preformed lithium tetraalkylborate with a styrenyl bromide, catalysed by Pd(PPh3)4.23,25 The yield of cross-coupled product was found to be only 9%; which was proposed as evidence against the boronate pathway. The possibility that quaternisation of boron with an alkyl group, rather than by a coordinating heteroatom, may have attenuated the transmetalating activity does not seem to have been considered at that time. Nevertheless, further stoichiometric studies employing a disiamylborane with palladium(II) trichlorovinyl complexes gave evidence in favour of the oxo–palladium pathway. The order of reactivity for the ligand (‘Y’, Scheme 4) on palladium was established as being OMe ≫ Cl based on yield of coupling product. It was thus concluded that formation of analogous species under catalytic conditions by a metathetical type displacement with, for example, sodium methoxide, was an important pathway. Analogous studies with catechol 1-octenylboronic ester confirmed this effect.
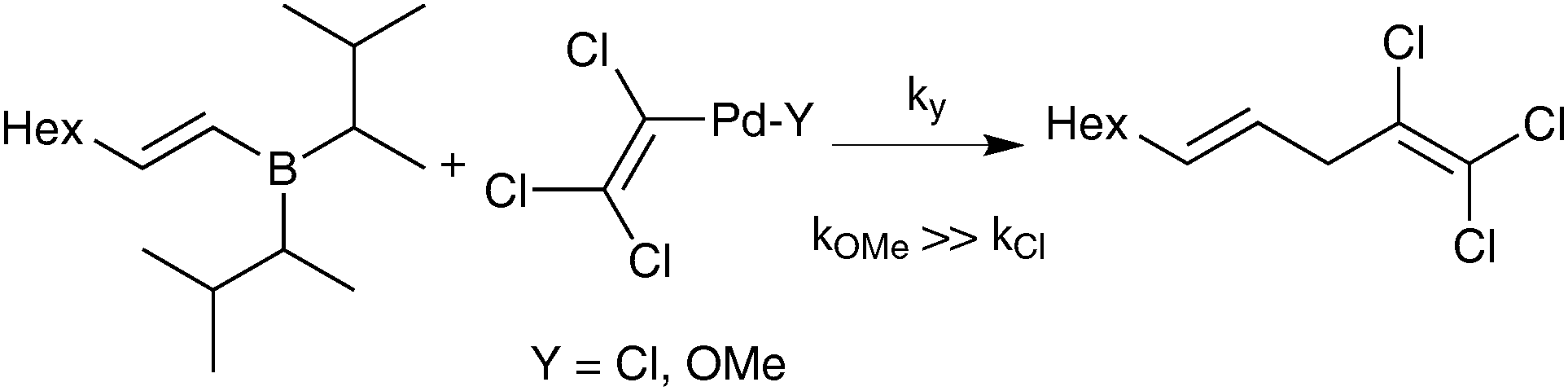 | ||
| Scheme 4 Suzuki and Miyaura's stoichiometric reactivity studies. | ||
Matos and Soderquist provided a more thorough mechanistic study on the transmetalation of organoboranes in SM coupling.26 They compared the coupling between two alkyl boron reagents with bromo- and iodobenzene in aqueous THF solutions. The study revealed that the transmetalation pathway taken depended on the boron species employed, Scheme 5. Lewis-acidic alkylboranes, e.g.1, readily formed (11B NMR) boranate complexes in the presence of base. In contrast, the association of hydroxide was undetectable in boron reagents of lower Lewis-acidity, e.g. alkylborinic ester 2. When 1 and 2 were competed for limiting bromobenzene it was found that the product originated solely from reagent 1. This was attributed to a fast transmetalation of 1 through the boronate pathway and a slower transmetalation of neutral borinic ester 2 through the oxo–palladium pathway. This was consistent with the independently measured rate of hydrolysis of [PdBr(Ar)L] to the oxo–palladium species.
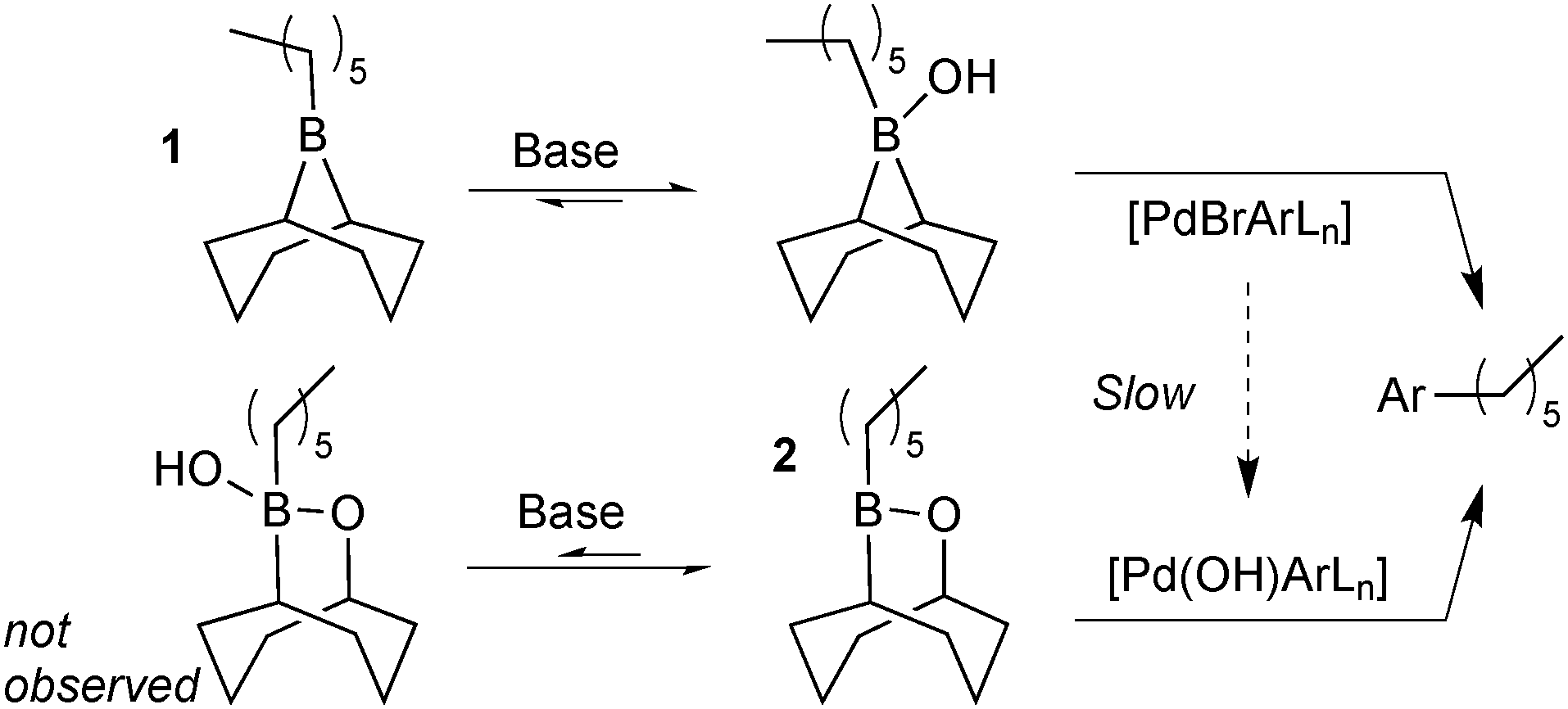 | ||
| Scheme 5 SM-coupling of borane 1 and borinic ester 2. | ||
2.2. Preparation
Dialkylboranes (HBR2) became the most popular reagents for hydroboration due to the greater regioselectivity in their addition to olefins. Disiamylborane, dicyclohexylborane and diisopinocamphenylborane are some of the most commonly employed reagents, but their thermal instability to dehydroboration means it is more efficient to prepare them in situ from BH3. 9-Borabicyclo[3,3,1]-nonane (9-BBN) does not dehydroborate and is thermally stable, rendering it the most useful reagent; especially as its steric bulk augments the anti-Markovnikov regioselectivity. Asymmetric induction can be achieved with chiral borane reagents. Reaction of α-pinene with BH3·THF generates the corresponding diisopinocamphenyl-borane, which can be combined with alkenes to provide highly diastereomerically enriched alkylboranes.38 Asymmetric induction by other moieties has also been achieved,39,40 and a chiral borabicyclodecane was found to be the most efficient hydroborating reagent for the more challenging 1,1-disubstituted alkenes.41,42
Functional group interconversion via C–B oxidation is the most common application of organoboranes, but their use in SM couplings has nonetheless been important. Hydroboration in the preparation of boronic esters is covered in Section 3.2.
2.3. Applications in SM coupling
Organoboranes have been used in a wide range of natural product syntheses. One example favours the use of a disiamylborane over the corresponding catechol boronic ester, due to a propargylic hydroxyl inhibiting hydroboration with the latter, Scheme 6.43 A leukotriene B4 precursor was prepared on a multi-gram scale, with the conjugated triene generated by coupling of a disiamylborane with an alkenyl iodide.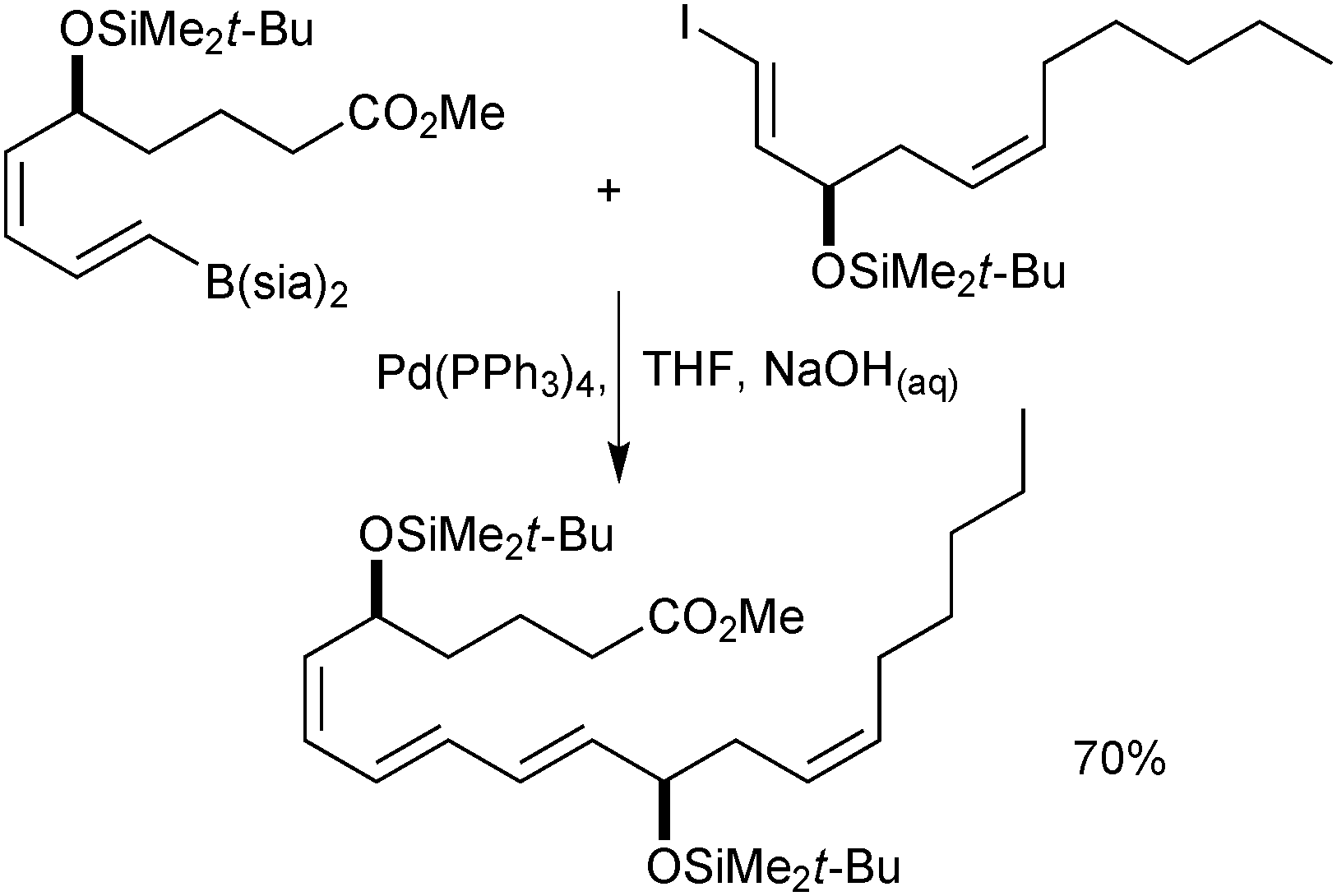 | ||
| Scheme 6 SM coupling in the synthesis of leukotriene B4. | ||
There are many examples of the 9-BBN-based boranes as aryl,44 alkenyl45 or allenyl46 coupling partners in the literature, but its most frequent use is in the delivery of an alkyl group, possibly due to the enhanced stability over alkylboronic acids and esters. A two stage hydroboration/cross-coupling of a terminal alkene with an alkenyl halide is a useful procedure for conjoining alkyl with alkenyl groups. Examples where this methodology has been employed include construction of the taxane skeleton47 or synthesis of dihydroxyserrulatic acid48 (3) and steroid204, Scheme 7.
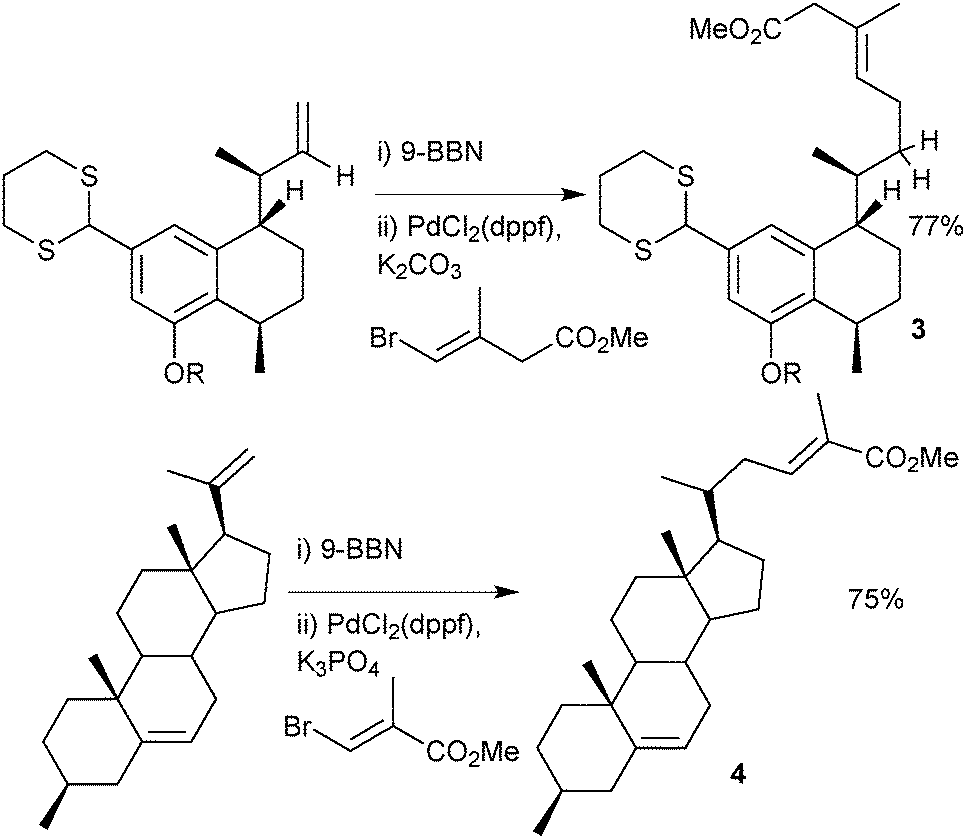
Alkene hydroboration with 9-BBN and subsequent cross-coupling under strictly anhydrous conditions can be helpful to protect water-sensitive functionality that would normally decompose under regular aqueous SM coupling conditions. For an example see Section 5.3, vide infra, on organotrifluoroborate salts.49
Soderquist50 and Fürstner51 both recognised that an alternative approach could be taken to assemble a 9-BBN reagent for SM coupling, and developed what is now known as the “9-MeO-9-BBN variant”. Rather than adding a base to the organoborane to form the reactive boronate species, a polar organometallic reagent was added to the commercially available B-methoxy-9-BBN, Scheme 8. Formation of the tetrahedral boranate complex was quantitative and rapid, and subsequent transmetalation with palladium proceeded well for a range of organic residues that cannot be coupled under conventional SM conditions. For example, the methodology is especially useful when the corresponding boronic acid or borane is unstable, such as alkynyl or methyl moieties.
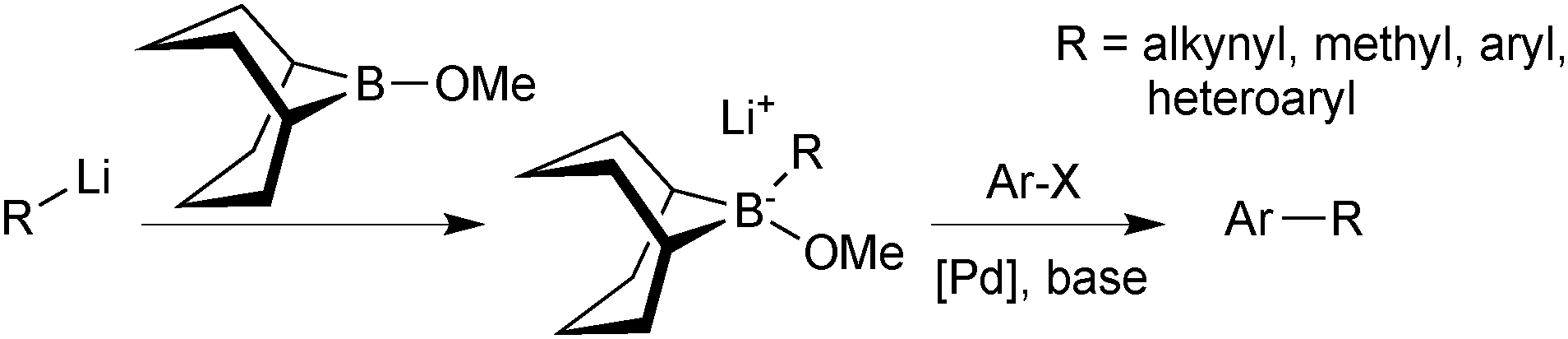 | ||
| Scheme 8 The “9-MeO-9-BBN variant”, wherein the boronate species is prepared in situ from 9-MeO-9-BBN and organometallic partner, e.g. RLi. | ||
In these reactions, the borane behaves as a shuttle, and thus preliminary attempts were made to render the reaction catalytic in boron and palladium, Scheme 9. Due to the incompatibility of lithium phenylacetylene with the palladium catalyst, slow addition of the organometallic reagent was necessary using 20 mol% boron to reach a yield almost as high as when stoichiometric quantities were employed.
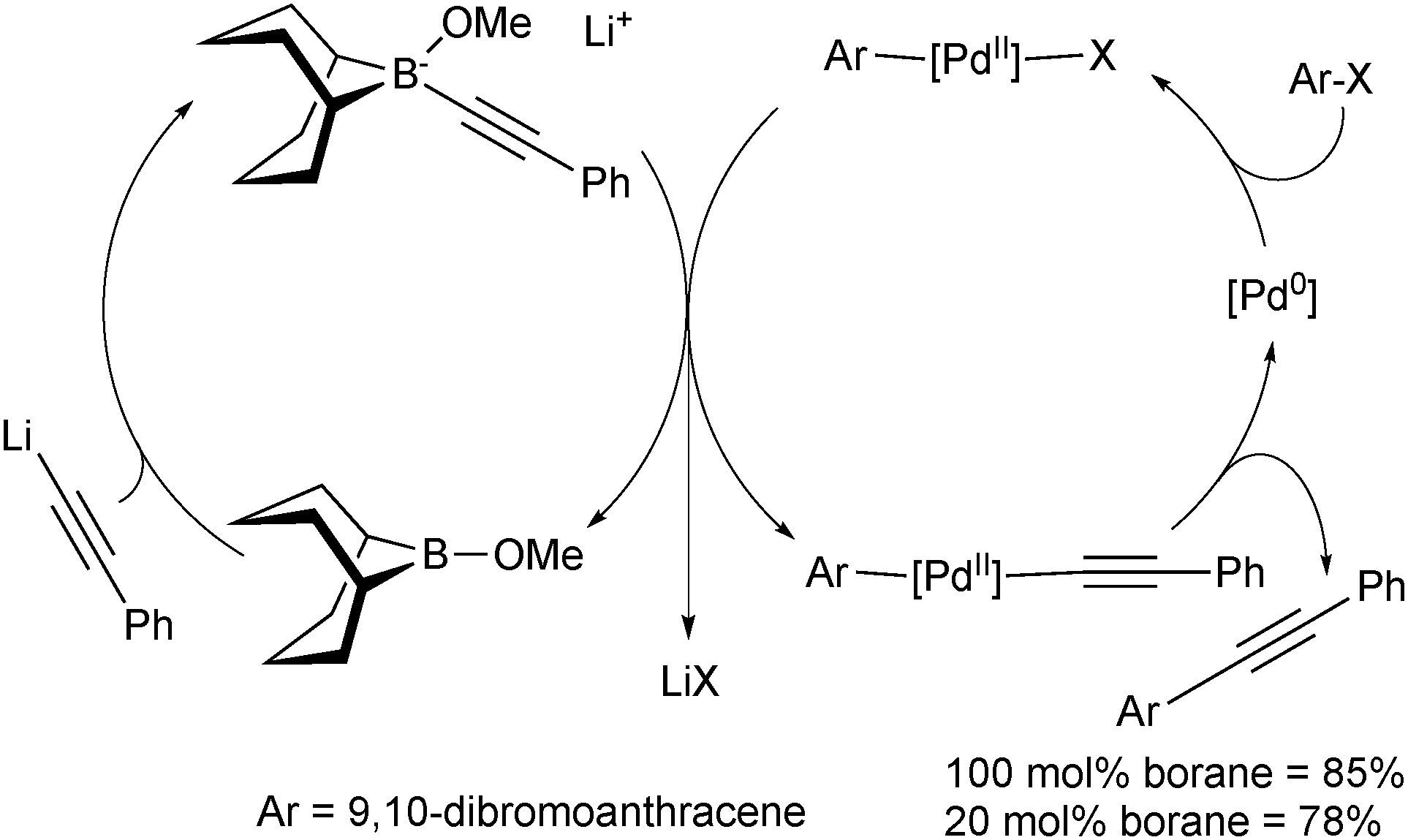 | ||
| Scheme 9 The mechanism of the SM coupling when catalytic in boron and palladium, using the 9-MeO-9-BBN variant. | ||
The stoichiometric borane cross-coupling variant has been useful in a number of natural product syntheses.52 Of particular note is in the synthesis of mycolactones A/B, Scheme 10, whereby it was demonstrated that sequential lithium–halogen exchange with t-BuLi and capture by B-methoxy-9-BBN is so rapid that there was no detectable degradation of a base sensitive lactone.53
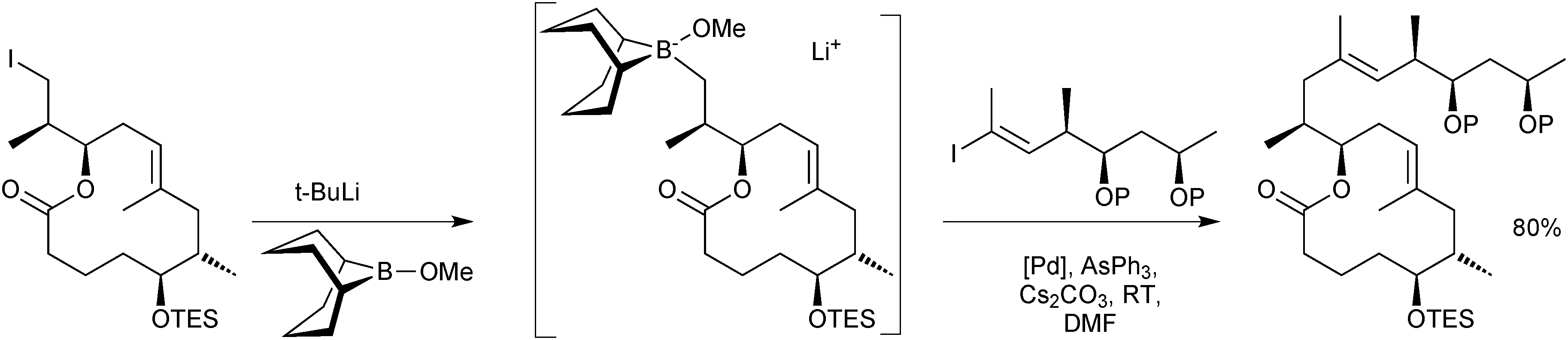 | ||
| Scheme 10 Synthesis of a precursor to mycolactones A/B using the stoichiometric 9-MeO-9-BBN variant methodology. | ||
3. Boronic esters
3.1. Properties and mechanism
The most commonly employed boronic esters for SM coupling are generally the pinacol, neopentyl- and catechol boronic esters. This is due to a combination of their relative cost, reactivity, stability and ease of preparation compared to a wide range of other boronic esters that are available.By virtue of the σ-donating ability of carbon, the lone pairs of oxygen in boronic esters are more readily conjugated into the electron deficient boron centre. This has the effect of reducing its Lewis-acidity, generally resulting in boronic esters being less reactive than boronic acids. In most cases they exhibit stability towards column chromatography, which aids in their isolation and purification. In addition, many are liquids at room temperature and can be easily distilled. Boronic esters dissolve readily in apolar solvents and unlike boronic acids, are not hydrogen bond donors, nor able to oligomerise, thus rendering them exclusively monomeric in nature.
A study to compare the stability of a range of boronic esters was conducted by analysing the transesterification equilibrium with the free diol and ethylene glycol boronic ester (5), Scheme 11.54 It was performed in the context of their deprotection and in particular from pinanediol boronic ester, which is considered to be one of the most stable.
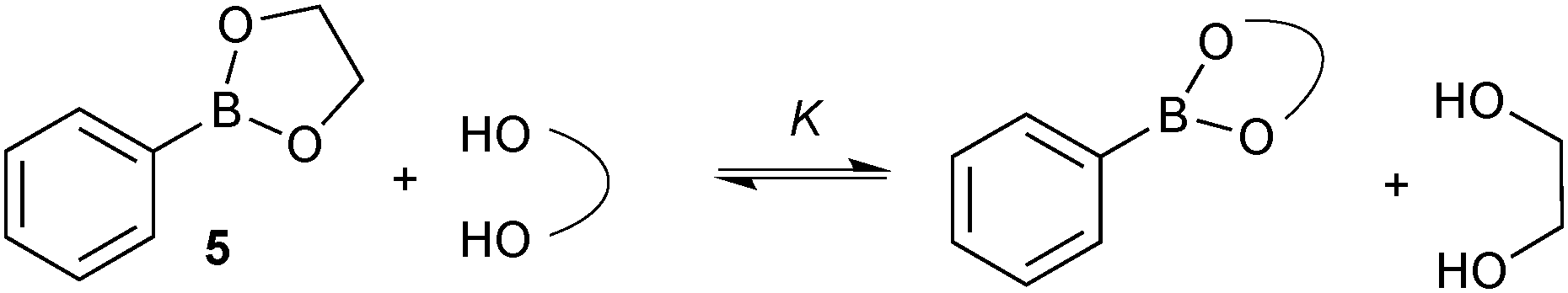 | ||
| Scheme 11 The equilibrium between the transesterification of glycol phenylboronic ester (2-(phenyl)-1,3,2-dioxaborolane) with diols. The extent of transesterification is used to assess the stability of the new boronic ester. | ||
A number of key points arose from the study, Fig. 4. The cis-stereochemistry of 5 and 6 membered saturated cyclic diols was found to be a prerequisite for transesterification; trans diols were completely unreactive. Six membered cyclic boronic esters (e.g.6) were found to be more thermodynamically stable than the corresponding five membered analogues (5), which is likely due to a more favourable orbital overlap between B and O for lone-pair donation. Methyl group substitution on the α-carbon of the diols led to further stabilisation (e.g.7 and 8). However, further substitution in the six membered ring, 9 both attenuated the extent, and reduced the rate, of transesterification. In contrast, further substitution in the five membered ring to the pinacol ester 10 induced greater stability. Of the boronic esters commonly employed for SM coupling, the pinacol and neopentylboronic esters were found to be of a similar stability. However, the stability of the catechol ester was substantially lower, which can be attributed to the decreased π-donating ability of oxygen to boron, due the competing conjugation with the phenyl ring.
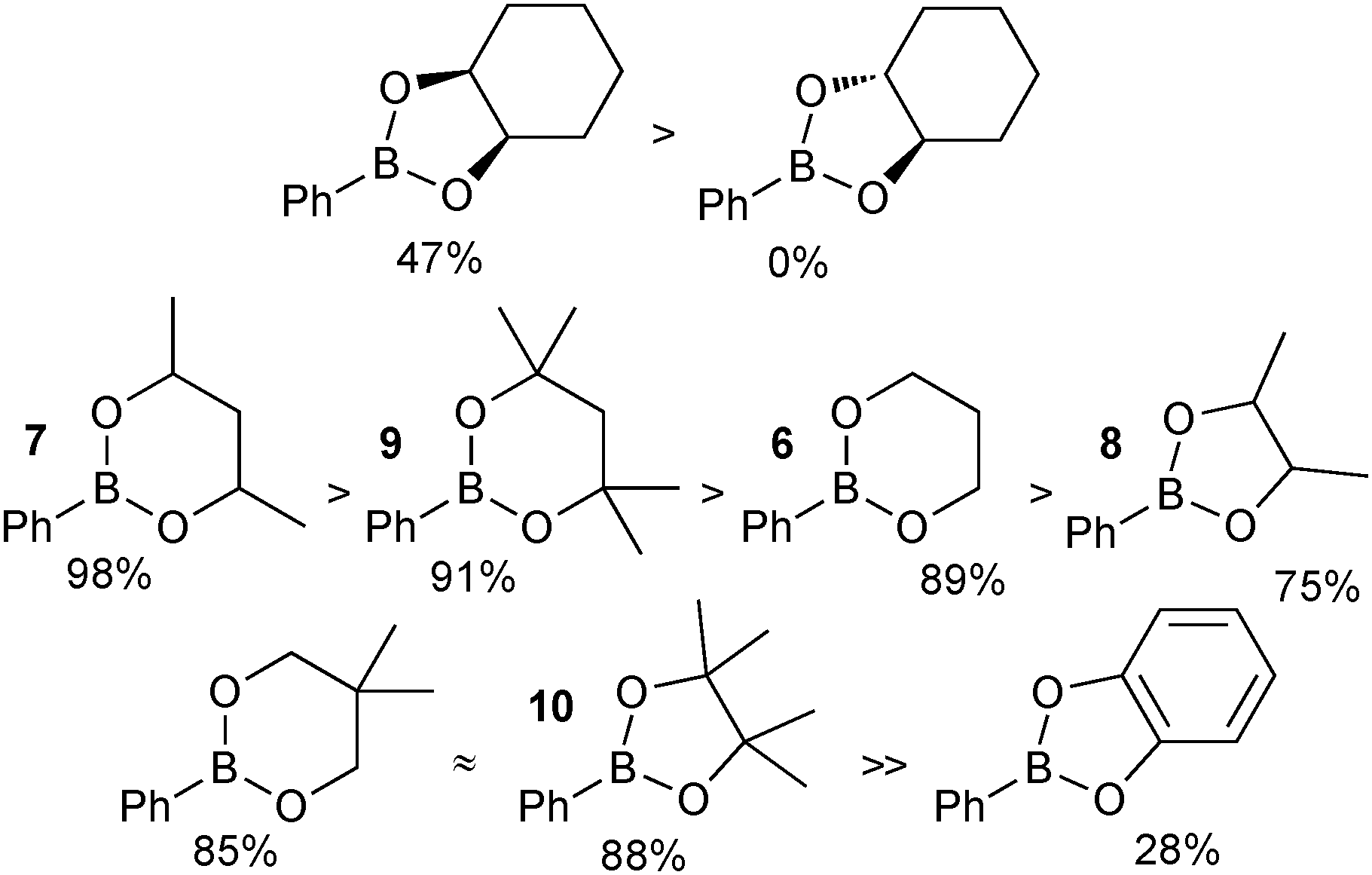 | ||
| Fig. 4 Stability sequences for a range of boronic esters, with percent transesterification from the glycol boronic ester indicated. | ||
A detailed study into the pH optimum for esterification of boronic acids by the diol moiety in sugars has been conducted by Springsteen,55 in which a simple correlation between Hammett sigma values (σ) and the pKa of arylboronic acids was determined (pKa = 2.06σ + 8.62; R2 = 0.94). A separate study compared the efficiency of cross-coupling of a neopentylboronic ester with that of a pinacol boronic ester in a nickel catalysed SM coupling reaction.56 It found, in competition experiments, that more of the neopentyl derivative was consumed than the corresponding pinacol ester, thereby implying its greater reactivity. However, both were found to be less efficient than the corresponding trifluoroborate and boronic acid; the latter being the most reactive overall.
Evidently boronic esters exhibit greater chemical stability than their corresponding boronic acids, but it is not clear what the active transmetalating species is during their SM coupling. Either the boronic ester directly reacts with an oxo–palladium species, or it undergoes complete or partial hydrolysis to form a more reactive species that can react via the oxo–palladium or boronate pathways. Conditions for the successful coupling of pinacol boronic esters without added water do exist,57 but the possibility for trace amounts of adventitious water present can often be high. It is more common for small proportions of water to be added into the reaction mixture,58,59 which is conducive towards both a prior hydrolysis of the boronic ester facilitating the reaction, or assistance in generation of the oxo–palladium(II) intermediate.
3.2. Preparation
There are numerous methods to prepare boronic esters; the following is a selection of some of the most relevant and interesting with respect to applications in SM coupling.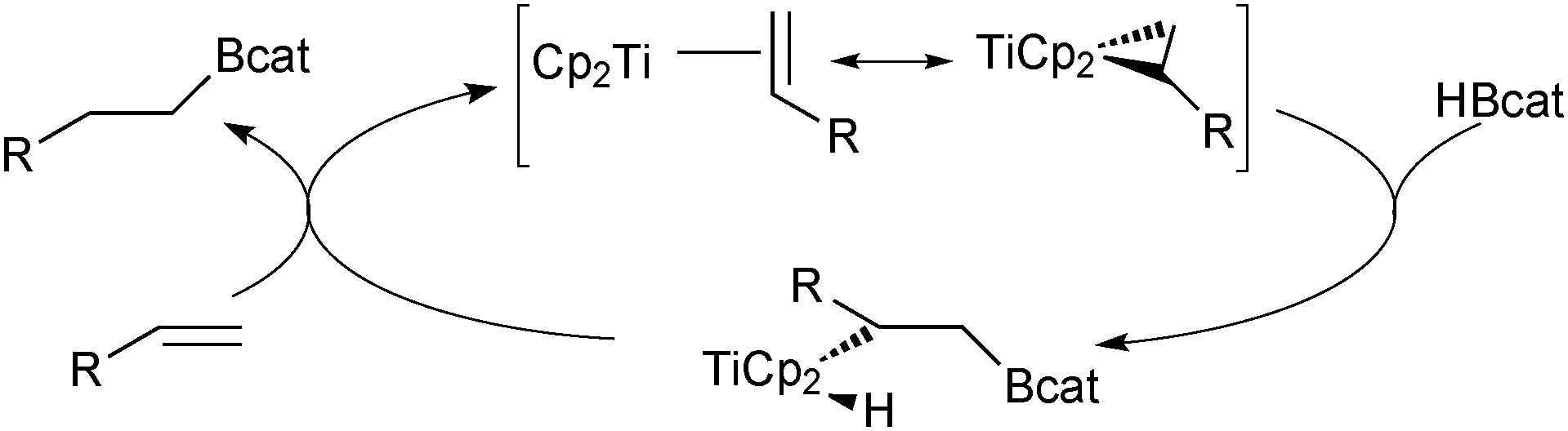 | ||
| Scheme 12 Titanocene catalysed cis hydroboration of an alkene with catecholborane. | ||
Soon after Nöth reported the rhodium catalysed hydroboration, much effort was directed towards developing an enantioselective variant. This was soon achieved with chiral ligands on rhodium,63,64 or with a chiral borane reagent.65
The development of non-precious metal catalyst systems has also been the subject of intense research. Electron rich iron PNN pincer complexes were found to be proficient catalysts for alkene hydroboration.66 Additionally, copper complexes with N-heterocyclic carbene (NHC) ligands can catalyse the regioselective hydroboration of internal alkynes.67 The use of chiral ligands for copper, such as NHCs68 or phosphines,69 can induce enantiocontrol. The NHC system was even successful for the difficult 1,1-disubstiuted alkenes. Similar copper catalysts are also proficient for terminal alkynes with high selectivity for internal borylation;70i.e. the opposite (Markovnikov) regioselectivity to that observed using all other methodologies. The resultant α-vinylboronic ester products are furnished in high yields and selectivities. The term ‘protoboration’ was proposed for the process in order to distinguish itself mechanistically from hydroboration, as there is formally no involvement of a hydridic species:71 Cu–B addition to the alkyne is followed by protonation of Cu–C. The protosilylation reaction is analogous and the term is thus used in the same vein. This copper/NHC protocol was also used to prepare α-vinylboronic esters from allenes.72
A completely transition metal-free procedure exists, whereby dicyclohexylborane is employed as catalyst for the cis-hydroboration of terminal alkynes. Stoichiometric quantities of either catecholborane73 or pinacolborane74 react rapidly with alkynes at room temperature in the presence of a catalytic quantity of the dialkylborane. Mechanistic proposals involve initial hydroboration of the alkyne with dicyclohexylborane to give an alkenyl dicyclohexylborane that was independently found to be a catalytically active intermediate, Scheme 13. The boronic ester is generated after alkenyl transfer from boron to boron in a four membered transition state, which concomitantly regenerates the dialkylborane catalyst.
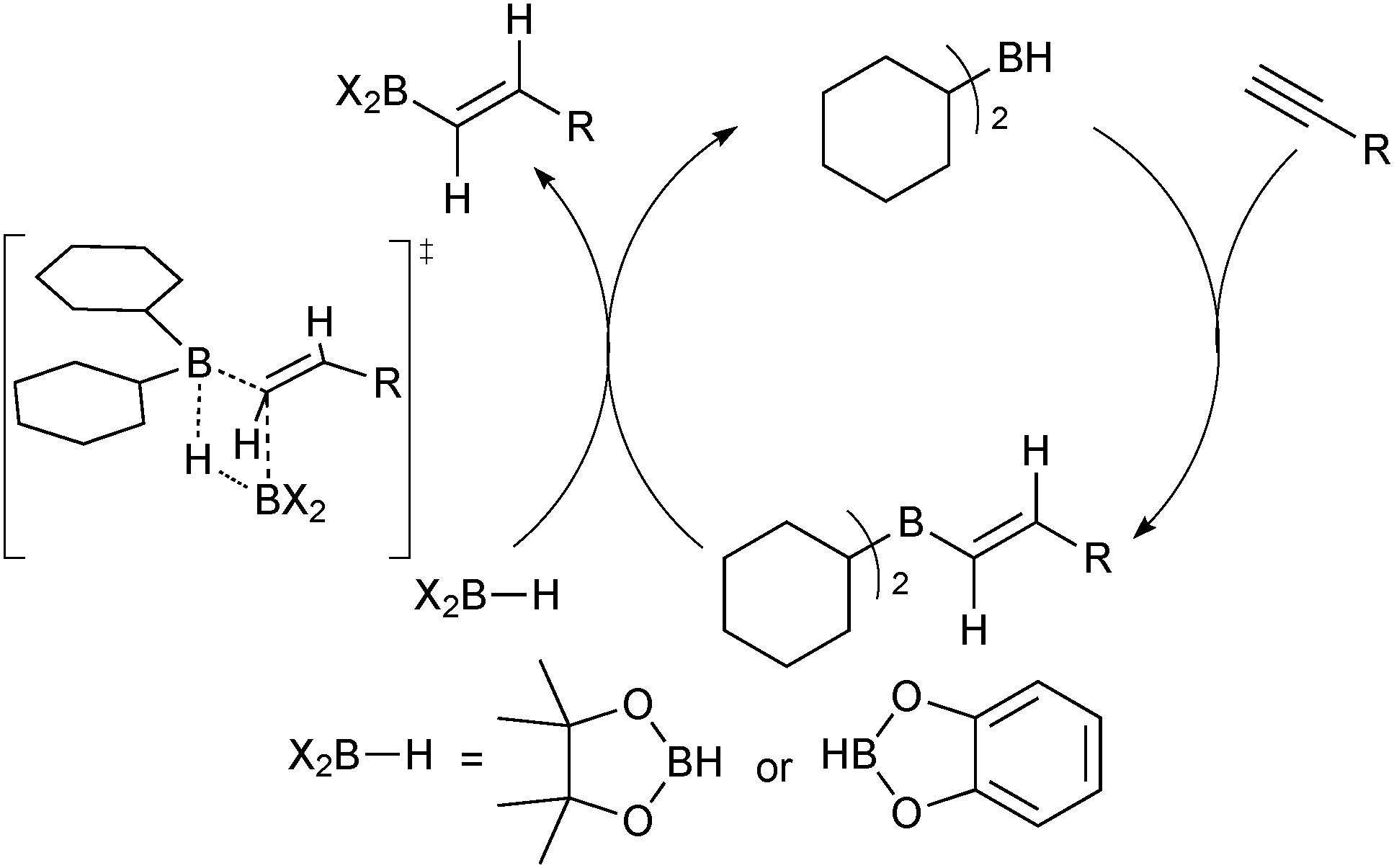 | ||
| Scheme 13 Dicyclohexylborane catalysed hydroboration of alkynes with pinacol or catechol borane. | ||
The majority of catalysed and uncatalysed alkyne hydroboration reactions proceed with syn addition and usually thus lead to a trans configured product. Selective preparation of the oppositely configured cis isomer is considerably more challenging. True anti-hydroboration to yield cis alkenes has been achieved with transition metal catalysis. Miyaura developed conditions wherein a rhodium complex successfully aided the anti-hydroboration of alkynes in the presence of an electron rich phosphine ligand and base.75 Catecholborane gave slightly better selectivities than pinacolborane, but the former could be employed in a two-step procedure with pinacol to give the pinacol boronic ester in high yields and selectivities. Leitner developed a ruthenium pincer complex that similarly led to cis alkenes in high yields and selectivities and without the need for additional base.76 Deuterium labeling experiments by both groups indicated the hydrogen from the borane ends up geminal to boron, which implies a hydride shift from the terminal alkyne, Scheme 14. The mechanisms only slightly contrasted, but both included the key step as migratory insertion of a vinylidene into the metal–boron bond. This step determines the product configuration through the stereoselective migration in the vinylidene complex.
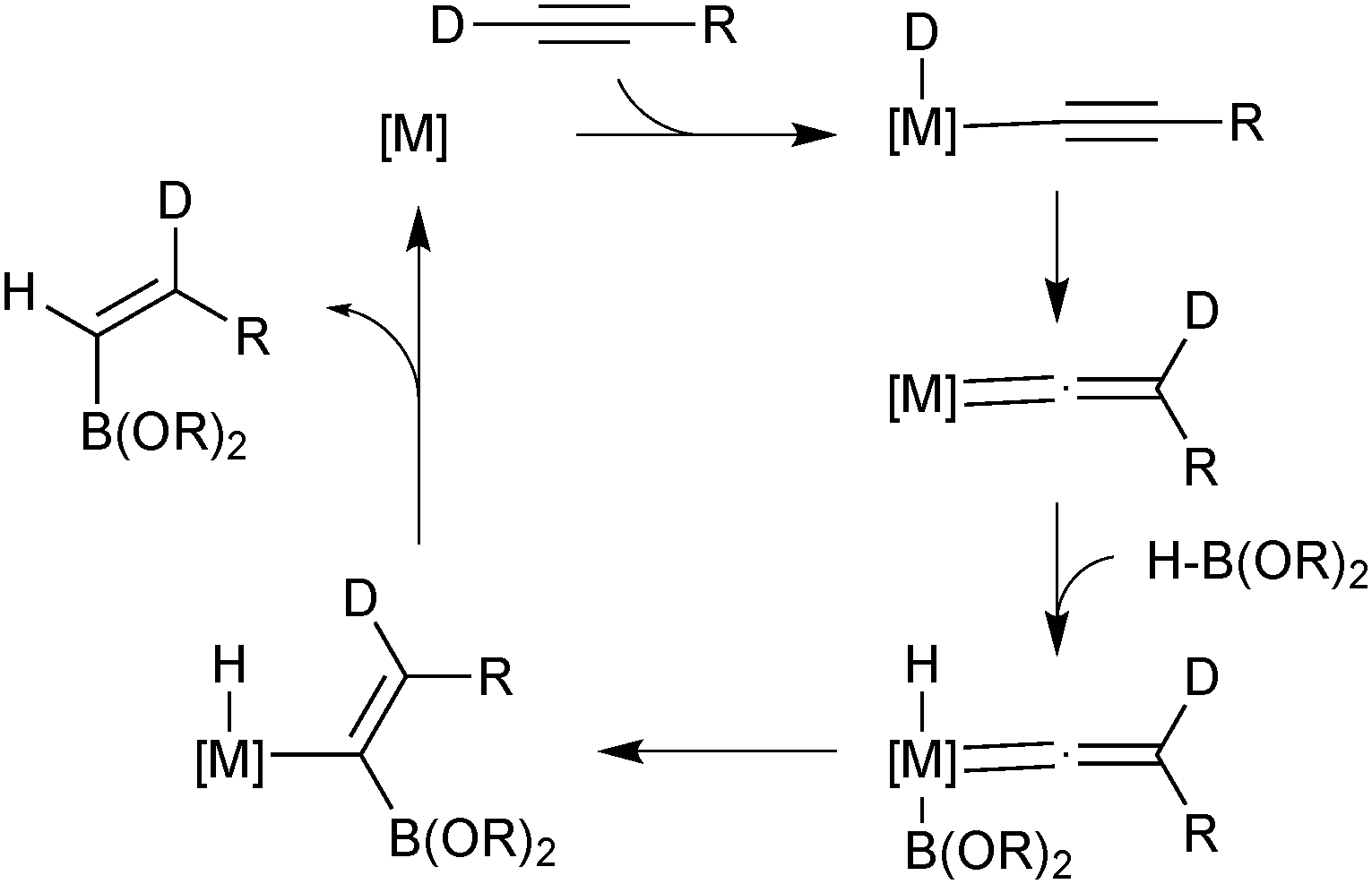 | ||
| Scheme 14 Miyaura's mechanism75 for the transition metal catalysed anti-hydroboration of alkynes. | ||
Diboration of unsaturated alkenes or alkynes with diboron tetrahalides generates useful bisfunctionalised building blocks after a subsequent boron ligation.77 However, the diboron tetrahalides are unstable and difficult to handle. Tetraalkoxy diboron reagents are more stable but require transition metal catalysis to break the B–B bond through oxidative addition.78 A simple platinum complex was first described to catalyse the diboration of alkynes to yield bis-borylated alkenes.79 For the diboration of alkenes, various transition metal complexes have been found to be effective, including a gold based catalyst system for the catechol diboration of alkenes that furnishes bis-borylated alkanes.80 The gold complex does not suffer from a competing β-hydride elimination reaction, which is prevalent in catalysts based on rhodium, Scheme 15.
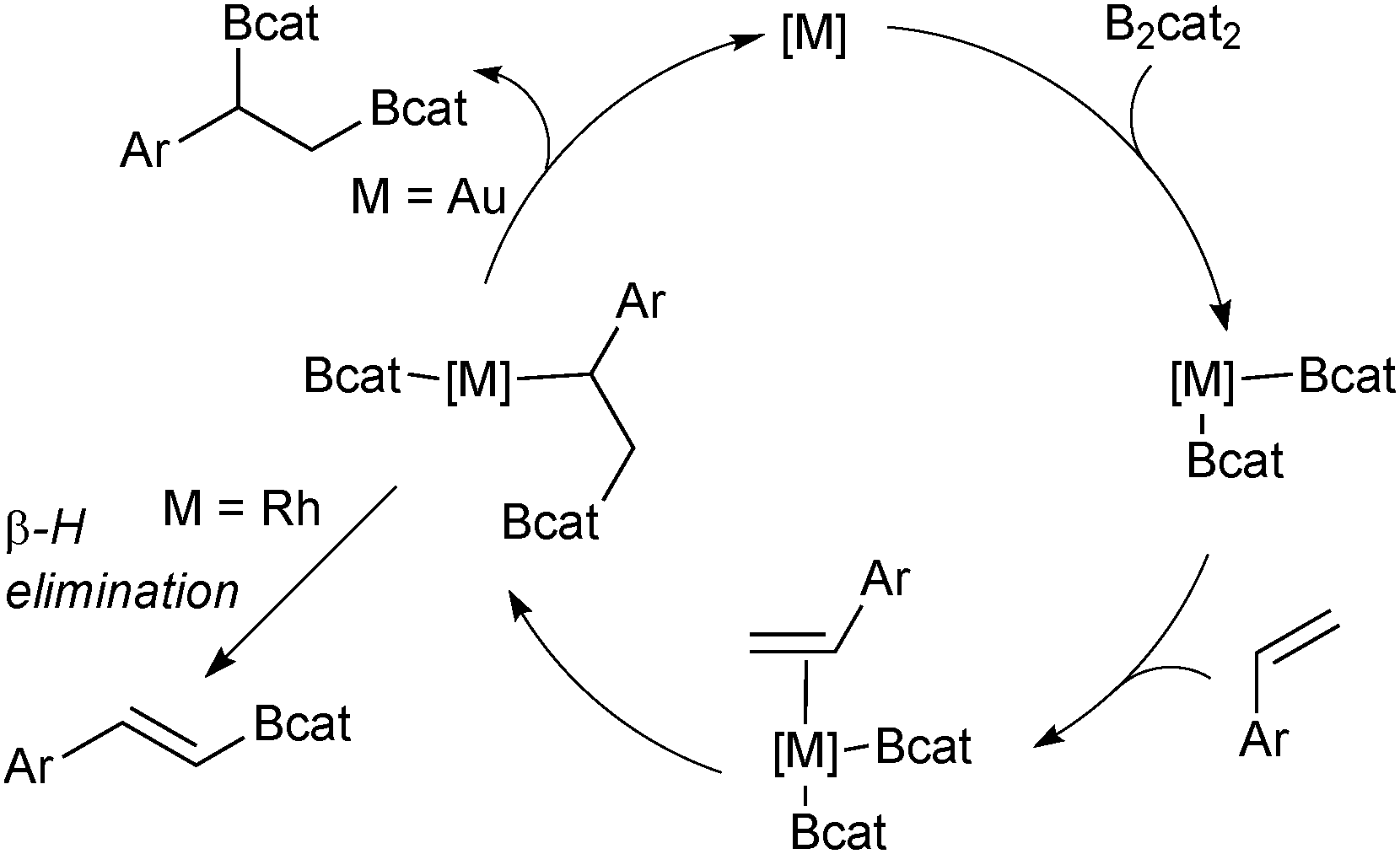 | ||
| Scheme 15 Metal-catalysed diboration of styrenes where gold based systems predominately lead to the desired product, but rhodium based systems suffer as competing β-hydride elimination. cat = catechol. | ||
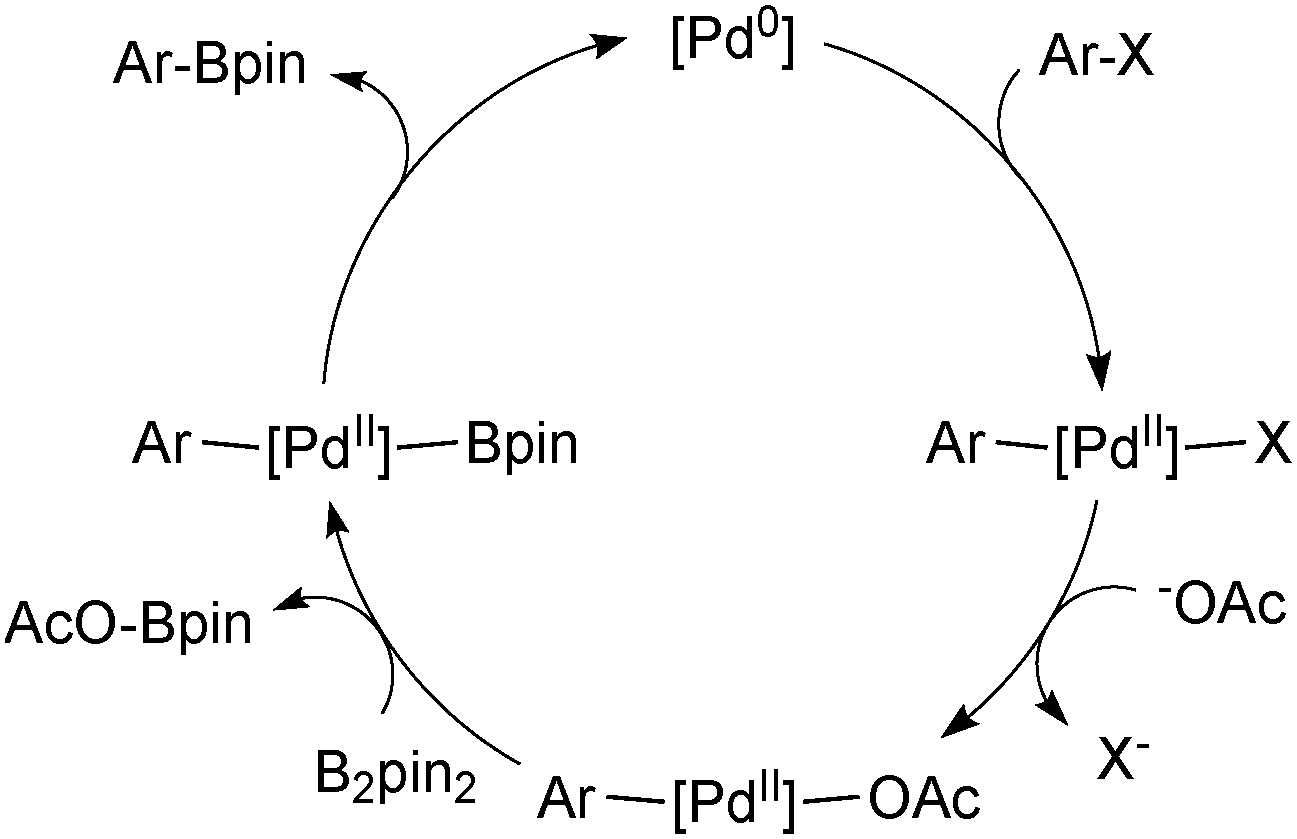 | ||
| Scheme 16 Mechanism for the Miyaura borylation of aryl halides. | ||
Following transmetalation of the diboron reagent, the co-generated acetoxy pinacol borate is not reactive, meaning that only half of the diboron reagent is converted to the boronic ester. However, the dialkoxyborane (HB(OR)2) can be directly employed, thus rendering it a more atom-economical procedure.84
Further atom economies can be achieved through the direct C–H borylation of arenes85 and alkanes,86 under remarkably mild conditions. An iridium based system successfully catalyses the mono-borylation, under steric control, of 1,2 and 1,4 symmetrically substituted arenes, and 1,3 asymmetric and symmetrically substituted arenes. The site selectivity of heteroarenes is largely governed by electronic effects.87 As the leaving group on the arene is essentially a hydride, the borylation employing diboron reagents (e.g. B2pin2) generates HBpin, which is an active borylating agent, Scheme 17. Thus, both equivalents of boron can be consumed, in contrast to when organohalides are employed. It should also be noted that other iridium88 and rhodium89 based catalyst systems can efficiently catalyse the borylation of unactivated alkanes and arenes respectively.
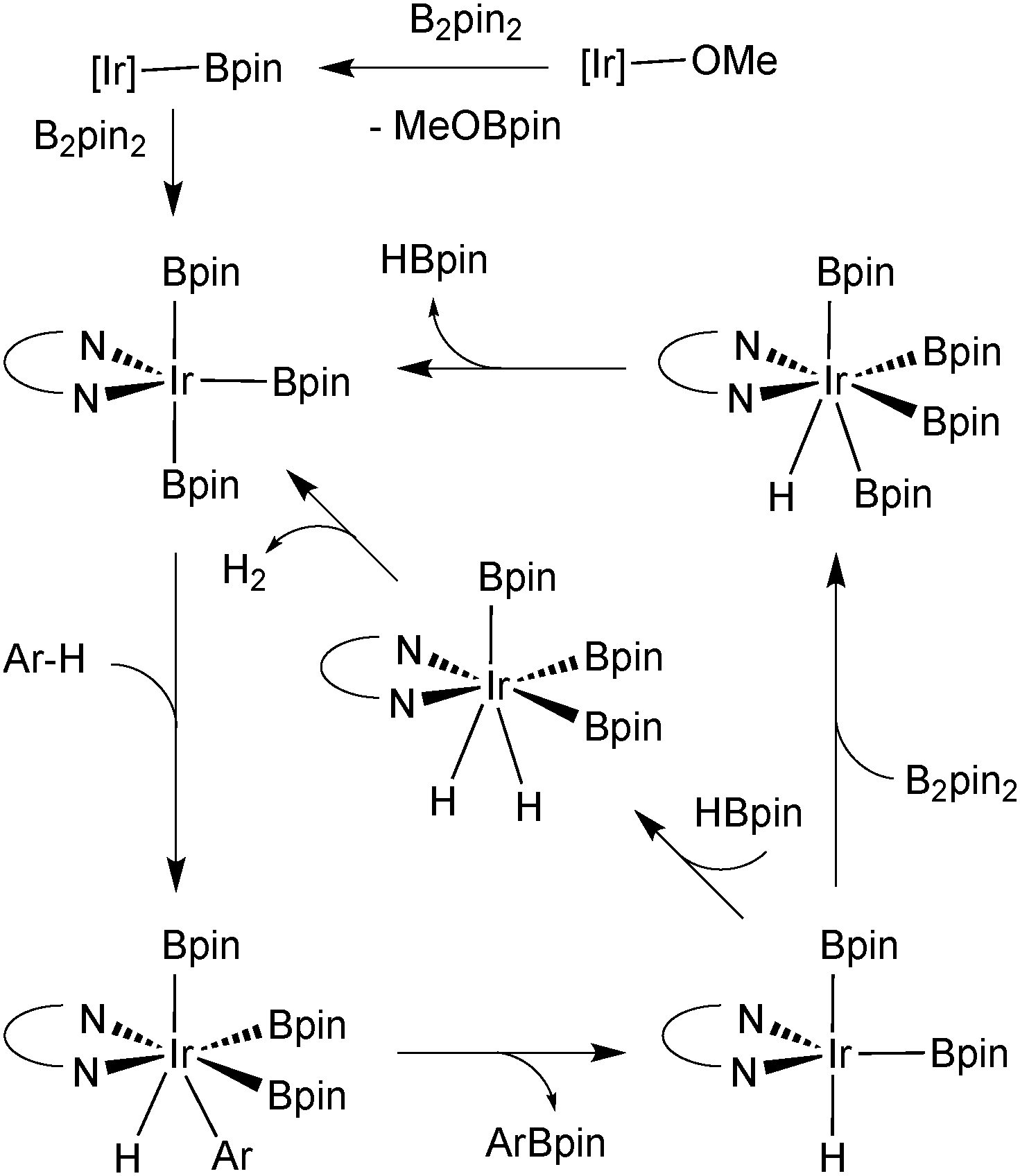 | ||
| Scheme 17 Mechanism for the Ir-catalysed borylation of arenes. | ||
A rhodium catalysed dehydrogenative borylation of alkenes gives products akin to the Miyaura-borylation of alkenylhalides, but without the requirement for the halide in the starting material, Scheme 18.90,91 Styrenyl and 1,1-disubstituted alkenes were suitable substrates for terminal mono-borylation to render vinylboronic esters with no competing hydrogenation of the alkene. A possible mechanism involves oxidative addition of bis(pinacolato) diboron, followed by alkene insertion into the Rh–B bond and β-hydride elimination. Recently the dehydrogenative borylation of terminal alkynes has been accomplished.92
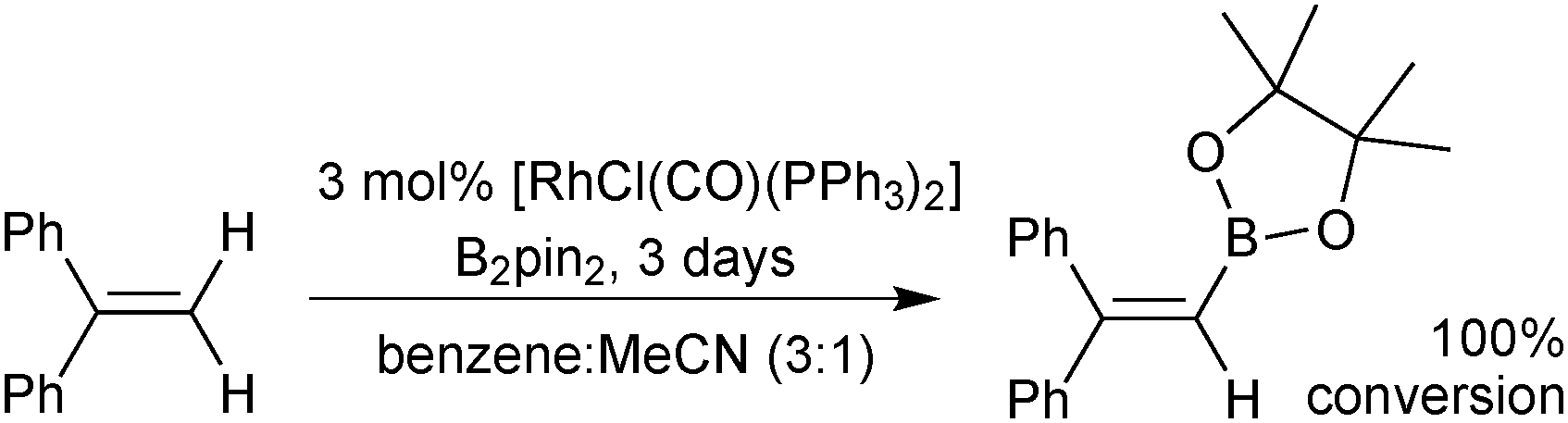 | ||
| Scheme 18 Rh-catalysed dehydrogenative borylation of alkenes. | ||
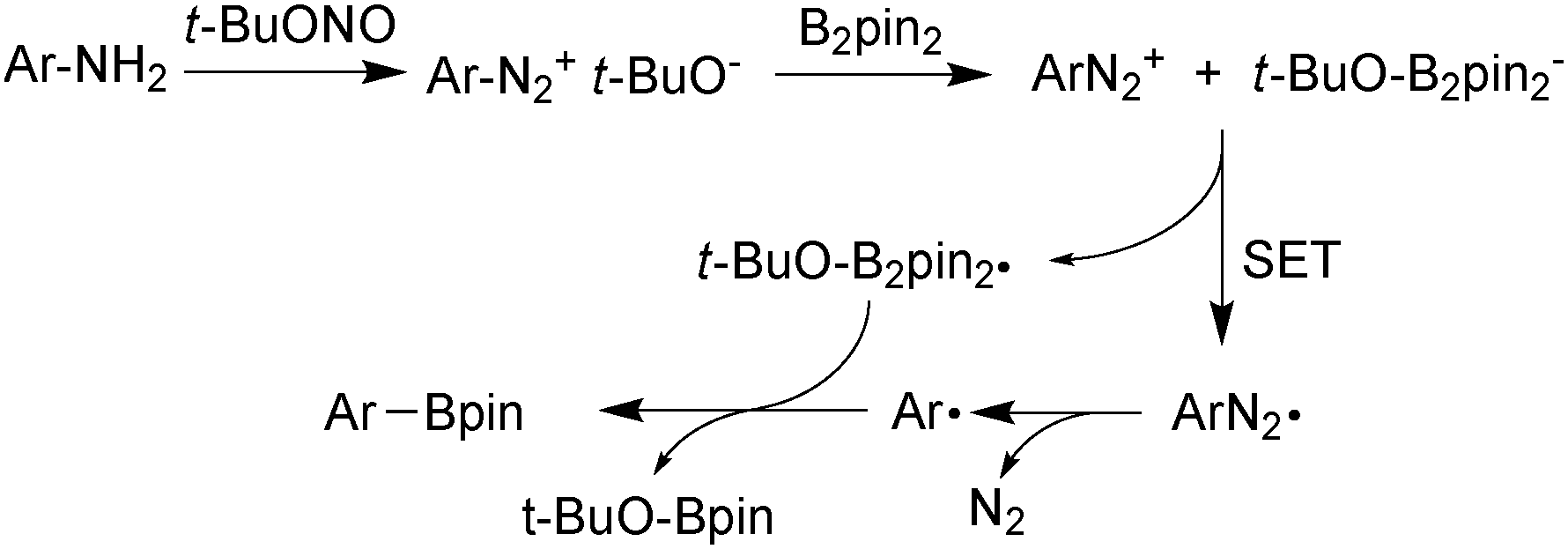 | ||
| Scheme 19 Borylation of anilines through a radical mechanism. | ||
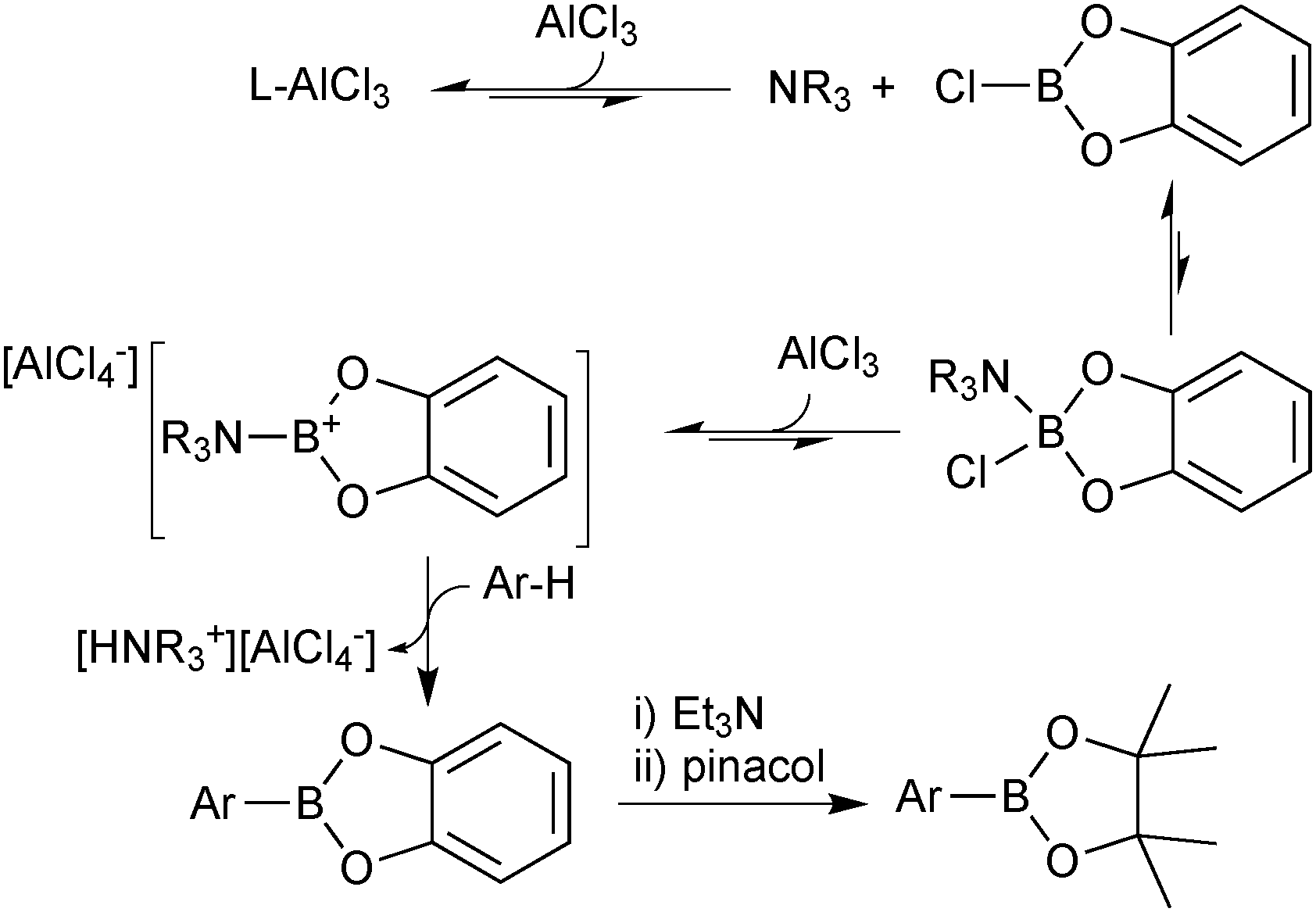 | ||
| Scheme 20 Mechanism for the Lewis-acid mediated direct electrophilic arene borylation. | ||
As the regioselectivity is determined by electronic factors, this methodology is complimentary to the iridium catalysed direct arene borylation, which operates primarily under steric control, or heteroatom direction. For example, the iridium catalysed borylation of N-methylindole predominately proceeds at C2,96 whereas this Lewis-acidic direct borylation selectively reacts at C3.97 Reaction with the corresponding pinacol borenium cation was not viable due to its lower electrophilicity, but transesterification of the catechol boronic ester with pinacol provides a viable alternative.
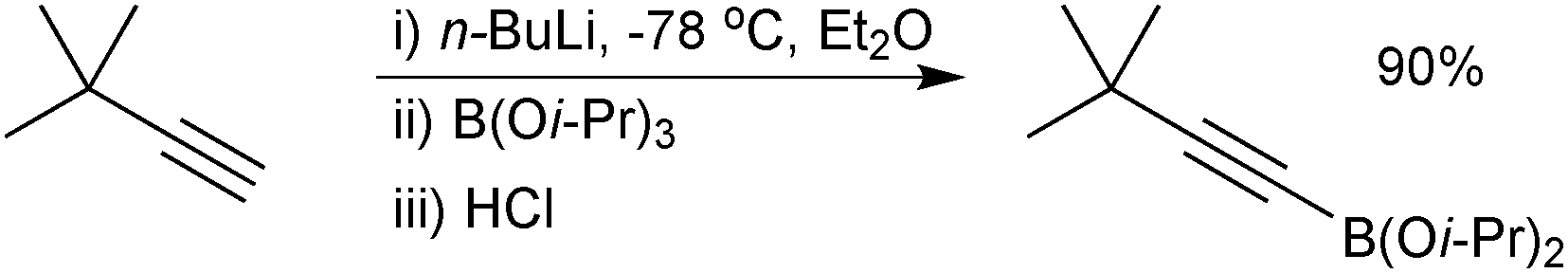 | ||
| Scheme 21 Preparation of an alkynylboronic ester via deprotonation/borylation. | ||
3.3. Applications in SM coupling
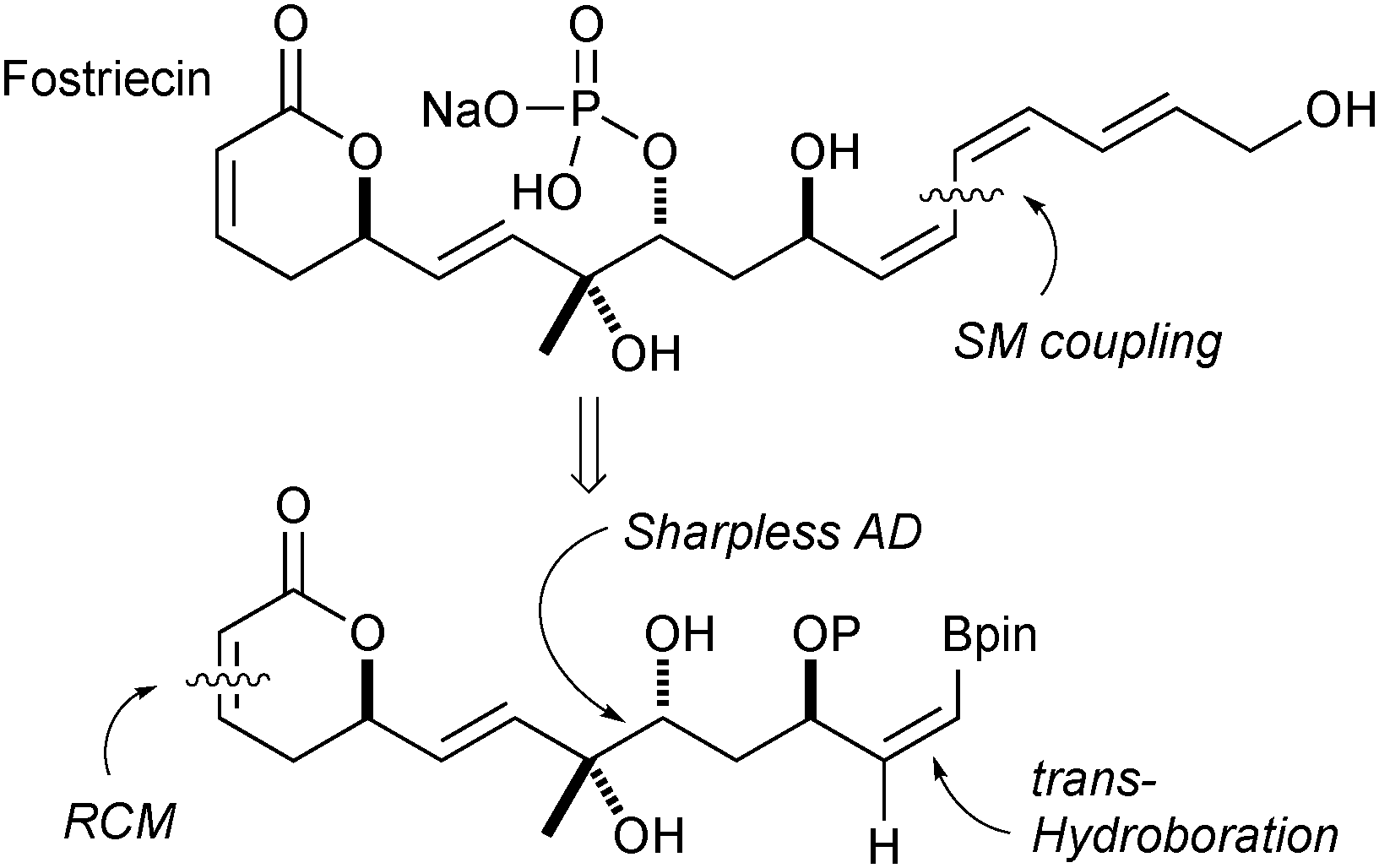 | ||
| Fig. 5 Retrosynthetic analysis of fostriecin. | ||
A catechol boronic ester was used in a convergent synthesis involving iterative SM couplings, to prepare new types of benzo-lipoxin A4 analogs, Scheme 22.100 These analogs were found to exhibit potent anti-inflammatory properties by in vivo suppression of neutrophil infiltration.
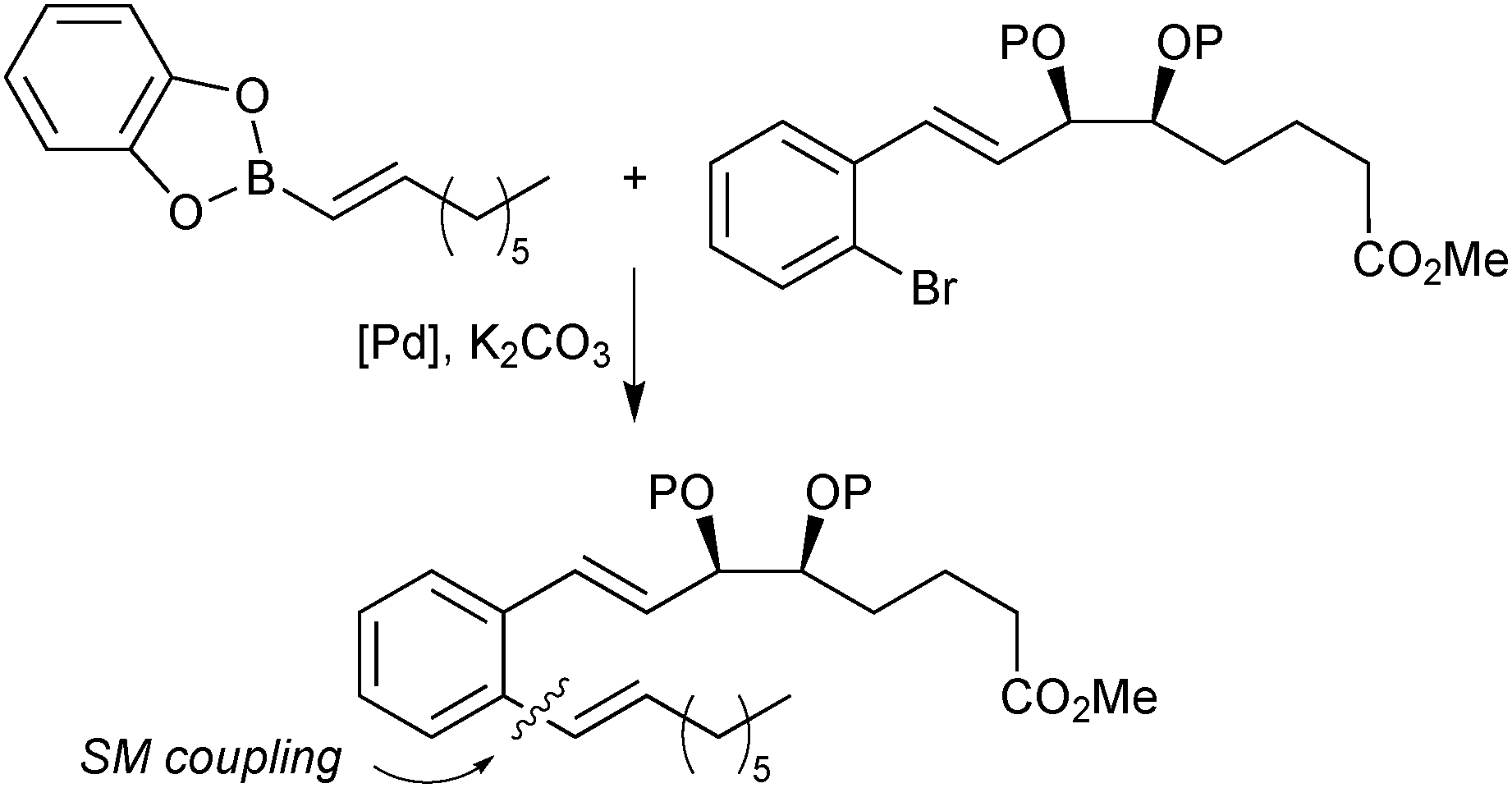 | ||
| Scheme 22 SM coupling of an alkenyl catechol boronic ester in the synthesis of a pharmaceutical target. | ||
A two-stage borylation/SM coupling of heteroarenes and polyfluoroarenes was demonstrated using an iridium catalyst.58 Borylation of the C–H bond was site-selective to the α-position of the heteroarene, Scheme 23, and the corresponding pinacol boronic ester was found to be stable for up to 60 days in air. Alternatively, the boronic esters could be used immediately in situ for the palladium catalysed SM coupling, which proceeded in good to excellent yields.
 | ||
| Scheme 23 Two-stage iridium catalysed C–H borylation/SM coupling. | ||
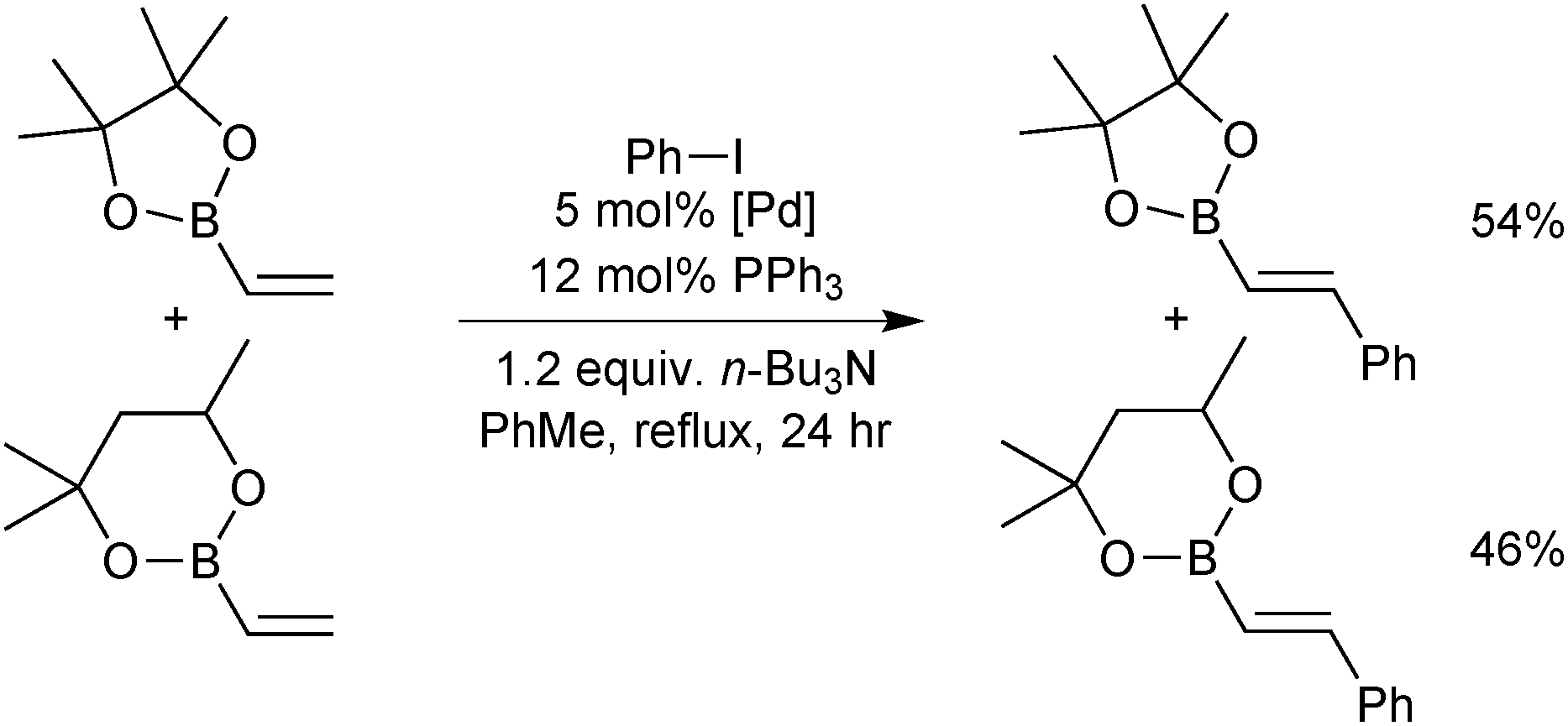 | ||
| Scheme 24 A competition between pinacol and hexylene-glycol vinylboronic esters in the Heck coupling with limiting phenyl iodide. | ||
4. Boronic acids
4.1. Properties and mechanism
Boronic acids were first employed for SM coupling in 1981,104 and continue to enjoy wide application. Their mode of Brønsted acidity depends on the medium. In anhydrous media, the hydroxyl group in the trigonal boronic acid species can act as the proton donor. However, in aqueous solution, the Lewis-acidic induced ionisation of water liberates a hydronium ion with concomitant generation of a trihydroxyboronate, Scheme 25.105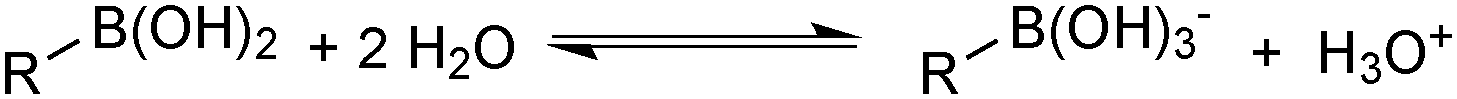 | ||
| Scheme 25 Lewis acid induced Brønsted acidity of boronic acids in aqueous solution. | ||
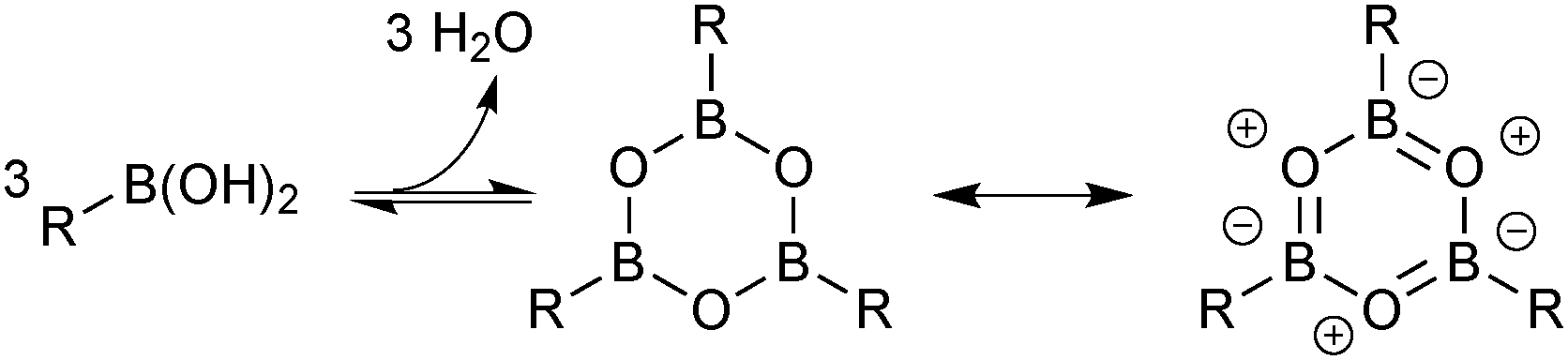
In general, boronic acids dissolve more readily in organic solvents than into neutral aqueous solutions. Under nominally anhydrous conditions, an equilibrium is established with the trimeric anhydride (boroxine); an entropically favoured process that liberates three equivalents of water, Scheme 26. In addition, boroxines are to some extent stabilised through partial aromatic character, albeit via triply zwitterionic mesomers. Setting the correct stoichiometry in a reaction can sometimes be non-trivial, as establishing this degree of dehydration is not straightforward, and it is common practice to add an excess of the reagent.
 | ||
| Scheme 26 Entropically favourable dehydration of boronic acids to form partially aromatic boroxines. | ||
However, three studies published in 2011 all provided convincing experimental evidence for the oxo–palladium pathway being the kinetically favoured pathway.110–112 The first study was reported by Amatore and Jutand, who employed electrochemical techniques to probe the mechanism and clarify the role of the base.110 The degradation or generation of palladium species gives a characteristic voltammogram and the resulting reduction or oxidation currents are proportional to the concentrations of the electroactive species. They considered the four possible transmetalation scenarios, wherein the base (a) plays no role, (b) reacts initially with the boronic acid, (c) reacts initially with the palladium(II) or (d) reacts with both the boronic acid and the palladium(II) species, Fig. 6.
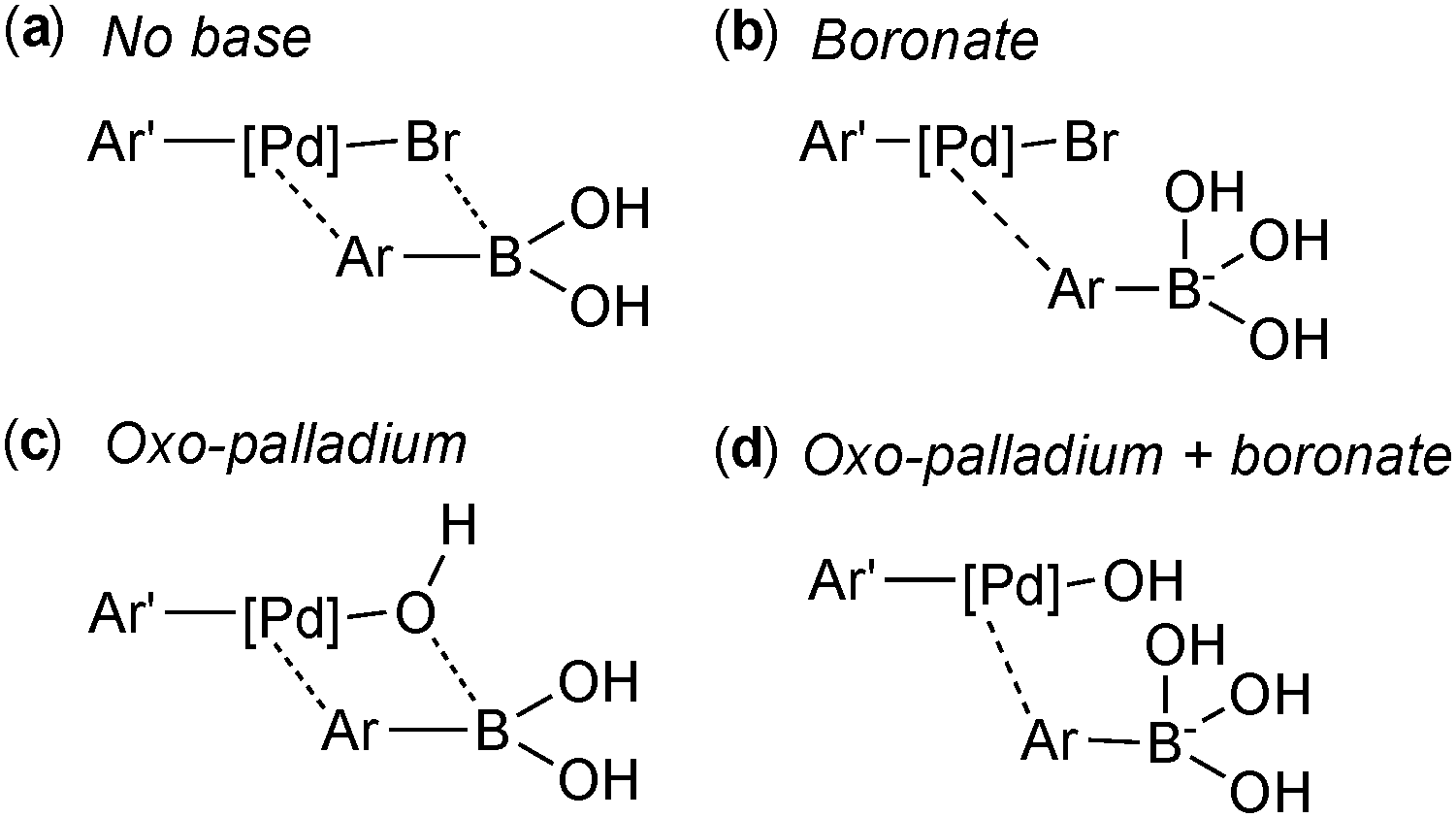 | ||
| Fig. 6 Four transmetalation scenarios, with the base playing three different roles. | ||
The kinetic data that was extracted indicated that the only reaction that occurs at a significant rate, is between the neutral boronic acid and oxo–palladium species, i.e. oxo–palladium pathway. The oxo–palladium species was readily formed from the halide complex, which is in direct contrast to the high barrier predicted in earlier theoretical work,107,108 which was unable to locate a pathway (DFT) for this hydrolysis. In the presence of a large excess of bromide ions (to bias the equilibrium away from the oxo palladium complex), reaction rates between the boronate species and the halide complex were found to be very slow indeed. Formation of the trihydroxyboronate was thus concluded to be detrimental to the coupling in that it sequesters the active transmetallating species: the boronic acid.
The second study was reported by Hartwig, who conducted a similar mechanistic study, but employing 31P NMR rather than electrochemical techniques.111 Rates of stoichiometric transmetalation were accurately measured between the halide complex [PdXAr(PPh3)2], and aryl trihydroxyboronate (boronate pathway), as well as between the oxo–palladium and boronic acid (oxo–palladium pathway), at low temperatures (−30 to −55 °C).111 The rate of transmetalation between the boronate and the bromide complex was found to be around four orders of magnitude slower than that between boronic acid and the oxo–palladium complex. Equilibrium studies were undertaken, and these confirmed ready access to the oxo–palladium species.
In the third study, Schmidt measured the stoichiometric rates of reaction between phenylboronic acid and an equilibrium mixture of [PdII(OAc)2] and base (NaOAc) using UV spectroscopy. This was compared to the rate between [PdII(OAc)2] with an equilibrium mixture of phenylboronic acid and base, Scheme 27.112 The formation of biphenyl was found to occur 1.3–2 times more rapidly under the conditions where neutral boronic acid was added to the pre-mixed solution of catalyst and base. This study thus indicated that the oxo–palladium pathway was also kinetically favoured under phosphine-free conditions.
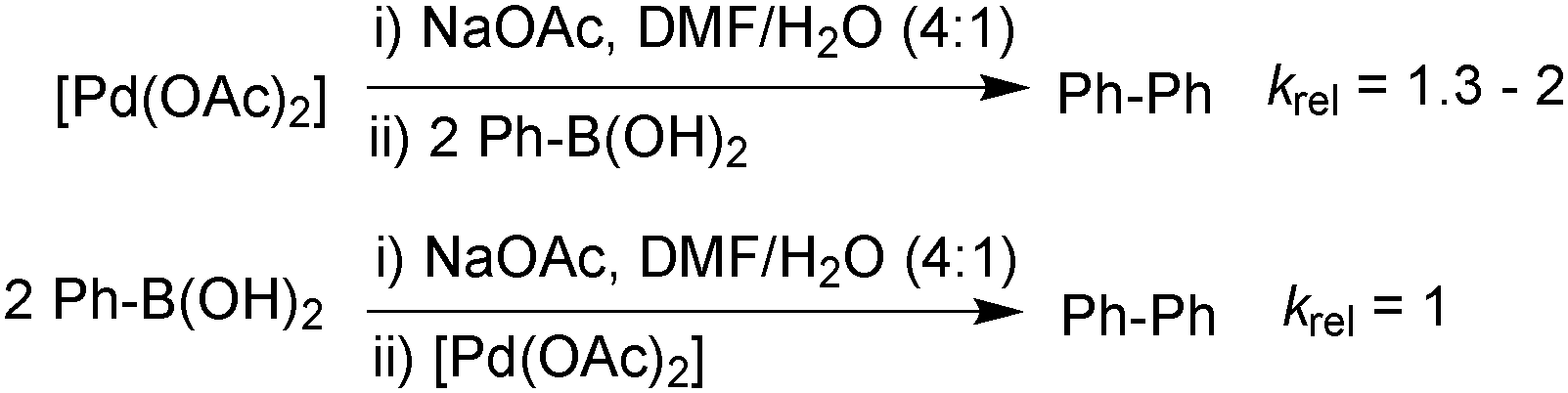 | ||
| Scheme 27 Relative rates of stoichiometric transmetalation in the homocoupling of phenylboronic acid, where the base is pre-equilibrated with either the boronic acid or palladium(II) catalyst. | ||
In a survey of almost forty thousand successful SM coupling reactions reported in the literature between 1981 and 2011, more than half were predicted to have had an aqueous biphase present.113 This suggests that the presence of a biphasic medium is important in these reactions. Such biphases can readily form upon the addition of an inorganic base to an initially homogeneous aqueous-organic solvent mixture.111 For example, in a study of an SM coupling in an aqueous THF medium (5M H2O), 11B NMR analysis of the distribution of boron species, indicated that boronic acid was present in the bulk organic phase, with only a small proportion of trihydroxyboronate present, and this was predominantly in the aqueous phase.113 Thus a biphasic medium appears well-primed for the oxo–palladium pathway because it limits accumulation of the unreactive trihydroxyboronate in the bulk phase, whilst still facilitating formation of the key catalytic intermediate, [Pd(OH)ArLn], via phase transfer of hydroxide between the aqueous-organic media. In contrast, homogeneous basic media appears better-primed for the boronate pathway, which according to recent mechanistic studies is slower, for boronic acids at least, than the oxo–palladium pathway.110–112
Detailed studies into the protodeboronation of arylboronic acids were conducted by Kuivila in the 1960s, well before the nascence of SM coupling. In addition to a direct uncatalysed reaction with water, three other mechanisms were identified: acid catalysed,115 base catalysed,116 and catalysis by a variety of metal salts.117 The base-catalysed process is obviously very pertinent to the conditions of SM coupling. However, although detailed kinetic analysis confirmed the base catalysis to be specific, not general, a rather limited pH range was explored (pH 5–7) due to competing oxidation processes above pH 7. In addition, the use of UV spectrophotometric techniques meant that reactions were conducted at much lower concentration than would normally be applied in an SM coupling. Nonetheless, a Hammett analysis (ρ = −2.32) suggested a small build-up of positive charge on the aryl ring, and the best correlation was obtained with regular σ values, rather than Brown's σ+ values, suggesting direct protonolysis,114 rather than cleavage via a Wheland intermediate, Scheme 28. Intriguingly, the simplest substrate, phenylboronic acid, was the slowest to protodeboronate and sat slightly off the line of best fit in the Hammett analysis. The two steps leading to protodeboronation (equilibrium generated boronate and rate limiting C–B cleavage) have opposing electronic demands, and thus any substituent in any position on the ring was reported to result in an increase in the rate of overall reaction.116
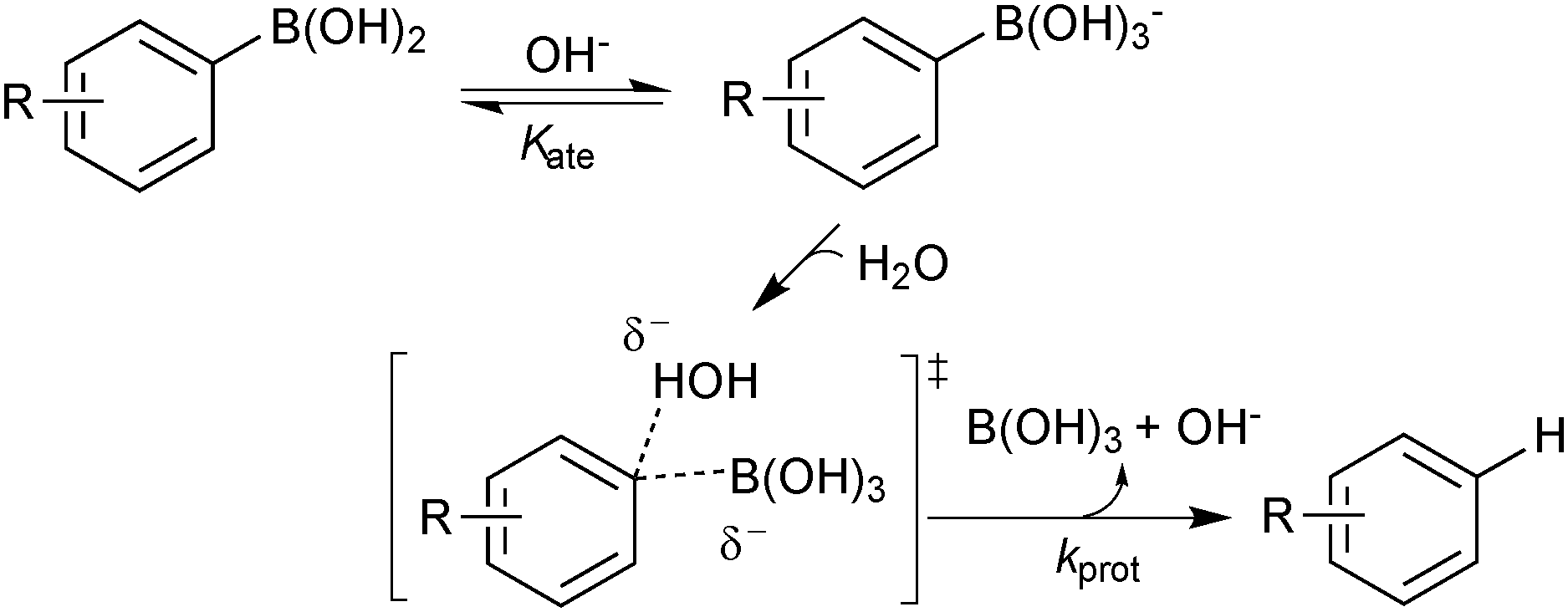 | ||
| Scheme 28 Base catalysed protodeboronation of arylboronic acids. | ||
Aerobically-generated peroxide-type oxidants can readily form in many ethereal solvents, and boronic acids are highly susceptible towards oxidation by these species under SM coupling conditions.118 Arylboronic acids form phenols following a 1,2-migration of the aryl moiety to an electrophilic oxygen atom, Scheme 29. Inhibitors or stabilisers such as butylhydroxytoluene (BHT) are sometimes added to attenuate the process but are removed through prior distillation of the solvent.
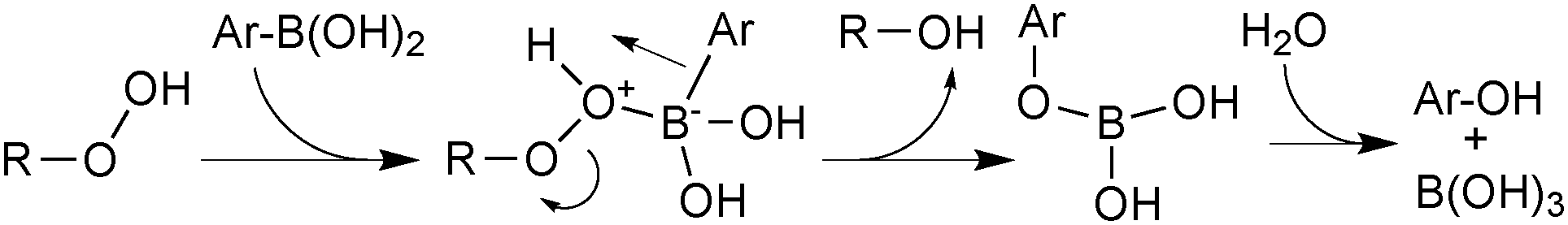 | ||
| Scheme 29 Oxidation of boronic acids from organic peroxides. | ||
There are two general conditions under which Pd(II) mediates boronic acid homocoupling. The first involves reductive activation of a Pd(II) precatalyst, consuming two boronic acid molecules, Scheme 30.
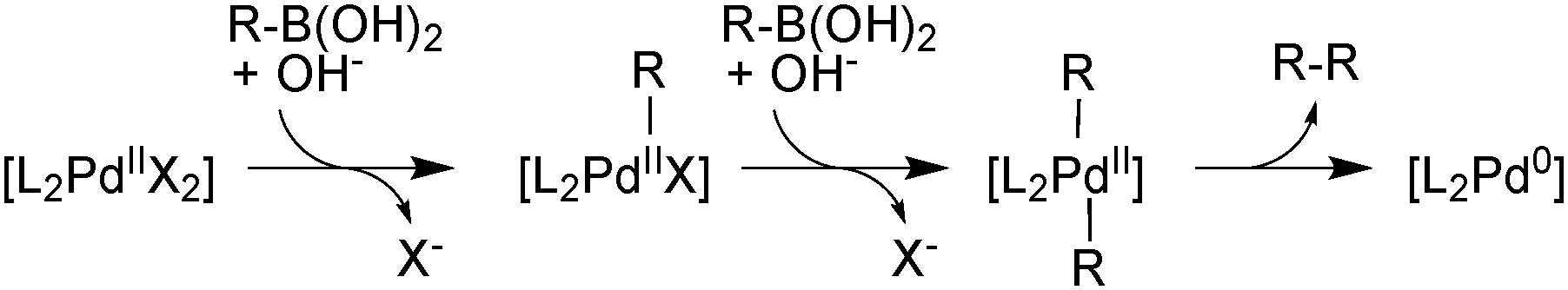 | ||
| Scheme 30 Reductive activation of a Pd(II) precatalyst resulting in homocoupling. | ||
The second common homocoupling process occurs when adventitious oxygen enters the system, for example, from the incomplete degassing of solvents or ingress of air through joints in the glassware. The mechanism for this catalytic side reaction, Scheme 31, was elucidated by Amatore and Jutand who again exploited electrochemical techniques.119 Palladium(0) reacts with oxygen to form a palladium(II) peroxo complex that consumes two molecules of boronic acid to form a homocoupled product.119,120 Perboric acid is a co-product, which, either itself or its hydrolysis product, e.g. hydrogen peroxide, oxidises a third molecule of boronic acid. For this reason, the two side products are formed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio throughout catalytic turnover. However, homocoupling without the accompanying oxidation to ROH is also reported to occur, especially in the presence of fluoride.118
:1 ratio throughout catalytic turnover. However, homocoupling without the accompanying oxidation to ROH is also reported to occur, especially in the presence of fluoride.118
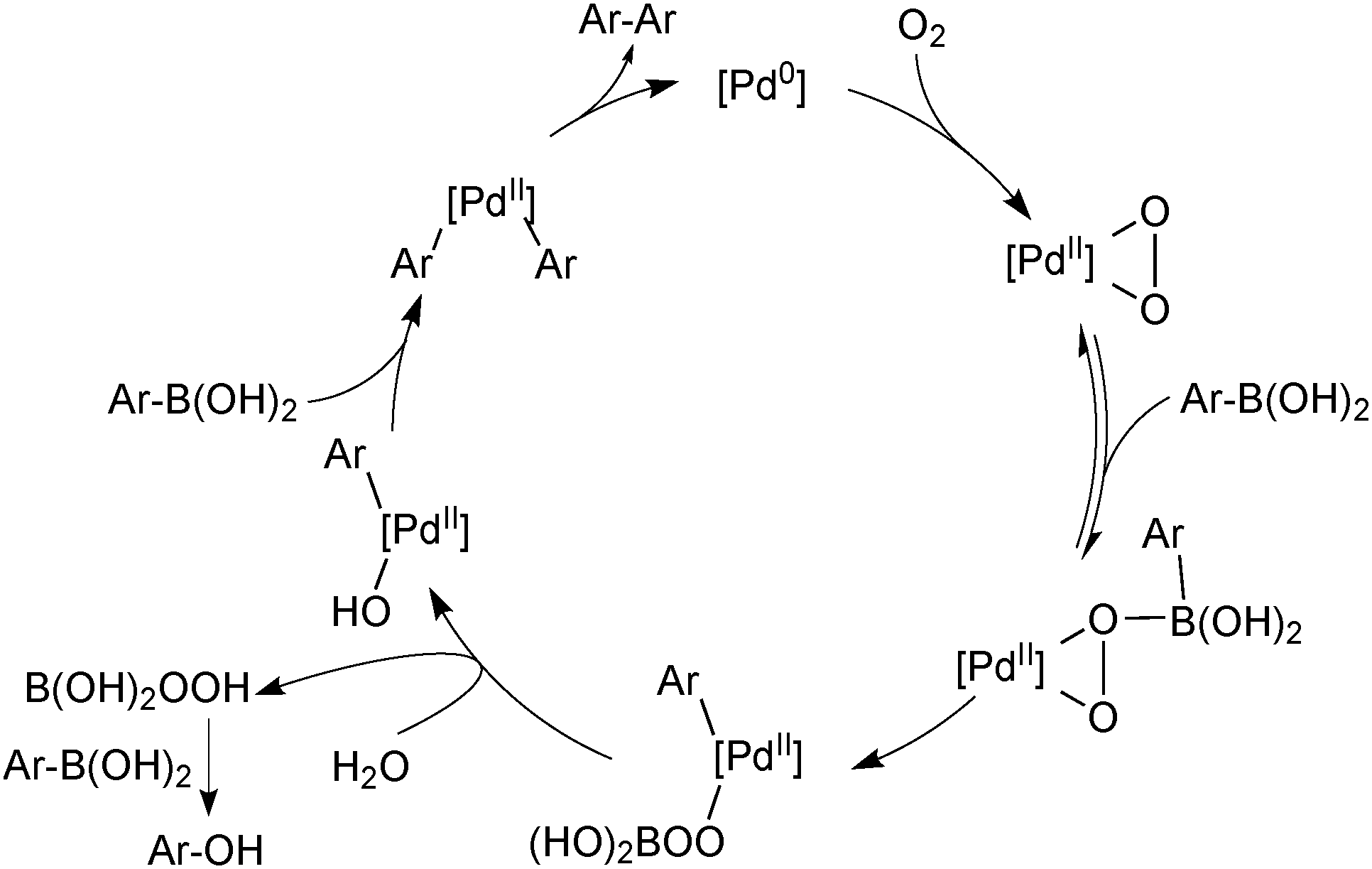 | ||
| Scheme 31 Mechanism of the oxidative homocoupling of arylboronic acids by palladium peroxo complex. | ||
4.2. Preparation
There are a wide range of methods developed for the preparation of boronic acids, but only the most relevant and useful routes for SM coupling are highlighted herein.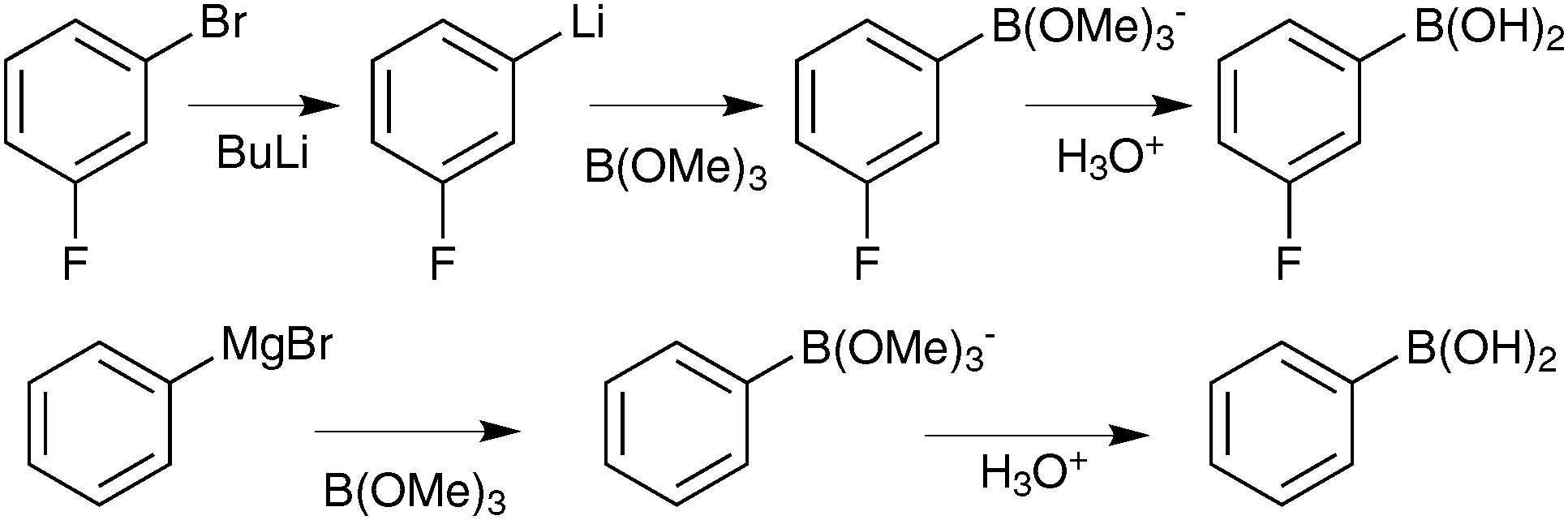 | ||
| Scheme 32 Boronic acid preparation via Mg and Li based reagents. | ||
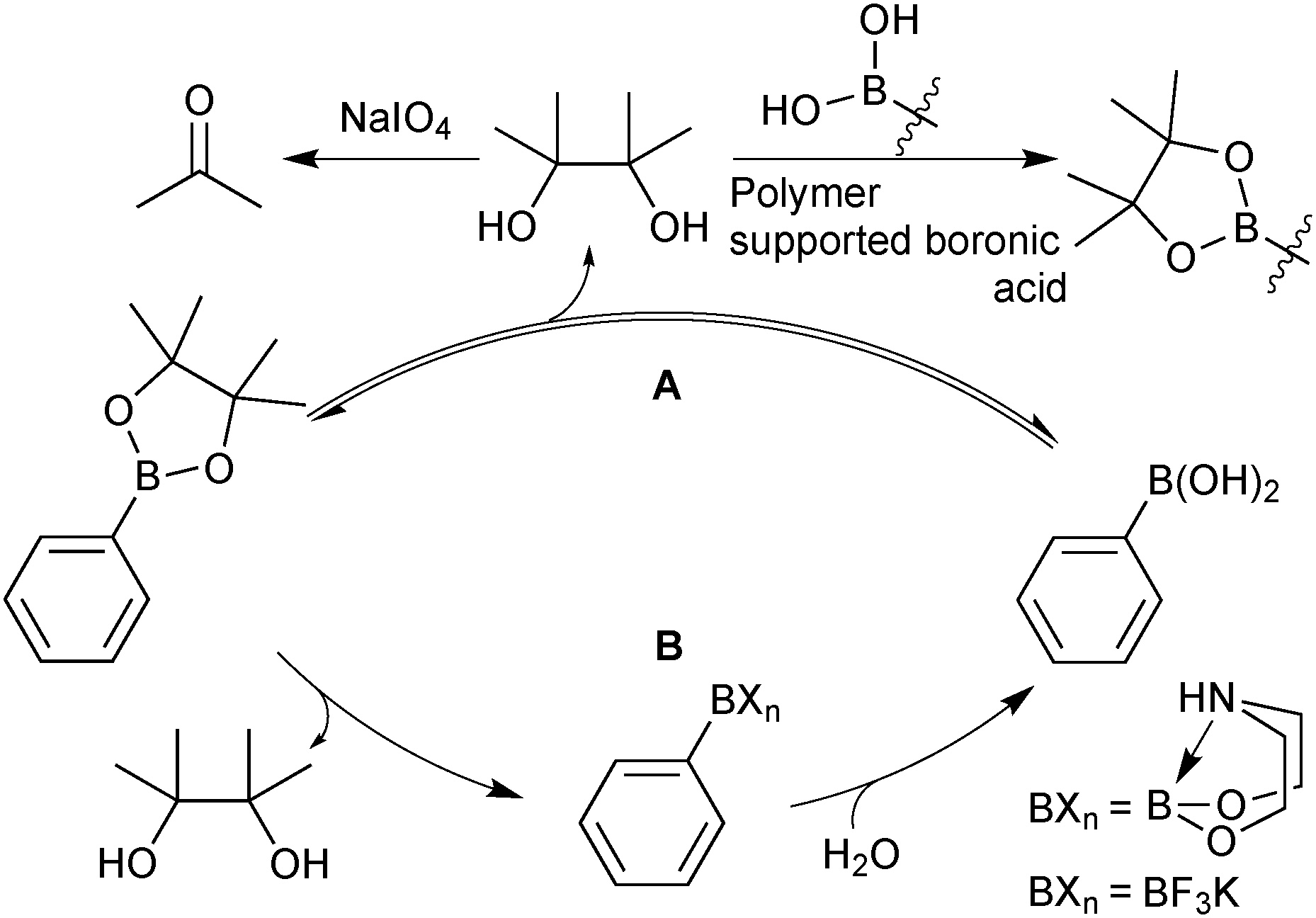 | ||
| Scheme 33 Two strategies for the hydrolysis of pinacol esters. | ||
The primary method under regime A, is to oxidise the pinacol to acetone, which can be easily removed under reduced pressure.124 This process has been applied to a wide range of systems and can be used in the presence of functionality sensitive to oxidation. However, conversions can often be unpredictable or poorly controlled due to the heterogeneous nature of the reaction. An alternative methodology utilises the transesterification of pinacol from the boronic ester to an excess of a polymer supported boronic acid.125 The solid polymer containing the pinacol can be physically separated, leaving the deprotected boronic acid in solution.
Under regime B, there are two major intermediates used to separate the boron reagent from pinacol; diethanolamine boronates and organotrifluoroborate salts. Diethanolamine undergoes transesterification with pinacol boronic esters and can be isolated via filtration.126,127 The diethanolamine complex readily hydrolyses under aqueous acidic conditions, leading to pure boronic acid, which does not recondense with the protonated form of the liberated diethanolamine. Organotrifluoroborates can be readily prepared from pinacol esters, the details of which are noted in Section 5.2. After purification from pinacol, hydrolysis can be performed in a number of ways. Under aqueous solvolytic conditions it has been shown that organotrifluoroborates undergo equilibration to form boronic acids, and fluoride.113 The preparative methods employ a variety of fluorophiles to “mop-up” fluoride, thereby pushing the equilibrium in the forward direction. Such fluorophiles include either those that form insoluble precipitates due to high lattice enthalpies, such as iron,128 and lithium129 salts, or those which form very strong bonds to fluoride such as silica-gel,130 silyl compounds129 and alumina,131Scheme 34. Reactions are generally conducted in water, which reduces reaction times, and favours equilibrium towards the boronic acid product. The stability of trifluoroborates with electron-withdrawing substituents is very high and elevated temperatures and reaction times are often required.
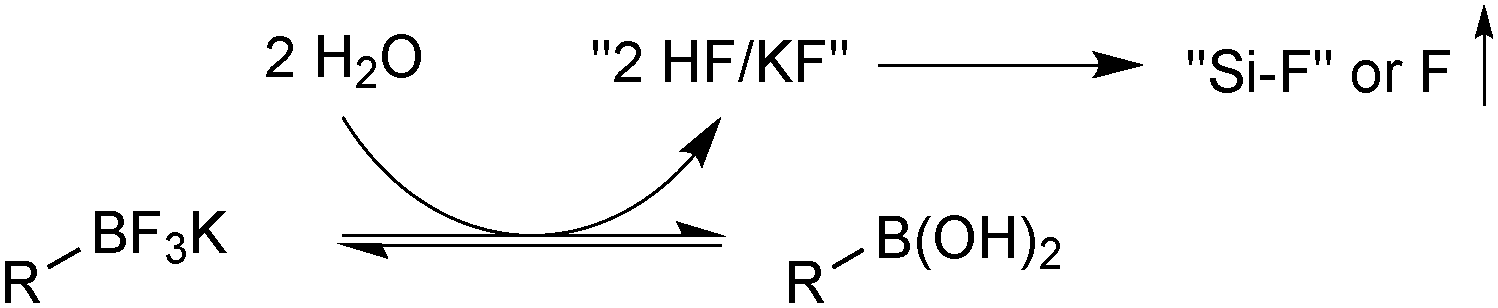 | ||
| Scheme 34 Hydrolysis of an organotrifluoroborate with removal of fluoride by a fluorophile, to reveal the parent boronic acid. | ||
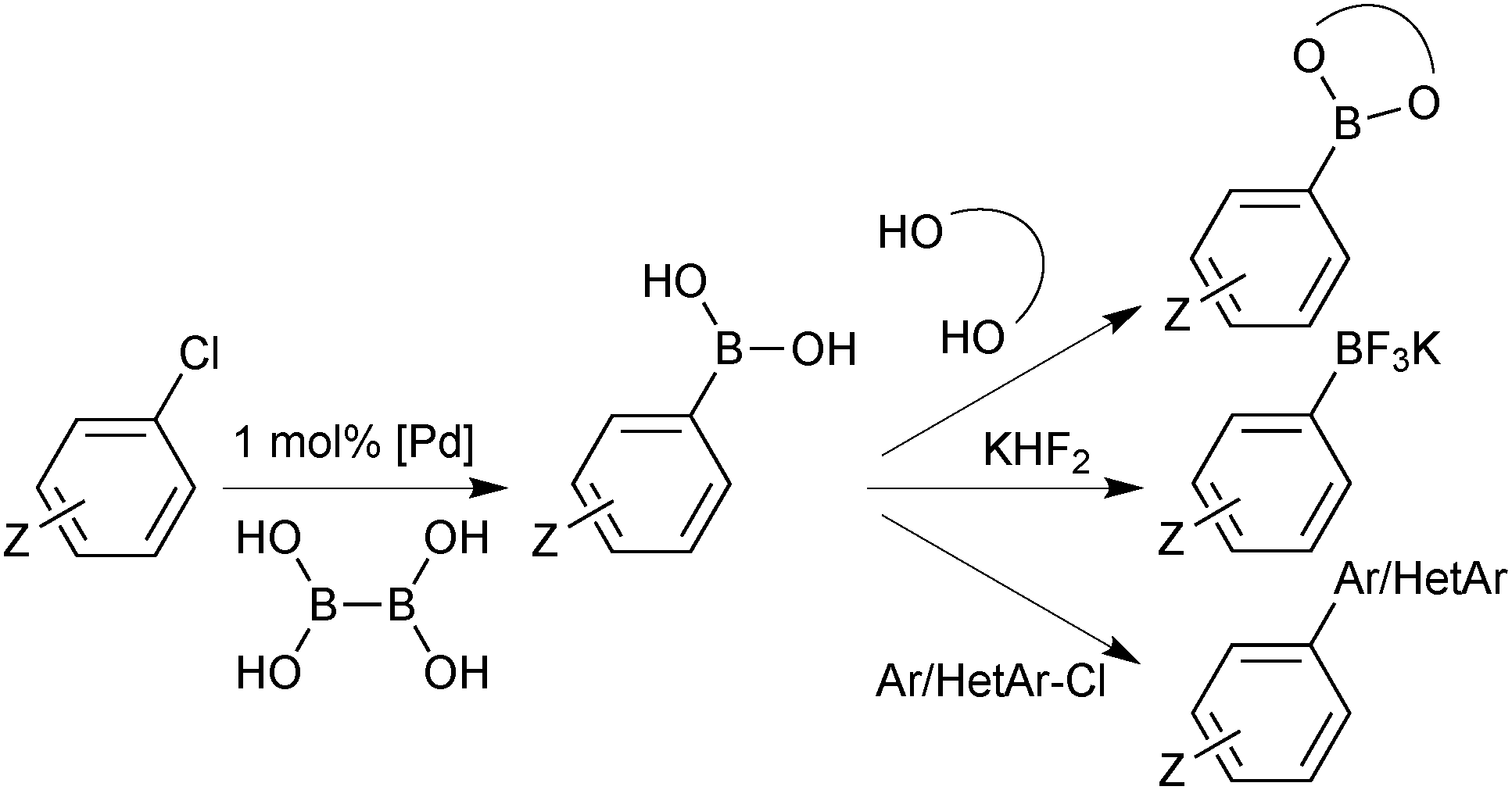 | ||
| Scheme 35 Pd catalysed borylation using bisboronic acid (BBA). | ||
A reasonable range of functionalities are tolerated, with the major exception being those susceptible to a competitive palladium catalysed hydride reduction, e.g. aldehydes or nitro groups. This side reaction was later exploited in a hydrogen transfer esterification methodology.134
A two-stage “one-pot” borylation/SM coupling protocol was also developed, through the subsequent addition of a carbonate base and organohalide coupling partner to the in situ formed boron reagent.135 Moderate to excellent yields of a variety of biaryl moieties were conveniently prepared.
The synthetic precursor to BBA, tetra(dimethylamino)diboron, was also shown to be an effective borylating reagent.136 Although not yet widely available, this procedure provides a more direct and atom efficient route from a range of aryl and heteroaryl bromides and chlorides.
4.3. Applications in SM coupling
The original reports from Suzuki and Miyaura employed organoboranes to cross-couple with aryl18 or alkenyl19 halides. Shortly after, it was revealed that organoboronic acids could also undergo transmetalation with palladium,104 and these have since become the standard reagent for the coupling due to their greater aerobic stability, ease of production and higher affinity for transmetalation. Accordingly, a very wide range of boronic acids are now commercially available.Boronic acids are employed in the synthesis of BASF's multi-purpose fungicide, Boscalid; undoubtedly the largest scale SM coupling reaction currently performed. More than 1000 tonnes per year are manufactured, with the arylboronic acid/aryl chloride coupling as a key step.137Merck's antihypertensive drug, Losartan, is another prominent example that has utilised arylboronic acids in SM coupling for the construction of the important biaryl motif.138 A multi-kilogram scale preparation of ABT-963, a potent and selective COX-2 inhibitor (non-steroidal anti-inflammatory drug), has been reported by Abbott Laboratories. The 4-step route includes a SM coupling with an arylboronic acid, giving the product in an 88% yield,139Fig. 7.
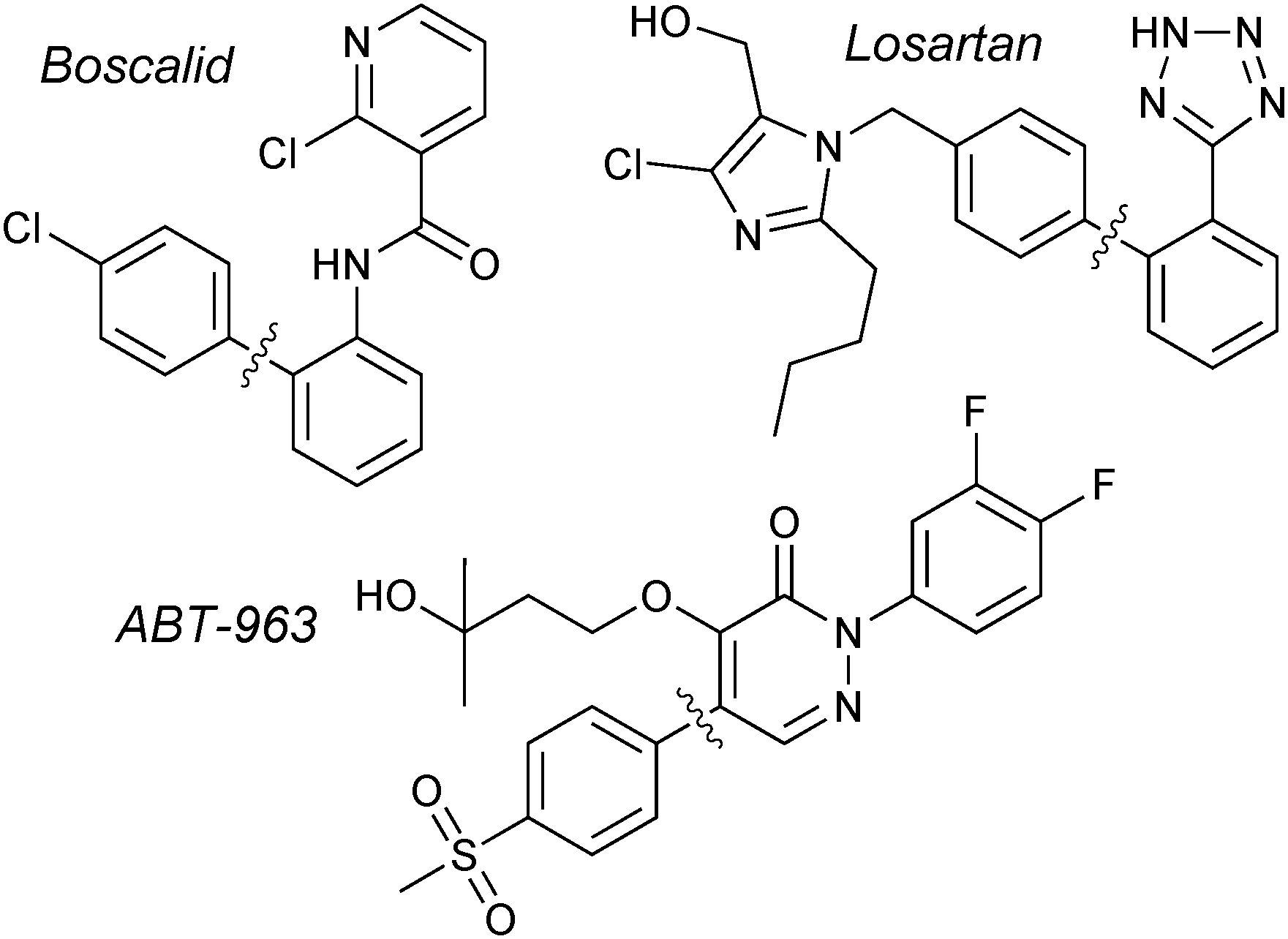 | ||
| Fig. 7 Large scale SM couplings employing boronic acids. | ||
Boronic acids are also regularly used as cross-coupling partners in natural product syntheses. One elegant example uses the SM reaction in the late stage coupling of two key fragments in the synthesis of (−)-FR182877, giving the product in an 84% isolated yield. A thallium base was employed due to the reported acceleration the counter cation effected on transmetalation, Scheme 36.140
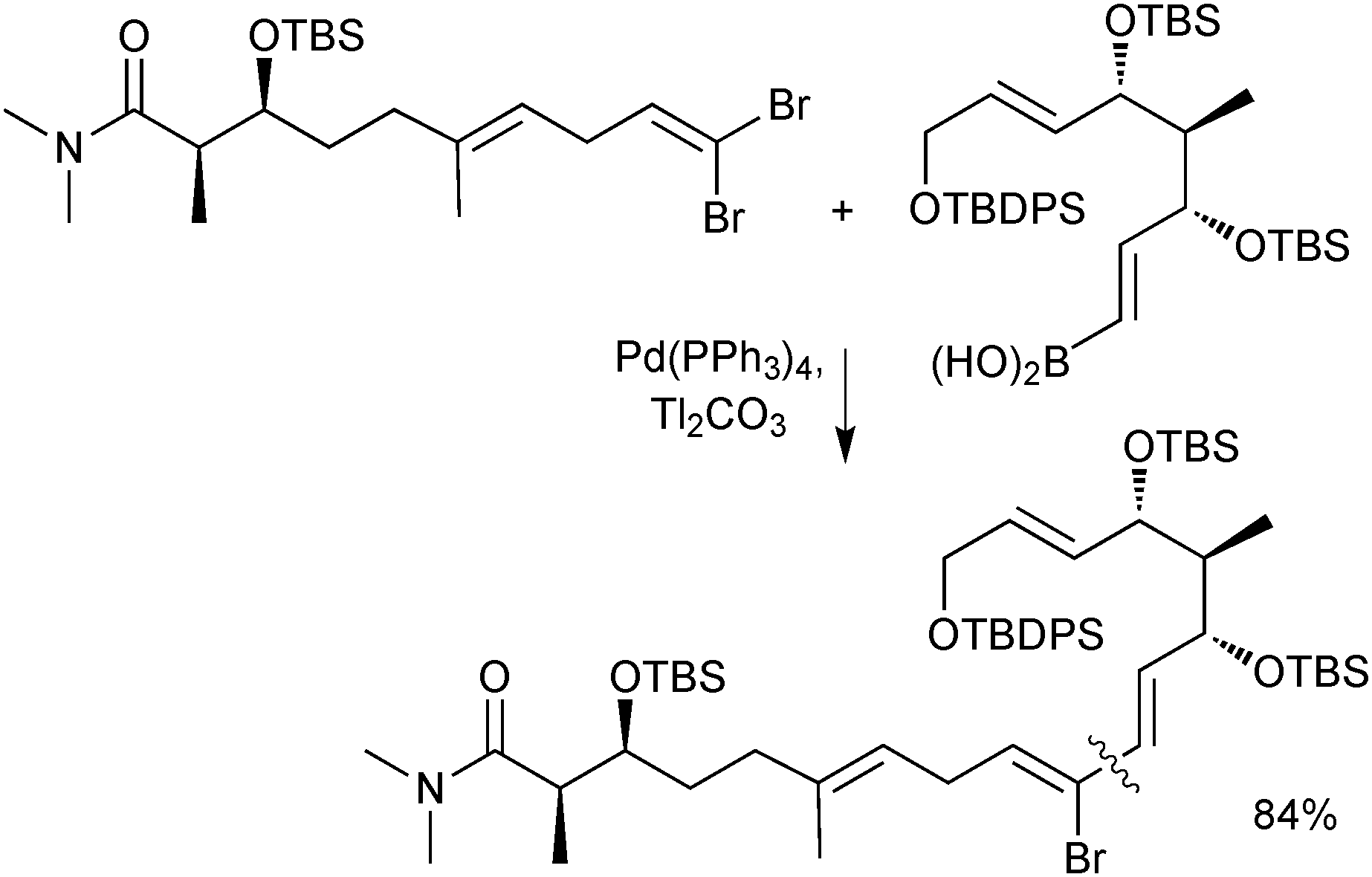 | ||
| Scheme 36 SM coupling in the synthesis of (−)-FR182877. | ||
Aryltriazenes, in combination with a Lewis acid was shown to be an effective system for the formation of unsymmetrical biaryl units.141 A good range of aryl moieties were successfully coupled under ligand141 and ligandless142 conditions, Scheme 37.
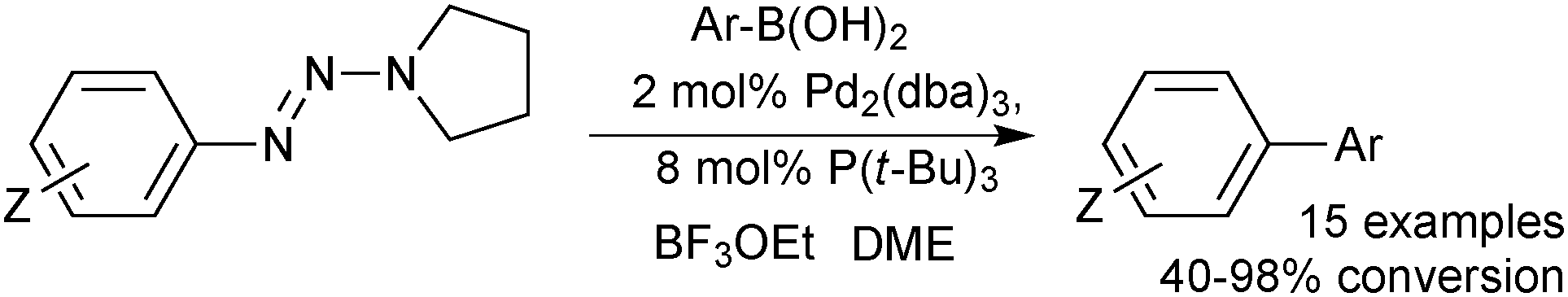 | ||
| Scheme 37 Conditions for the coupling between aryltriazenes and arylboronic acids. | ||
This unusual system has the advantage that the electrophilic component is easily formed from the corresponding arylamine. BF3·OEt was found to be the most effective Lewis acid. This was proposed to serve two roles: firstly, to activate the aryltriazene towards reaction with palladium(0) and secondly the resulting aminotrifluoroborate species serves as a fluoride source to activate the boronic acid towards transmetalation, Scheme 38.
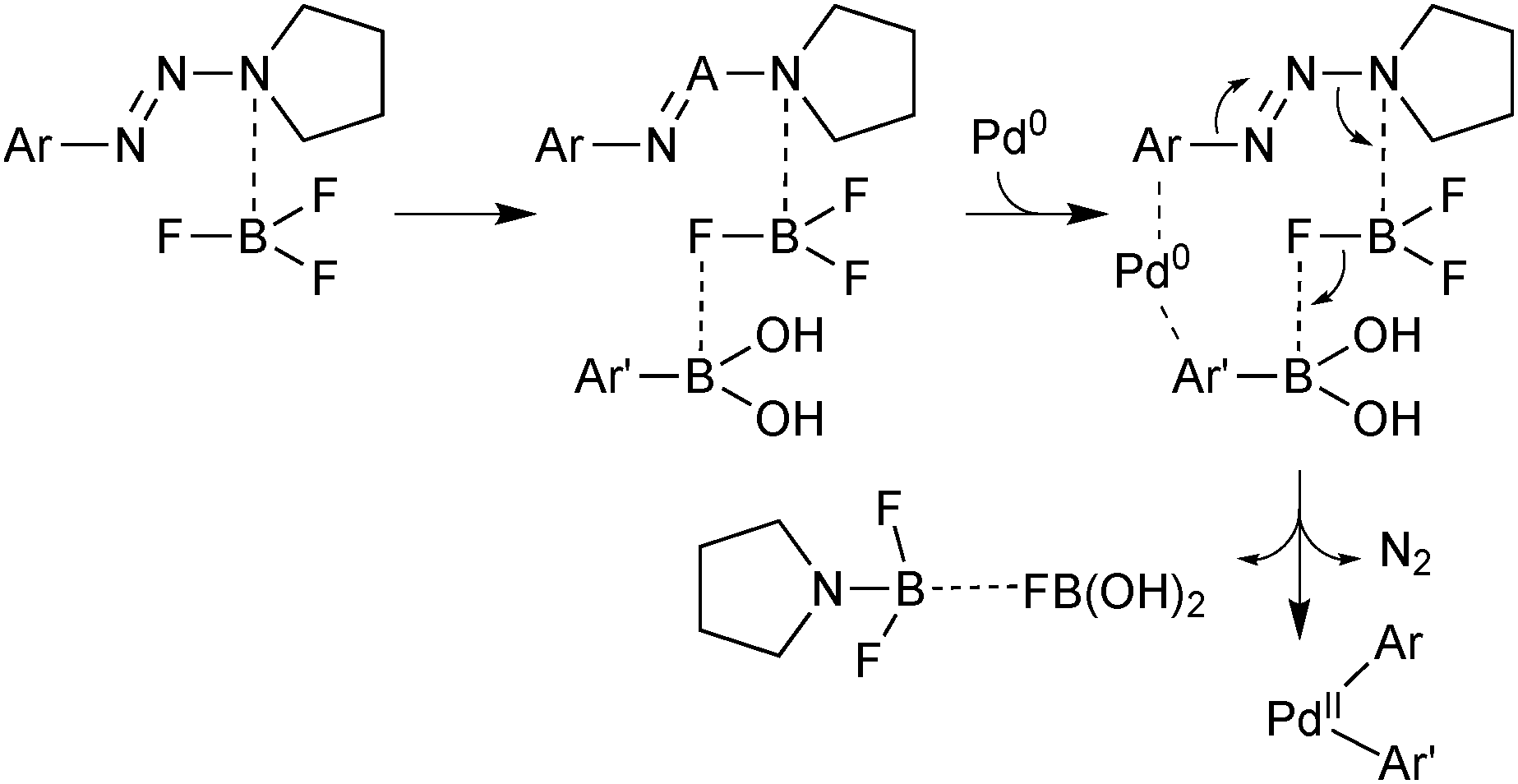 | ||
| Scheme 38 Mechanism proposed for the coupling between aryltriazenes and arylboronic acids. When considering this proposal, the entropic cost of such an assembly should be noted. | ||
5. Organotrifluoroborate salts
5.1. Properties and mechanism
Potassium organotrifluoroborate salts (R-BF3K) were first characterised in 1960 by Chambers,143 but the following three decades witnessed only a handful of further publications. However, since the mid-1990s, when their utility began to be recognised, they have steadily become established as a very widely used class of organoboron reagent.In contrast to boronic acids and esters, organotrifluoroborates are tetrahedral in geometry and not Lewis acidic, due to the additional ligand bound to the boron centre. This quarternisation with exceptionally strong B–F bonds, together with their salt-like structure, gives them favourable physical characteristics of being free-flowing crystalline solids, which tend to melt and decompose only at very high temperatures. As well as being monomeric in nature they are stable to air and aerobic moisture. These factors render them easy reagents to handle, unlike, for example, certain boronic acids, e.g. cyclobutylboronic acid decomposes in air,144 or pinacol boronic esters, many of which are liquids or low melting solids.
In solution, the trifluoroborate moiety is stable under anhydrous conditions, but when subjected to aqueous or protic media they hydrolyse, via equilibrium, to form the corresponding boronic acid or ester, Scheme 39.113 Upon hydrolysis, HF is formally liberated, which in aqueous conditions can cause etching of glassware if it is not rapidly quenched by base or an alternative sacrificial fluorophile. Nonetheless, R-BF3K salts are still considered to be chemically robust materials, capable of withstanding a number of standard organic reaction conditions. As such, they can be used as intermediates for a range of synthetic pathways; popularised by the stability exhibited towards distal manipulation of various functional groups.
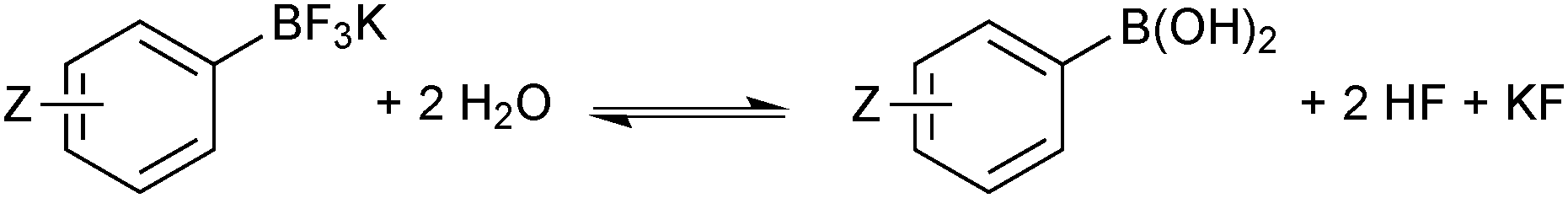 | ||
| Scheme 39 Hydrolysis of aryltrifluoroborates, which liberates arylboronic acid with HF/KF co-products. | ||
R-BF3K salts are tolerant to the conditions employed in a range of common synthetic transformations, including Swern/Dess–Martin oxidations,145 ozonolysis,146 Wittig and Horner–Wadsworth–Edmonds olefinations,147 condensation reactions,148 and 1,3-dipolar cycloadditions (“click” chemistry).149 However, for certain transformations, e.g. reductive amination150 and lithium–halogen exchange,151 KHF2 is employed during work up, suggesting that the trifluoroborate functionality is not always maintained throughout the procedure. Moreover, the greatest disadvantage to R-BF3K salts is in their instability to silica-gel and their insolubility in many apolar solvents. Nonetheless, they are easily purified through crystallisation techniques, which can be especially beneficial on scale-up.
Functional group interconversion of the trifluoroborate functionality is possible and expands the realm of application accessed through these reagents. Switching the “R-group synthon” from being nucleophilic to electrophilic is readily achieved by transformation to an organohalide. This halodeboronation has been demonstrated with the use of electrophilic sources of iodide,152 chloride,153 bromide154,155 and fluoride.156,157 In addition, it has been shown that Ar-BF3K salts are oxidised to phenols,158 and nitrosated at the ipso position,159 from which a whole plethora of chemical transformations are available, Scheme 40.
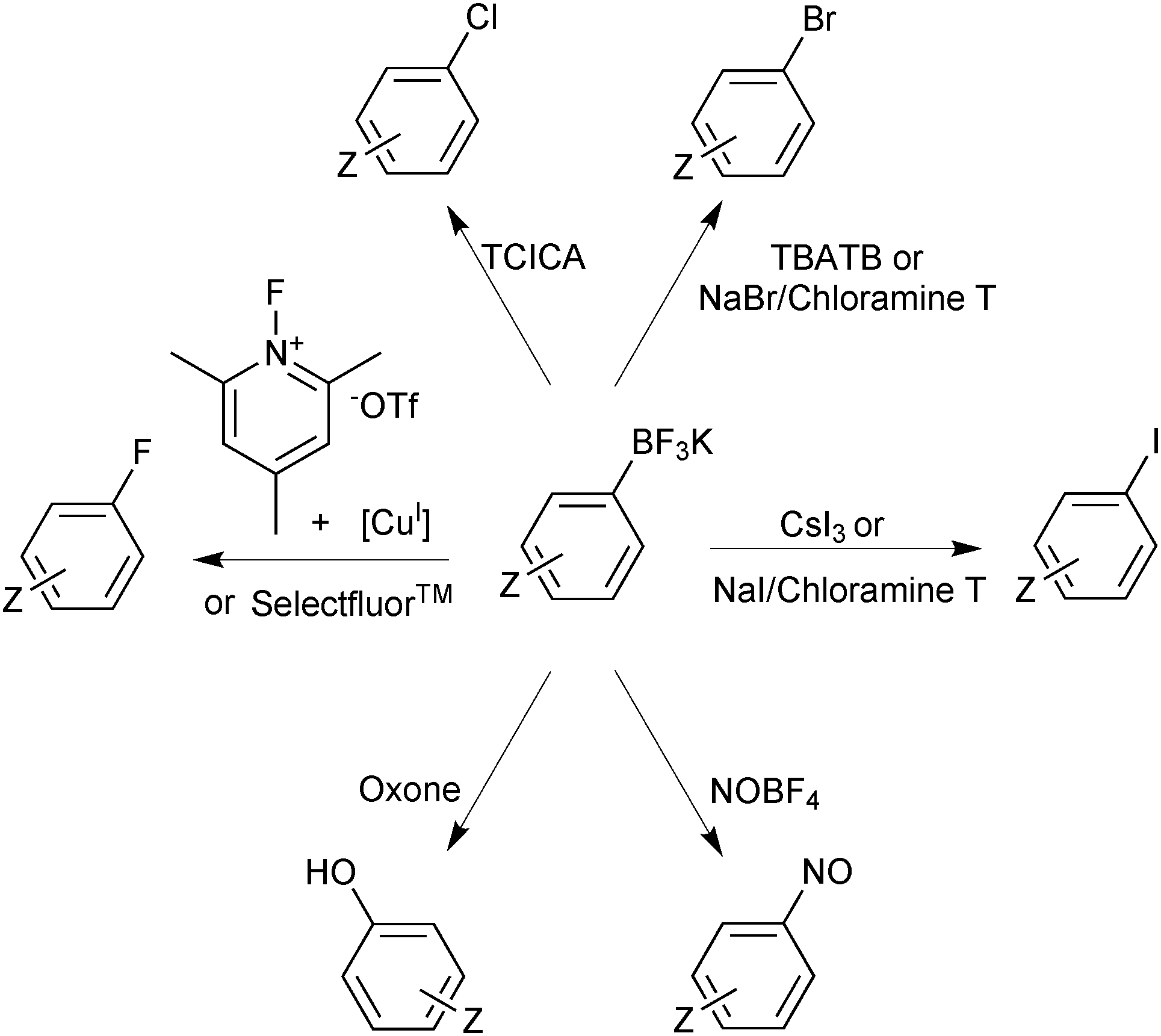 | ||
| Scheme 40 Functional group interconversion of aryltrifluoroborates. | ||
In SM coupling, superior reaction outcomes, in terms of yield of product, have been widely reported when employing organotrifluoroborate salts in place of the corresponding boronic acid.160 Initial mechanistic investigations found that the organotrifluoroborate salt was not the active transmetalating species.4,161 To rationalise the superior behaviour it was proposed that partial hydrolysis to a more active mixed fluoro/hydroxy boronate intermediate occurred, Scheme 41. Base titrations of the trifluoroborate and observations of the mixed ligated species by ESI MS provided evidence in support of this proposal.129,162 However, a later investigation demonstrated that complete hydrolysis to the boronic acid took place, and that transmetalation primarily occurred through this species.118
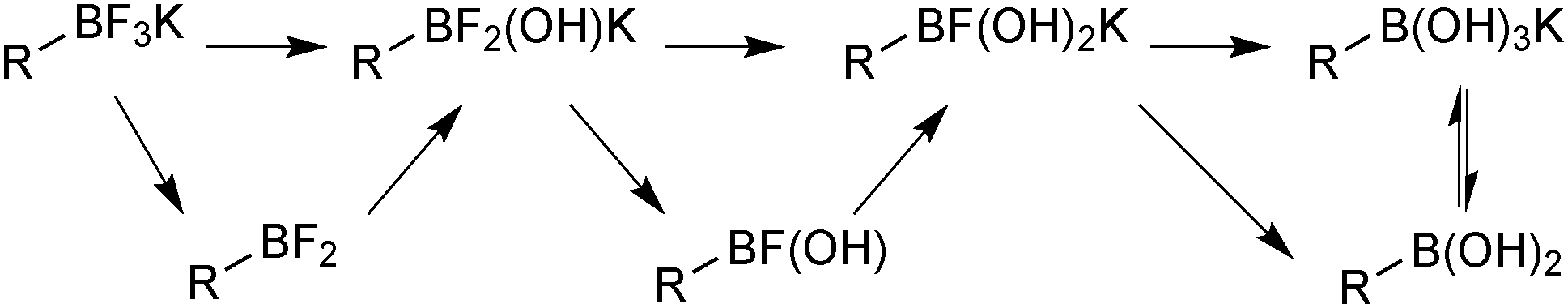 | ||
| Scheme 41 Hydrolysis of organotrifluoroborates to boronic acids via mixed ligated species. | ||
DFT calculations of the barrier height for the process showed the lowest, most favourable pathway for transmetalation to be when all ligands on boron were hydroxide and not fluoride.118 This is consistent with a reduction in the nucleophilicity of the organic fragment, when ligated by the highly electronegative fluoride, as well as a reduction in the ability of the ligand to bridge the metal species. Kinetic analysis of a competition conducted between [2H4]-11 and [2H0]-12 for limiting 13, demonstrated experimentally that the boronic acid was the most reactive species. Even when the proportion of boronic acid [2H4]-11 was very small in comparison to [2H0]-12, the product contained the labelled ring during the initial stages of reaction, Scheme 42.
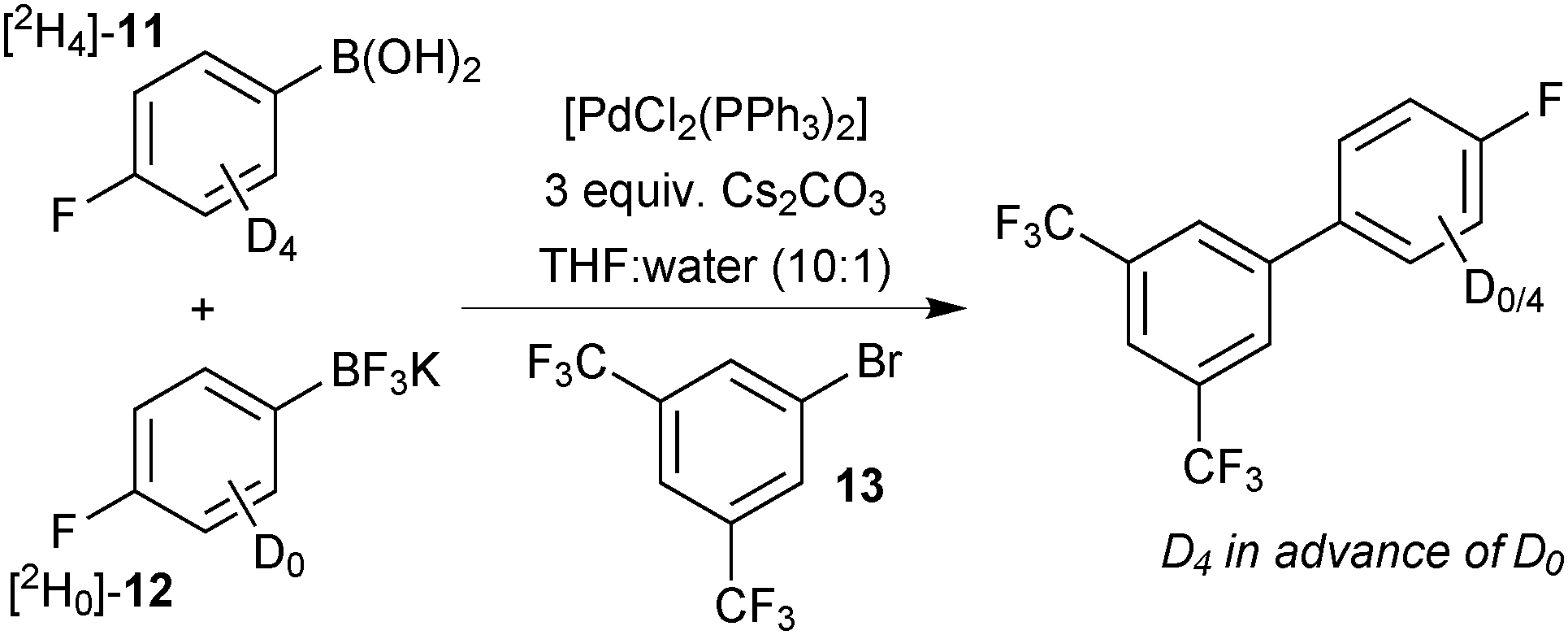 | ||
| Scheme 42 SM coupling competition between boronic acid [2H4]-11 and trifluoroborate [2H0]-12 for limiting arylbromide 13.118 | ||
The superior reaction outcome employing trifluoroborates thus originated, not from a more rapid transmetalation that would out-compete the side-reactions, but from a suppression of side-product formation.118 Boronic acid is consumed to form a homocoupled biaryl and phenol, through three separate side reactions: palladium precatalyst activation (I), oxidation (II) and oxidative homocoupling (III), Scheme 43. The use of organotrifluoroborate salts was shown to suppress all three. The endogenous fluoride liberated from the hydrolysis (“F−”), and the slow release of boronic acid from R-BF3K, were both important features that contributed to the attenuation of these side-products. The slow release rate of the active boronic acid allowed it to stay in low concentration, which led to a favourable partitioning between cross-coupling and oxidative homo-coupling. The low concentration of boronic acid also reduces the absolute rate of protodeboronation, which is highly useful for the coupling of unstable substrates.163
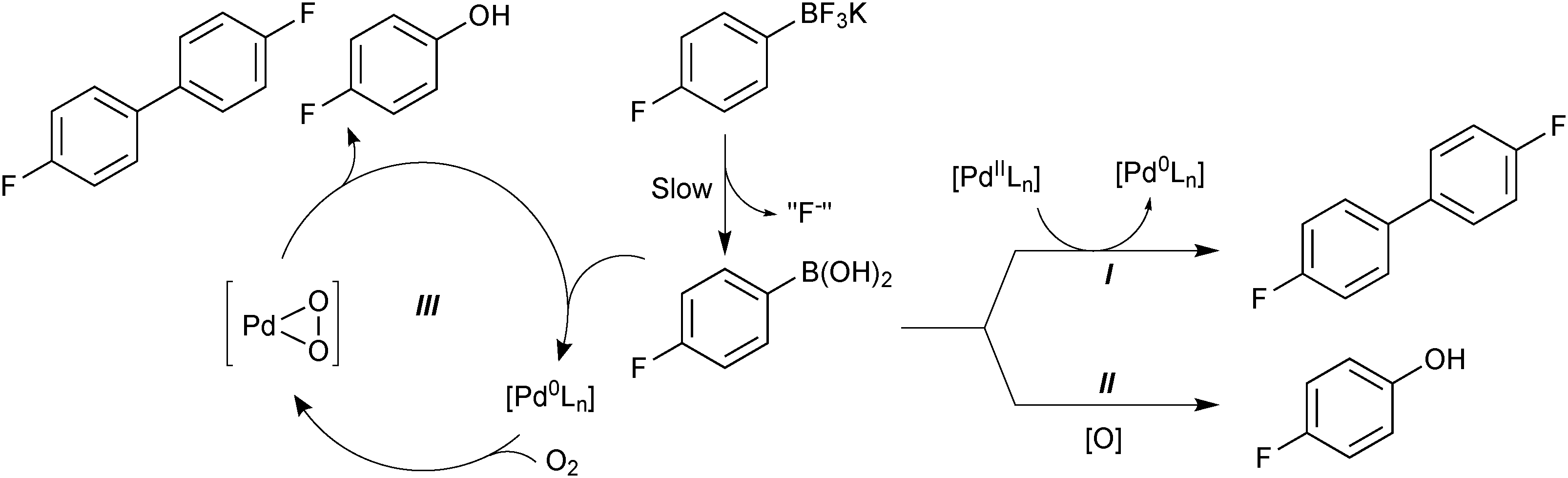 | ||
| Scheme 43 Degradation pathways (I, II and III) for 4-fluorophenylboronic acid, which are reduced through its slow release from the trifluoroborate, and also by the fluoride that is co-liberated. | ||
Further mechanistic investigations on the hydrolysis of R-BF3K salts under SM coupling conditions led to a number of key findings.113 An acid catalysed pathway gave rise to rapid rates of hydrolysis to the corresponding boronic acid. The rates were found to be vessel-dependent under the basic conditions of SM coupling. Under these conditions (THF:water 10:1, Cs2CO3), a biphase exists with a very basic minor aqueous phase and a much less basic organic bulk phase, Fig. 8.
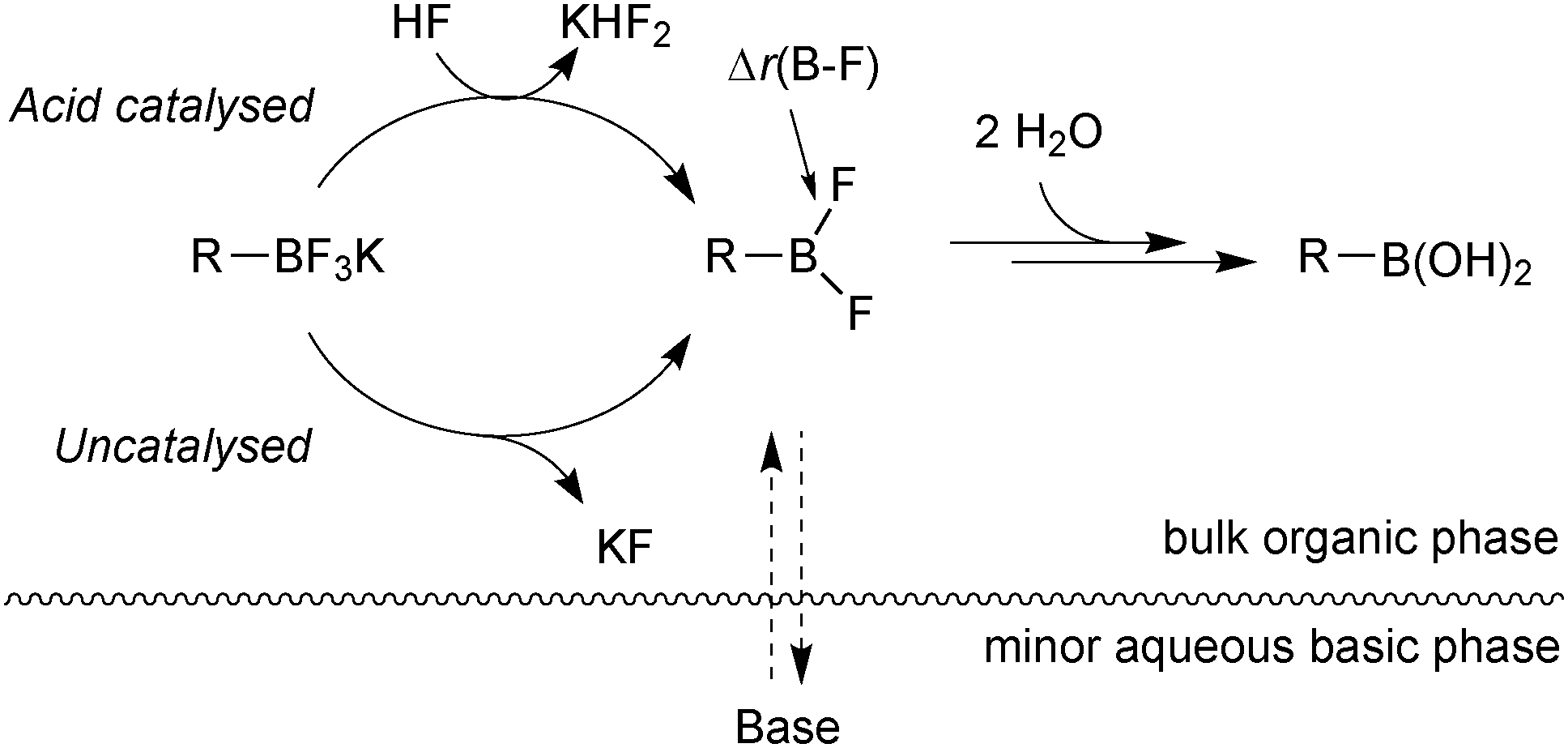 | ||
| Fig. 8 Hydrolysis of trifluoroborates: the acid catalysed pathway can be attenuated by efficient mixing with the basic minor phase. | ||
Access to the acid catalysed pathway was found to be dependent on the mixing efficiency of the phases. Systems that induced good mixing led to a disabling of the acid-catalysed hydrolysis. The low concentration of boronic acid from the slow hydrolysis led to fewer side-products than in systems with poor mixing, which gave fast rates of hydrolysis and thus high concentrations of boronic acid.
 | ||
| Scheme 44 First preparation of an organotrifluoroborate salt. | ||
Rates of hydrolysis were measured for a range of R-BF3K salts, under carefully controlled biphasic conditions, and found to span five orders of magnitude, with half-lives ranging from minutes to months. A background uncatalysed pathway dominated under efficient phase mixing conditions. This rate was found to correlate well to the DFT derived B–F bond length of the intermediate difluoroborane (r(B–F)) that was sensitive to the structural characteristics that dominate hydrolysis rates, Fig. 8. Alternatively, the more easily sourced Swain–Lupton resonance value in combination with a weighted Charton steric parameter (ℜSL − 0.09ν) also correlated well with relative rates of hydrolysis. Thus both parameters provide a rapid and simple tool for the prediction of hydrolytic propensity, and therefore give an indication of the mode of application in a particular SM coupling.
5.2. Preparation
This strategy, of tin displacement by boron to form the C–B bond, was further developed by Stafford in 1963,164 who synthesised the first potassium vinyl and methyltrifluoroborates.
Shortly after, Chambers and Chivers prepared potassium pentafluorophenyltrifluoroborate,165 and in 1970 Chivers synthesised the 2-(trifluoromethyl)phenyltrifluoroborate salt.166
An alternative strategy involves heteroatom/fluoro exchange on boron, which was first realised by Kaufmann in 1988,167Scheme 45. A dibromoborane camphenyl derivative was treated with potassium fluoride to give isopinocamphenyltrifluoroborate salt in good yield, although this was the only example reported.
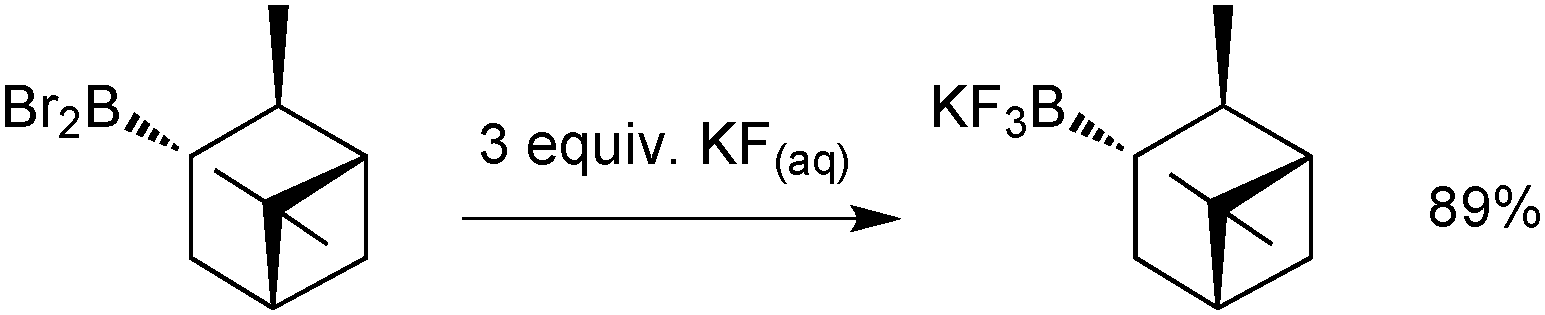 | ||
| Scheme 45 Ligand exchange at boron to generate a trifluoroborate. | ||
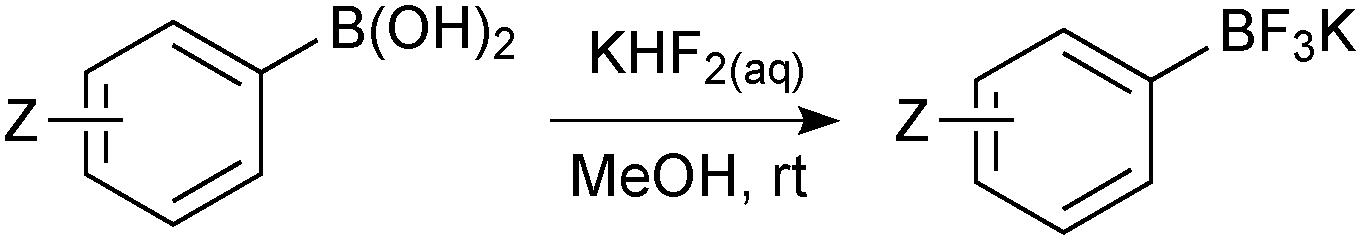 | ||
| Scheme 46 Preparation of aryltrifluoroborate salts with KHF2. | ||
The methodology also cleanly converts boronic esters, such as pinacol boronic esters, to a mixture of organotrifluoroborate and pinacol. There are then two general methods to purify the product from pinacol. Firstly, Hartwig170 demonstrated that it could be removed in vacuo (6 mTorr, 60 °C) from the mixture, and secondly, Aggarwal171 demonstrated that pinacol formed an azeotrope with methanol and water. Through repetitive addition and evaporation of the solvent mixture, pinacol could be removed from the R-BF3K salt and excess KHF2. The number of cycles varied from 1–9 and depended on the substrate and precise make-up of the azeotrope.
Genet described a “one-pot” method to prepare organotrifluoroborate salts from organometallic intermediates, e.g. organolithium reagents.172 Following treatment with borate to yield the intermediate boronate, an acidic work-up gives boronic acids (see Section 4.2), however, employing KHF2 leads directly to the RBF3K salt, thus eliminating one step. KHF2 was also shown to cleanly convert crude intermediate boronic ester mixtures generated from, for example, copper catalysed β-borylation of α,β-saturated ketones,173 or palladium catalysed borylation of alkyl174 or aryl bromides.133
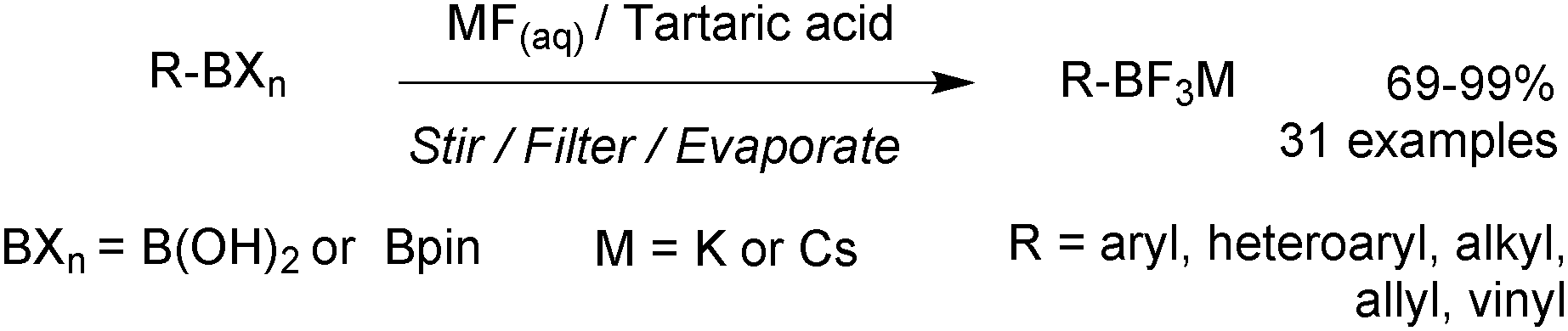 | ||
| Scheme 47 Preparation of RBF3 M salts using MF/tartaric acid. | ||
5.3. Applications in SM coupling
An extensive optimisation regime of SM coupling conditions for the use of organotrifluoroborates has been undertaken by Molander.160 Conditions for the cross-coupling of a wide range of (hetero)aryl,161,176,177 alkenyl178,179 and alkynyl180 moieties have been developed, and give good yields in most cases. Due to the necessity for prior hydrolysis,113 conditions normally employ water as a co-solvent. Occasionally alcoholic rather than hydrous media are employed, and in such cases ‘bench grade’ alcohol has been reported to give superior yields, presumably the traces of water present in such solvent grades, aids the hydrolysis.181The cross-coupling of sp3 systems has long-been seen as problematic, due to an inherently slower transmetalation, instability towards protodeboronation and competitive β-hydride elimination side reactions. Employing R-BF3K substrates, which show relative resistance to protodeboronation, and a suitable catalyst system to outcompete β-hydride elimination with reductive elimination, these problems could be attenuated, and a number of successes have been reported, Scheme 48. For example, the pharmacologically important aminomethyl182/ethyl183 and alkoxymethyl184/ethyl185 motifs were found to be suitable nucleophilic partners for couplings with a range of aryl/heteroaryl bromides and chlorides. Additionally, conditions for the incorporation of 3-oxoalkyl186 and cyclobutyl/propyl187 functionalities have been optimised.
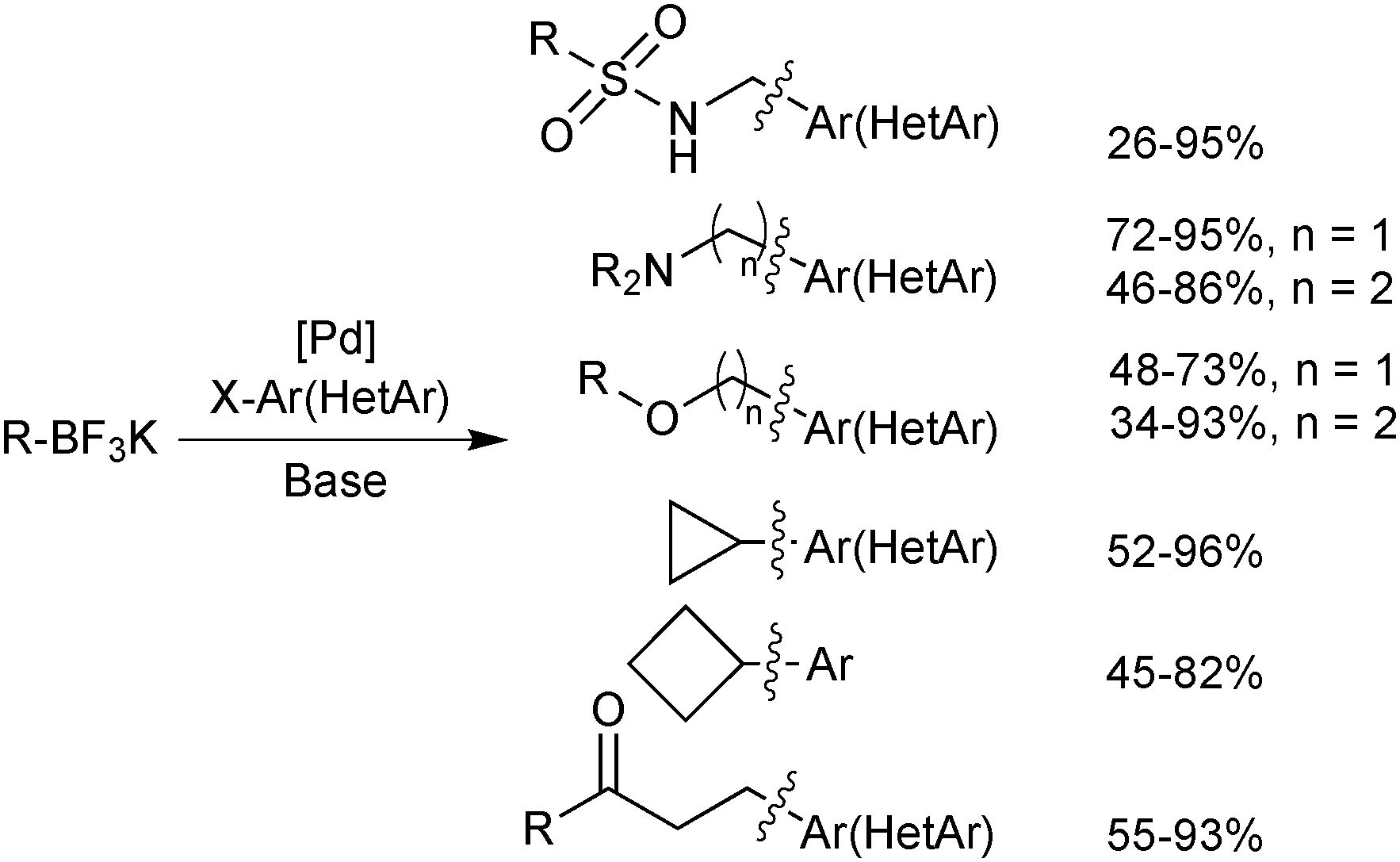 | ||
| Scheme 48 Cross-coupling of pharmacologically important sp3 building blocks. | ||
Due to the stability of trifluoroborates under anhydrous conditions, orthogonal and iterative one-pot chemistry can be conducted. Chemoselective coupling was demonstrated between two boryl groups, exhibiting opposing reactivity in specific solvent systems. Organoboranes undergo smooth coupling in anhydrous solvent systems where trifluoroborates are completely inert. Switching to an aqueous or alcoholic system, which is able to hydrolytically reveal the boronic acid, allows subsequent cross-coupling, all in “one-pot”. The procedure is initiated by hydroboration of an olefin, leading to the organoborane. The trifluoroborate moiety participates in the sequence in two general forms: either it is attached to the aryl halide component of the first cross-coupling (1), or it is appended to the olefin that is initially hydroborated (2), e.g. potassium vinyltrifluoroborate, Scheme 49.49,188
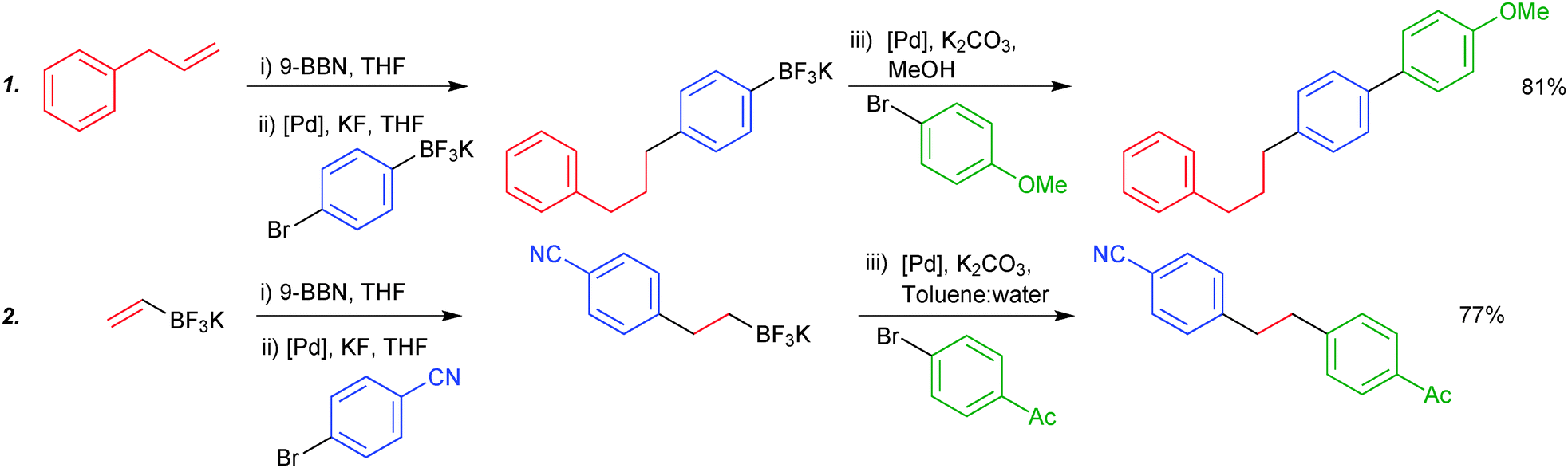 | ||
| Scheme 49 Two stage SM coupling of an organoborane and trifluoroborate, using solvent to control reactivity of the boron functionalities. | ||
The transmetalation of alkenyltrifluoroborate salts was found to proceed with retention of configuration when a THF:water mixture was employed.178 Interestingly, when the solvent was switched to an alcohol (with a different catalyst precursor) the stereoselectivity was attenuated. This stereoselectivity in transmetalation was exploited in the sequential cross-coupling of 1,1-dibromoalkenes, Scheme 50.189 Palladium(0) complexes are most reactive towards E-alkenyl bromides in oxidative addition and thus excellent yields of stereo-defined conjugated dienes were achieved in “one-pot”.
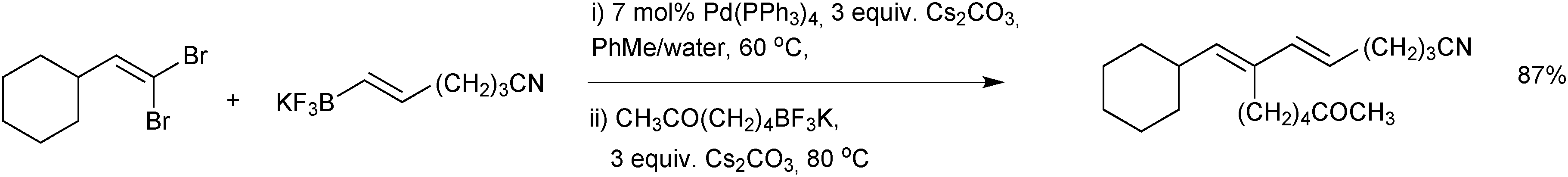 | ||
| Scheme 50 SM coupling of an alkenyl halide with alkenyltrifluoroborate salts, where the E-alkenyl bromide is selectively reacted first. | ||
The cross-coupling of alkenyltrifluoroborates was applied to a formal total synthesis of the natural product, oximidine II, Fig. 9.190 It was demonstrated that employing a trifluoroborate gave excellent results for the construction of the highly strained polyunsaturated 12-membered macrolactone.
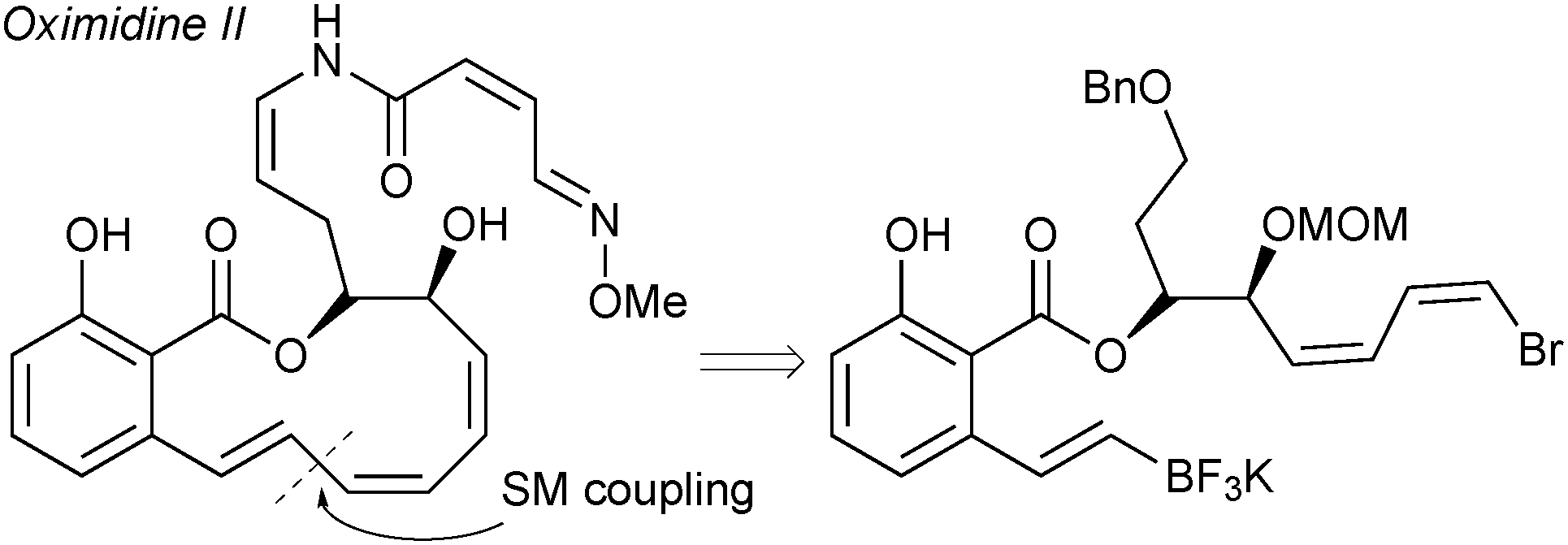 | ||
| Fig. 9 Intramolecular SM coupling in the synthesis of oximidine II. | ||
The use of organotrifluoroborate salts in the SM coupling with arene diazonium salts has been shown to give superior yields to the corresponding coupling with boronic acids.191 As a cationic palladium complex is formed, it is possible that it reacts via the ‘boronate pathway’,192Scheme 51.
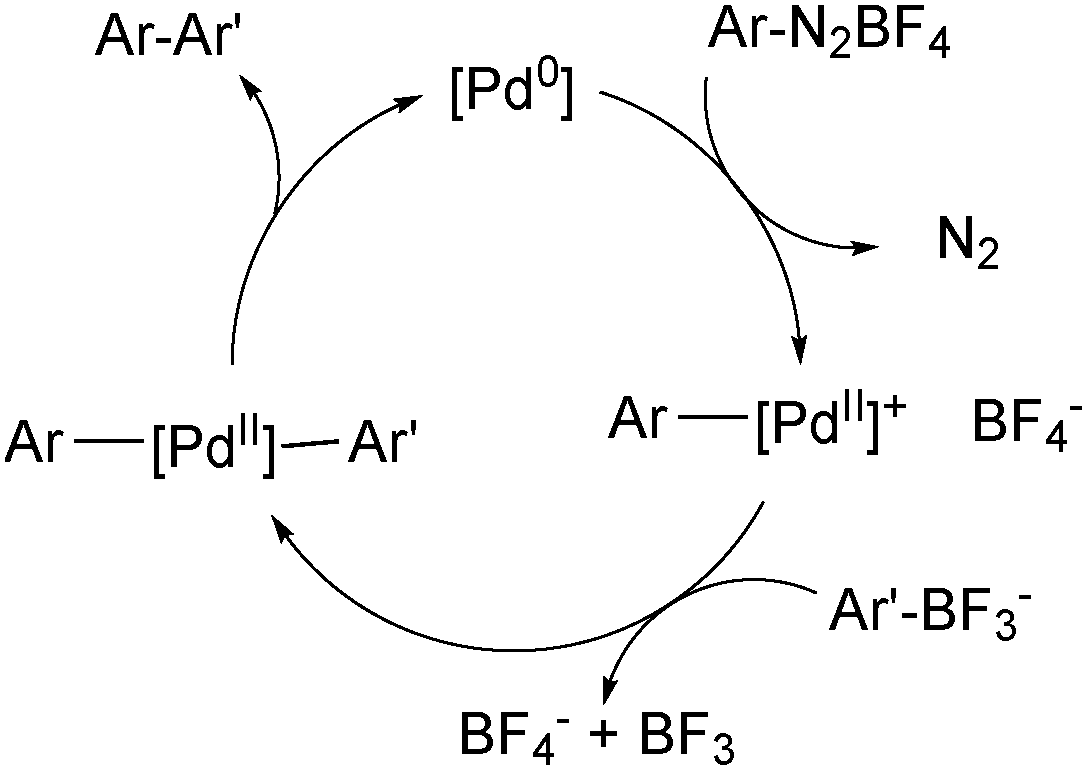 | ||
| Scheme 51 Mechanism for the SM coupling with arene diazonium salts, which proceeds via cationic palladium intermediates. | ||
Hydroxide has been shown to bridge the metals during transmetalation more proficiently than fluoride,118 and thus it is anticipated that the use of arene diazonium salts in combination with a preformed trihydroxyboronate may result in a particularly efficient process.
6. N-Coordinated boronates
6.1. Properties and mechanism
This group of compounds is characterised by a nitrogen atom contained in a cyclic boronic ester backbone. The most popular ligands used in the context of SM coupling are diethanolamine (14), N-methyldiethanolamine (15), N-phenyldiethanolamine (16) and N-methyliminodiacetic acid (MIDA) (17), Fig. 10. There are formally two B–O covalent bonds plus a dative bond that forms from donation of the Lewis basic lone pair on nitrogen to the Lewis-acidic boron atom. This donation hybridises boron from sp2 to sp3, whilst weakening the B–O bonds and forcing the boron into a tetrahedral geometry. The coordinatively saturated boron centre does not facilitate trans-ligation of hemi-labile ligands, therefore, these boronates are all monomeric in nature. | ||
| Fig. 10 N-Coordinated boronates used in SM coupling reactions. | ||
The trivalent, heteroatomic N-methyliminodiacetic acid (MIDA) ligand condenses with boronic acids to form MIDA boronates (17). This quaternisation renders them free-flowing, crystalline solids, which are indefinitely stable to air, moisture and silica-gel chromatography, and they can thus be stored without precaution “on the bench top”.193 These properties extend to all MIDA boronates reported to date, including the troublesome 2-heterocyclic surrogates; although it is noted that some commercially available samples can be far from crystalline. The MIDA boronates were first prepared and characterised in the early 1980s,194 and later pioneered in iterative SM cross-couplings by Burke.195 Their stability towards SM coupling conditions, yet ready hydrolysis when required was paramount to their success in this context. Due to the effective removal of the vacant p-orbital required for transmetalation, under anhydrous SM coupling conditions, the MIDA boronate functionality (17) was found to remain intact.195a It did not undergo any competing cross-coupling when in the presence of a reactive boronic acid functionality, unlike 15 that underwent competing transmetalation. Variable temperature 1H NMR has previously shown that diethanolamine (14) and N-methyldiethanolamine boronates (15) undergo conformational flipping,194a which transiently exposes the reactive p-orbital. However, even at high temperatures, the signals arising from the protons of the MIDA backbone in 17 remain as a pair of sharp doublets,196 in contrast to broadening and shifting of peaks with 15, Scheme 52. This confirmed that the MIDA boronates are conformationally rigid, at the NMR timescale at least, consistent with their enhanced stability in SM coupling conditions. Kinetically, a greater barrier height for the ring flipping process may originate from a combination of increased strain induced in the transition state by the carbonyl groups and the greater Lewis-acidity at boron, due to the more electron withdrawing carboxylate groups, enhancing binding of the MeN-group.
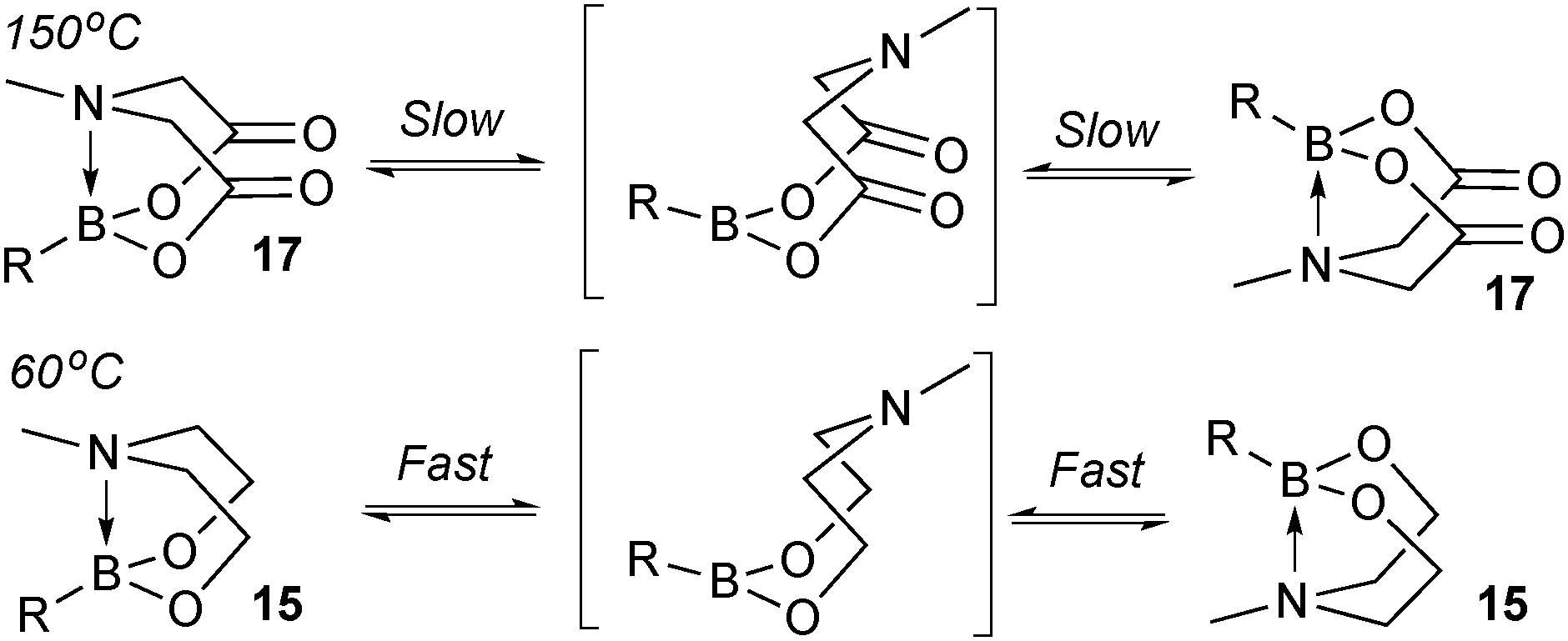 | ||
| Scheme 52 Conformational rigidity is observed at the NMR timescale in MIDA boronates 17, unlike diethanolamine boronates 15. | ||
Hydrolysis occurs slowly when MIDA boronates are subjected to protic or alcoholic solvents, a process that is substantially accelerated by heat or base. MIDA boronates are also incompatible with hard nucleophiles such as LiAlH4, DIBAL, TBAF or metal alkoxides.196 However, they are resistant to oxidising conditions, such as those of Swern, Dess–Martin and the highly acidic Jones oxidation.196 MIDA boronates additionally displayed stability in iodination, Evans aldol and reductive amination reactions, Horner–Wadsworth–Evans and Takai olefinations, mild reductions and a range of common work-up conditions and salts such as NH4Cl(aq) and NaHCO3(aq). As well as being inert under anhydrous SM coupling conditions, they are also stable to Stille, Heck, Negishi and Sonogashira couplings, Grubbs metathesis and Miyaura borylation reactions.195b
6.2. Preparation
Boronic acids readily condense with diethanolamine based ligands, with concomitant extrusion of water. Thus diethanolamine boronates (14) can be prepared from boronic acids197 or in a one-pot procedure analogous to Genet's process that converts organolithium reagents to organotrifluoroborate salts. Lithium–halogen exchange of an aryl bromide followed by borylation and diethanolamine addition leads to high yields of 14.198 The preparation of N-methyldiethanolamine boronates (15) also occurs readily via condensation of (hetero)arylboronic acids and the diol ligand.199 Eliminated water was removed with anhydrous MgSO4. The synthesis of substituted200 and unsubstituted201 2-pyridyl N-phenyldiethanolamine boronates (16) was achieved via the one-pot lithiation/borylation protocol. The corresponding boronic acid is not stable enough to be employed as a starting material.In line with their growing popularity, a very large range of MIDA boronates have now become commercially available. The MIDA ligand is comparatively expensive, but with economies of scale and increased demand, many of the complexed boronates have become well priced. Nonetheless, a number of straightforward procedures exist to prepare them, albeit in DMSO which requires a somewhat troublesome in vacuo removal.
The preparation of MIDA boronates from simple boronic acid substrates involves refluxing with the MIDA ligand under Dean–Stark conditions, to evict the water liberated upon condensation, Scheme 53.193
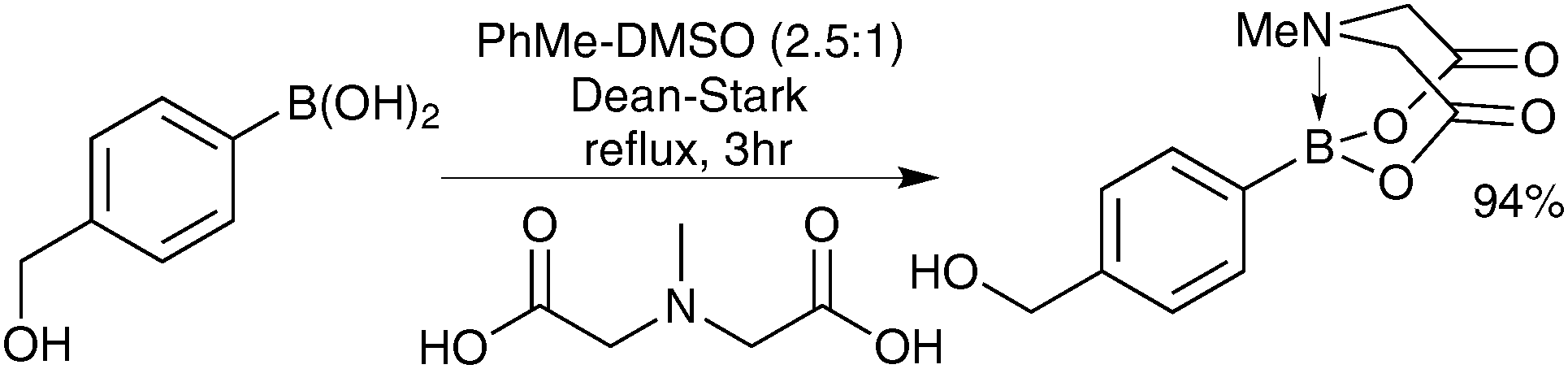 | ||
| Scheme 53 Preparation of a MIDA boronate from a boronic acid. | ||
As with 14, preparation of 2-heterocyclic MIDA boronates (17) are more challenging, in part due to the instability of the parent boronic acid. The organometallic ‘one-pot’ process gave good to excellent yields on the gram scale,202Scheme 54. This protocol was also shown to be effective for the synthesis of ethynyl MIDA boronate, whose precursor, ethynyl magnesium bromide, was used on a 45 gram scale.203
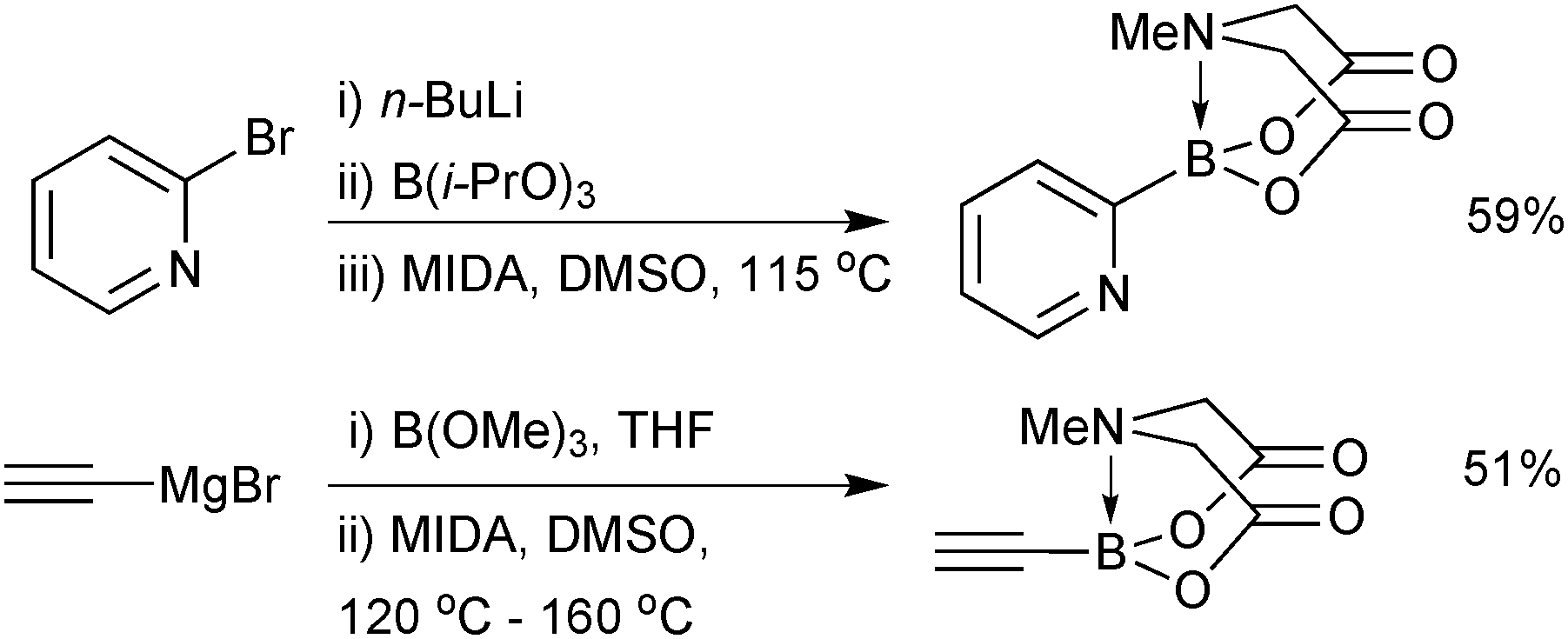 | ||
| Scheme 54 Preparation of MIDA boronates via Li or Mg reagents. | ||
Alternative approaches were required for the preparation of the small unsaturated MIDA boronates, as again, the corresponding boronic acid or boronates are not stable as starting materials or intermediates. Bromoborylation of acetylene, followed by trapping with MIDA in the presence of a base successfully led to bromovinyl MIDA boronate, Scheme 55.195b A transmetalation approach between vinyl TMS and BBr3 and subsequent trapping with the bis-sodium salt of MIDA, gave excellent yields of the vinyl MIDA boronate.204
 | ||
| Scheme 55 Preparation of small MIDA boronate building blocks. | ||
6.3. Applications in SM coupling
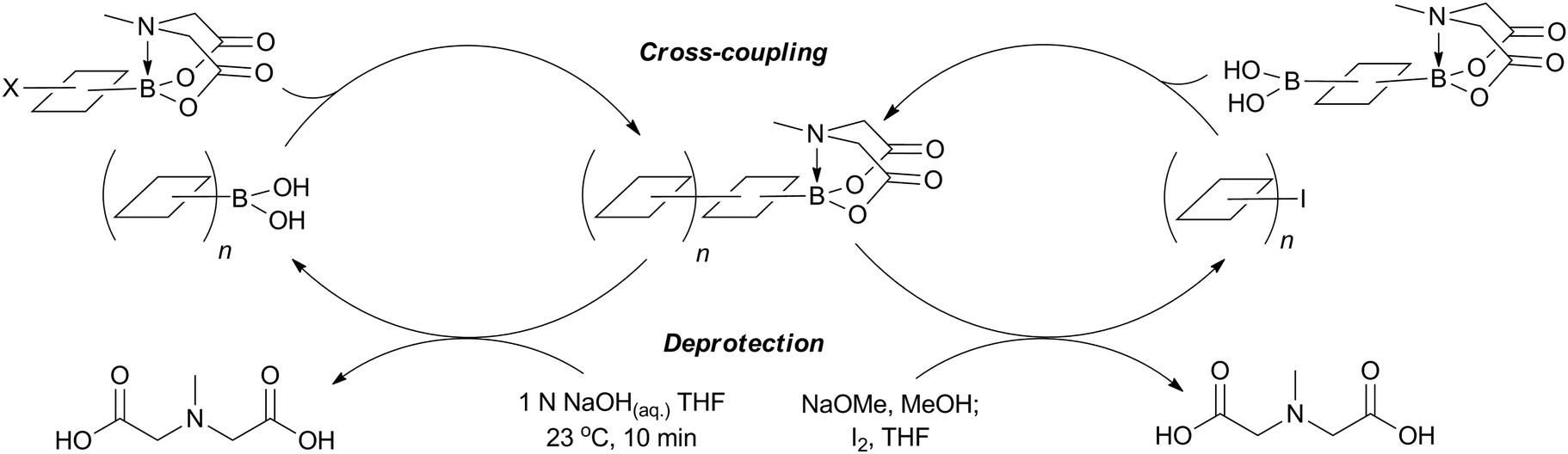 | ||
| Scheme 56 Iterative cross-coupling (ICC) of MIDA boronates. | ||
Due to the stereospecific nature of SM coupling, the iterative cross-coupling strategy is well suited to the generation of complex polyene frameworks. Complete stereochemical information of the modular alkenyl building blocks can be transferred and maintained throughout the coupling. This has been elegantly demonstrated in a number of examples. A modular total synthesis of the carotenoid synechoxanthin was performed through ICC, whereby MIDA boronates acted as both masked alkenyl iodides and alkenylboronic acids, Fig. 11. SM coupling was the only reaction used to join the building blocks.
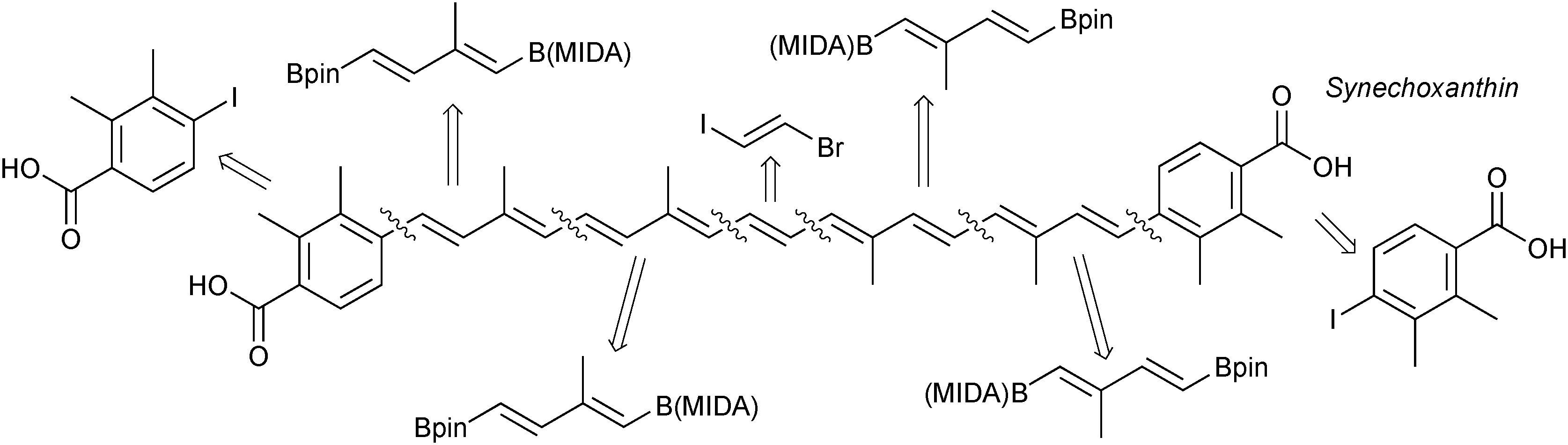 | ||
| Fig. 11 Retrosynthetic strategy for synechoxanthin. | ||
The synthesis of the antifungal heptaene macrolide, amphotericin B, Fig. 12,195b the light-harvesting carotenoid, (−)-peridinin,207 and a complex (E,E,E,Z,Z,E,E)-heptaene motif are other impressive examples.208
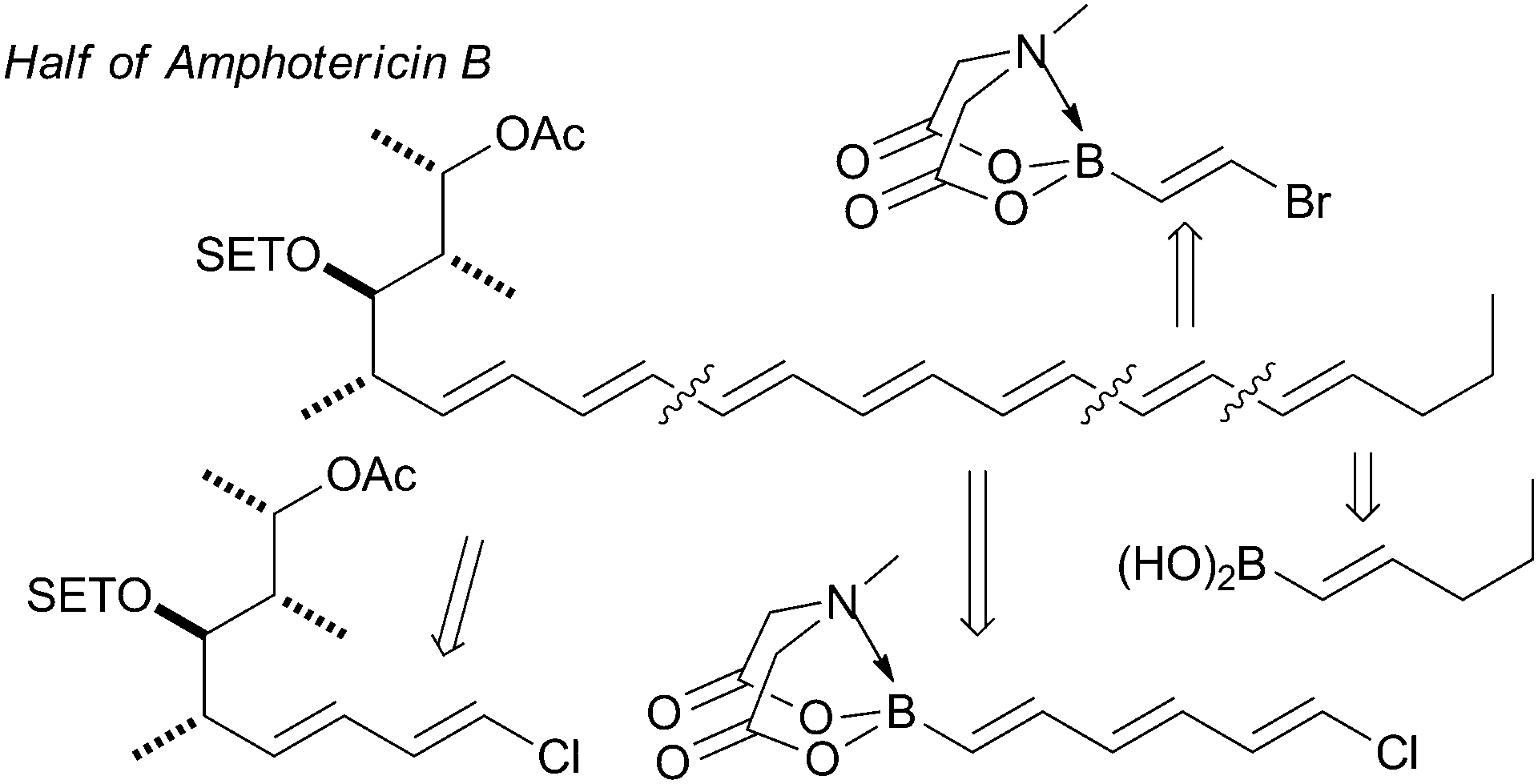 | ||
| Fig. 12 Retrosynthetic strategy for half of amphotericin B. | ||
In a seminal publication by Burke, it was shown that under optimised SM coupling conditions MIDA boronates could slowly hydrolyse, with catalytic turn-over of the resulting unstable boronic acids remaining rapid, Scheme 57. This slow-release mechanism, analogous to that occurring with organotrifluoroborates, Section 5.1 vide supra,114 ensures the boronic acid concentration is kept low and leads to a favourable partitioning between productive cross-coupling and competitive side-reactions, such as protodeboronation. Using this strategy, a number of unstable boronic acids were cross-coupled in very high yields when subjected to the slow-release conditions from their MIDA boronates.210
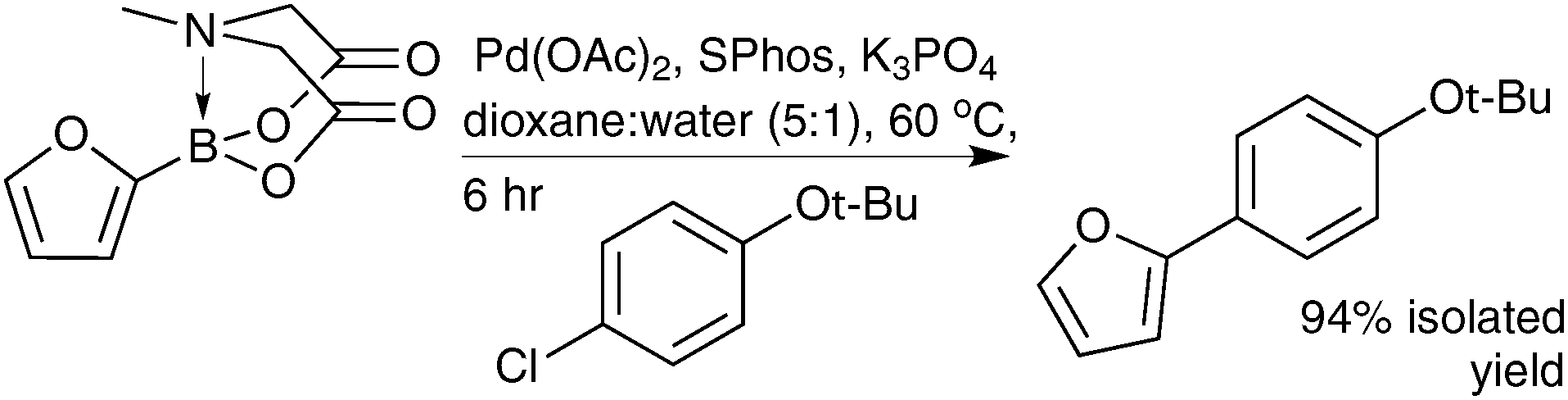 | ||
| Scheme 57 SM coupling of heteroaryl MIDA boronate under “slow-release” conditions. | ||
Under conditions that effect a rapid release of the boronic acid, the 2-furyl substrate underwent coupling in comparable yield to the corresponding freshly prepared 2-furylboronic acid (68% vs. 59% respectively). To further confirm that slow-release and thus low concentration of boronic acid was responsible for the increased yields, a slow, syringe-pump addition of the boronic acid restored the yield of cross-coupled product to be comparable to that achieved with the 2-furyl MIDA boronate (94%).
This methodology was appropriately applied in the total synthesis of (+)-dictyosphaeric acid A. A vinylic MIDA boronate, whose boronic acid can be unstable towards polymerization at high concentrations, crossed coupled with an alkenyl iodide in an isolated yield of 82%.211
Arguably one of the most difficult substrates to cross-couple is the 2-pyridyl moiety, as the corresponding boronic acid is notoriously unstable towards protodeboronation. However, conditions were developed for the coupling of a range of substituted and unsubstituted 2-pyridyl moieties, which afforded 2-aryl pyridines in moderate to excellent isolated yields, Scheme 58.212
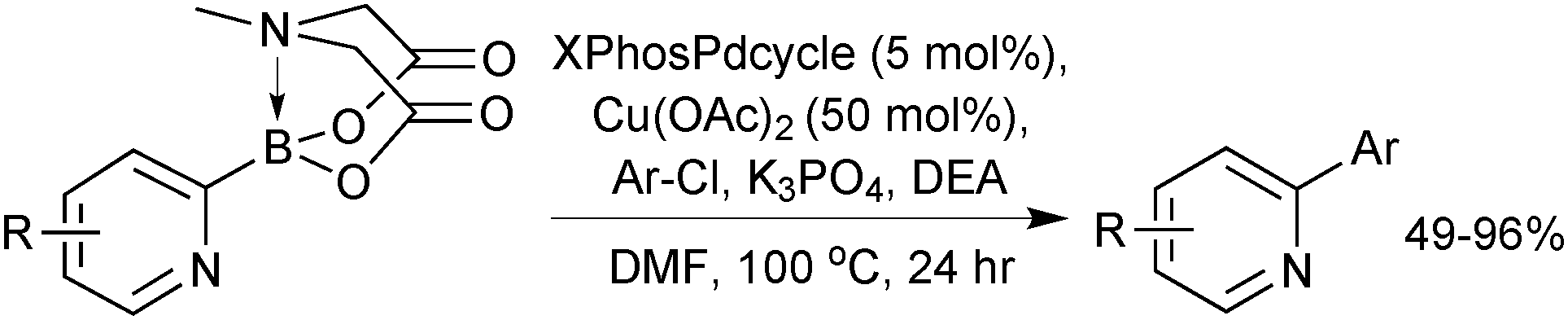 | ||
| Scheme 58 SM coupling of 2-pyridyl MIDA boronate with arylchlorides. | ||
Four strategies that are commonly used to mitigate side reactions of the boron reagent in SM coupling have been identified.114 Cross-coupling of the 2-pyridyl moiety with MIDA boronates utilised all four of these, Scheme 59.
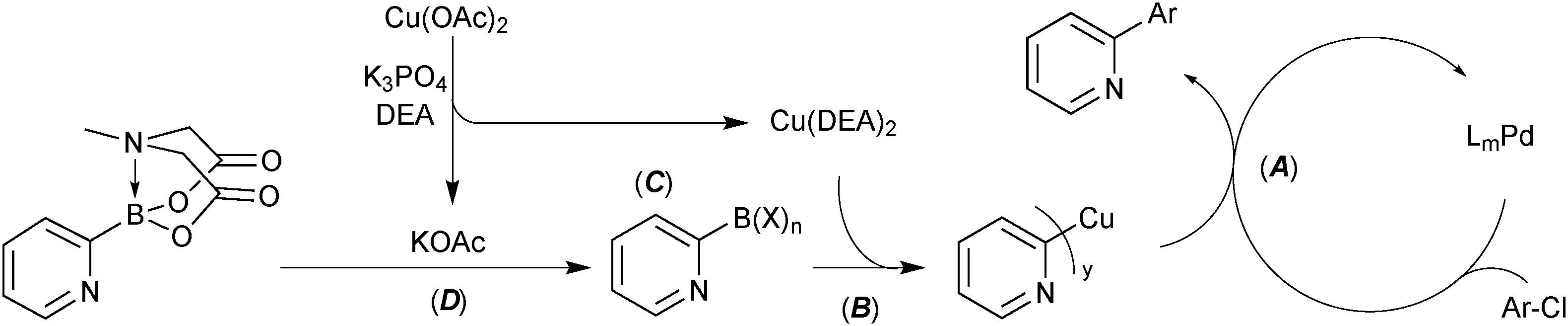 | ||
| Scheme 59 Four strategies (A – active catalyst, B – boron reagent activation, C – boron reagent masking and D – slow-release) identified for the successful coupling of the 2-pyridyl moiety. | ||
(A) Active catalyst. A precatalyst was employed that undergoes rapid activation under the reaction conditions to directly form a highly active mono-coordinated, XPhos ligated palladium(0) complex.213 Due to high electron density about palladium, these Buchwald catalyst systems are especially proficient in oxidative addition. This aids in shifting the turnover limiting step towards transmetalation, thereby increasing the concentration of palladium(II) available for transmetalation. The resulting increase in turnover frequency reduces the time that the boronic acid is exposed to the reaction conditions from which it can degrade.
(B) Boron reagent activation. The addition of activating reagents such as silver214,215 or copper101 salts have been shown to increase the rate of transmetalation to palladium. Silver aids in the halogen–hydroxide exchange on palladium and copper effects a more efficient pre-transmetalation with boron. Copper acetate in combination with diethanolamine was found to substantially increase yields in the 2-pydridyl MIDA boronate system. Mechanistic studies elucidated that a Cu(DEA)2 species is likely formed.
(C) Boron reagent masking. Success in the cross-coupling of unstable substrates has been achieved through masking of the Lewis-acidic boronic acids with more Lewis-basic ligands, e.g. alkoxides.58 Although not mechanistically confirmed, it is likely that diethanolamine will coordinate to boron following hydrolysis of the MIDA boronate. This intermediate, or one involving acetate, can then undergo transmetalation with copper, prior to reaction with palladium(II).
(D) Slow-release. The 2-pyridyl MIDA boronate, which itself is not sensitive to protodeboronation, steadily hydrolyses throughout the SM coupling, thereby reducing the exposure of the liberated boronic acid to potential protodeboronation.
Through single crystal X-ray analysis and variable temperature 1H NMR of MIDA boronates, it was established that the MIDA ligand is conformationally rigid with the N-methyl group close in proximity to the organic group appended to boron. Therefore, it was postulated that stereoselective transformations might be induced in distal functionality through the use of a chiral auxiliary in place of the methyl group, Scheme 60. Of the bulky chiral groups tested, α-pinene (PIDA) led to the greatest transfer of stereochemical information in the epoxidation of styrenyl boronates.217 This was then exemplified on a range of substrates. This key finding was included in a short modular synthesis of a glucagon receptor antagonist, where the PIDA ligand induced excellent stereocontrol for the epoxidation reaction and stability towards further manipulations. For the subsequent SM coupling and recovery of PIDA ligand, transesterification to the pinacol ester was evidently necessary, as hydrolysis to the more atom-economic boronic acid was not undertaken.
7. Boronates
7.1. Properties and mechanism
Pre-formed tetrahedral boronates have been shown to be useful coupling partners in SM-couplings. The three most common are trihydroxyboronates,218,219 cyclic triol boronates220 and triisopropylboronates, Fig. 13.221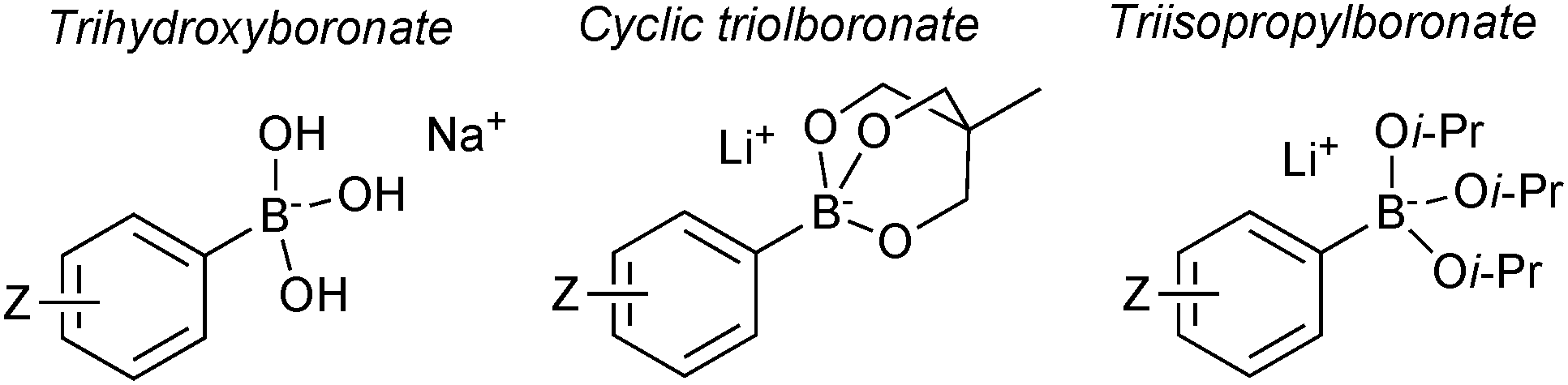 | ||
| Fig. 13 Three most common boronates used in SM coupling. | ||
Sodium aryl and alkyl trihydroxyboronate salts are solid and crystalline tetrahedral complexes. Like MIDA boronates and organotrifluoroborates, populating the vacant p-orbital renders them monomeric and stable to air. They undergo clean SM coupling under nominally base-free conditions.218 This suggests that reaction involves direct transmetalation with the palladium(II) complex, i.e. the boronate pathway. However, the solubility of the reagent in dry toluene is low, unlike the corresponding boronic acids that could be liberated through equilibrium, along with an equivalent of sodium hydroxide. Furthermore, under these conditions, dehydration of the boronic acids to the corresponding boroxines liberates one equivalent of water per boron unit, and this can potentially solubilise the sodium hydroxide thus facilitating coupling via the oxo–palladium pathway. Sodium aryl trihydroxyboronate salts have also been utilised under aqueous conditions,219 where preceding liberation of an equivalent of base is even more likely. Triisopropylboronate and cyclic triol boronate salts also undergo efficient SM couplings without base, but with water present in the solvent mixture.220,222
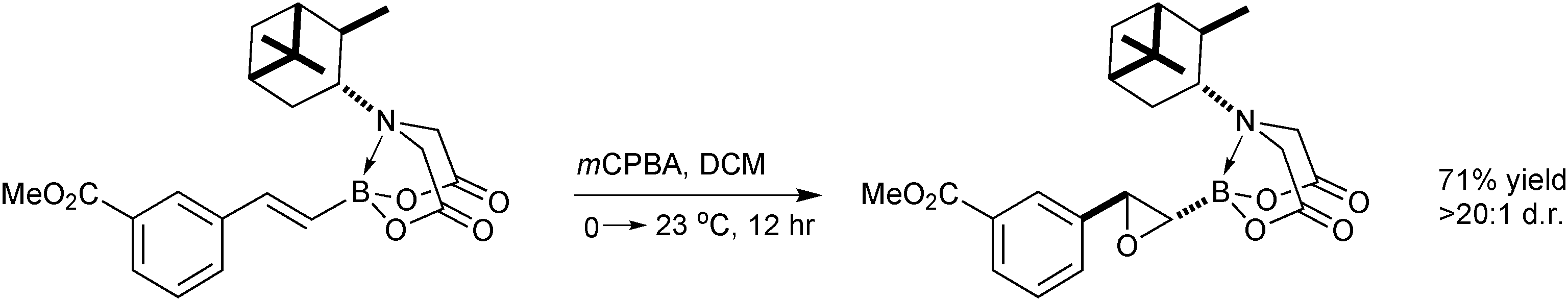 | ||
| Scheme 60 Asymmetric induction from the PIDA ligand in the epoxidation of a distal alkene. | ||
Cyclic triol boronates are stable reagents, as boron is doubly chelated by a triol-derived trialkoxide. The organic group appended to the bridging carbon dictates the solubility properties of the salt. When it is a methyl group the reagents are more soluble in organic solvents than organotrifluoroborate salts. Matteson demonstrated that the solubility in aqueous solutions could be raised by appending a polar sulfonate group to this bridged position.223 In the same study it was shown that such triols were good reagents for transesterification, and thus deprotection, of the stable pinanediol boronic esters. Recovery of free boronic acid was achieved hydrolytically under aqueous acidic conditions. Potassium cyclic triol boronates can also undergo functional group interconversion to the corresponding aryl iodide after treatment with sodium iodide and Chloroamine-T, Scheme 61.224
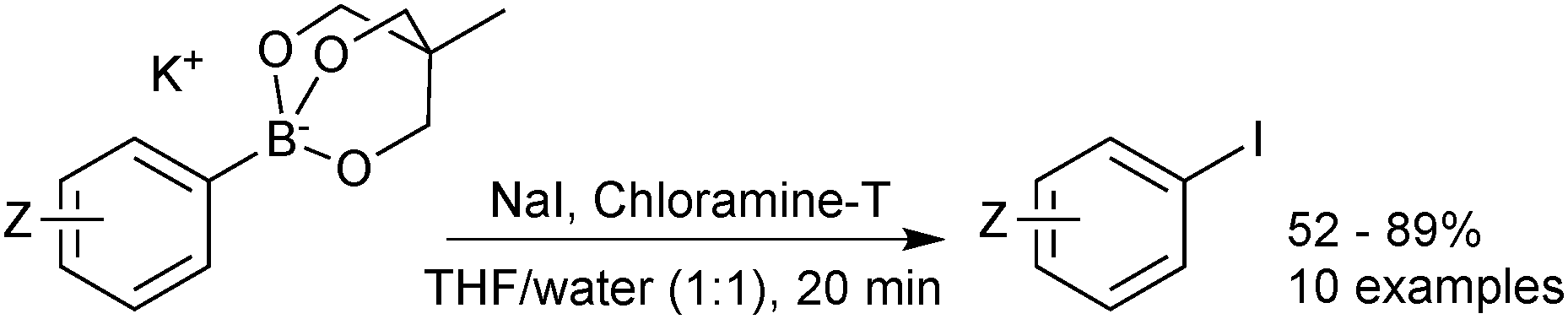 | ||
| Scheme 61 Functional group interconversion of aryl cyclic triolboronate to an aryl iodide. | ||
Lithium triisopropylboronates are stabilised by additional Lewis-base coordination to boron. In the solid state these tetrahedral species have been found to be more resistant to protodeboronation than regular boronic acids, especially for the 2-heteroaryl substrates.222 For example, 2-furanylboronic acid lost 90% of its activity when used for SM coupling after 15 days storage at ambient temperature. In contrast, the triisopropylboronate gave comparable yields in SM coupling to a freshly prepared sample, even after having been stored for four months in air.
7.2. Preparation
Sodium trihydroxyboronate salts are prepared from their parent boronic acids, simply by dissolution in toluene, and then drop-wise addition of a saturated aqueous sodium hydroxide solution, Scheme 62.218 The product precipitates and can be isolated through filtration. Potassium and barium salts are prepared similarly. | ||
| Scheme 62 Preparation of aryl trihydroxyboronate salts. | ||
Cyclic triolboronates can be prepared from the parent organoboronic acids and the triol. Water liberated through the condensation is removed azeotropically with toluene, to afford the trivalent boronic ester.220 On subsequent addition of KOH, quaternisation of boron occurs, and the potassium salt of the cyclic triolboronate precipitates from toluene as a white solid, Scheme 63. Replacing KOH with n-Bu4NOH leads to the corresponding n-Bu4N+ salt. Alternatively, the lithium salt can be directly prepared from the alkylation of B(OMe)3 or B(Oi-Pr)3 with R–Li, followed by transesterification with the triol. This anhydrous method is more suited to the preparation of substrates sensitive to protodeboronation, e.g. 2-pyridyl.
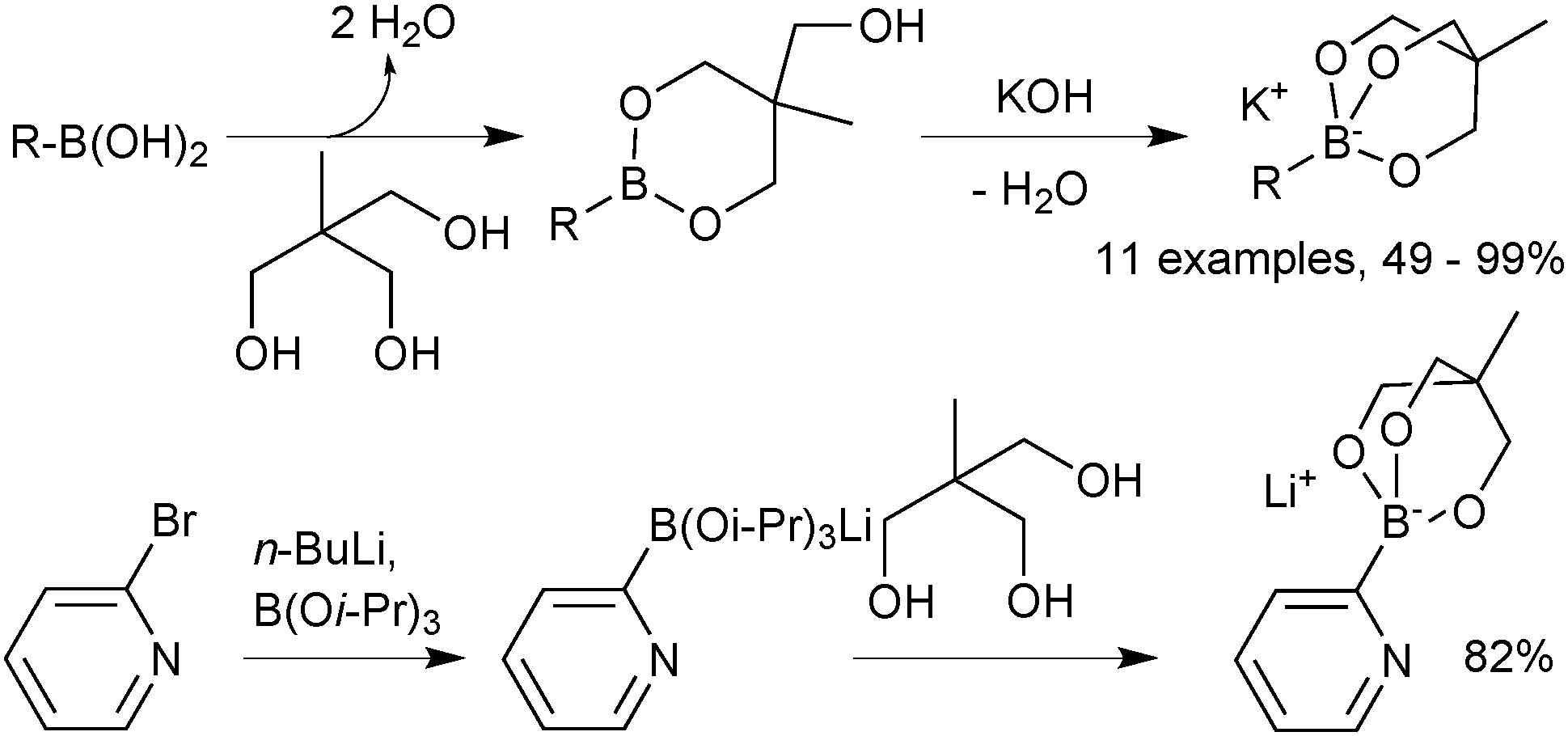 | ||
| Scheme 63 Preparation of cyclic triolboronates via esterification of a boronic acid or transesterification. | ||
Lithium triisopropylboronate salts are an intermediate when preparing any boronic acid, ester, MIDA boronate or trifluoroborate via the organometallic pathway. Lithium–halogen exchange from the corresponding aryl halide reveals the reactive nucleophilic arene, which is rapidly quenched in situ with triisopropylborate to form the lithium salt of the boronate ester,221Scheme 64. When the intention is to isolate the triisopropylboronate salt, isolation is simply achieved through removal of the solvent and bromobutane in vacuo. The order of addition of base and borate is reversed in the preparation of other heteroaryl boronates.222
 | ||
| Scheme 64 Preparation of lithium 2-pyridyl triisopropylboronate salts. | ||
7.3. Applications in SM coupling
Sodium aryl trihydroxyboronates have been illustrated to be useful coupling partners in an environmentally friendly procedure. Reactions are conducted “on-water” at room temperature and require no ligand for palladium, which is only employed in low loadings.219 The methodology accommodated aryl iodides and bromides at room temperature but elevated temperatures were required for the coupling of chlorides. A heterogeneous polymer supported palladium catalyst was also shown to work well for the coupling, which aids in recovery of the expensive metal. Good to excellent yields of biaryls were provided by both catalyst systems, Scheme 65. | ||
| Scheme 65 Green SM coupling employing trihydroxyboronate salts. | ||
Cyclic triol boronates are suitable cross-coupling partners in rhodium catalysed conjugate additions,225 copper catalysed arylation of amines,220 as well as SM coupling reactions.220 They have led to high yields of isolated products in a range of aryl–aryl couplings, Scheme 66. Cyclic triol boronate salts seem to be particularly effective coupling partners for sterically congested systems. Tetra-ortho-substituted biaryls226 and diaryl substituted planar frameworks227 have both been successfully prepared using these substrates in combination with a copper co-catalyst.
 | ||
| Scheme 66 SM coupling of cyclic triol boronate salts with aryl halides. | ||
They were directly compared to boronic acids in the double cross-coupling of dibromo arenes, and found to provide superior yields, Scheme 67.227 The best results for the generation of the sterically congested aryl systems were again obtained by reaction conducted in the presence of a copper salt (CuCl). Interestingly, the addition of base to the boronate (K2CO3, 2 equiv.) also improved yields compared to when no additive was present, possibly inferring prior hydrolysis is necessary. However, in the preparation of the tetra-ortho-substituted biaryls,226 an anhydrous/base-free DMF system was used, which suggests that prior hydrolysis does not takes place.
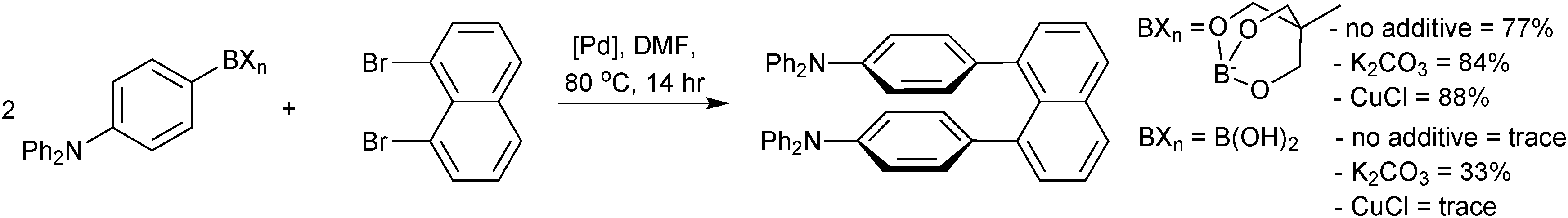 | ||
| Scheme 67 SM coupling of a cyclic triolate and a boronic acid with dibromonaphthalene. | ||
The lithium triisopropylboronates have been used in the SM coupling reactions of unstable heteroarylboronic acids. They have shown particular promise in the coupling of the notoriously difficult substituted and unsubstituted 2-pyridyl moieties. Phosphine oxide ligands were originally employed,221 but use of the X-Phos precatalyst, expanded the substrate scope leading to general conditions for the coupling of heteroaryl boronates, Scheme 68.222 Under anhydrous conditions, no coupling was observed, from which it can be inferred that a hydrolysis event is required prior to transmetalation. The pH of a typical SM coupling in THF–water, without added base, Scheme 68, was reported as being between 12 and 13. This evidence suggests liberation of isopropoxide, which would make the solution basic. SM coupling without added base was shown to be effective for base-sensitive organohalide coupling partners, such as methyl esters or oxazoles.222 However, with the addition of potassium phosphate, superior yields were then observed. A separate study found a beneficial effect with the addition of CuCl in combination with ZnCl2, the reasons of which were stated as unknown.228
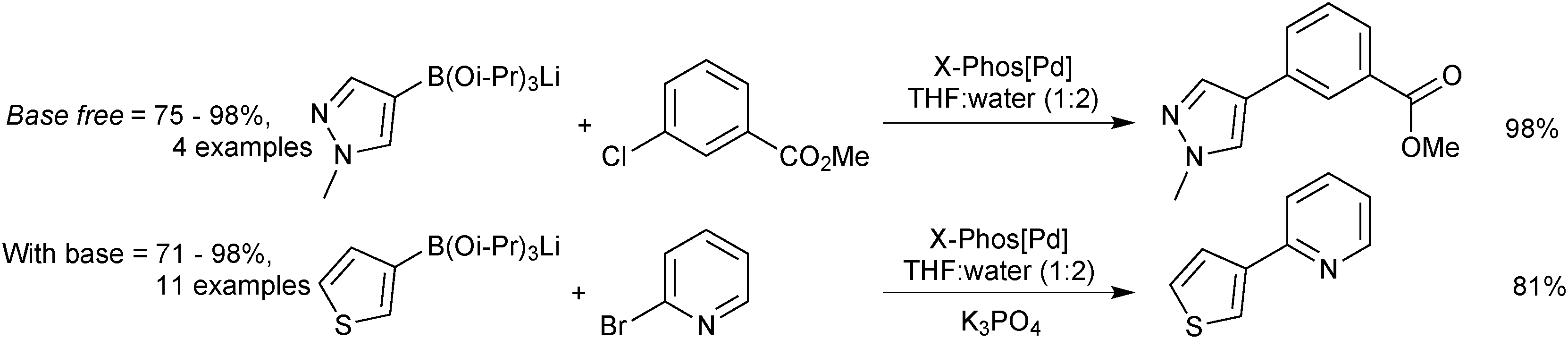 | ||
| Scheme 68 SM coupling of lithium heteroaryl trisopropylboronate salts with aryl and heteroaryl halides. | ||
A “one-pot” protocol was developed by Buchwald for the preparation of the lithium triisopropylborate salts and their immediate SM coupling, thus negating the necessity for intermediate isolation, Scheme 69.222 A good range of heteroaryl/aryl halide and heteroaryl/aryl boronate couplings with varying electronic properties were illustrated. The procedure gave similar yields to those when the intermediate boronate salt was isolated. Further simplifications were made to the “one-pot” protocol for substrates that undergo ortho-lithiation.
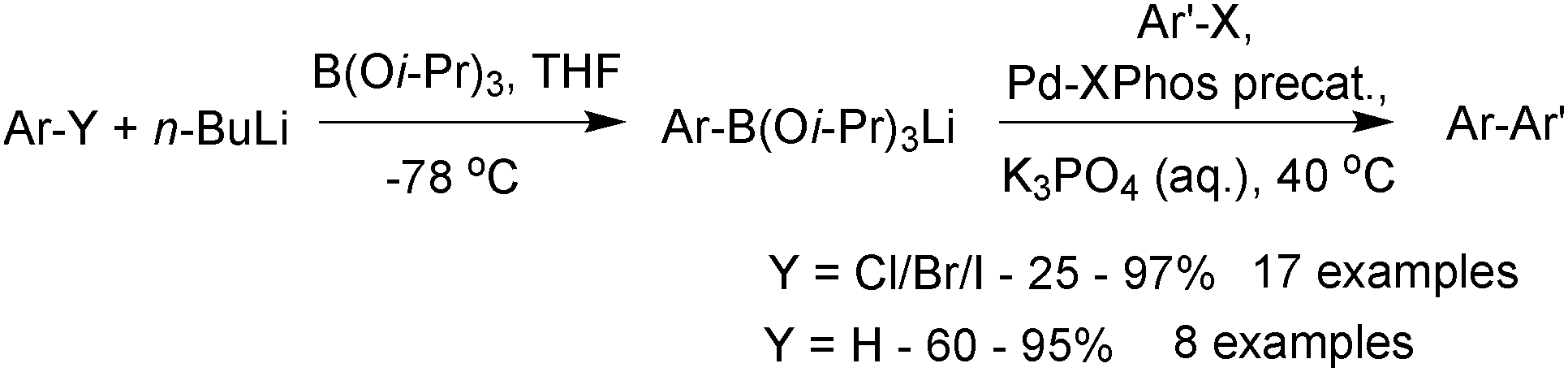 | ||
| Scheme 69 “One-pot” procedure for the borylation and SM coupling of aryl halides. | ||
8. Boronamides
8.1. Properties and mechanism
Boronamides are neutral species whose sp2 hybridised boron is bonded to two amide moieties. This class of reagent has primarily been developed by Suginome over the last decade and has enjoyed particular application in iterative cross-coupling (ICC). Of the three ligands reported in this class, the 1,8-diaminonaphthalene (DAN) ligand was shown to exhibit superior stability towards hydrolysis than the anthranilamide (AAM) and 2-(pyrazol-5-yl)aniline (PZA) analogs,229Scheme 70. Lone-pair donation from the Lewis-basic nitrogen to boron in the DAN ligand reduces the Lewis acidity at boron, making it very stable. Carbonyl conjugation and nitrogen aromaticity reduces this lone pair donation in the case of AAM and PZA respectively.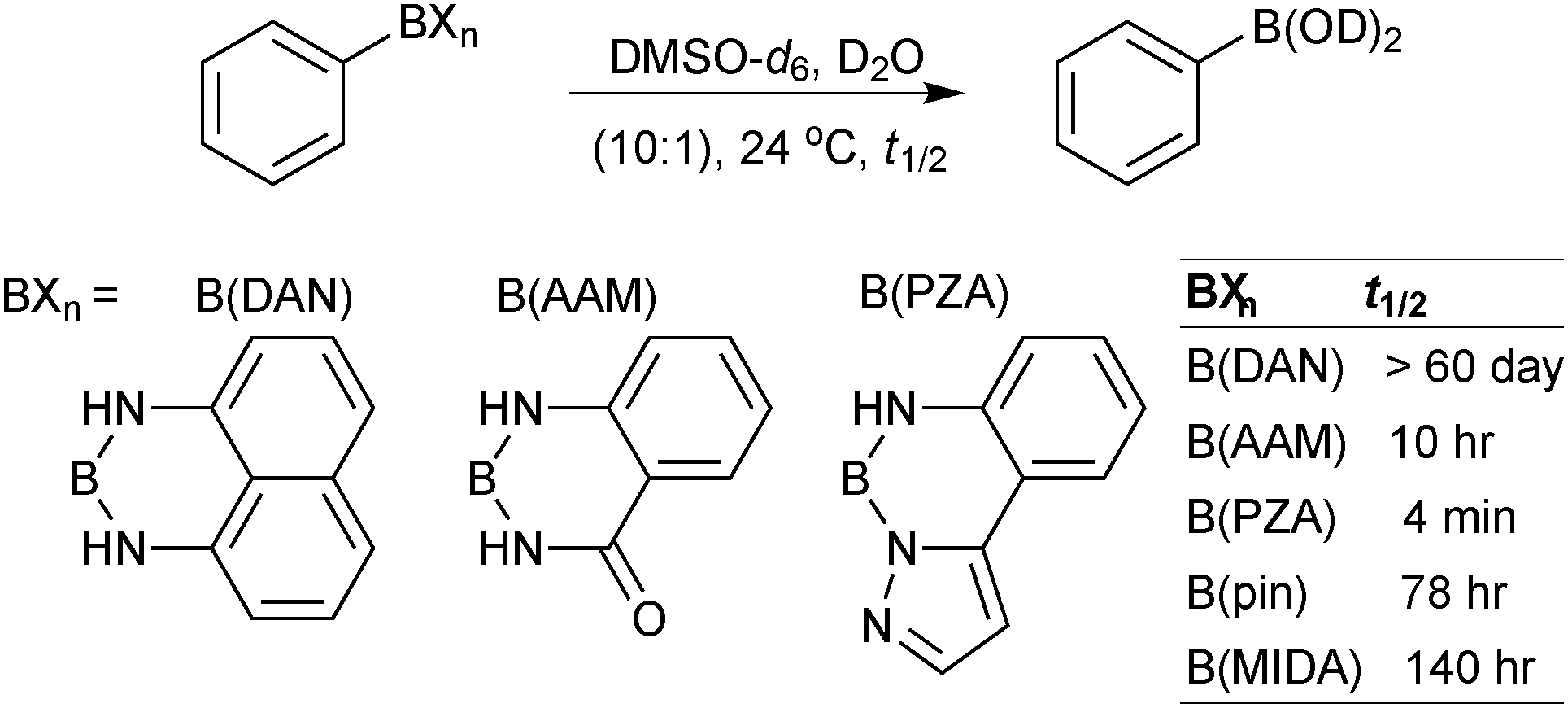 | ||
| Scheme 70 A comparison of boronamide stability. | ||
The first protecting group developed for boronic acids in SM coupling was the DAN ligand. The boron centre is very unreactive, which makes them suitable towards aqueous work-up and column chromatography. They are stable towards basic SM coupling conditions, but are readily deprotected with mild acidic treatment. Presumably protonation of nitrogen is necessary to weaken the B–N bond and liberate the p-orbital on boron for hydrolytic attack; equilibrium is then driven to the boronic acid via protonation of the liberated DAN ligand. This acidic deprotection makes them chemically distinct from MIDA boronates that activate under basic conditions.
AAM and PZA boronamides exhibit dual functionality as they are boron protecting groups and ortho-directing groups.229,230 The AAM derivative was found to be stable towards column chromatography but the PZA derivative less so. As is the case for the DAN ligand, the AAM and PZA groups are also removed upon acidic treatment.
8.2. Preparation
1,8-Diaminonaphthylboronamides are prepared through a condensation reaction between the corresponding boronic acid and 1,8-diaminonaphthalene, whereby water is azeotropically removed in toluene, Scheme 71.231 The reaction was also shown to proceed in the solid state with ball-milling at 0 °C.232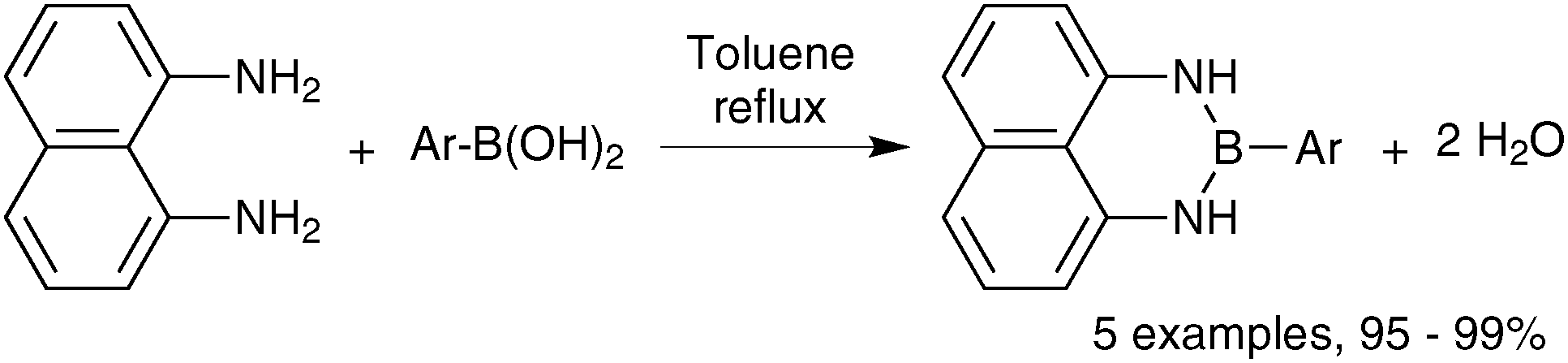 | ||
| Scheme 71 Preparation of DAN boronamides from boronic acids. | ||
AAM and PZA boronamides were prepared in an analogous manner, whereby refluxing the boronic acid with the free amine ligand in toluene led to high yields of product.
Recognising the inefficiency of using intermediate boronic acids, a procedure was developed whereby 1,8-naphthalenediaminatoborane (DANBH) was employed in an iridium catalysed borylation of aromatic C–H bonds.233 Moderate to excellent yields were demonstrated in both unsubstituted arenes as well as halo-containing arenes, which are chemically primed for ICC. DANBH was also shown to efficiently hydroborate alkynes under iridium catalysis. A broad range of terminal alkynes were transformed into E-alkenes in good to excellent yields.234 Finally, a differentially protected diboron reagent was shown to diborate terminal alkynes, Scheme 72.235 Again, an iridium catalyst provided the best regioselectivity, with the DANB functionality being delivered exclusively to the terminal position.
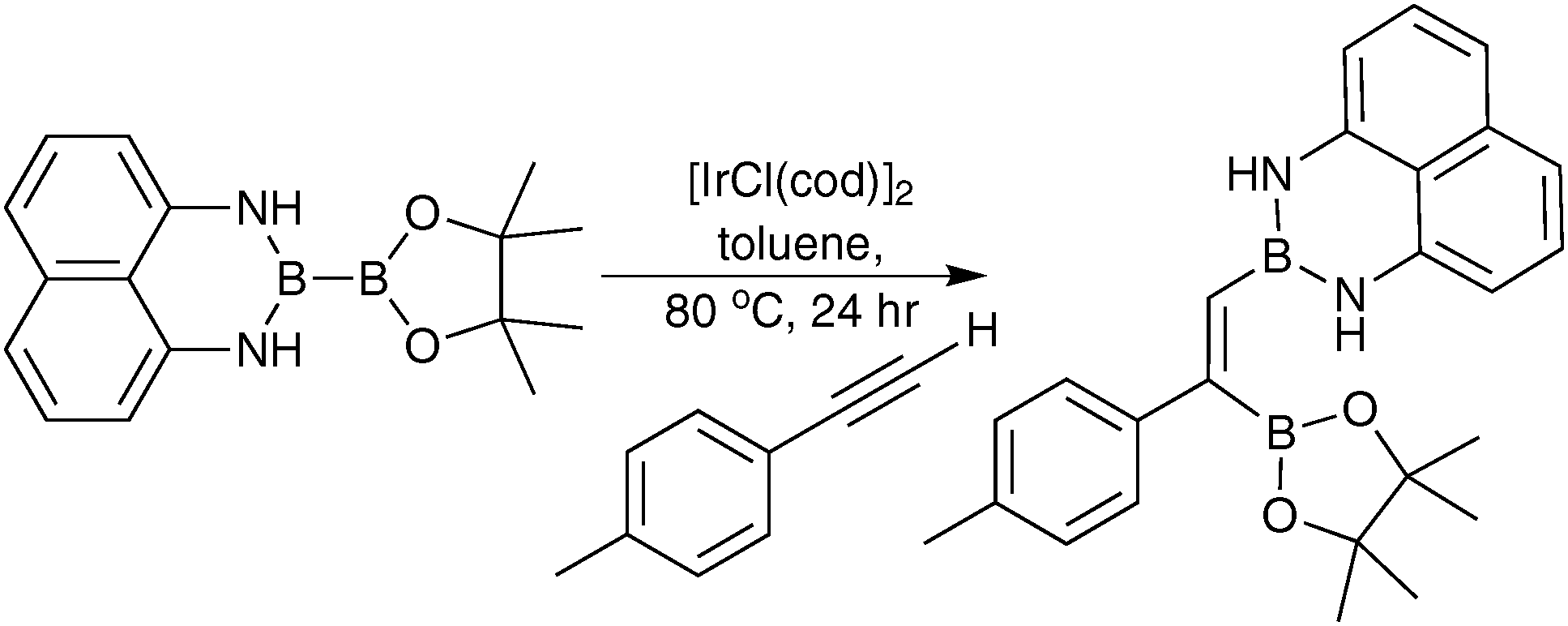 | ||
| Scheme 72 Diboration of a terminal alkyne with a differentially protected boron reagent. | ||
8.3. Applications in SM coupling
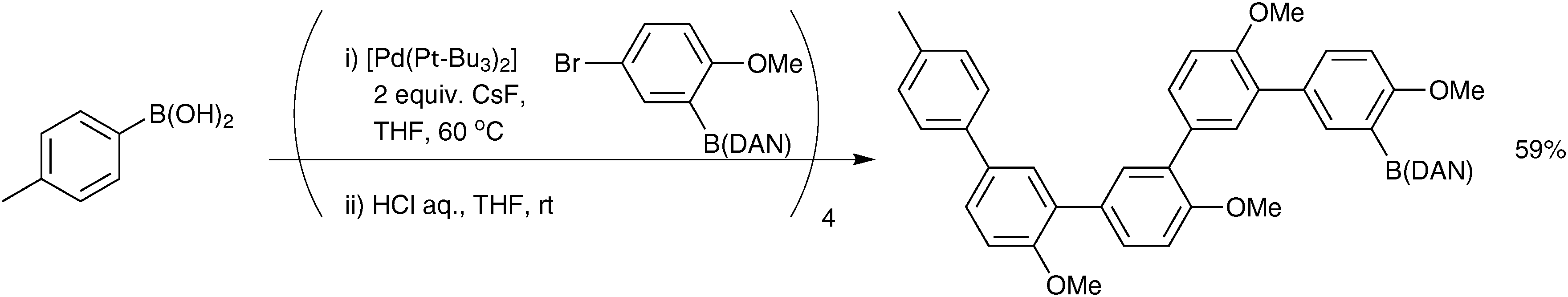 | ||
| Scheme 73 Iterative cross-coupling to give a polyaromatic compound. | ||
Anthranilamide (AAM) boronamides readily facilitate ortho-silylation that can then undergo halo-desilylation to set up the required bisfunctionalised substrate.236 Acidic deprotection of AAM reveals the masked boronic acid that subsequently couples with another bisfunctionalised substrate. Three cycles of iterative coupling were demonstrated.
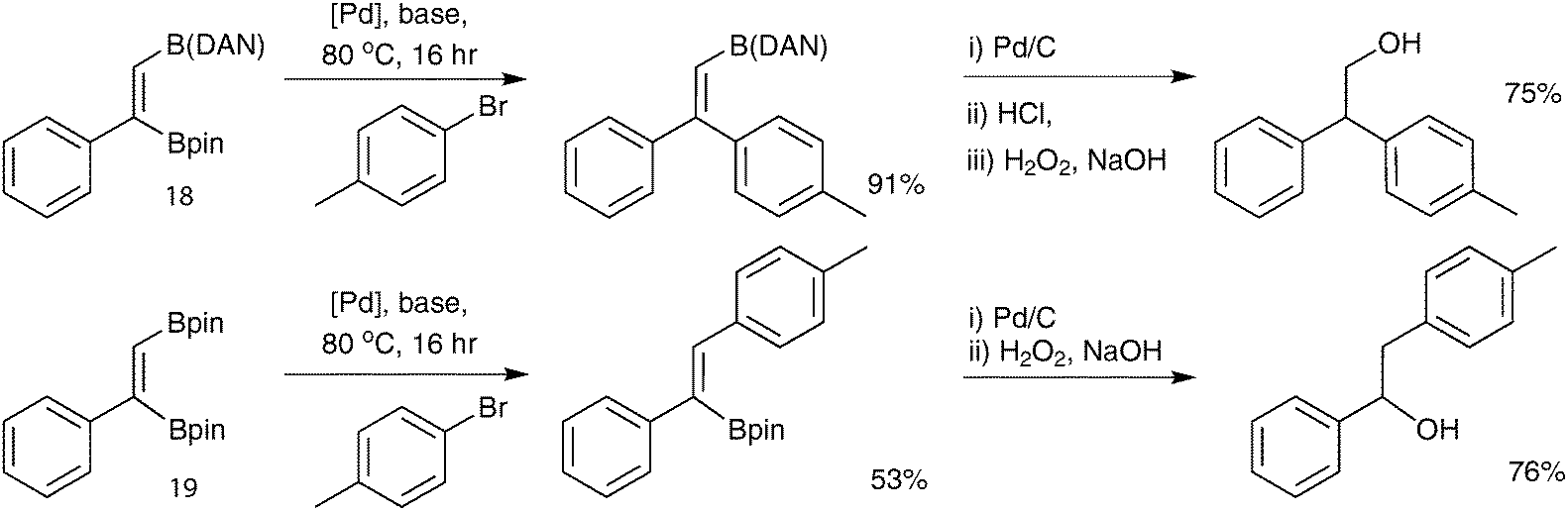 | ||
| Scheme 74 Chemoselective SM coupling leading to a primary alcohol, and a regioselective SM coupling, followed by oxidation. | ||
9. Summary and outlook
As is the case for all synthetic reactions that display the potential for genuine widespread utility, the Suzuki–Miyaura coupling reaction has steadily been improved and expanded in scope. Over a period of over three decades, in addition to the design and development of new catalysts and organo halides or pseudohalides, there has been significant effort applied to the development and diversification of the suite of boron reagents. Perhaps of all three, it is the latter that has made the biggest impact in terms of facilitating the application of the reaction in a much wider range of contexts. Central to this theme is that each of the seven major classes of boron reagent exhibits a unique set of physical and chemical characteristics that align it to particular applications (Table 1). For example, pinacol boronic esters and MIDA boronates can be used to couple unstable substrates, organotrifluoroborates have been shown to couple a wide variety of organic groups, powerful Iterative Cross-Coupling (ICC) has been successfully demonstrated with MIDA boronates and with DAN boronamides, chemoselective orthogonal couplings can be performed with organotrifluoroborates and 9-BBN boranes, and the use of PIDA boronates allows enantiocontrol in distal functional group manipulations.The physical and practical properties exhibited also differentiates the boron reagents from one another. For example, organotrifluoroborates only dissolve in polar media whereas boronic acids dissolve better in apolar media. Boronic acids are the cheapest and most atomic economic reagent and together with organoboranes are simple to prepare, but they are not always the easiest to purify. Pinacol boronic esters and MIDA boronates on the other hand can be purified easily by column chromatography, and organotrifluoroborates purified by recrystallisation.
An appreciation of these differences between boron reagents will naturally allow for more rapid reaction optimisation by aiding the correct choice of reagent and the best use of it. Therefore, it is hoped that this review has accurately collected the key reactivity, mechanistic and practical attributes of each boron reagent in SM coupling and that their identities are better established. Nonetheless, there will probably never be one single boron species that is the ‘reagent of choice’ for all SM couplings, and future developments of specific classes of reagent will likely focus on specific applications. In addition, improving atom economy, ease of preparation and reducing their cost, will also be key features of any new reagents or further developments of existing ones.
| Class | Structure | Preparation | 11B NMR δa/ppm | Pros | Cons | Reactivity | |
|---|---|---|---|---|---|---|---|
| a Relative to BF3·OEt2; s = singlet, q = quartet, br = broad. Impurities commonly encountered include, B(OH)3 (δB = 18–19 ppm), B(OR)3 (δB = 17–19 ppm), BF4K (δB = 0 to −2 ppm), BF3OH− M+ (δB ≈ −0.3 ppm). | |||||||
| Organoboranes |
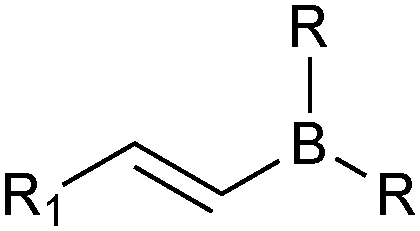
|
– Hydroboration | 75–90, br | – Easily prepared | – Prone to oxidation | – Boronate or oxo–palladium pathway, depending on boron Lewis acidity | |
| – Less reactive in transmetalation | |||||||
| Boronic esters |
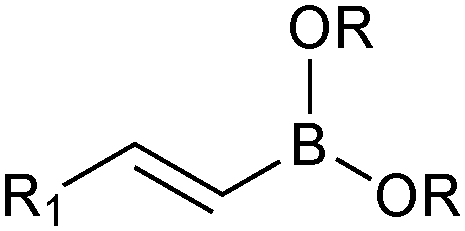
|
– Miyaura borylation | 25–33, br | – Easily prepared and purified | – Less reactive in transmetalation | – Mechanism of transmetalation uncertain | |
| – Hydroboration | – Monomeric | ||||||
| – Stable to silica gel | |||||||
| Boronic acids |
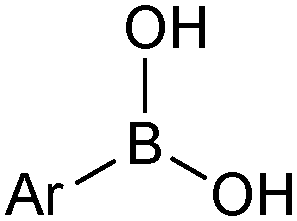
|
– Organometallic | 27–33, br | – Easily prepared | – Susceptible to protodeboronation, oxidation and homocoupling | – Oxo–palladium pathway is kinetically the most likely pathway for transmetalation | |
| – Very atom-efficient | |||||||
| – Highly reactive in transmetalation | |||||||
| Trifluoroborates |
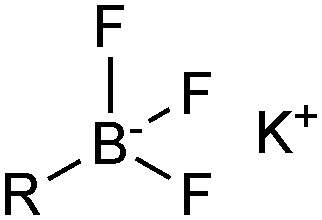
|
– KHF2 | 2–7, q | – Stable solids | – Can cause etching of glassware | – Prior hydrolysis to boronic acid necessary | |
| – KF/tartaric acid | – Monomeric | – Hydrolysis rate and thus transmetalation rate is variable – dependent on large number of factors | – Electron poor = slow hydrolysis | ||||
| – Free-flowing powder | – Electron rich = fast hydrolysis | ||||||
| N-Coordinated boronates |
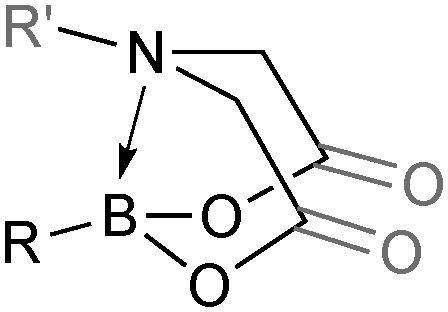
|
– Condensation | 8–12, br | – Stable solids | – Low atom efficiency | – Prior hydrolysis to boronic acid necessary for transmetalation | |
| – Organometallic | – Monomeric | ||||||
| – Hydrolysis easily controlled | |||||||
| – ICC | |||||||
| Boronates |
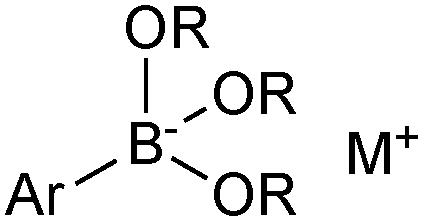
|
– Organometallic | 5–7, s | – Stable | – Not yet commercially available | – Boronate pathway possible, but in situ liberation of base also likely | |
| – Condensation | – Monomeric | ||||||
| – Base-free SM coupling | |||||||
| Boronamides |
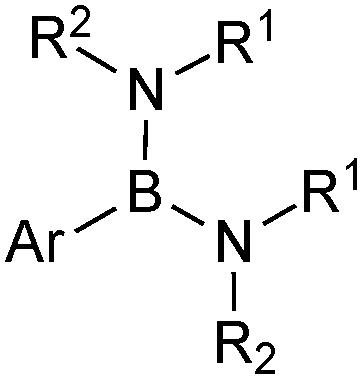
|
– Condensation | 27–29 | – Monomeric | – Low atom efficiency | – Prior hydrolysis to boronic acid necessary for transmetalation | |
| – Stable to aqueous SM coupling, silica-gel & work-up | |||||||
| – ICC | |||||||
Acknowledgements
We thank Dr Allan Watson (University of Strathclyde) for valuable comments and suggestions during the preparation of this review, and AstraZeneca (AJJL) and the Royal Society (Wolfson Research Merit award to GCLJ) for support.Notes and references
- T. W. J. Cooper, I. B. Campbell and S. J. F. Macdonald, Angew. Chem., Int. Ed., 2010, 49, 8082–8091 CrossRef CAS PubMed.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS.
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195–15201 CrossRef CAS PubMed.
- G. C. Fu, A. F. Littke and C. Dai, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef.
- D. Zim, A. S. Gruber, G. Ebeling, J. Dupont and A. L. Monteiro, Org. Lett., 2000, 2, 2881–2884 CrossRef CAS PubMed.
- J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS.
- A. Alimardanov, L. Schmieder-van de Vondervoort, A. H. M. De Vries and J. G. De Vries, Adv. Synth. Catal., 2004, 346, 1812–1817 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419–2440 CAS.
- H. Doucet, Eur. J. Org. Chem., 2008, 2013–2030 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- N. Streidl, B. Denegri, O. Kronja and H. Mayr, Acc. Chem. Res., 2010, 43, 1537–1549 CrossRef CAS PubMed.
- G. Berionni, B. Maji, P. Knochel and H. Mayr, Chem. Sci., 2012, 3, 878 RSC.
- IUPAC Principles of Chemical Nomenclature, ed. G. J. Leigh, RSC Publishing, Cambridge, 2011 Search PubMed.
- IUPAC Nomenclature of Organic Chemistry, ed. J. Rigaudy and S. P. Klesney, Pergamon Press, Oxford, 1979 Search PubMed.
- S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544–4568 CrossRef CAS.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866 RSC.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- N. Miyaura, T. Ishiyama, H. Sasaki, M. Ishikawa, M. Sato and A. Suzuki, J. Am. Chem. Soc., 1989, 111, 314–321 CrossRef CAS.
- H. C. Brown and G. A. Molander, J. Org. Chem., 1986, 51, 4512–4514 CrossRef CAS.
- N. Miyaura, M. Satoh and A. Suzuki, Tetrahedron Lett., 1986, 27, 3745–3748 CrossRef CAS.
- N. Miyaura, K. Yamada, H. Suginome and A. Suzuki, J. Am. Chem. Soc., 1985, 107, 972–980 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- K. Matos and J. A. Soderquist, J. Org. Chem., 1998, 63, 461–470 CrossRef CAS PubMed.
- R. S. Dhillion, Hydroboration and Organic Synthesis, Springer, 2007 Search PubMed.
- H. C. Brown, Hydroboration, Wiley Interscience, New York, 1962 Search PubMed.
- I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS.
- P. C. Keller, Synth. React. Inorg. Met.-Org. Chem., 1976, 6, 77–78 CrossRef.
- A. Pelter, K. Smith and H. C. Brown, Borane Reagents, Academic Press, New York, 1988 Search PubMed.
- K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS.
- E. R. Burkhardt and K. Matos, Chem. Rev., 2006, 106, 2617–2650 CrossRef CAS PubMed.
- H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1956, 78, 5694–5695 CrossRef CAS.
- H. C. Brown and B. C. S. Rao, J. Am. Chem. Soc., 1956, 78, 2582–2588 CrossRef CAS.
- J. C. Colberg, A. Rane, J. Vaquer and J. A. Soderquist, J. Am. Chem. Soc., 1993, 115, 6065–6071 CrossRef CAS.
- H. Brown and B. C. Rao, J. Org. Chem., 1957, 22, 1136–1137 CrossRef CAS.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1961, 83, 486–487 Search PubMed.
- S. Masamune, B. M. Kim, J. S. Petersen, T. Sato, S. J. Veenstra and T. Imai, J. Am. Chem. Soc., 1985, 107, 4549–4551 CrossRef CAS.
- H. C. Brown, J. R. Schwier and B. Singaram, J. Org. Chem., 1978, 43, 4395–4397 CrossRef CAS.
- S. P. Thomas and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 1896–1898 CrossRef CAS PubMed.
- A. Z. Gonzalez, J. G. Román, E. Gonzalez, J. Martinez, J. R. Medina, K. Matos and J. A. Soderquist, J. Am. Chem. Soc., 2008, 130, 9218–9219 CrossRef CAS PubMed.
- Y. Kobayashi, T. Shimazaki and F. Sato, Tetrahedron Lett., 1987, 28, 5849–5852 CrossRef CAS.
- M. Lee, J. B. Rangisetty, M. R. Pullagurla, M. Dukat, V. Setola, B. L. Roth and R. A. Glennon, Bioorg. Med. Chem. Lett., 2005, 15, 1707–1711 CrossRef CAS PubMed.
- J. A. Soderquist and J. C. Colberg, Synlett, 1989, 25–27 CAS.
- K. Radkowski, G. Seidel and A. Fürstner, Chem. Lett., 2011, 950–952 CrossRef CAS.
- M. Utsugi, Y. Kamada, H. Miyamoto and M. Nakada, Tetrahedron Lett., 2007, 48, 6868–6872 CrossRef CAS PubMed.
- M. Uemura, H. Nishimura, T. Minami and Y. Hayashi, J. Am. Chem. Soc., 1991, 113, 5402–5410 CrossRef CAS.
- G. A. Molander and D. L. Sandrock, Org. Lett., 2009, 11, 2369–2372 CrossRef CAS PubMed.
- J. A. Soderquist, K. Matos, A. Rane and J. Ramos, Tetrahedron Lett., 1995, 36, 2401–2402 CrossRef CAS.
- A. Fürstner and G. Seidel, Tetrahedron, 1995, 51, 11165–11176 CrossRef.
- G. Seidel and A. Fürstner, Chem. Commun., 2012, 48, 2055–2070 RSC.
- P. Gersbach, A. Jantsch, F. Feyen, N. Scherr, J.-P. Dangy, G. Pluschke and K.-H. Altmann, Chem.–Eur. J., 2011, 17, 13017–13031 CrossRef CAS PubMed.
- C. D. Roy and H. C. Brown, J. Organomet. Chem., 2007, 692, 784–790 CrossRef CAS PubMed.
- J. Yan, G. Springsteen, S. Deeter and B. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS PubMed.
- N. Zhang, D. J. Hoffman, N. Gutsche, J. Gupta and V. Percec, J. Org. Chem., 2012, 77, 5956–5964 CrossRef CAS PubMed.
- S. Fujii, S. Y. Chang and M. D. Burke, Angew. Chem., Int. Ed., 2011, 50, 7862–7864 CrossRef CAS PubMed.
- D. W. Robbins and J. F. Hartwig, Org. Lett., 2012, 14, 4266–4269 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- D. Mannig and H. Nöth, Angew. Chem., Int. Ed. Engl., 1985, 24, 878–879 CrossRef.
- S. Pereira and M. Srebnik, Organometallics, 1995, 14, 3127–3128 CrossRef CAS.
- X. He and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 1696–1702 CrossRef CAS.
- C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS.
- A.-M. Carroll, T. O'Sullivan and P. Guiry, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef CAS.
- J. M. Brown and G. C. Lloyd-Jones, Tetrahedron: Asymmetry, 1990, 1, 869–872 CrossRef CAS.
- L. Zhang, D. Peng, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2013, 52, 3676–3680 CrossRef CAS PubMed.
- H. R. Kim, I. G. Jung, K. Yoo, K. Jang, E. S. Lee, J. Yun and S. U. Son, Chem. Commun., 2010, 46, 758–760 RSC.
- R. Corberán, N. W. Mszar and A. H. Hoveyda, Angew. Chem., Int. Ed., 2011, 50, 7079–7082 CrossRef PubMed.
- X. Feng, H. Jeon and J. Yun, Angew. Chem., Int. Ed., 2013, 52, 3989–3992 CrossRef CAS PubMed.
- H. Jang, A. R. Zhugralin, Y. Lee and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7859–7871 CrossRef CAS PubMed.
- F. Meng, H. Jang and A. H. Hoveyda, Chem.–Eur. J., 2013, 19, 3204–3214 CrossRef CAS PubMed.
- F. Meng, B. Jung, F. Haeffner and A. H. Hoveyda, Org. Lett., 2013, 15, 1414–1417 CrossRef CAS PubMed.
- A. Arase, M. Hoshi, A. Mijin and K. Nishi, Synth. Commun., 1995, 25, 1957–1962 CrossRef CAS.
- K. Shirakawa, A. Arase and M. Hoshi, Synthesis, 2004, 1814–1820 CAS.
- T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990–4991 CrossRef CAS.
- C. Gunanathan, M. Hölscher, F. Pan and W. Leitner, J. Am. Chem. Soc., 2012, 134, 14349–14352 CrossRef CAS PubMed.
- P. Ceron, A. Finch, J. Frey, J. Kerrigan, T. Parsons, G. Urry and H. I. Schlesinger, J. Am. Chem. Soc., 1959, 81, 6368–6371 CrossRef CAS.
- T. Marder and N. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS.
- T. Ishiyama, N. Matsuda, N. Miyaura and A. Suzuki, J. Am. Chem. Soc., 1993, 115, 11018–11019 CrossRef CAS.
- R. T. Baker, P. Nguyen, T. B. Marder and S. A. Westcott, Angew. Chem., Int. Ed. Engl., 1995, 34, 1336–1338 CrossRef CAS.
- T. Ishiyama, M. Murata and N. Miyaura, J. Org. Chem., 1995, 60, 7508–7510 CrossRef CAS.
- T. Ishiyama, K. Ishida and N. Miyaura, Tetrahedron, 2001, 57, 9813–9816 CrossRef CAS.
- J. Takagi, K. Takahashi, T. Ishiyama and N. Miyaura, J. Am. Chem. Soc., 2002, 124, 8001–8006 CrossRef CAS PubMed.
- M. Murata, S. Watanabe and Y. Masuda, J. Org. Chem., 1997, 62, 6458–6459 CrossRef CAS.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed.
- H. Chen and J. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391–3393 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed.
- C. N. Iverson and M. R. Smith, J. Am. Chem. Soc., 1999, 121, 7696–7697 CrossRef CAS.
- S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168–2171 CrossRef CAS.
- R. B. Coapes, F. E. S. Souza, R. L. Thomas, J. J. Hall and T. B. Marder, Chem. Commun., 2003, 614–615 RSC.
- I. A. I. Mkhalid, R. B. Coapes, S. N. Edes, D. N. Coventry, F. E. S. Souza, R. L. Thomas, J. J. Hall, S.-W. Bi, Z. Lin and T. B. Marder, Dalton Trans., 2008, 1055–1064 RSC.
- C.-I. Lee, J. Zhou and O. V Ozerov, J. Am. Chem. Soc., 2013, 135, 3560–3566 CrossRef CAS PubMed.
- F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846–1849 CrossRef CAS PubMed.
- D. Qiu, L. Jin, Z. Zheng, H. Meng, F. Mo, X. Wang, Y. Zhang and J. Wang, J. Org. Chem., 2013, 78, 1923–1933 CrossRef CAS PubMed.
- M. J. Ingleson, Synlett, 2012, 1411–1415 CrossRef CAS PubMed.
- J. Takagi, K. Sato, J. F. Hartwig, T. Ishiyama and N. Miyaura, Tetrahedron Lett., 2002, 43, 5649–5651 CrossRef CAS.
- A. Del Grosso, M. D. Helm, S. A. Solomon, D. Caras-Quintero and M. J. Ingleson, Chem. Commun., 2011, 47, 12459–12461 RSC.
- H. C. Brown, N. G. Bhat and M. Srebnik, Tetrahedron Lett., 1988, 29, 2631–2634 CrossRef CAS.
- D. Gao and G. A. O'Doherty, Org. Lett., 2010, 12, 3752–3755 CrossRef CAS PubMed.
- N. A. Petasis, R. Keledjian, Y.-P. Sun, K. C. Nagulapalli, E. Tjonahen, R. Yang and C. N. Serhan, Bioorg. Med. Chem. Lett., 2008, 18, 1382–1387 CrossRef CAS PubMed.
- J. Z. Deng, D. V Paone, A. T. Ginnetti, H. Kurihara, S. D. Dreher, S. A. Weissman, S. R. Stauffer and C. S. Burgey, Org. Lett., 2009, 11, 345–347 CrossRef CAS PubMed.
- A. P. Lightfoot, S. J. R. Twiddle and A. Whiting, Synlett, 2005, 529–531 CAS.
- A. P. Lightfoot, G. Maw, C. Thirsk, S. J. R. Twiddle and A. Whiting, Tetrahedron Lett., 2003, 44, 7645–7648 CrossRef CAS PubMed.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS.
- D. G. Hall, in Boronic Acids. Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley-VCH, 2011, pp. 1–109 Search PubMed.
- A. A. C. Braga, G. Ujaque and F. Maseras, Organometallics, 2006, 25, 3647–3658 CrossRef CAS.
- A. A. C. Braga, N. H. Morgon, G. Ujaque and F. Maseras, J. Am. Chem. Soc., 2005, 127, 9298–9307 CrossRef CAS PubMed.
- A. A. C. Braga, N. H. Morgon, G. Ujaque, A. Lledós and F. Maseras, J. Organomet. Chem., 2006, 691, 4459–4466 CrossRef CAS PubMed.
- R. Glaser and N. Knotts, J. Phys. Chem. A, 2006, 110, 1295–1304 CrossRef CAS PubMed.
- C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2011, 17, 2492–2503 CrossRef CAS PubMed.
- B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2116–2119 CrossRef CAS PubMed.
- A. F. Schmidt, A. A. Kurokhtina and E. V. Larina, Russ. J. Gen. Chem., 2011, 81, 1573–1574 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431–7441 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Isr. J. Chem., 2010, 50, 664–674 CrossRef CAS.
- H. G. Kuivila and K. V. Nahabedian, J. Am. Chem. Soc., 1961, 83, 2159–2163 CrossRef CAS.
- H. G. Kuivila, J. F. Reuwer Jr. and J. A. Mangravite, Can. J. Chem., 1963, 41, 3081–3090 CrossRef CAS.
- H. G. Kuivila, J. F. Reuwer and J. A. Mangravite, J. Am. Chem. Soc., 1964, 86, 2666–2670 CrossRef CAS.
- M. Butters, J. N. Harvey, J. Jover, A. J. J. Lennox, G. C. Lloyd-Jones and P. M. Murray, Angew. Chem., Int. Ed., 2010, 49, 5156–5160 CrossRef CAS PubMed.
- C. Adamo, C. Amatore, I. Ciofini, A. Jutand and H. Lakmini, J. Am. Chem. Soc., 2006, 128, 6829–6836 CrossRef CAS PubMed.
- M. Moreno-Mañas, M. Pérez and R. Pleixats, J. Org. Chem., 1996, 61, 2346–2351 CrossRef.
- (a) S. O. Lawesson, Acta Chem. Scand., 1957, 11, 1075–1076 CrossRef CAS PubMed; (b) W. Li, D. P. Nelson, M. S. Jensen, R. S. Hoerrner, D. Cai, R. D. Larsen and P. J. Reider, J. Org. Chem., 2002, 67, 5394–5397 CrossRef CAS PubMed.
- E. Khotinsky and M. Melamed, Ber. Dtsch. Chem. Ges., 1909, 42, 3090–3096 CrossRef CAS.
- D. C. Gerbino, S. D. Mandolesi, H.-G. Schmalz and J. C. Podestá, Eur. J. Org. Chem., 2009, 3964–3972 CrossRef CAS.
- H. Nakamura, M. Fujiwara and Y. Yamamoto, J. Org. Chem., 1998, 63, 7529–7530 CrossRef CAS.
- T. E. Pennington, C. Kardiman and C. A. Hutton, Tetrahedron Lett., 2004, 45, 6657–6660 CrossRef CAS PubMed.
- J. Sun, M. T. Perfetti and W. L. Santos, J. Org. Chem., 2011, 76, 3571–3575 CrossRef CAS PubMed.
- M. E. Jung and T. I. Lazarova, J. Org. Chem., 1999, 64, 2976–2977 CrossRef CAS.
- D. W. Blevins, M.-L. Yao, L. Yong and G. W. Kabalka, Tetrahedron Lett., 2011, 52, 6534–6536 CrossRef CAS PubMed.
- C. A. Hutton and A. K. L. Yuen, Tetrahedron Lett., 2005, 46, 7899–7903 CrossRef PubMed.
- G. A. Molander, L. N. Cavalcanti, B. Canturk, P.-S. Pan and L. E. Kennedy, J. Org. Chem., 2009, 74, 7364–7369 CrossRef CAS PubMed.
- G. W. Kabalka and V. Coltuclu, Tetrahedron Lett., 2009, 50, 6271–6272 CrossRef CAS PubMed.
- G. A. Molander, S. L. J. Trice and S. D. Dreher, J. Am. Chem. Soc., 2010, 132, 17701–17703 CrossRef CAS PubMed.
- G. A. Molander, S. L. J. Trice, S. M. Kennedy, S. D. Dreher and M. T. Tudge, J. Am. Chem. Soc., 2012, 134, 11667–11673 CrossRef CAS PubMed.
- B. a. Tschaen, J. R. Schmink and G. a. Molander, Org. Lett., 2013, 15, 500–503 CrossRef CAS PubMed.
- G. A. Molander, S. L. J. Trice and S. M. Kennedy, J. Org. Chem., 2012, 77, 8678–8688 CrossRef CAS PubMed.
- G. A. Molander, S. L. J. Trice and S. M. Kennedy, Org. Lett., 2012, 14, 4814–4817 CrossRef CAS PubMed.
- T. N. Glasnov and C. O. Kappe, Adv. Synth. Catal., 2010, 352, 3089–3097 CrossRef CAS.
- R. D. Larsen, A. O. King, C. Y. Chen, E. G. Corley, B. S. Foster, F. E. Roberts, C. Yang, D. R. Lieberman and R. A. Reamer, J. Org. Chem., 1994, 59, 6391–6394 CrossRef CAS.
- F. A. J. Kerdesky, M. R. Leanna, J. Zhang, W. Li, J. E. Lallaman, J. Ji and H. E. Morton, Org. Process Res. Dev., 2006, 10, 512–517 CrossRef CAS.
- J. Uenishi, J. M. Beau, R. W. Armstrong and Y. Kishi, J. Am. Chem. Soc., 1987, 109, 4756–4758 CrossRef CAS.
- T. Saeki, E.-C. Son and K. Tamao, Org. Lett., 2004, 6, 617–619 CrossRef CAS PubMed.
- G. Nan, F. Zhu and Z. Wei, Chin. J. Chem., 2011, 29, 72–78 CrossRef CAS.
- R. D. Chambers, H. C. Clark and C. J. Willis, J. Am. Chem. Soc., 1960, 82, 5298–5301 CrossRef CAS.
- D. G. Hall, Structure, Properties, Preparation of Boronic Acid Derivatives. Overview of their Reactions and Applications, Wiley-VCH, Weinheim, 2005 Search PubMed.
- G. A. Molander and D. E. Petrillo, J. Am. Chem. Soc., 2006, 128, 9634–9635 CrossRef CAS PubMed.
- G. A. Molander and D. J. Cooper, J. Org. Chem., 2007, 72, 3558–3560 CrossRef CAS PubMed.
- G. A. Molander and R. Figueroa, J. Org. Chem., 2006, 71, 6135–6140 CrossRef CAS PubMed.
- G. A. Molander, W. Febo-Ayala and L. Jean-Gérard, Org. Lett., 2009, 11, 3830–3833 CrossRef CAS PubMed.
- G. A. Molander and J. Ham, Org. Lett., 2006, 8, 2767–2770 CrossRef CAS PubMed.
- G. A. Molander and D. J. Cooper, J. Org. Chem., 2008, 73, 3885–3891 CrossRef CAS PubMed.
- G. A. Molander and N. M. Ellis, J. Org. Chem., 2006, 71, 7491–7493 CrossRef CAS PubMed.
- G. W. Kabalka and A. R. Mereddy, Tetrahedron Lett., 2004, 45, 343–345 CrossRef CAS PubMed.
- G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 7195–7203 CrossRef CAS PubMed.
- G. W. Kabalka and A. R. Mereddy, Organometallics, 2004, 23, 4519–4521 CrossRef CAS.
- M.-L. Yao, M. S. Reddy, L. Yong, I. Walfish, D. W. Blevins and G. W. Kabalka, Org. Lett., 2010, 700–703 CrossRef CAS PubMed.
- N. A. Petasis, A. K. Yudin, I. A. Zavialov, G. K. S. Prakash and G. A. Olah, Synlett, 1997, 606–608 CrossRef CAS PubMed.
- Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 4648–4651 CrossRef CAS PubMed.
- G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 623–630 CrossRef CAS PubMed.
- G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2012, 77, 4402–4413 CrossRef CAS PubMed.
- G. A. Molander and B. Canturk, Angew. Chem., Int. Ed., 2009, 48, 9240–9261 CrossRef CAS PubMed.
- G. A. Molander and B. Biolatto, J. Org. Chem., 2003, 68, 4302–4314 CrossRef CAS PubMed.
- R. A. Batey and T. D. Quach, Tetrahedron Lett., 2001, 42, 9099–9103 CrossRef CAS.
- W. Ren, J. Li, D. Zou, Y. Wu and Y. Wu, Tetrahedron, 2012, 68, 1351–1358 CrossRef CAS PubMed.
- S. L. Stafford, Can. J. Chem., 1963, 41, 807–808 CrossRef CAS.
- R. D. Chambers, T. Chivers and D. A. Pyke, J. Chem. Soc., 1965, 5144–5145 RSC.
- T. Chivers, Can. J. Chem., 1970, 48, 3856–3859 CrossRef CAS PubMed.
- D. Kaufmann, G. Bir and W. Schacht, J. Organomet. Chem., 1988, 340, 267–271 CrossRef.
- E. Vedejs, R. W. Chapman, S. C. Fields, S. Lin and M. R. Schrimpf, J. Org. Chem., 1995, 60, 3020–3027 CrossRef CAS.
- D. Thierig and F. Umland, Naturwiss. Unterr. Chem., 1967, 54, 563 CAS.
- J. M. Murphy, C. C. Tzschucke and J. F. Hartwig, Org. Lett., 2007, 9, 757–760 CrossRef CAS PubMed.
- V. Bagutski, A. Ros and V. K. Aggarwal, Tetrahedron, 2009, 65, 9956–9960 CrossRef CAS PubMed.
- J.-P. Genet, S. Darses and G. Michaud, Eur. J. Org. Chem., 1999, 1875–1883 Search PubMed.
- G. A. Molander and S. A. McKee, Org. Lett., 2011, 13, 4684–4687 CrossRef CAS PubMed.
- A. Joshi-Pangu, X. Ma, M. Diane, S. Iqbal, R. J. Kribs, R. Huang, C.-Y. Wang and M. R. Biscoe, J. Org. Chem., 2012, 77, 6629–6633 CrossRef CAS PubMed.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2012, 51, 9385–9388 CrossRef CAS PubMed.
- G. A. Molander and B. Biolatto, Org. Lett., 2002, 4, 1867–1870 CrossRef CAS PubMed.
- G. A. Molander, D. E. Petrillo, N. R. Landzberg, J. C. Rohanna and B. Biolatto, Synlett, 2005, 1763 CrossRef CAS PubMed , 1766.
- G. A. Molander and L. A. Felix, J. Org. Chem., 2005, 70, 3950–3956 CrossRef CAS PubMed.
- G. A. Molander and M. R. Rivero, Org. Lett., 2002, 4, 107–109 CrossRef CAS PubMed.
- G. A. Molander, B. W. Katona and F. Machrouhi, J. Org. Chem., 2002, 67, 8416–8423 CrossRef CAS PubMed.
- S. L. Buchwald and T. E. Barder, Org. Lett., 2004, 6, 2649–2652 CrossRef PubMed.
- G. A. Molander, P. E. Gormisky and D. L. Sandrock, J. Org. Chem., 2008, 73, 2052–2057 CrossRef CAS PubMed.
- G. A. Molander and L. Jean-Gérard, J. Org. Chem., 2007, 72, 8422–8426 CrossRef CAS PubMed.
- G. A. Molander and B. Canturk, Org. Lett., 2008, 10, 2135–2138 CrossRef CAS PubMed.
- N. Fleury-Brégeot, M. Presset, F. Beaumard, V. Colombel, D. Oehlrich, F. Rombouts and G. a. Molander, J. Org. Chem., 2012, 77, 10399–10408 CrossRef PubMed.
- G. A. Molander and L. Jean-GeÌrard, J. Org. Chem., 2009, 74, 1297–1303 CrossRef CAS PubMed.
- G. A. Molander and P. E. Gormisky, J. Org. Chem., 2008, 73, 7481–7485 CrossRef CAS PubMed.
- G. A. Molander and D. L. Sandrock, J. Am. Chem. Soc., 2008, 130, 15792–15793 CrossRef CAS PubMed.
- G. A. Molander and Y. Yokoyama, J. Org. Chem., 2006, 71, 2493–2498 CrossRef CAS PubMed.
- G. A. Molander and F. Dehmel, J. Am. Chem. Soc., 2004, 126, 10313–10318 CrossRef CAS PubMed.
- S. Darses, J.-P. Genêt, J.-L. Brayer and J.-P. Demoute, Tetrahedron Lett., 1997, 38, 4393–4396 CrossRef CAS.
- P. Mastrorilli, N. Taccardi, R. Paolillo, V. Gallo, C. F. Nobile, M. Räisänen and T. Repo, Eur. J. Inorg. Chem., 2007, 4645–4652 Search PubMed.
- E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17–27 CAS.
- (a) R. Contreras, C. García, T. Mancilla and B. Wrackmeyer, J. Organomet. Chem., 1983, 246, 213–217 CrossRef CAS; (b) T. Mancilla, R. Contreras and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 1–6 CrossRef CAS; (c) B. Garrigues and M. Mulliez, J. Organomet. Chem., 1986, 314, 19–24 CrossRef CAS; (d) B. Garrigues, M. Mulliez and A. Raharinirina, J. Organomet. Chem., 1986, 302, 153–158 CrossRef CAS.
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed; (b) S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 466–468 CrossRef CAS PubMed.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2008, 130, 14084–14085 CrossRef CAS PubMed.
- M. Reilly and S. Rychnovsky, Synlett, 2011, 2392–2396 CAS.
- H. Bonin, D. Delbrayelle, P. Demonchaux and E. Gras, Chem. Commun., 2010, 46, 2677–2679 RSC.
- A. Bouillon, J.-C. Lancelot, J. Sopkova De Oliveira Santos, V. Collot, P. R. Bovy and S. Rault, Tetrahedron, 2003, 59, 10043–10049 CrossRef CAS PubMed.
- C. Gütz and A. Lützen, Synthesis, 2010, 85–90 Search PubMed.
- P. B. Hodgson and F. H. Salingue, Tetrahedron Lett., 2004, 45, 685–687 CrossRef CAS PubMed.
- G. R. Dick, D. M. Knapp, E. P. Gillis and M. D. Burke, Org. Lett., 2010, 12, 2314–2317 CrossRef CAS PubMed.
- J. R. Struble, S. J. Lee and M. D. Burke, Tetrahedron, 2010, 66, 4710–4718 CrossRef CAS PubMed.
- B. E. Uno, E. P. Gillis and M. D. Burke, Tetrahedron, 2009, 65, 3130–3138 CrossRef CAS PubMed.
- M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2009, 48, 3565–3568 CrossRef CAS PubMed.
- C. Wang and F. Glorius, Angew. Chem., Int. Ed., 2009, 48, 5240–5244 CrossRef CAS PubMed.
- E. M. Woerly, A. H. Cherney, E. K. Davis and M. D. Burke, J. Am. Chem. Soc., 2010, 132, 6941–6943 CrossRef CAS PubMed.
- S. J. Lee, T. M. Anderson and M. D. Burke, Angew. Chem., Int. Ed., 2010, 49, 8860–8863 CrossRef CAS PubMed.
- N. A. Jones, J. W. Antoon, A. L. Bowie, J. B. Borak and E. P. Stevens, J. Heterocycl. Chem., 2007, 44, 363–367 CrossRef CAS.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed.
- A. R. Burns, G. D. McAllister, S. E. Shanahan and R. J. K. Taylor, Angew. Chem., Int. Ed., 2010, 49, 5574–5577 CrossRef CAS PubMed.
- G. R. Dick, E. M. Woerly and M. D. Burke, Angew. Chem., Int. Ed., 2012, 51, 2667–2672 CrossRef CAS PubMed.
- T. Kinzel, Y. Zhang and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14073–14075 CrossRef CAS PubMed.
- J. Chen and A. Cammers-Goodwin, Tetrahedron Lett., 2003, 44, 1503–1506 CrossRef CAS.
- (a) N. Y. Adonin, D. E. Babushkin, V. N. Parmon, V. V Bardin, G. A. Kostin, V. I. Mashukov and H.-J. Frohn, Tetrahedron, 2008, 64, 5920–5924 CrossRef CAS PubMed; (b) T. Korenaga, T. Kosaki, R. Fukumura, T. Ema and T. Sakai, Org. Lett., 2005, 7, 4915–4917 CrossRef CAS PubMed.
- H. C. Brown and A. K. Gupta, J. Organomet. Chem., 1988, 341, 73–81 CrossRef CAS.
- J. Li and M. D. Burke, J. Am. Chem. Soc., 2011, 133, 13774–13777 CrossRef CAS PubMed.
- A. N. Cammidge, V. H. M. Goddard, H. Gopee, N. L. Harrison, D. L. Hughes, C. J. Schubert, B. M. Sutton, G. L. Watts and A. J. Whitehead, Org. Lett., 2006, 8, 4071–4074 CrossRef CAS PubMed.
- B. Basu, K. Biswas, S. Kundu and S. Ghosh, Green Chem., 2010, 12, 1734–1738 RSC.
- Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928–931 CrossRef CAS PubMed.
- K. L. Billingsley and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 4695–4698 CrossRef CAS PubMed.
- M. A. Oberli and S. L. Buchwald, Org. Lett., 2012, 14, 4606–4609 CrossRef CAS PubMed.
- D. S. Matteson and H.-W. Man, J. Org. Chem., 1996, 61, 6047–6051 CrossRef CAS.
- M. R. Akula, M.-L. Yao and G. W. Kabalka, Tetrahedron Lett., 2010, 51, 1170–1171 CrossRef CAS PubMed.
- X.-Q. Yu, Y. Yamamoto and N. Miyaura, Synlett, 2009, 994–998 CAS.
- G.-Q. Li, Y. Yamamoto and N. Miyaura, Synlett, 2011, 1769–1773 CAS.
- G.-Q. Li, Y. Yamamoto and N. Miyaura, Tetrahedron, 2011, 67, 6804–6811 CrossRef CAS PubMed.
- K. Chen, R. Peterson, S. K. Math, J. B. LaMunyon, C. A. Testa and D. R. Cefalo, Tetrahedron Lett., 2012, 53, 4873–4876 CrossRef CAS PubMed.
- H. Ihara, M. Koyanagi and M. Suginome, Org. Lett., 2011, 13, 2662–2665 CrossRef CAS PubMed.
- H. Ihara and M. Suginome, J. Am. Chem. Soc., 2009, 131, 7502–7503 CrossRef CAS PubMed.
- H. Noguchi, K. Hojo and M. Suginome, J. Am. Chem. Soc., 2007, 129, 758–759 CrossRef CAS PubMed.
- G. Kaupp, M. R. Naimi-Jamal and V. Stepanenko, Chem.–Eur. J., 2003, 9, 4156–4161 CrossRef CAS PubMed.
- N. Iwadate and M. Suginome, J. Organomet. Chem., 2009, 694, 1713–1717 CrossRef CAS PubMed.
- N. Iwadate and M. Suginome, Org. Lett., 2009, 11, 1899–1902 CrossRef CAS PubMed.
- N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548–2549 CrossRef CAS PubMed.
- M. Koyanagi, N. Eichenauer, H. Ihara, T. Yamamoto and M. Suginome, Chem. Lett., 2013, 541–543 CrossRef CAS.
- H. Noguchi, T. Shioda, C.-M. Chou and M. Suginome, Org. Lett., 2008, 10, 377–380 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |