What molecular assembly can learn from catalytic chemistry†
Yu
Wang
ab,
Hai-Xin
Lin
b,
Liang
Chen
b,
Song-Yuan
Ding
ab,
Zhi-Chao
Lei
b,
De-Yu
Liu
b,
Xiao-Yu
Cao
*b,
Hao-Jun
Liang
ac,
Yun-Bao
Jiang
ab and
Zhong-Qun
Tian
*ab
aCollaborative Innovation Center of Chemistry for Energy Materials, Xiamen University, Xiamen, 361005, China. E-mail: zqtian@xmu.edu.cn; Fax: +86-592-2085349; Tel: +86-592-2182536
bState Key Laboratory of Physical Chemistry of Solid Surfaces and College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: zqtian@xmu.edu.cn; xcao@xmu.edu.cn
cCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China
First published on 24th September 2013
Abstract
One important objective of molecular assembly research is to create highly complex functional chemical systems capable of responding, adapting, and evolving. Compared with living systems, the synthetic systems are still rather primitive and are far from realizing those features. Nature is by far the most important source of inspiration for designing and creating such systems. In this critical review, we summarize an alternative approach, inspired by catalysis, to examine and describe some molecular assembly processes. A new term, “catassembly,” is suggested to refer to the increase in the rate and control of a molecular assembly process. This term combines the words “catalysis” and “assembly,” and identifiably retains the Greek root “cat-” of catalysis. The corresponding verb is “catassemble” and the noun is “catassembler”, referring to the “helper” species. Catassembly in molecular assembly is a concept that is analogous to catalysis in chemical synthesis. After using several examples to illustrate the characteristics of catassembly, we discuss future methodological and theoretical developments. We also emphasize the significance of the synergy between chemical synthesis and molecular assembly, especially for hierarchical assembly systems. Because most efforts in the field of molecular assembly have been devoted to the design and synthesis of molecular building blocks, we wish to stress the apparently missing yet critical link to complex chemical systems, i.e., the design and utilization of molecular catassemblers to facilitate the formation of functional molecular assemblies from building blocks with high efficiency and selectivity. This rational control and accelerated method will promote the systems chemistry approach, and may expand the spectrum of molecular assembly from basic science to applications.
 From left to right: Yun-Bao Jiang, Yu Wang, Xiao-Yu Cao, Zhi-Chao Lei, Hai-Xin Lin, Song-Yuan Ding and Zhong-Qun Tian | Yu Wang received his BS degree in chemistry from Nanjing University in 2009. He is currently a PhD candidate with Professor Zhong-Qun Tian at Xiamen University. He is currently investigating the controllable assembly of π-conjugated molecules and photo-regulated assembly on surfaces. |
Hai-Xin Lin received his BS degree in chemistry from Xiamen University in 2010. Then he became a PhD candidate in the same university under the supervision of Professor Zhong-Qun Tian. His research focuses on the synthesis and controllable assembly of nanoparticles. |
Liang Chen received his BS degree from Xiamen University in 2013. Now he is pursuing his PhD in the group of Professor Zhong-Qun Tian. His research focuses on the synthesis and assembly of nanoparticles for preparing functional materials. |
Song-Yuan Ding received a PhD from Xiamen University in 2012, under the supervision of Professor Zhong-Qun Tian. He is currently an iChEM fellow. His research focuses on the control theory of molecular assembly and the development of active substrates for surface-enhanced infrared absorption spectroscopy. |
Zhi-Chao Lei received his BS degree from Xiamen University in 2012. He is now pursuing his PhD degree with Professor Zhong-Qun Tian. His research focuses on the controllable assembly of DNA nano-structures. |
De-Yu Liu received his BS degree from Xiamen University in 2009. He is currently a PhD candidate of Professor Galen D. Stucky in the University of California, Santa Barbara. His research focuses on fabricating heterogeneous structures of metals and semiconductors with special properties. |
Xiao-Yu Cao is currently an associate professor in Xiamen University. He obtained his PhD from Université Louis Pasteur, Strasbourg I, France in 2009, under the supervision of Professor Jean-Marie Lehn. He worked with Prof. E. W. (Bert) Meijer in Eindhoven University of Technology, the Netherlands, as a Marie-Curie Intra-European fellowship holder before he joined Xiamen University in 2011. His research focuses on the design, synthesis, controllable assembly and application of novel aromatic molecules. |
Hao-Jun Liang is a full professor at University of Science and Technology of China. He won the Chinese Young Chemists Award in 2005. Research in his group focuses on assembly and molecular devices in experiment and numerical simulation in condensed matter physics and biophysics. |
Yun-Bao Jiang is a full professor at Xiamen University. He started his career after obtaining a PhD in 1990 from the same university. Dr Jiang did his postdoc research with Professor Klaas A. Zachariasse at the Max-Planck Institute for biophysical Chemistry, Germany, and with Professor Chi-Ming Che at the University of Hong Kong, one of which was supported by the prestigious Alexander von Humboldt research fellowship. Photophysics and structures of aggregates for sensing applications are among the research interests of his group. |
Zhong-Qun Tian is a full professor at Xiamen University. He is a Member of Chinese Academy of Sciences, Fellow of the Royal Society of Chemistry and a Member of the International Society of Electrochemistry and a member of the advisory board of eleven international journals. His main research interests are surface enhanced Raman spectroscopy, spectro-electrochemistry, nanochemistry and catassembly. |
1. Introduction
Chemistry is an advanced science, and one of its most distinctive objectives is to provide new functions by creating new molecules and new materials. Using chemical reactions (many of which involve catalysis), chemists have synthesized materials that shaped our modern life, such as polymers, medicines, pesticides, energy materials, etc. Nonetheless, most current functional materials use only covalent bonds, and covalent synthesis has its limitations in terms of the size, complexity and functionality of the products.Molecular assembly provides an elegant approach to functional materials beyond molecules with more biomimetic elements.1–10 Using molecules or molecule-modified nanoparticles as building blocks, many complex materials11–15 with unique functions (such as self-healing materials,16–19 biomimetic materials,20,21 smart molecular devices,22–28etc.29–32) have been fabricated by molecular assembly through various noncovalent interactions (van der Waals,10 hydrogen bonding,33–35 hydrophobic effects,36,37 weak coordination,38–42etc.43).
To achieve more advanced functions like what nature does, an artificial cell for instance, one has to construct complex functional chemical systems based on a deep understanding of molecular assembly. Such research is also essential to answer the big question for chemists: how does matter become complex?44
The current investigations on molecular assembly processes mainly focus on building blocks (through tailoring their sizes, shapes, solubilities, functional groups, or non-covalent interactions) and their interplay with the environment (through the variations of solvent, temperature, concentration or solid interface). The implementation of molecular information45 into the structural motifs of each building block has indeed realized certain controllability of the molecular assembly process, in many cases giving a notion that these components build up the ordered assemblies by themselves.
Nonetheless, most artificial chemical systems pale in comparison to life systems in terms of complexity and efficiency.46 Importance should also be attached to increasing the efficiency of artificial systems. Especially, in complex assembly systems with hierarchical assembly processes, efficiency and controllability of each sub-process become significant for maintaining the overall efficiency. Therefore, developing strategies to enhance the assembly efficiency and controllability is crucial.
In this review, we attempt to import the concept of catalysis, which is well known as the most efficient control method of chemical reactions, into molecular assembly. Examples are chosen to demonstrate the superiority of this concept in increasing assembly efficiency and controllability. We further propose to introduce the research methodology of catalysis (e.g., various types of catalytic methods, theories and characterization techniques) into molecular assembly and attempt to answer the question “what else can be learned from catalysis?” which could be vitally important in expanding the scope of molecular assembly from basic research to applications.
2. Can molecular assembly learn from catalysis?
Molecular assembly and chemical reaction are the two most important approaches to creating new materials. The well-established field of chemical synthesis has developed many effective control methods in past centuries. Beginning only a few decades ago, the field of molecular assembly is much younger than that of chemical synthesis, thus the former can learn some control methods from the latter.For chemical synthesis, field-assisted reaction (e.g., photo-reaction and electro-reaction) and catalysis47–51 (e.g. photo-catalysis, electro-catalysis, and supramolecular catalysis52–54) have been developed as effective control methods. Especially, catalysis plays an indispensable role in chemical engineering to meet the demands of high product selectivity and high yield.49 With respect to molecular assembly, although methods of self-assembly and field-assisted assembly (e.g., photo-assisted assembly,23,55–57 and electro-assisted assembly43,57–59) have been well developed, so far the “catalysed-assembly” method is almost unknown (question marks in Table 1). Although some experimental research studies may be applied (as shown in Section 2.2), this concept of catalysed-assembly has not yet drawn great attention, nor has it been addressed explicitly.60
| Chemical synthesis | Molecular assembly |
|---|---|
| Spontaneous reaction | Self-assembly |
| Field-assisted reaction | Field-assisted assembly |
| Photo-reaction | Photo-assisted assembly |
| Electro-reaction | Electro-assisted assembly |
| … | … |
| Catalysis | ? |
| Photo-catalysis | ? |
| Electro-catalysis | ? |
| Supramolecular catalysis | ? |
| … | ? |
2.1 Definition of catassembly
Because this phenomenon has barely been recognized, conceptualization and specialized terminology need to be established before starting to discuss the examples and further develop the methodology. Initially, we tried to use the term catalysed-assembly to describe this phenomenon. However, we cannot find a corresponding term or phrase to appropriately describe the substance that accelerates a catalysed-assembly process, because “supramolecular catalysts”52–54 and “noncovalent catalysts”61 have been restricted to substances that accelerate a chemical reaction, instead of an assembly process. Besides, this phenomenon in molecular assembly differs from catalysis in essence (see ESI† for detailed explanation).Therefore, we suggest “catassembly” as an abbreviation of catalysed-assembly,60 as it combines the words “catalysed” and “assembly”, has a set of syllables that can be read easily, and identifiably retains the Greek root “cat-” of catalysis. In addition, the noun form could be defined as: a catassembler is a substance (or a group of substances working synergistically) that increases the rate of an assembly process without modifying the overall standard Gibbs energy change; the process is called catassembly. The corresponding verb and adjective forms are catassemble and catassembled. This series of terms will completely avoid any confusion with supramolecular catalysts and noncovalent catalysts.
Through an analogy to building a floating bridge, Fig. 1 illustrates why catassembly is more efficient than molecular self-assembly in the formation of complex systems. At a narrow site in a river, plates with specific concave and convex sites could accumulate and collide with each other, subsequently adjust the interfaces to implement embedment and could finally form a bridge. This process is analogous to molecular self-assembly: the river water corresponds to the solvent, the plates correspond to molecular building blocks, and the interactions at the concave and convex sites of plates correspond to the noncovalent interactions between building blocks. However, this self-assembly process can be estimated to require a substantial amount of time to form the bridge. In contrast, an alternative approach can significantly increase the efficiency by conveying and assembling the plates with porters and builders respectively. In addition, these porters and builders can build more bridges. This process is analogous to catassembly: the workers correspond to catassemblers, and the interactions between the catassemblers and building blocks are also noncovalent.
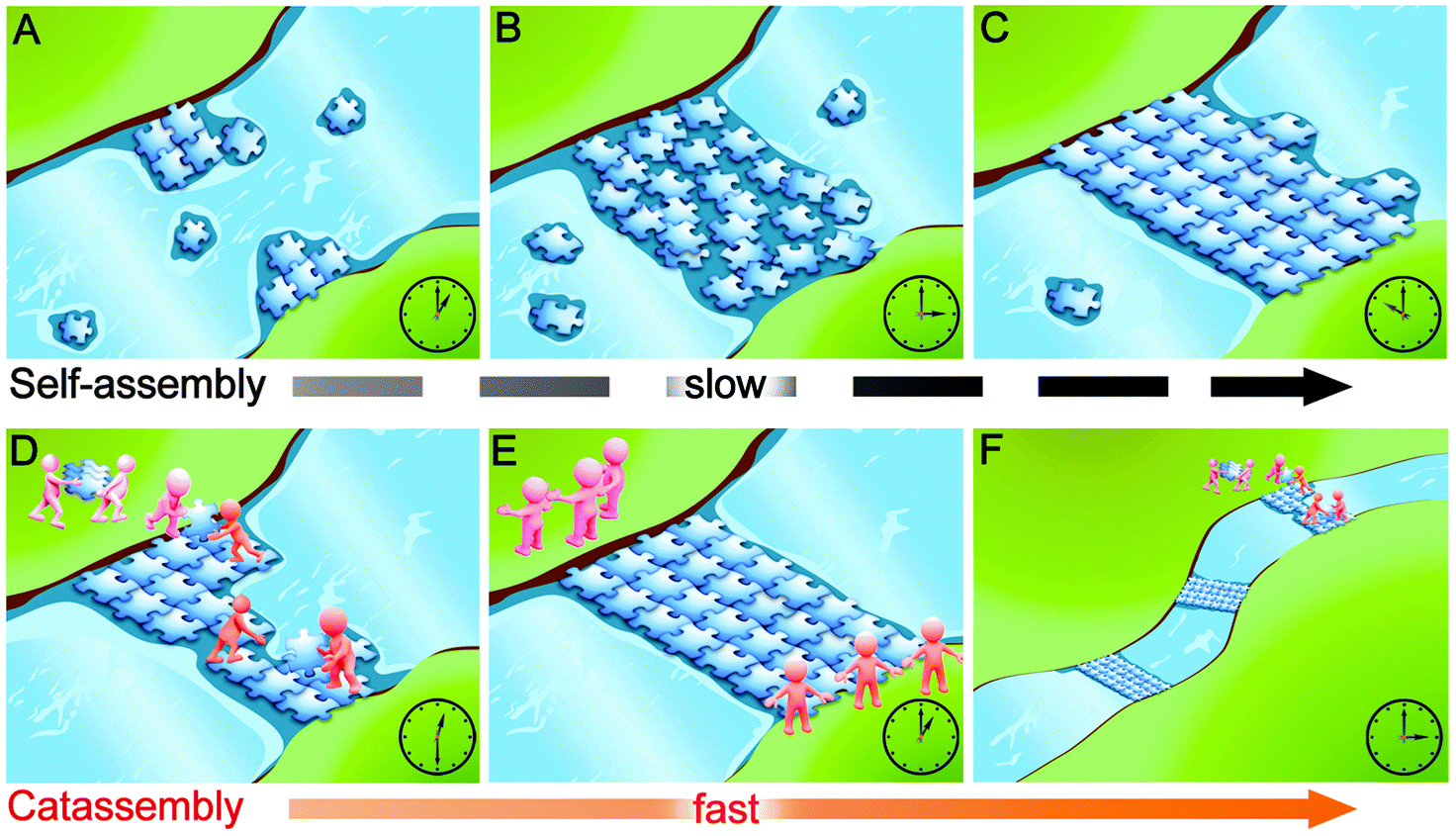 | ||
| Fig. 1 Cartoons of the construction of a floating bridge that illustrate why catassembly is more efficient than self-assembly. Panels A to C represent a self-assembly process, which requires many hours to form a floating bridge. In contrast, with the assistance of porters and builders (corresponding to two kinds of catassembler), a bridge can be built in 1 hour (panels D and E). After the first bridge is completed, the workers leave it and build other bridges somewhere else (panel F). | ||
2.2 Primitive examples of catassembly
Small molecules, such as organic compounds, oligomers and related coordination complexes, can be assembled into supramolecular structures through noncovalent interactions under certain conditions. The catassembly processes are regulated by catassemblers through noncovalent interactions as well. Catassemblers may transport building blocks to the appropriate assembly sites, regulate the configurations and orientations of building blocks, and promote molecular recognition at multiple interaction sites.Reinhoudt et al. reported a type of asymmetric catassembly.62 Achiral calix[4]arenedimelamine (CA) and cyanurate (CYA) self-assembled into a chiral supramolecular structure through multiple cooperative hydrogen bonds. The (P)-enantiomer and (M)-enantiomer of the assembly could undergo interconversion through a “disassembly and reassembly” process (Fig. 2) and racemize the chiral assembly. Fortunately, however, because the energy barrier was relatively high, the interconversion was sufficiently slow for the enantiomers to be isolated. Interestingly, the addition of chiral barbiturate (BAR) significantly accelerated the racemization by lowering the energy barrier. BAR facilitated the assembly processes through the formation and cleavage of hydrogen bonds with the building blocks but ultimately departed from the assemblies without modifying the overall Gibbs energy change. Therefore, BAR could be regarded as a small-molecule catassembler in this catassembly process.
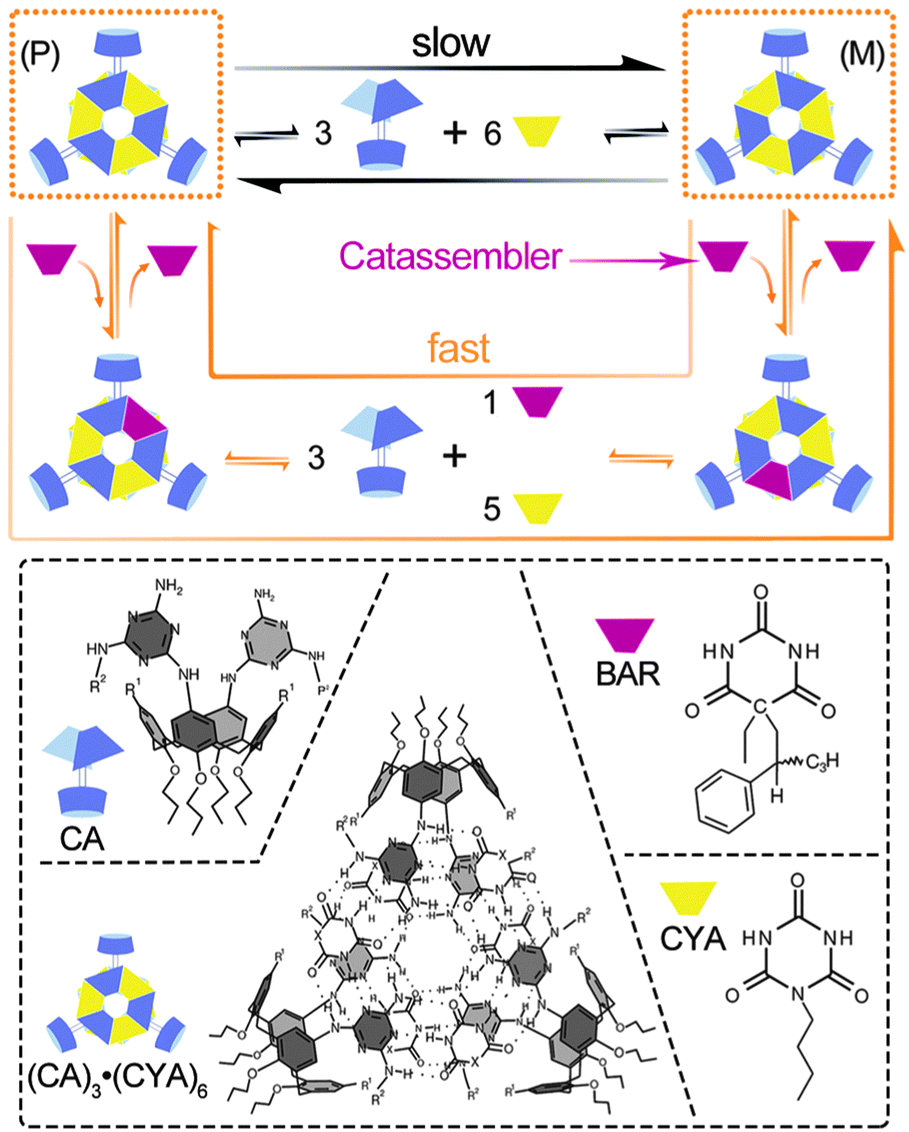 | ||
| Fig. 2 Racemization of the enantiomers (P) and (M) under the disassembly–reassembly and catassembly processes. In the normal disassembly–reassembly racemization process, which is represented by the long black arrow, the (P)- or (M)-enantiomer of (CA)3·(CYA)6 first disassembles into the building blocks CA and CYA; then, the building blocks reassemble into the two enantiomers. In the catassembled racemization process represented by the long orange arrow, one of the CYA building blocks in the (P)- or (M)-enantiomer of (CA)3·(CYA)6 is first displaced by the chiral catassembler BAR and forms a ternary complex (CA)3·(BAR)·(CYA)5, which further changes into the corresponding ternary enantiomer through a disassembly–reassembly process. The BAR in the ternary enantiomer is finally displaced by CYA and forms the other enantiomer of (CA)3·(CYA)6. The molecular structures of CYA, BAR, CA and enantiomer (P) are presented in the dashed box. Structural images adapted from ref. 62 (Copyright (2000), with permission from Nature Publishing Group). | ||
Another type of asymmetric catassembly has been achieved by using chiral molecules to control the nucleation processes of one-dimensional assembly of π-conjugated systems, usually followed by further self-replication processes. One example is Purrello et al. used a chiral amino acid phenylalanine (Phe) to promote the asymmetric selectivity of the co-assemblies of two opposite-charged achiral porphyrins.63 These enantiospecific co-assemblies could be separated from the system by physical means (centrifugation and filtration), without containing a trace of Phe. Thus, Phe could be considered as an asymmetric catassembler in this process.
Along this line, we reported a strategy for the creation of isolable chiral assemblies from a variety of π-conjugated carboxylic acids (CCAs) with a catassembler of carboxymethyl cellulose (CMC).64 For example, the assembly process for N,N′-di(1-carboxylethyl)-3,4,9,10-perylenetetracarboxylbisimide (CPTCI) was controlled at a slow rate by the addition of the water-soluble CPTCI conjugate base into an acidic aqueous solution that contained chiral CMC (Fig. 3A). Under these conditions the CPTCI assembly could be isolated from the solution by simple centrifugation. No CMC was found in the isolated final chiral assembly. However, either the absence of the catassembler CMC or the use of a small chiral molecule, such as tartaric acid, instead of CMC resulted in achiral assembly of CPTCI. The chiral induction by CMC was ascribed to the weak intermolecular COOH–COOH hydrogen-bonding interaction between CMC and CPTCI. This intermolecular interaction was subsequently replaced by the intra-assembly “layer-to-layer” COOH–COOH interaction between the CPTCI molecules; and this replacement released the catassembler CMC from the assemblies (Fig. 3B). In other similar catassembly processes, chiral supramolecular materials have been created by the assembly of achiral building blocks into isolable chiral homo- and hetero-aggregates in aqueous solutions.65,66
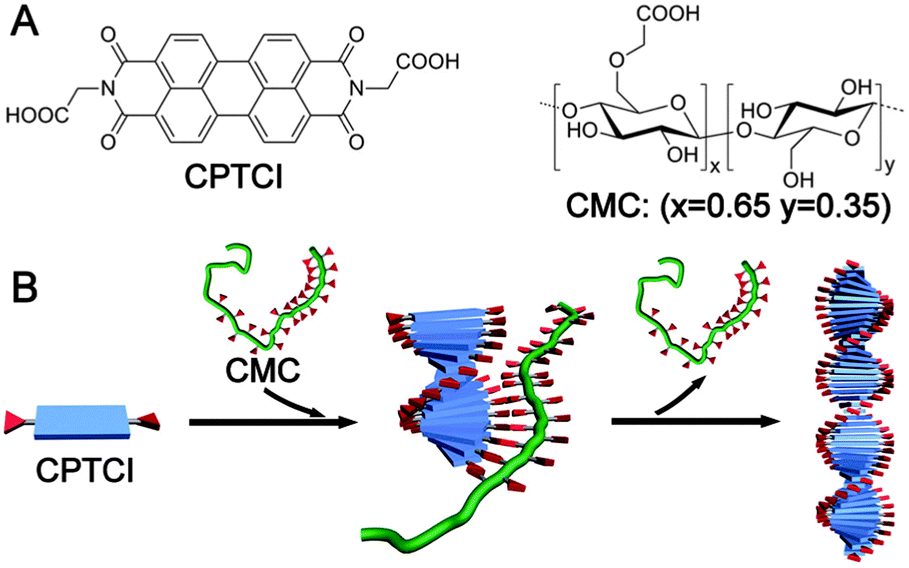 | ||
| Fig. 3 (A) Molecular structures of CPTCI and CMC. (B) Schematic representation of the catassembled pathway of CPTCI. Chiral assembly of CPTCI is induced by the formation of weak intermolecular COOH–COOH hydrogen bonds between CPTCI and the chiral polymer CMC. After the CPTCI molecules are well assembled, the intra-assembly “layer-to-layer” COOH–COOH interactions replace the interactions between CPTCI and CMC; hence, the assemblies are released from the catassembler CMC. | ||
Hybridization processes of DNA strands, known as “displacement reactions,” are central to both natural biological systems67,68 and artificial DNA nanotechnology.69,70 The processes are essentially the assembly and disassembly processes for DNA strands because the interactions between complementary base pairs are noncovalent hydrogen bonds. A sophisticated catassembly process for the displacement of DNA strands has been designed by Turberfield et al.71 Simple hybridization between complementary strands L (70 bases) and L′ would occur rapidly. Nonetheless, a protective strand S′ (30 bases) was designed to hybridize 15-base sections at either end of strand L to form a loop (Fig. 4), which slowed the hybridization process between L and L′ 100-fold. The catassembler DNA strand M (21 bases) could hybridize strand L from an external toehold, thus displacing half of S′ from L and opening the loop. After the loop was open, L′ could easily hybridize L and displace strand S′ and the catassembler (Fig. 4). This catassembly process was approximately 30 times faster than the non-catassembled process. This pioneering work has triggered several studies on catassembled displacement of DNA strands.72–75 For example, in a similar system, Winfree et al. reported a catassembled efficiency enhancement of approximately 5000-fold over the non-catassembled process.73
 | ||
| Fig. 4 Scheme for kinetic control of DNA hybridization by a catassembler strand. The thin and thick line segments with the same colours represent the complementary base pairs. The black arrow represents the self-assembly process, whereas the orange arrow represents the catassembly process. Adapted from ref. 71 (Copyright (2003), with permission from The American Physical Society). | ||
Unlike the assembly of unmodified inorganic nanoparticles (NPs), the assembly of modified NPs depends on the interactions of the adsorbed molecules. Therefore, molecular catassembly systems could be transplanted onto the surface of NPs to allow the catassembly of NPs. For example, we have successfully transplanted the catassembled displacement of DNA strands onto the surface of gold NPs.76 The assembly of two types of modified NPs was driven by a toehold-mediated DNA strand displacement process, which was catassembled by an oligonucleotide. As shown in Fig. 5, NP-1 was modified with oligomer-1, and NP-2 was modified with a complex of oligomer-2, a linker-oligomer, and a protector-oligomer. The displacement of the protector-oligomer by oligomer-1 would associate the two NPs, but the direct occurrence of this process was difficult. The DNA strand displacement on the surface of the NPs would occur only if the toehold was at least eight bases long, whereas a five-base toehold was long enough to achieve a sufficiently high rate in pure DNA systems. The displacement of the protector-oligomer by the catassembler-oligomer occurs through a five-base toehold-mediated process, which would elongate the toehold at the linker-oligomer terminal. Hence, the assembly rate of NP-1 and NP-2 was significantly enhanced by the catassembler-oligomer.
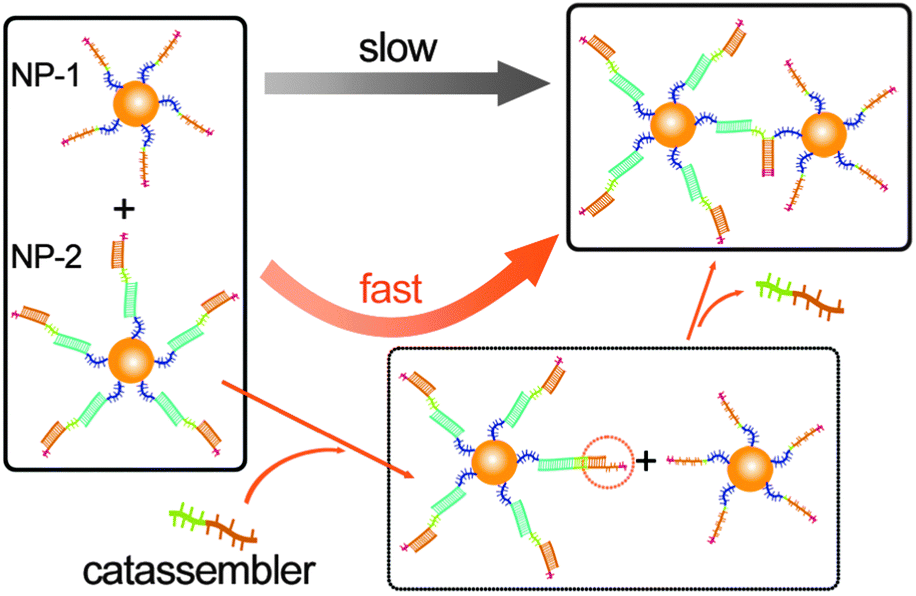 | ||
| Fig. 5 Graphical representation of self-assembly and catassembly processes of DNA-modified Au NPs. | ||
Note that molecular assembly for functional materials is just in its infancy; hence, only primitive examples are provided in this review. However, it can be expected that more advanced materials with high efficiency and selectivity could be achieved with the development of catassembly.
2.3 Features of catassembly
From the definition and examples above, we summarize the features of catassembly: (1) the catassembly processes are regulated by catassemblers through multi-site synergistic noncovalent interactions, (2) a catassembler increases the efficiency and/or selectivity of an assembly process by dividing it into a cascade of sub-processes, (3) a catassembler is capable of assisting many building block molecules in a single-step procedure, as it would leave the assembly voluntarily when finishing its mission, and is not present in the final structure.60With these distinct characteristics, a catassembler can be distinguished from the similar or relative concepts (Table 2), such as a coassembled linker,77–79 a template15,38,80–82 (which can be considered co-assembled species because a template does not voluntarily leave the assembly when finishing its mission as a helper60,83), a catalyst (including the traditional catalyst, enzyme, and supramolecular catalyst52–54), and a chaperone (which is usually a multi-component protein that promotes polypeptide folding with the role of preventing incorrect folding or oligomerization,84–86 see more in Section 5).
| Catassembler | Catalyst | Supramolecular catalyst | Template | |
|---|---|---|---|---|
| Region of applicability | Molecular assembly | Chemical reaction | Chemical reaction | From atomic to macro level |
| Interactions | Noncovalent | Covalent | Noncovalent | Covalent and noncovalent |
| Active sites | Several/multiple | One or two | Several/multiple | From one to multiple |
| Self-releasable | Yes | Yes | Yes | No |
| Selectivity | High | High | High | From low to high |
Note that some features of catassembly have been pointed out in the definition (Section 2.1), but were not revealed in the examples above (Section 2.2), such as a porter-like catassembler and the synergy of a group of catassemblers. This is because we could only provide primitive artificial examples at the present stage. However, molecular assembly has complexity at different levels, as shown in Fig. 6. At level 1, assembly involves relatively simple noncovalent interactions between one or two kinds of molecules, and thus can be accomplished through a spontaneous pathway (self-assembly) easily. However, at level 2, hierarchical assembly involves multi-components and complex interactions, thus catassemblers tend to be useful, as the builders in Fig. 1 will become more important when the bridge turns complex. And at higher levels, supramolecular systems involve multiple associated assembly processes at multiple time and space scales, the synergy of a group of catassemblers will be important, and the effect of porters starts to emerge gradually. For instance, the synergy of a group of catassemblers can be found in life science as shown in Session 5. And although porter-like molecules are seldom created in chemical reactions87 or “low-level” assembly processes, they play an important role in life science, such as tRNA46 and kinesin.88
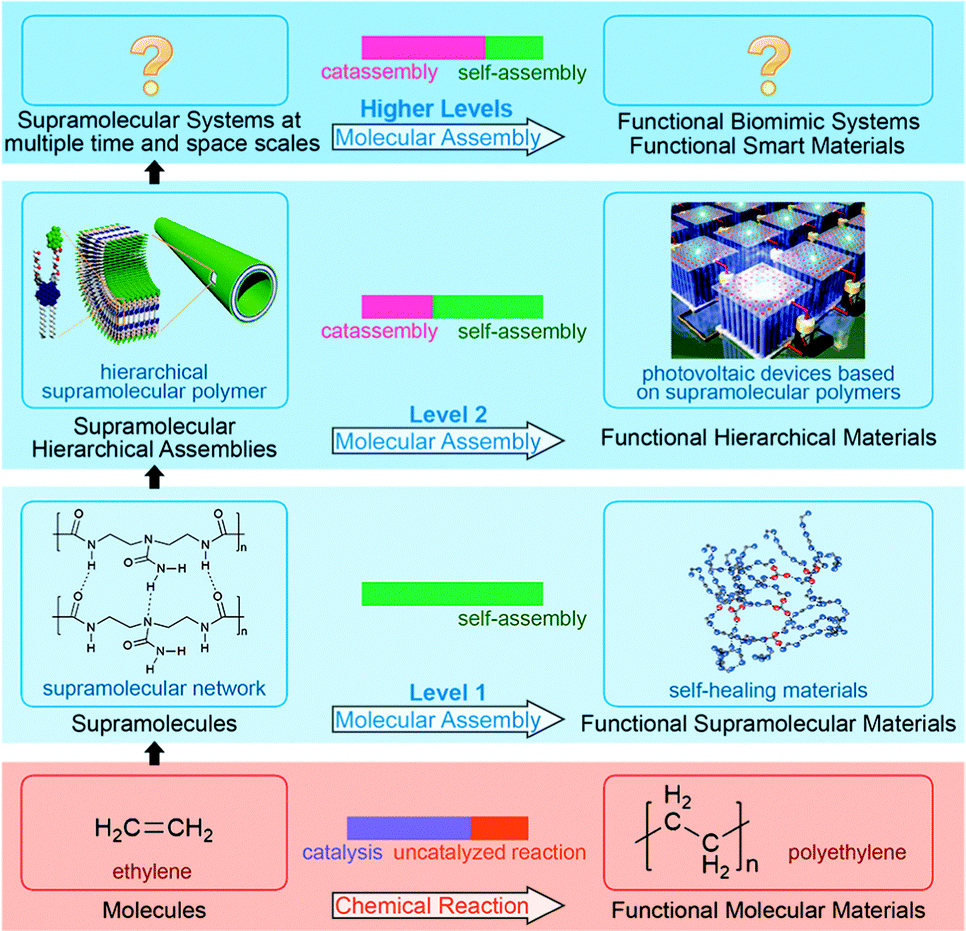 | ||
| Fig. 6 Fabrication of functional materials through chemical reaction and molecular assembly with complexity at different levels. Hierarchical material images adapted from ref. 5 (Copyright (2012), with permission from the American Association for the Advancement of Science). Self-healing material image adapted from ref. 16 (Copyright (2008), with permission from Nature Publishing Group). | ||
3. What else can catassembly learn from catalysis?
Although the previously discussed cases show some features of catassembly, well-developed systems remain rare, and controllability of these systems is expected to be further improved. In contrast, catalytic developments are effectively promoted through methodological studies, including various experimental methods, theories and characterization techniques. Can we find additional inspiration from catalysis to promote the development of methodology for catassembly research?3.1 Catassembly could be homogeneous or heterogeneous
Catalysts can either be homogeneous or heterogeneous, depending on whether a catalyst exists in the same phase as the substrate.47–49 The previously presented catassembly examples are essentially homogeneous catassembly because both the building blocks and the catassemblers exist in the same solutions. In contrast, a heterogeneous catassembler that accelerates an assembly process should be in a different phase from the building blocks, as in the following example.Wu et al. reported that assembly at a liquid/solid interface could be efficiently accelerated by protons in the solution phase.89 A highly ordered self-assembled structure of a neutral bipyridyl compound (P3) was formed at the heptanoic acid/HOPG interface within 4–6 hours, whereas the same structure was formed within minutes in the presence of protons (Fig. 7A). Protons were not present in the final assembly. The proposed catassembled mechanism (Fig. 7B) suggests that a proton first converts a pyridine segment of the first P3 molecule into a cationic pyridinium, thus enhancing the ability of the first P3 molecule to form hydrogen bonds with the second P3 molecule. Upon the formation of these hydrogen bonds, the proton is more susceptible to the third P3 molecule because the pyridine segment in the hydrogen-bonded first P3 molecule becomes less basic. This proton activates the α-H and β-H sites of the third P3 molecule through the formation of pyridinium, which then again forms hydrogen bonds with the second P3 molecule. The protons in the solution phase accelerate the assembly process of the P3 molecules at the interface without staying in the final assembly; this proton-assisted assembly process can be regarded as an example of heterogeneous catassembly, in which the proton is the heterogeneous catassembler.
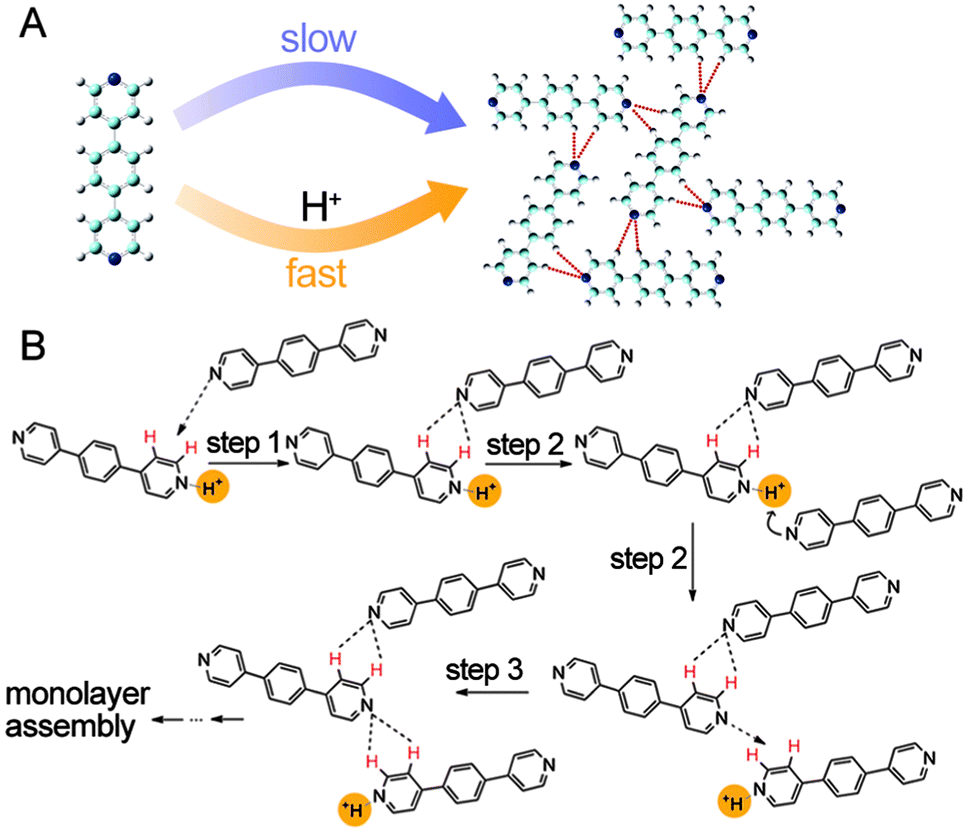 | ||
| Fig. 7 (A) Self-assembly and catassembly processes of P3. (B) Proposed mechanism for the catassembly process of P3. The red-coloured H indicates the activated H atom that serves as a hydrogen donor. The dashed arrows indicate the approach of the pyridyl N atom to the activated H atom. A catassembler proton is highlighted in yellow. Adapted from ref. 89 (Copyright (2012), with permission from The American Chemical Society). | ||
In this example, the catassembly occurs at the liquid/solid interface, and the catassembler is in the solution phase. This situation differs from that observed for many examples of heterogeneous catalysis at liquid/solid interfaces in which the reactions occur in the solution phase and the solid surfaces are the catalysts. The development of functional surfaces to be catassemblers for molecular assembly in the solution phase remains a challenge. To realize this type of heterogeneous catassembly, one possible approach is to transplant typical homogeneous catassemblers onto solid surfaces, such as electrodes or NPs.60 The decorated heterogeneous catassemblers would have features that differ distinctly from those of the original homogeneous catassemblers. For example, the catassemblers could easily be separated and could allow spatial control of catassembly.
3.2 Development of theories and characterization methods
Theoretical investigations and the development of characterization techniques have effectively promoted both basic research and industrial applications of catalysis. Although the concept of catalytic chemistry is well defined and the theory and characterization methods for the study of thermodynamics and kinetics are quite rich, these methods remain far from describing well all the phenomena and processes of catassembly in which synergetic intermolecular interactions play a central role. The developments in the theory and characterization methods for molecular assembly can be reasonably expected to pave the way for a more comprehensive understanding of both self-assembly and catassembly.A catalyst accelerates the rate of a reaction by selectively reducing the activation energies of certain pathways.49,90 For general catalytic reactions, the active centre is localized, and enthalpy is the main driving force; therefore, high temperatures and/or high pressures are necessary for many catalytic reactions. In contrast, an assembly process usually occurs under mild conditions and with a smaller enthalpy change. When building blocks are bound by multi-site noncovalent interactions, the reduction in the degrees of freedom of molecular translations and rotations and the increase in the degrees of freedom of molecular vibrations may result in an increase in entropy, which is the main driving force of most assembly processes.91,92 During a catassembly process, the catassembler primarily decreases the entropy barrier by regulating the assembly pathways.
The introduction of a catassembler could divide an assembly process into associated sub-processes and thus break a large energy barrier into small barriers. In these sub-processes, the catassembler may co-assemble with two or more building blocks to form a large assembly, where a facile intra-assembly pathway replaces the original inter-assembly pathway. Therefore, an understanding of these sub-processes and the roles of the catassembler in the sub-processes is significant for the design of efficient catassemblers.
A more comprehensive description and modeling based on the cooperative entropic and enthalpic contributions are highly desirable. From the perspective of kinetics, a universal theoretical model for catassembly, analogous to the transition-state theory for catalysis, would be essential. More importantly, catassembly is primarily based on multi-site noncovalent interactions with distinct synergistic characteristics; the research into catassembly should not only learn from catalysis but also establish its own domain by taking advantage of knowledge from the life and physical sciences.
It has been well known that in addition to theories and simulations, the establishment and development of in situ characterization techniques have promoted catalysis research significantly.49 However, these powerful methods of catalysis remain far from well probing all processes of catassembly in which synergetic intermolecular interactions play a central role. Accordingly, insightful study of catassembly would similarly require corresponding new characterization techniques. As catassembly is primarily based on multi-site noncovalent interactions with distinct synergistic characteristics, the characterization of the dynamic noncovalent interactions over multiple time and length scales remains a great challenge.
With the strength of high energetic resolution, in situ NMR,93–95 circular dichroism (CD),96–98 vibrational CD (VCD),99,100 Raman optical activity (ROA) spectroscopies,101 and their combined time-resolved methods will most likely play an important role in this direction. Regarding the techniques with high spatial resolution, in situ transmission electron microscope (TEM) that can obtain atomic imaging of specimens in liquid will be a highly promising technique.102 Besides, an alternative approach is the strategic hybridization of existing techniques, such as neutron scattering,103 inelastic X-ray scattering (IXS),104 small-angle X-ray scattering (SAXS),105,106 glancing angle X-ray diffraction (GAXRD),105,106 X-ray photon correlation spectroscopy (XPCS),107etc.103,108
4. Synergy between molecular assembly and chemical reaction
As previously discussed, the field of molecular assembly can learn from the well-developed field of chemical synthesis; the synergy between the fields will also be significant. Chemical reactions are essential for molecular assembly because they produce not only the various building blocks but also the catassemblers. In contrast, the features of molecular assembly (reversibility and dynamic characters) have been imported into chemical synthesis and have led to the emergence of dynamic covalent chemistry.45,109–111Furthermore, reactions and assembly could be combined to develop various multi-process methods for the fabrication of highly complex materials. For example, in “chemical reaction-induced assembly” systems, a chemical reaction and the subsequent assembly of the reaction products occur synchronously under the same conditions.112–117 Thus, the assembly could be triggered by the preceding reaction, whereas the kinetic assembly process could be controlled through regulation of the rate of the chemical reaction. Various types of chemical reactions, such as heat-induced reactions,112 photo-induced reactions,113 electro-induced reactions114 and enzyme-catalysed reactions,115–117 have been used in this associated procedure. In contrast, most of these associated processes involve only self-assembly. More efficient methods would be realized through the introduction of catassembly and field-assisted assembly into this “chemical reaction–molecular assembly” process. For example, we developed a “dehydration reaction–asymmetric catassembly” procedure to fabricate chiral J-aggregates of perylene dianhydride (PDA).118 The PDA building block was generated from the dehydration reaction of its precursor in an acidic solution of cetyltrimethylammonium bromide (CTAB). The subsequent co-assembly of PDA and CTAB was catassembled by chiral tartaric acid. By modulating the concentration of tartaric acid, we could controllably fabricate the chiral and achiral J-aggregates of PDA.
Likewise, a “molecular assembly–chemical reaction” procedure has been widely reported,119–121e.g., the “covalent capture” method.119,120 Although materials fabricated by molecular assembly have unique characteristics, such as good reversibility and self-repairability,16–18 many materials require greater stability than the typical assembled materials can provide. An effective approach is to introduce covalent bonds into an assembly process at appropriate sites and time. This associated process of “molecular assembly–chemical reaction” would inherit the advantages of both molecular assembly, e.g., well-defined structures on a large scale, and chemical reactions, such as robustness. Various types of chemical reactions, such as photoreactions122 and the click reaction,123 have been used to reinforce stability in self-assembled structures. Nonetheless, the combined “catassembly–reaction” process has seldom been used.
One analogous example was reported by Michl et al.124 (Fig. 8), who first formed a quadrate intermediate assembly by the co-assembly of pyridine-terminated connectors and molecular rods that contained Pt cations at each end (Fig. 8B). They then subjected the assembly to Cu(I)-catalysed replacement of all the pyridine-terminated connectors for analogous ethynyl-terminated connectors; this replacement converted the noncovalent coordinate bonds (N → Pt+) into covalent C–Pt bonds. In this single-step “covalent stabilization” process, the pyridine-terminated connectors served as an intermediate template to enhance the selectivity of the product, whereas the Cu(I) catalysed the covalent stabilization sub-process by increasing the efficiency of the oxidation reaction. The pyridine-terminated connector functioned as a catassembler because it assisted the process through noncovalent interactions and was absent from the final products. Therefore, this procedure not only combined the facility of reversible assembly and the robustness of covalent synthesis but also combined the high selectivity and efficiency of both catassembly and catalysis.
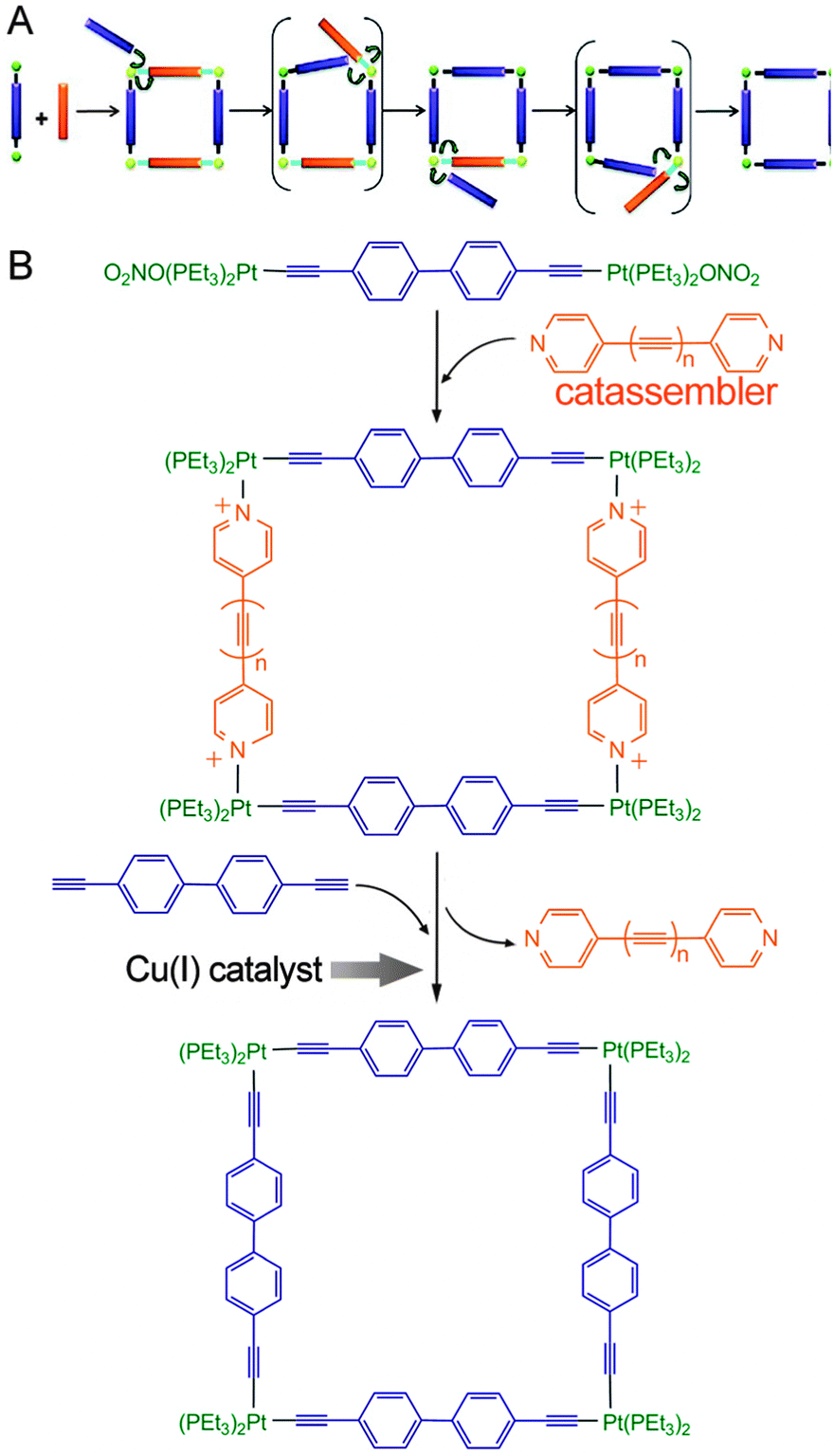 | ||
| Fig. 8 (A) Two-step synthesis of a covalently stabilized molecular square. (B) Molecular structures in this process. Blue and orange rod connectors represent molecular connectors terminated with ethynyls and pyridines, respectively. Green balls represent the coordinated Pt cations, which are covalently attached to the ethynyls and noncovalently attached to the pyridines. Adapted from ref. 124 (Copyright (2012), with permission from The American Chemical Society). | ||
Molecular assembly could also be associated with disassembly, which is analogous to chemical decomposition. The associated process of “assembly–disassembly” has been widely employed as a two-step noncovalent synthetic method.125–127 In this kind of process, transient template functions similarly to the catassembler but with a clear difference: removal of the template must be accomplished through an extra disassembly process in the second step; thus, the template only functions once every two steps, whereas a catassembler could work repeatedly in a single step. In addition to the two-step associated methods, a more complicated multi-process association (e.g., molecular assembly–chemical reaction–disassembly128) has also been reported.
In summary, chemical reactions are essential to the fabrication of molecular building blocks and catassemblers. Both chemical reactions and molecular assembly can be used to fabricate functional materials; however, molecular assembly, especially processes that involve catassembly, is more capable of fabricating high-complexity materials. When the targeted materials become more functional, the procedures to make them will be more sophisticated, and catassembly will play a more important role. These processes are analogous to the construction of cable-stayed bridges, which are much more complicated than the floating bridge shown in Fig. 1. The workers become essential and must form a team with comprehensive skills. The association of chemical reactions with molecular assembly (including self-assembly, field-assisted assembly, and catassembly) in a variety of ways is important to the development of multi-step processes (Fig. 9). If catassembly can be applied to these associated processes, more efficient methods might be established for the fabrication of functional materials with remarkably high complexities.
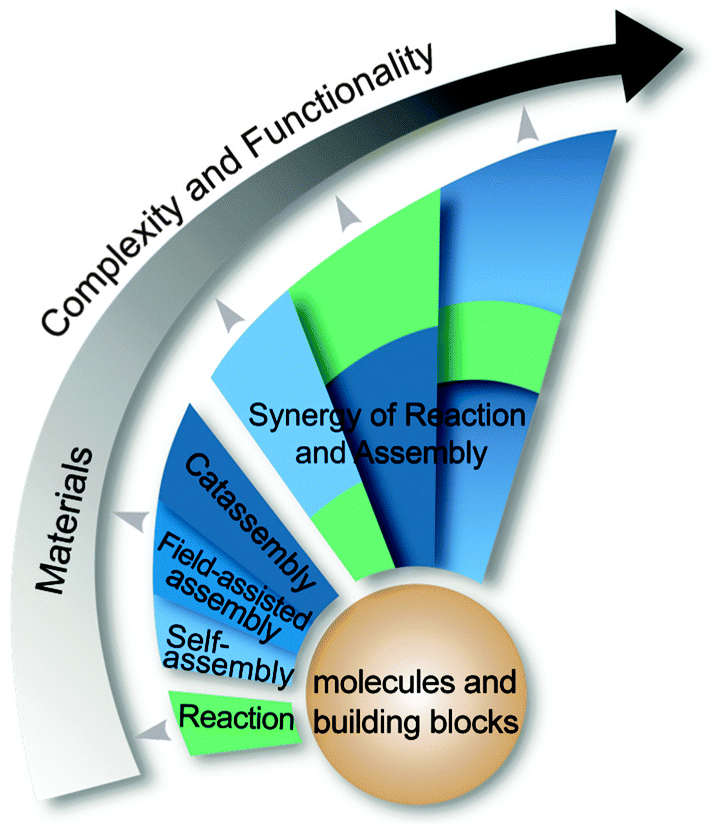 | ||
| Fig. 9 Synergy between chemical reactions and molecular assembly. | ||
5. Catassembly can learn from life science
The study of molecular assembly has two main purposes: to develop new functional materials and to understand how matter becomes complex. From a certain perspective, biological systems are functional systems with the highest degree of complexity, as reflected in the intricate associations of multiple reactions and assembly processes in the life sciences. Some of these assembly processes are closely related to catassembly. For example, the active 70S initiation complex (70SIC) in Fig. 10, which is a ribosome in prokaryotic cells, is formed through the co-assembly of mRNA, fmet-tRNA (fmet) and two ribosomal subunits (the 50S subunit and the 30S subunit).129–131 During this process, three initiation factors (IFs)—IF1, IF2, and IF3—synergistically assist the combination of the mRNA, tRNA and 30S subunits to form the 30S initiation complex (30SIC). The 30SIC then engages the 50S subunit, and the removal of the IFs completes the assembly of the 70SIC. The IFs are not present in the final structure and can continue to assist the assembly by starting a new cycle. The efficiency has been shown to be decreased by seven-fold, 600-fold or nine-fold if IF1, IF2 or IF3 is, respectively, absent from this process.130 Therefore, IFs play the role of catassemblers in the assembly of 70SIC.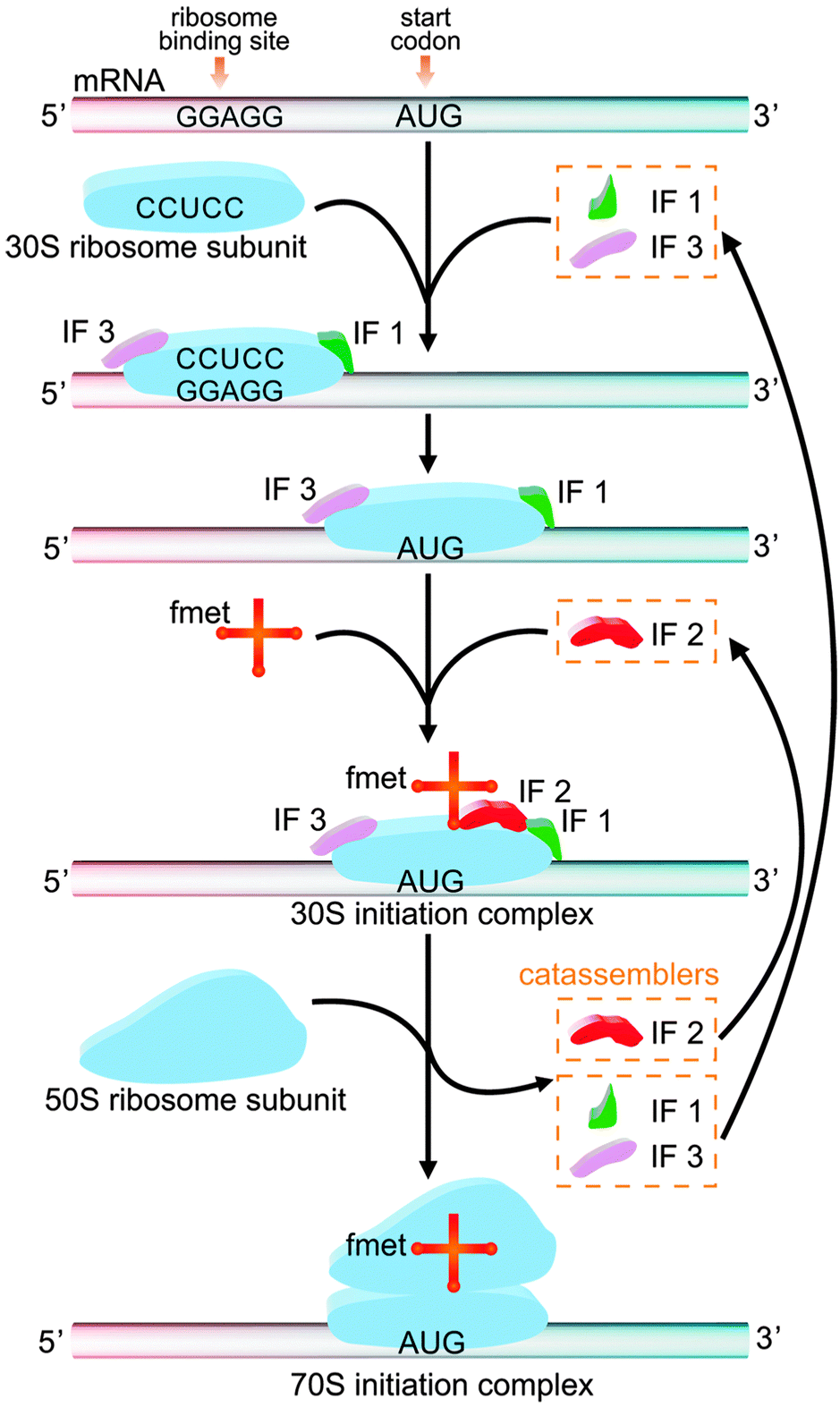 | ||
| Fig. 10 The formation of 70S initiation complex through the co-assembly of mRNA, fmet, the 30S ribosomal subunit and the 50S ribosomal subunit, with the assistance of IF 1, 2, and 3. | ||
Protein folding and assembly processes assisted by molecular chaperones can also be considered to be a catassembly process to some extent. The assistant effect of the chaperone has its own characteristics: (1) molecular chaperones are usually multi-component molecular machines that promote folding through ATP- and cofactor-regulated binding and release cycles; (2) molecular chaperones have relatively low selectivity for polypeptide chains; one type of molecular chaperone can assist the folding of various polypeptide chains of different amino acid sequences; and (3) the role of the molecular chaperones is to prevent incorrect polypeptide folding or oligomerization—not to convey steric information for assembly.84–86
Catassembler researchers in materials science and the life sciences could learn from each other in at least the following three aspects: (1) the particular catassemblers in the life sciences or their derivatives may be used as catassemblers for catassembled fabrication of functional materials, and (2) the mechanisms of catassemblers in the life sciences could be extended into the design of catassemblers for materials science. For example, chaperonin, a typical molecular chaperone, assists protein folding through ATP-driven cyclic changes of its inner cage between hydrophilicity and hydrophobicity.84 Inspired by this mechanism, researchers have designed artificial chaperones based on fuel-driven cyclic changes of a NP surface between hydrophilicity and hydrophobicity.132 (3) Biological catassemblers could be modified chemically to regulate their assistant effects in the life sciences. For example, in E. coli, chaperonin can promote the folding of at least 250 types of proteins;84 thus, chaperonin exhibits low selectivity. If the selectivity of chaperonin could be improved through chemical modification (e.g., the binding of a special peptide segment to chaperonin), specific folding and proteome maintenance functions might be achieved.
Finally, systematic research and discovery of new catassembled processes could enhance the perception of the life sciences. Because cellular environments are highly crowded with macromolecules, molecular assembly in biology is much more complex than that in chemistry.133 Therefore, a deep understanding of catassembly in chemistry would facilitate the discovery of more complex catassembled processes in the life sciences. In this way, catassembled phenomena might be found to be even more fundamental and important in life sciences than we have realized to date.
6. Conclusions and outlook
In this review, we have proposed a different strategy, based on catalytic chemistry, to examine and describe molecular-assembly processes for the creation of functional chemical systems with high efficiency and selectivity. Because chemical synthesis is generally much more developed than molecular assembly, the latter field can learn a substantial amount from the former field. Therefore, the concept of catalysis, which is widely used in chemical synthesis, could be extended to assembly. Accordingly, we propose the term catassembly, which refers to increased rates and better control of assembly processes.To address the key question of “can molecular assembly learn from catalytic chemistry?” we used some primitive examples of catassembly to present their distinct characteristics and their difference from template-assisted self-assembly. These highly efficient assembly processes involve small molecules, macromolecules and molecule-modified NPs and demonstrate the characteristics and superiority of catassembly. In some of these examples, the strategy of catassembly has apparently been applied unknowingly. More importantly, because most efforts have focused on how to rationally design and synthesize molecular building blocks, and the creation of functional chemical systems relying on the self-assembly of these components, we wish to emphasize the seemingly missing yet critical consideration in this field: the design and utilization of molecular catassemblers for the construction of new functional chemical systems with significantly high efficiency and selectivity. This approach was the main motivation for this review.
To this end, we predict that the broad knowledge of catalysis will inspire the development of catassembly methodologies, e.g., heterogeneous catassembly. These new approaches to the control and acceleration of processes will expand the spectrum of molecular assembly from basic science to applications.
In the construction of functional chemical systems, molecular assembly is hierarchical and occurs at different levels with multiple length scales. As the targeted systems become more functional, the construction procedures become more complicated. In this regard, a single self-assembly process is insufficient, at least not to a large extent. Instead, catassembly could be a more efficient approach. In addition, to ensure the stability of the final product, the interplay and even synergy between chemical reactions and molecular assembly could endow the target systems with both high stability and high functionality.
Although the concept of catalytic chemistry is well defined and the theory and characterization methods for the study of thermodynamics and kinetics are quite rich, these methods remain far from describing well all the phenomena and processes of catassembly in which synergetic intermolecular interactions play a central role. A great challenge is the development of new techniques capable of in situ characterization of the synergetic and dynamic noncovalent interactions at multiple time and length scales to unravel molecular assembly and especially catassembly processes. The phenomenal free energy approach of thermodynamics does not appear to be very useful in controllable molecular assembly; a more comprehensive description and modeling based on the cooperative enthalpic and entropic contributions are highly desirable. From the perspective of kinetics, a theoretical method for catassembly, analogous to the transition-state theory for catalysis, would be useful. More importantly, catassembly is primarily based on multi-site noncovalent interactions with distinct synergistic characteristics; the field of catassembly should not only learn from catalysis but also establish its own domain by taking advantage of knowledge from the life and physical sciences.
Overall, we are optimistic that, with the development of catassembly, numerous opportunities will emerge in the field of molecular assembly and thus enhance our understanding of the assembly mechanisms. This approach will promote the systems chemistry approach,134–136 and may pave the way toward new discoveries of highly complex functional chemical systems and result in new contributions to the chemical, materials and life sciences.
Acknowledgements
We thank Xi Zhang, E.W. (Bert) Meijer, Jia-Rui Wu, Li-Jun Wan, Jean-Marie Lehn, Richard Zare, Feng-Ru Fan, Ming-Hua Liu, Xian-En Zhang, He Tian, Zhi-Xiang Wei, Zhi-Yong Tang, Hai-Bo Yang, Dong-Sheng Liu, Zeng-Yi Chang, Thomas M. Hermans, Kyle Biship and Severin T. Schneebeli for helpful discussions. This work is supported by the NSF of China (91227111, 21021002, 20923004, 91127019, 91127046).References
- P. Alivisatos, P. F. Barbara, A. W. Castleman, J. Chang, D. A. Dixon, M. L. Klein, G. L. McLendon, J. S. Miller, M. A. Ratner, P. J. Rossky, S. I. Stupp and M. E. Thompson, Adv. Mater., 1998, 10, 1297–1336 CrossRef.
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1155–1196 CrossRef CAS.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 1–96 CrossRef.
- Y. S. Lee, Self-Assembly and Nanotechnology: A Force Balance Approach, Wiley, Hoboken, 2008 Search PubMed.
- J. A. Pelesko, Self-Assembly: The Science of Things that Put Themselves Together, Chapman & Hall/CRC, New York, 2007 Search PubMed.
- B. A. Grzybowski, C. E. Wilmer, J. Kim, K. P. Browne and K. J. M. Bishop, Soft Matter, 2009, 5, 1110–1128 RSC.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef PubMed.
- J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed.
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- T. Wang, J. Q. Zhuang, J. Lynch, O. Chen, Z. L. Wang, X. R. Wang, D. LaMontagne, H. M. Wu, Z. W. Wang and Y. C. Cao, Science, 2012, 338, 358–363 CrossRef CAS PubMed.
- H. Zhang, Y. Liu, D. Yao and B. Yang, Chem. Soc. Rev., 2012, 41, 6066–6088 RSC.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- S. Otto, Acc. Chem. Res., 2012, 45, 2200–2210 CrossRef CAS PubMed.
- M. Burnworth, L. M. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- P. B. Dervan, Bioorg. Med. Chem., 2001, 9, 2215–2235 CrossRef CAS.
- H. Cui, T. Muraoka, A. G. Cheetham and S. I. Stupp, Nano Lett., 2009, 9, 945–951 CrossRef CAS PubMed.
- C. P. Collier, E. W. Wong, M. Belohradsky, F. M. Raymo, J. F. Stoddart, P. J. Kuekes, R. S. Williams and J. R. Heath, Science, 1999, 285, 391–394 CrossRef CAS.
- S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77–92 RSC.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- X. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214–218 CrossRef CAS PubMed.
- Y. P. Wang, H. P. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS.
- H. Y. Fan, K. Yang, D. M. Boye, T. Sigmon, K. J. Malloy, H. Xu, G. P. López and C. J. Brinker, Science, 2004, 304, 567–571 CrossRef CAS PubMed.
- Y. Yan, R. Wang, X. H. Qiu and Z. X. Wei, J. Am. Chem. Soc., 2010, 132, 12006–12012 CrossRef CAS PubMed.
- L. G. Xu, W. Ma, L. B. Wang, C. L. Xu, H. Kuang and N. A. Kotov, Chem. Soc. Rev., 2013, 42, 3114–3126 RSC.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- T. Tjivikua, P. Ballester and J. Rebek, J. Am. Chem. Soc., 1990, 112, 1249–1250 CrossRef CAS.
- R. Madueno, M. T. Raisanen, C. Silien and M. Buck, Nature, 2008, 454, 618–621 CrossRef CAS PubMed.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- S. S. Li, B. H. Northrop, Q. H. Yuan, L. J. Wan and P. J. Stang, Acc. Chem. Res., 2009, 42, 249–259 CrossRef CAS PubMed.
- B. H. Northrop, Y. R. Zheng, K. W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- G. Seeber, B. E. F. Tiedemann and K. N. Raymond, Top. Curr. Chem., 2006, 265, 147–183 CrossRef CAS.
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS PubMed.
- D. A. Walker, B. Kowalczyk, M. O. de la Cruz and B. A. Grzybowski, Nanoscale, 2011, 3, 1316–1344 RSC.
- J.-M. Lehn, C. R. Chimie, 2011, 14, 348–361 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, New York, 2008 Search PubMed.
- G. Ertl, H. Knözinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 1997 Search PubMed.
- G. A. Somorjai and Y. M. Li, Introduction to Surface Chemistry and Catalysis, Wiley, New York, 2010 Search PubMed.
- M. Beller, A. Renken and R. A. van Santen, Catalysis: from Principle to Applications, Wiley, Weinheim, 2012 Search PubMed.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2 edn, 2007 Search PubMed.
- Y. Wu, Catalytic Chemistry (in Chinese), Science, Beijing, 1998 Search PubMed.
- P. W. N. M. van Leeuwen, Supramolecular Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- K. M. Sanders, Chem.–Eur. J, 1998, 4, 1378–1383 CrossRef.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- H. G. Cui, E. T. Pashuck, Y. S. Velichko, S. J. Weigand, A. G. Cheetham, C. J. Newcomb and S. I. Stupp, Science, 2010, 327, 555–559 CrossRef CAS PubMed.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, K. R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart, M. Venturi and S. Wenger, Chem.–Eur. J, 2000, 6, 3558–3574 CrossRef CAS.
- T. Kato and K. Tanabe, Chem. Lett., 2009, 38, 634–639 CrossRef CAS.
- A. O'Riordan, P. Delaney and G. Redmond, Nano Lett., 2004, 4, 761–765 CrossRef CAS.
- Y. Wang, H. X. Lin, S. Y. Ding, D. Y. Liu, L. Chen, Z. C. Lei, F. R. Fan and Z. Q. Tian, Sci. Sin.: Chim., 2012, 42, 525–547 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- J. Prins, F. De Jong, P. Timmerman and D. N. Reinhoudt, Nature, 2000, 408, 181–184 CrossRef PubMed.
- R. Lauceri, A. Raudino, L. M. Scolaro, N. Micali and R. Purrello, J. Am. Chem. Soc., 2002, 124, 894–895 CrossRef CAS PubMed.
- J. S. Zhao, J. H. Wang, W. B. He, Y. B. Ruan and Y. B. Jiang, Chem.–Eur. J, 2012, 18, 3631–3636 CrossRef CAS PubMed.
- X. Zeng, Y. J. He, Z. F. Dai, J. Wang, Q. Cao and Y. L. Zhang, ChemPhysChem, 2009, 10, 954–962 CrossRef PubMed.
- L. Zhang, Y. Tian and M. H. Liu, Phys. Chem. Chem. Phys., 2011, 13, 17205–17209 RSC.
- B. E. Tropp, Molecular Biology: Genes to Proteins, Jones & Bartlett, Sudbury, 2011 Search PubMed.
- V. A. Bloomfield, D. M. Crothers and I. Tinoco, Nucleic Acids: Structures, Properties and Functions, Univ Science, Sausalito, 2000 Search PubMed.
- C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- J. Turberfield, J. C. Mitchell, B. Yurke, A. P. Mills, M. I. Blakey and F. C. Simmel, Phys. Rev. Lett., 2003, 90, 118102 CrossRef.
- S. J. Green, D. Lubrich and A. J. Turberfield, Biophys. J., 2006, 91, 2966–2975 CrossRef CAS PubMed.
- G. Seelig, B. Yurke and E. Winfree, J. Am. Chem. Soc., 2006, 128, 12211–12220 CrossRef CAS PubMed.
- Y. Z. Xing, Z. Q. Yang and D. S. Liu, Angew. Chem., Int. Ed., 2011, 50, 11934–11936 CrossRef CAS PubMed.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- T. J. Song and H. J. Liang, J. Am. Chem. Soc., 2012, 134, 10803–10806 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- M. P. Nikitin, T. A. Zdobnova, S. V. Lukash, O. A. Stremovskiy and S. M. Deyev, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5827–5832 CrossRef CAS PubMed.
- S. J. Hurst, E. K. Payne, L. D. Qin and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 2672–2692 CrossRef CAS PubMed.
- P. Broekmann, K.-H. Dötz and C. A. Schalley, Templates in Chemistry III, Springer-Verlag, Heidelberg, 2009 Search PubMed.
- Y. Lei, S. K. Yang, M. H. Wu and G. Wilde, Chem. Soc. Rev., 2011, 40, 1247–1258 RSC.
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH, Weinheim, 1995, p. 141 Search PubMed.
- F. U. Hartl, A. Bracher and M. Hayer-Hartl, Nature, 2011, 475, 324–332 CrossRef CAS PubMed.
- R. J. Ellis, Trends Biochem. Sci., 2006, 31, 395–401 CrossRef CAS PubMed.
- B. Bukau, J. Weissman and A. Horwich, Cell, 2006, 125, 443–451 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- R. D. Vale and R. A. Milligan, Science, 2000, 288, 88–95 CrossRef CAS.
- H. Li, X. G. Xu, J. Shang, J. L. Li, X. Q. Hu, B. K. Teo and K. Wu, J. Phys. Chem. C, 2012, 116, 21753–21761 CAS.
- E. Pollak and P. Talkner, Chaos, 2005, 15, 026116 CrossRef PubMed.
- C.-A. Palma, M. Cecchini and P. Samori, Chem. Soc. Rev., 2012, 41, 3713–3730 RSC.
- H. X. Zhou and M. K. Gilson, Chem. Rev., 2009, 109, 4092–4107 CrossRef CAS PubMed.
- M. Pons and O. Millet, Prog. Nucl. Magn. Reson. Spectrosc., 2001, 38, 267–324 CrossRef CAS.
- R. Sprangers and L. E. Kay, Nature, 2007, 445, 618–622 CrossRef CAS PubMed.
- A. Pastor and E. Martinez-Viviente, Coord. Chem. Rev., 2008, 252, 2314–2345 CrossRef CAS PubMed.
- G. Gottarelli, S. Lena, S. Masiero, S. Pieraccini and G. P. Spada, Chirality, 2008, 20, 471–485 CrossRef CAS PubMed.
- W. Cai, G. T. Wang, P. Du, R. X. Wang, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2008, 130, 13450–13459 CrossRef CAS PubMed.
- P. Iavicoli, H. Xu, L. N. Feldborg, M. Linares, M. Paradinas, S. Stafstrom, C. Ocal, B. L. Nieto-Ortega, J. Casado, J. T. L. Navarrete, R. Lazzaroni, S. De Feyter and D. B. Amabilino, J. Am. Chem. Soc., 2010, 132, 9350–9362 CrossRef CAS PubMed.
- L. Nafie, Vibrational Optical Activity: Principles and Applications, John Wiley & Sons, New York, 2011 Search PubMed.
- V. Setnicka, M. Urbanova, K. Volka, S. Nampally and J. M. Lehn, Chem.–Eur. J, 2006, 12, 8735–8743 CrossRef CAS PubMed.
- F. J. Zhu, N. W. Isaacs, L. Hecht and L. D. Barron, Structure, 2005, 13, 1409–1419 CrossRef CAS PubMed.
- H. G. Liao, L. k. Cui, S. Whitelam and H. M. Zheng, Science, 2012, 336, 1011–1014 CrossRef CAS PubMed.
- V. G. Sakai, C. Alba-Simionesco and S. H. Chen, Dynamics of Soft Matter: Neutron Applications, Springer, Heidelberg, 2011 Search PubMed.
- F. Sette, M. H. Krisch, C. Masciovecchio, G. Ruocco and G. Monaco, Science, 1998, 280, 1550–1555 CrossRef CAS.
- G. Renaud, R. Lazzari and F. Leroy, Surf. Sci. Rep., 2009, 64, 255–380 CrossRef CAS PubMed.
- N. Pienack and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 2014–2034 CrossRef CAS PubMed.
- K. A. Nugent, Adv. Phys., 2010, 59, 1–99 CrossRef.
- R. Borsali and R. Pecora, Soft-Matter Characterization, Springer, Berlin, 2008 Search PubMed.
- J.-M. Lehn, in Constitutional Dynamic Chemistry, ed. M. Barboiu, 2012, vol. 322, pp. 1–32 Search PubMed.
- T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef PubMed.
- F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed.
- C. Wu, A. Z. Niu, L. M. Leung and T. S. Lam, J. Am. Chem. Soc., 1999, 121, 1954–1955 CrossRef CAS.
- Y. P. Wang, N. Ma, Z. Q. Wang and X. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2823–2826 CrossRef CAS PubMed.
- M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, M. Montalti, N. Spencer, J. F. Stoddart and M. Venturi, Chem.–Eur. J, 1997, 3, 1992–1996 CrossRef CAS.
- J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef PubMed.
- Z. M. Yang, G. L. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS PubMed.
- M. E. Hahn and N. C. Gianneschi, Chem. Commun., 2011, 47, 11814–11821 RSC.
- J. S. Zhao, Y. B. Ruan, R. Zhou and Y. B. Jiang, Chem. Sci., 2011, 2, 937–944 RSC.
- D. Clark and M. R. Ghadiri, J. Am. Chem. Soc., 1995, 117, 12364–12365 CrossRef.
- L. J. Prins and P. Scrimin, Angew. Chem., Int. Ed., 2009, 48, 2288–2306 CrossRef CAS PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- J. Q. Sun, T. Wu, F. Liu, Z. Q. Wang, X. Zhang and J. C. Shen, Langmuir, 2000, 16, 4620–4624 CrossRef CAS.
- R. Vestberg, M. Malkoch, M. Kade, P. Wu, V. V. Fokin, K. B. Sharpless, E. Drockenmuller and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2835–2846 CrossRef CAS.
- G. L. Olive, K. Parkan, C. Givelet and J. Michl, J. Am. Chem. Soc., 2011, 133, 20108–20111 CrossRef PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–497 CrossRef CAS PubMed.
- R. W. Sinkeldam, F. J. M. Hoeben, M. J. Pouderoijen, I. De Cat, J. Zhang, S. Furukawa, S. De Feyter, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2006, 128, 16113–16121 CrossRef CAS PubMed.
- H. N. Miras, G. J. T. Cooper, D. L. Long, H. Bogge, A. Muller, C. Streb and L. Cronin, Science, 2010, 327, 72–74 CrossRef CAS PubMed.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef CAS PubMed.
- L. Gold, Annu. Rev. Biochem., 1988, 57, 199–233 CrossRef CAS PubMed.
- A. Antoun, M. Y. Pavlov, M. Lovmar and M. Ehrenberg, EMBO J., 2006, 25, 2539–2550 CrossRef CAS PubMed.
- S. Laursen, H. P. Sorensen, K. K. Mortensen and H. U. Sperling-Petersen, Microbiol. Mol. Biol. Rev., 2005, 69, 101–123 CrossRef PubMed.
- R. Villalonga, R. Cao and A. Fragoso, Chem. Rev., 2007, 107, 3088–3116 CrossRef CAS PubMed.
- B. van den Berg, R. Wain, C. M. Dobson and R. J. Ellis, EMBO J., 2000, 19, 3870–3875 CrossRef CAS PubMed.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS PubMed.
- N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cs60212e |
| This journal is © The Royal Society of Chemistry 2014 |