Imaging lipid order changes in endosome membranes of live cells by using a Nile Red-based membrane probe†
Zeinab Darwich,
Andrey S. Klymchenko*,
Denis Dujardin and
Yves Mély*
Laboratoire de Biophotonique et Pharmacologie, UMR 7213 CNRS, Université de Strasbourg, Faculté de Pharmacie, 74, Route du Rhin, 67401 ILLKIRCH Cedex, France. E-mail: andrey.klymchenko@unistra.fr; yves.mely@unistra.fr; Fax: +33 368 85 43 13; Tel: +33 368 85 42 55 Tel: +33 368 85 42 63
First published on 15th January 2014
Abstract
During maturation of endosomes, in addition to the decrease in their internal pH and the modifications of their proteins, changes are thought to occur at the level of their lipid membranes. In the present work, we used the recently developed environment-sensitive membrane probe NR12S to monitor in living cells the lipid order changes that accompany the maturation of endosomes. Internalization studies in HeLa cells using two-photon fluorescence microscopy in the presence of endocytosis markers and inhibitors show that the probe is endocytosed through different energy-dependent pathways. While marginal changes in the probe colour occur during the initial steps of endocytosis, dramatic changes in colour are observed in the membranes of late endosomes and lysosomes. This remarkable colour change, which takes place 2 h after initial binding of NR12S to cell plasma membranes, suggests a loss of lipid ordered phase during endosome maturation. This change of lipid phase likely results from the decrease in the cholesterol content and the hydrolysis of sphingomyelin occurring in the membranes of late endosomes and lysosomes. In comparison to the common endocytosis marker FM4-64, NR12S is many-fold brighter, it can monitor in situ changes in the lipid organization of endosome membranes and its fluorescence at the plasma membrane can be selectively switched off by sodium dithionite. The present work proposes thus a new powerful tool for endocytosis research that enables monitoring changes in the lipid composition of endosome membranes.
1. Introduction
Cell plasma membranes allow the transport of material from the outside to the inside of the cells through endocytosis.1,2 This mechanism is essential for cell life and is also central in internalization of drugs and nanoscale delivery vehicles.3,4 Many events occur during the endocytosis process,5,6 starting by the formation of vesicles called early endosomes, which can be recycled to the cell plasma membrane,7–9 or maturate into late endosomes,10 and then transform into lysosomes, characterized by low pH and reductive environment.4,11 It is well-established that endosomes in their different stages differ by their content in Rab proteins,2,12 which participate in the maturation process.13 Lysosomes exhibit a perinuclear localization and an acidic pH, resulting from their high content in hydrolases.14 In contrast, much less is known about the lipid composition and its changes during the maturation of endosomes. Previous reports showed that early endosomes have almost the same composition as the plasma membrane, while after maturation, late endosomes exhibit lower cholesterol content15 due to its extraction by NPC1 and NPC2 proteins.7,16 Remarkably, mutant forms of these proteins cause Niemann–Pick disease, a lysosomal storage disorder in which cholesterol accumulates in late endosomes and lysosomes.17–19 Cholesterol extraction from late endosomes is also associated with sphingomyelin (SM) hydrolysis into ceramide.20 Cholesterol and SM are known to interact together in the plasma membrane to form packed domains of liquid ordered (Lo) phase, also called rafts,21,22 that are thought to play important biological functions, including cholesterol distribution and transport.23 Therefore, decrease in the cholesterol and SM contents may drastically affect the lipid order of endosomal membranes. Moreover, it was suggested that further conversion of endosomes into lysosomes results in an almost complete loss of cholesterol.24However, it remains a challenge to monitor in situ the membrane changes during endosome maturation, because the well-established methods in this field are based on suborganellar fractionation15 or require cellular fixation for subsequent electron microscopy or immunofluorescence analysis. To overcome these limitations, a potential approach is to monitor endosomal membranes using membrane probes, as they could directly label plasma membranes and then be internalized through endocytosis. Though typical membrane probes, such as FM4-64, can be used as endosomal markers,25,26 they do not allow monitoring the lipid composition of the endosomes. This goal could be reached by applying a recently developed membrane probe based on Nile Red, NR12S (Scheme 1), which binds specifically cell plasma membranes and is highly sensitive to lipid composition.27,28 Its fluorescence emission is sensitive to the lipid order, with a shift toward shorter wavelengths when SM/cholesterol-rich Lo phase is compared to a liquid disordered (Ld) phase composed of unsaturated lipids. Moreover, due to its zwitterionic group and long alkyl chain, this probe stains selectively the outer leaflet of model and cell plasma membranes with very slow flip-flop kinetics even at 37 °C.27 In the present work, internalization of NR12S probe into live cells was monitored by two-photon and confocal fluorescence microscopy. As the probe should remain at the leaflet it was initially bound, the probe is expected to be entrapped at the inner leaflet of the endosomes. This internal location of the probe should ensure its long residence time in the vesicles. Our data show that NR12S can be used not only as an endocytosis marker but also to monitor long-term changes in the lipid order of endosomal membranes. Indeed, late endosomes and lysosomes were found to be characterized by much lower lipid order, compared to plasma membranes, validating the previously proposed decrease in cholesterol content and SM hydrolysis in these compartments.
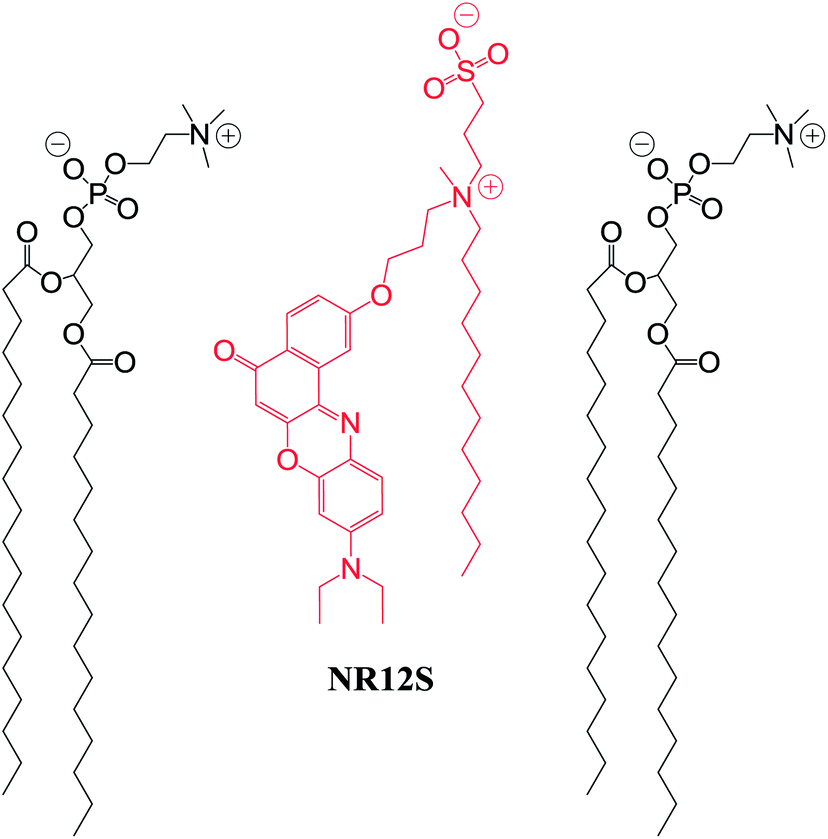 | ||
| Scheme 1 Probe NR12S (in red) and its hypothetic localization with respect to lipids (in black) in membranes. | ||
2. Materials and methods
All chemicals and solvents for synthesis were from Sigma-Aldrich. NR12S was synthesized as described elsewhere.272.1. Cell lines, culture conditions and treatment
HeLa cells were cultured in Dulbecco's modified Eagle medium (D-MEM, high glucose, Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS, Lonza), 1% antibiotic solution (penicillin–streptomycin, Invitrogen) in a humidified incubator with 5% CO2/95% air atmosphere at 37 °C.Alexa Fluor 488 conjugate of transferrin from human serum, Lysotracker Green DND-26, ER-Tracker Green (BODIPY FL glibenclamide) and FM4-64 probe were from Invitrogen. Chlorpromazine and methyl-β-cyclodextrin were from Sigma-Aldrich. To follow NR12S uptake, HeLa cells were stained with 0.2 μM NR12S in Opti-MEM for 5, 15, 30 min, 2 h and 4 h at 37 °C, then were visualized under two-photon microscope with or without sodium dithionite. Cells were also incubated with the same concentration of NR12S in Opti-MEM at 4 °C for 2 h and 4 h. Inhibition of NR12S internalization was performed by pre-incubation of cells for 30 min with chlorpromazine (10 μg mL−1), then NR12S was added in the presence of this inhibitor for 30 min at 37 °C. Alternatively, cells were pre-treated with methyl-β-cyclodextrin (5 mM) for 2 h and then stained with NR12S for 30 min at 37 °C.
For colocalization with transferrin, cells were stained with NR12S for 5 min and then the Alexa Fluor 488 conjugate of transferrin (20 μg mL−1) was added for 5, 30 and 60 min at 37 °C, before the measurements. For colocalization with Lysotracker or ER-tracker, cells were stained with each marker (50 nM for Lysotracker and 0.75 μM for ER-tracker) for 30 min at 37 °C and then with NR12S for 5 min. For 30 min incubation time with NR12S, cells were stained with NR12S together with each marker for 30 min. For longer incubation times, cells were stained with NR12S, then 30 min before observation with the microscope, Lysotracker or ER-tracker was added. To compare the uptake of NR12S to FM4-64, cells were incubated with 1 μM of either NR12S or FM4-64 for 5 min, 30 min and 60 min at 37 °C. After staining, cells were washed gently twice with HBSS (Hank's Buffered Salt Solution) before visualization on the microscope.
2.2. Fluorescence microscopy
3. Results
HeLa cells were stained with the Nile Red derivative dye, NR12S for 5 min, 15 min, 30 min, 2 h and 4 h at 37 °C. A probe concentration of 50 to 200 nM was chosen based on our previous works.27,28 At short incubation times, the fluorescence of the probe was mainly located at the plasma membrane (Fig. 1A–C). After longer incubation times (≥30 min), intracellular fluorescence of NR12S became clearly visible (Fig. 1D and E). After 2 h of incubation, this internalization was accompanied by a change in the colour (Fig. 1D), which represents the ratio of the green to the red parts of NR12S emission spectrum split at 585 nm.27,28 Based on previous work with model vesicles and live cells,27 this colour change can be interpreted as a decrease in the fraction of the liquid ordered (Lo) phase in the labelled intracellular compartments. An important property of NR12S probe is its ability to be bleached by sodium dithionite (Na2S2O4). As this salt is not efficiently internalized in cells, it could be used to bleach exclusively the fluorescence of NR12S in the outer leaflet of the plasma membrane, while preserving the fluorescence in the intracellular compartments. Indeed, it can be seen that after addition of 20 mM sodium dithionite, the fluorescence of NR12S totally disappears at the plasma membrane, so that only fluorescent dots inside the cells could be observed, probably corresponding to endosomes stained with NR12S (Fig. 1F–J). The use of dithionite improved thus the visualization of the probe internalization and allowed us confirming that the change in the emission colour of the probe is related to the intracellular compartments.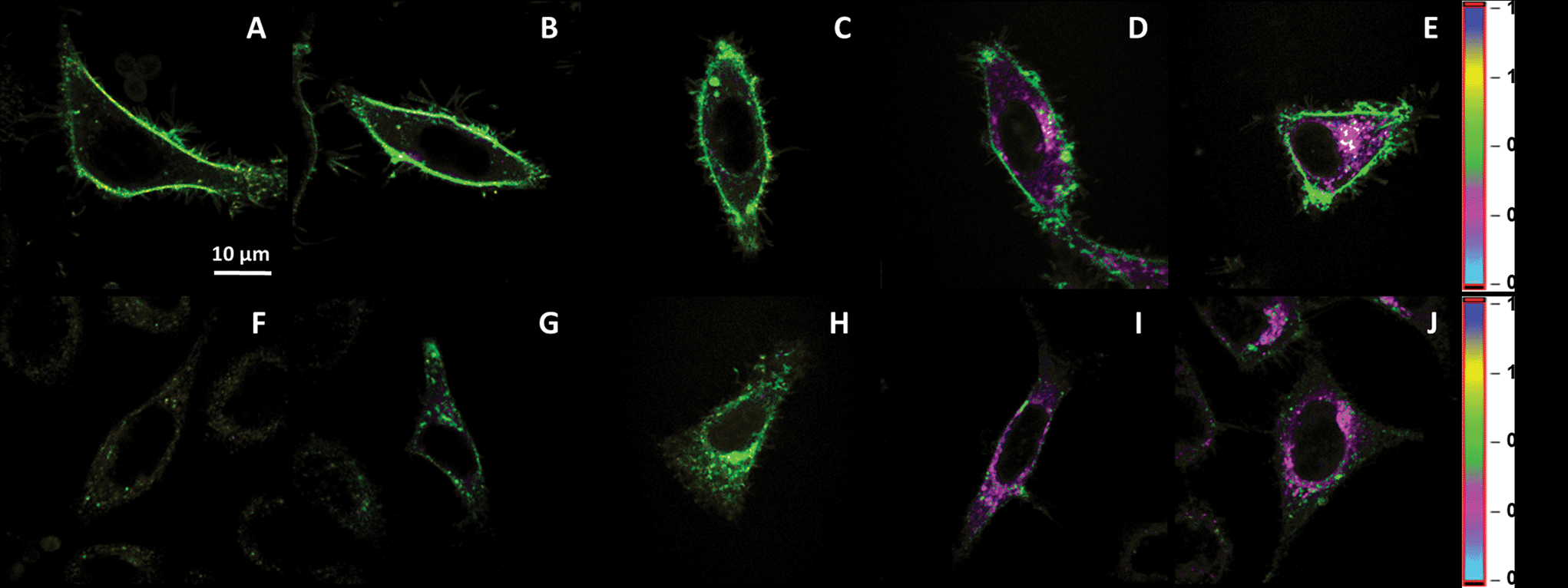 | ||
| Fig. 1 Fluorescence imaging of NR12S internalization in HeLa cells using two-photon microscopy. Internalization of NR12S after 5 min (A and F), 15 min (B and G), 30 min (C and H), 2 h (D and I) and 4 h (E and J) at 37 °C without (A–E) and with 20 mM of sodium dithionite (F–J). The pseudo-colour scale corresponds to the ratio of Green/Red regions. The green colour coding for a high ratio (0.7) and the pink coding for a low ratio (0.4) correspond to Lo and Ld phases, respectively.27 NR12S concentration was 0.2 μM. Two-photon excitation (830 nm) was used. | ||
To determine whether NR12S enters into cells through endocytosis, cells were incubated with NR12S at 4 °C, in order to inhibit the active endocytosis-based transport of molecules.30,31 Our fluorescence images using two-photon microscopy show that NR12S remains at the plasma membrane even after 2 and 4 hours of incubation at 4 °C (Fig. 2). This observation suggests that NR12S cannot diffuse directly through the cell plasma membrane, so that at 37 °C it is mainly internalized via an energy-dependent pathway.
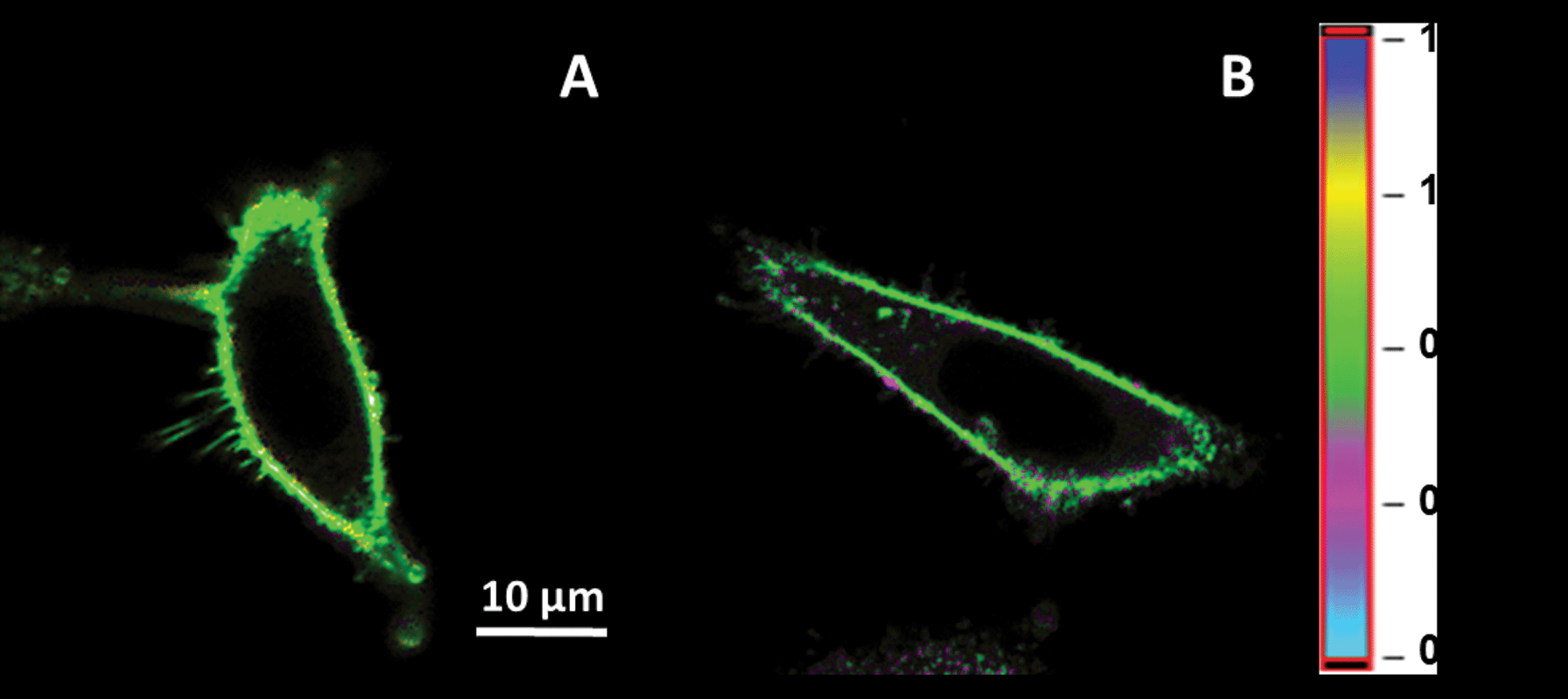 | ||
| Fig. 2 Fluorescence ratiometric images of HeLa cells incubated with NR12S at 4 °C for 2 hours (A) and 4 hours (B). NR12S concentration was 0.2 μM. Two-photon excitation at 830 nm was used. | ||
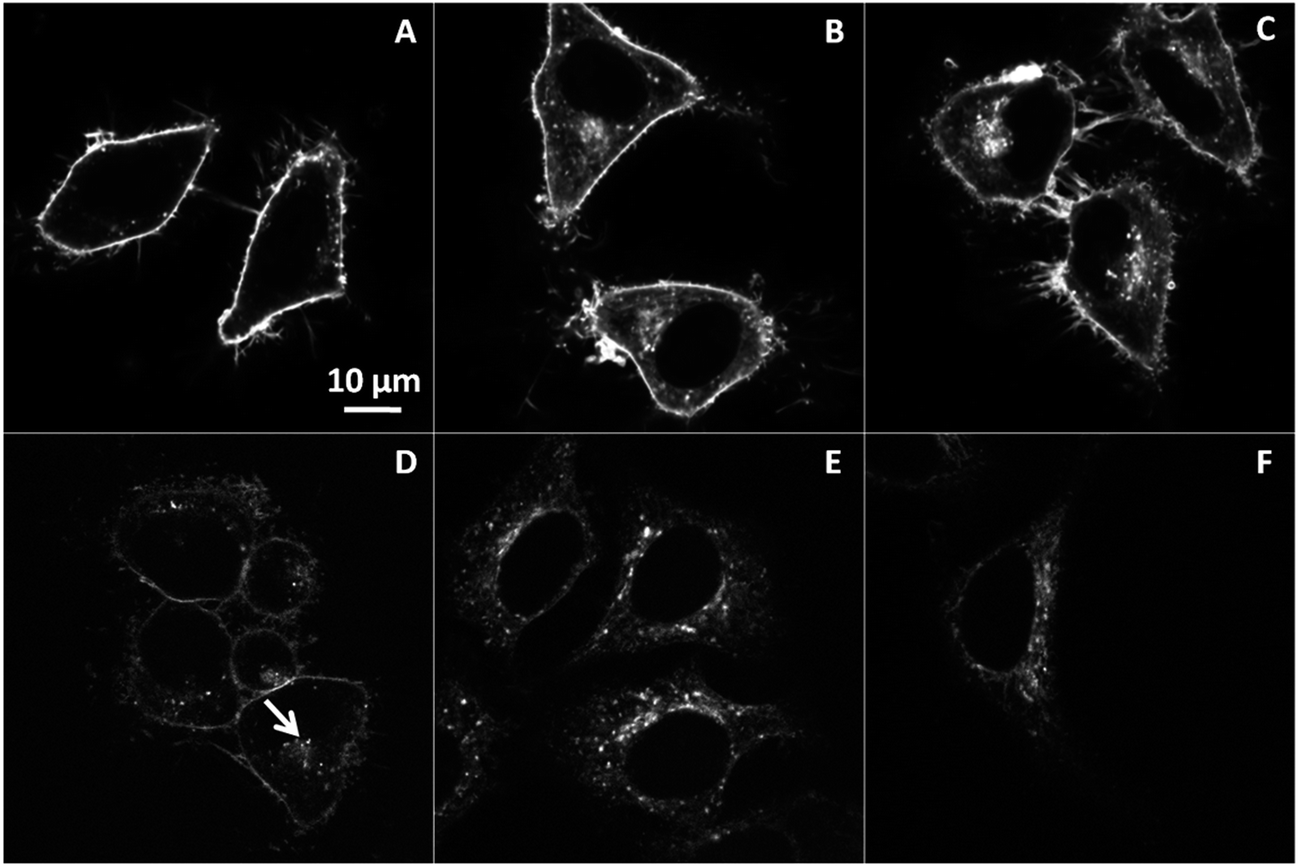
We further applied different endocytosis inhibitors, i.e. chlorpromazine, and methyl-β-cyclodextrin, in order to identify the pathway of the endocytotic uptake of NR12S (Fig. 3). Chlorpromazine is an inhibitor of clathrin-mediated endocytosis,32,33 while methyl-β-cyclodextrin extracts cholesterol from plasma membranes and thus, mainly inhibits caveolin-associated endocytosis.34,35 In our experiments, NR12S was incubated with the cells for 30 min at 37 °C with and without the corresponding inhibitor. Fluorescence imaging showed that the accumulation of the internalized probe next to nucleus, which can be assigned to lysosomes and/or late endosomes (Fig. 3A, arrow), disappears after chlorpromazine treatment (Fig. 3B). Thus, the internalization of NR12S is probably slowed down by chlorpromazine but is not totally inhibited as intracellular fluorescence of the probe can be still observed. Cells treated with methyl-β-cyclodextrin also showed some internalization of NR12S, after 30 min of incubation (Fig. 3C) but much less than in the case of non-treated cells, since patches of internalized probe were also absent. The visible effects of chlorpromazine and methyl-β-cyclodextrin indicate that NR12S is internalized both through clathrin- and caveolin-dependent endocytosis. This result was expected, since this probe binds rather homogeneously to the outer leaflet of the cell plasma membrane, and should thus be present in the different types of endosomes.
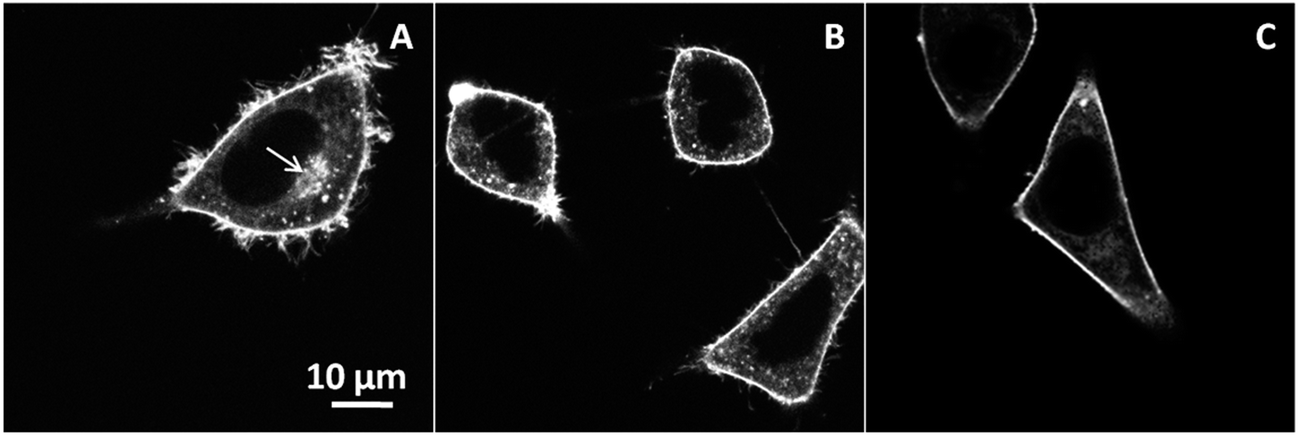 | ||
| Fig. 3 Inhibition of NR12S internalization. Confocal microscopy images of NR12S alone for 30 min in non-treated cells (A), in presence of 10 μg mL−1 chlorpromazine for 30 min (B), or 5 mM methyl-β-cyclodextrin for 2 h (C). NR12S concentration was 0.05 μM. Arrow shows a patch of internalized NR12S molecules in non-treated cells. | ||
To further characterize the mechanism of NR12S entry in cells, we analysed its colocalization with known endosomal markers. Primarily, we used fluorescently labelled transferrin, which is a marker for clathrin-mediated endocytosis due to receptor-specific binding.36–38 The endocytosis process of NR12S was directly compared with that of transferrin by colocalization experiments in HeLa cells using confocal microscopy. To this end, NR12S and Alexa 488-labeled transferrin were added simultaneously to the cells and imaged after 5, 30 and 60 min incubation. The colocalization between NR12S and transferrin was not easily seen after 5 min, but clearly appeared after 30 min and 60 min (Fig. 4H and I), confirming that NR12S is partly internalized through clathrin-dependent endocytosis. Noticeably, the limited colocalization observed at 5 min is likely related to the much stronger fluorescence of NR12S in cell plasma membranes as compared to early endosomes, so that the fluorescence of this probe is barely detectable in the latter at this early time (Fig. 4D).
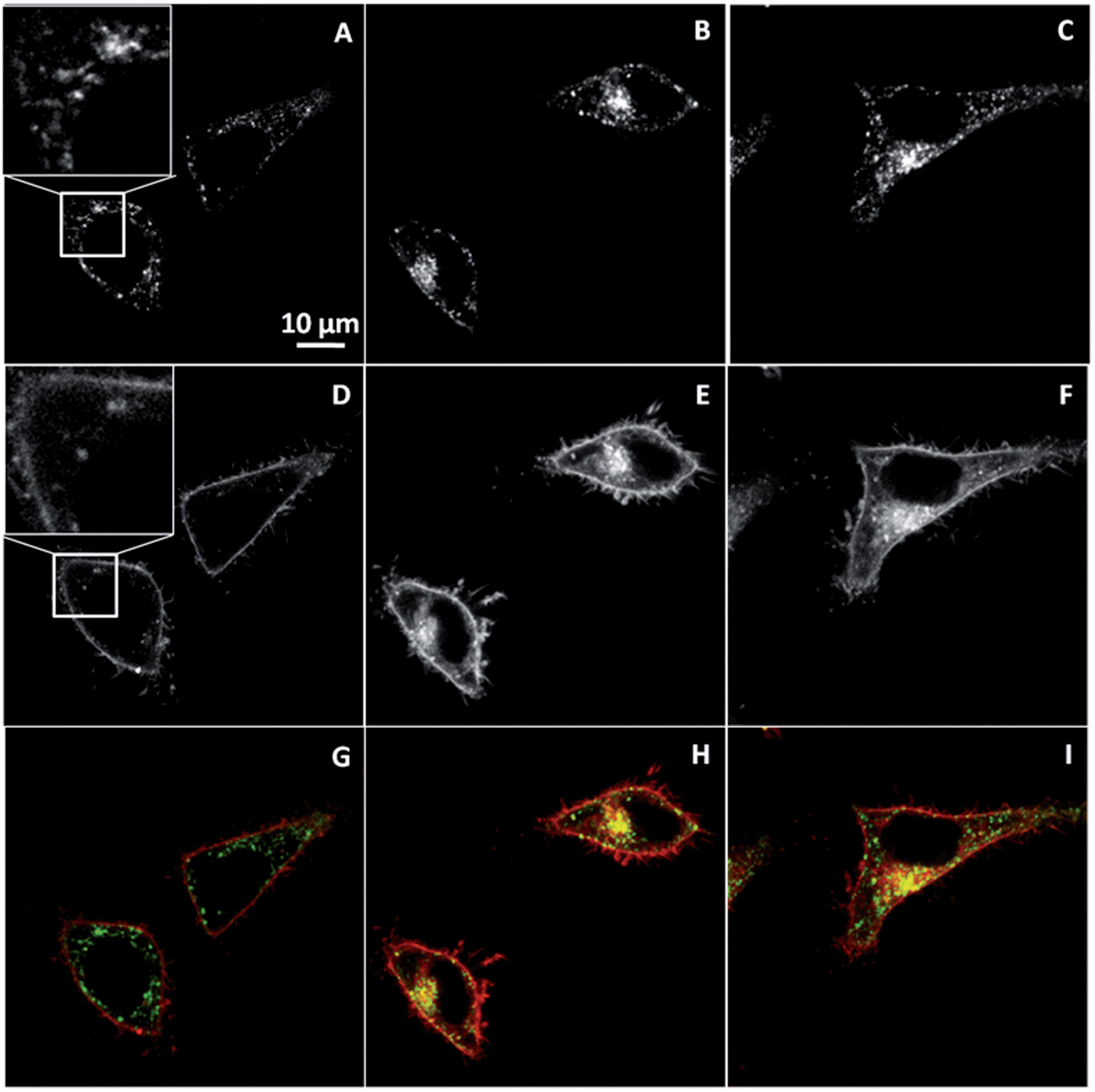 | ||
| Fig. 4 Colocalization of NR12S (0.05 μM) with transferrin (20 μg mL−1) after 5 min (A, D and G), 30 min (B, E and H), or 60 min (C, F and I). Confocal microscopy images corresponding to staining with Alexa Fluor-488 transferrin (upper panels), NR12S (middle panels) and their merge (bottom panels) are shown. In the merge figures, NR12S is in red, transferrin is in green, and their colocalization is in yellow. | ||
The fate of NR12S inside the cytoplasm was further monitored by colocalization experiments with LysoTracker® Green DND-26, a marker of lysosomes. Fluorescence confocal images revealed that NR12S does not colocalize with the lysosome tracker below 30 min of incubation (Fig. 5A, F and K), while their colocalization improves drastically for longer incubation times, so that after 2 h both probes are concentrated close to the nucleus (Fig. 5N and O), where the lysosomes are mainly localised. These data show that it takes about 2 h for the dye to go from the plasma membrane up to the lysosome. This time fits well with the described time frame of the endocytosis process, starting from formation of the endosomal vesicle up to its conversion into late endosomes and lysosomes.24
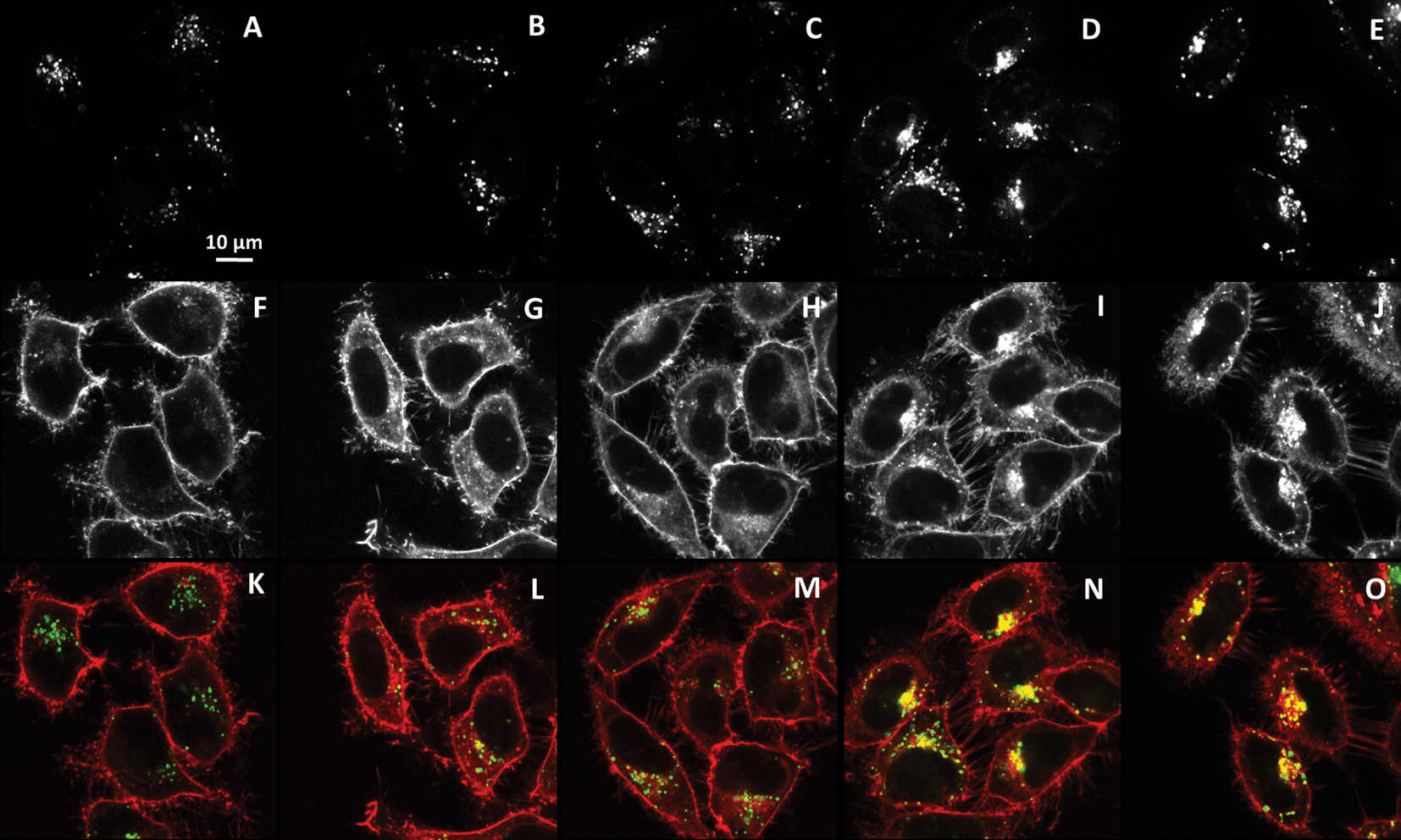 | ||
| Fig. 5 Colocalization of NR12S probe with lysotracker (50 nM) after 5 min (A, F and K), 30 min (B, G and L), 1 h (C, H and M), 2 h (D, I and N), and 4 h (E, J and O) of incubation with NR12S (0.05 μM). Confocal microscopy images correspond to staining with Alexa488-lysotracker (upper panels), NR12S (middle panels) and their merge (bottom panels). In the merge figures, NR12S is in red, lysotracker is in green, and their colocalization is in yellow. | ||
Moreover, these results shed light on our two-color imaging data (Fig. 1), where NR12S probe showed changes in its emission ratio also after 2 h of incubation. We can thus suggest that the probe evidenced changes in the lipid composition and order in late endosomes and lysosomes. We could easily exclude an effect of pH, as in control experiments, the fluorescence of NR12S does not change in the pH range between 3.5 and 8.4 (Fig. S1 in ESI†). Thus, the NR12S probe data suggest that late endosomes and especially lysosomes present a lower fraction of Lo phase as compared to plasma membranes and early endosomes. This observation supports the hypothesis that the contents of cholesterol and sphingomyelin in endosomal compartments decreased during maturation.17,20,24
However, it cannot be excluded that after 2 h, some part of the internalized dye is localized in the ER or other cell compartments, where the content of cholesterol and SM is also rather low,24 thus explaining the observed decrease in the lipid order. To address this point, colocalization of NR12S with ER marker was further tested. After 5 min (Fig. 6A), the plasma membrane stained by NR12S probe appears in red, while the ER appears separately as green cytoplasmic filaments. After 2 h and 4 h, NR12S fluorescence is mainly observed either at the membranes or inside the cells in form of dots, which is again clearly different from the staining profile by the ER marker (Fig. 6B and C). Thus, after 2 and 4 h of incubation, the major part of the probe likely remains in the late endosomes and lysosomes with no significant localization within the ER. This could be explained by the fact that the NR12S probe stains selectively the outer leaflet of the plasma membrane with nearly no flip-flop.27 Therefore, it gets into the inner leaflet of the endosomes and predominantly remains there for the whole endosome lifetime (Fig. 7), until it converts into lysosome and/or fuses with other intracellular compartments. Taken together, our data strongly suggest that the changes in NR12S emission which accompany endosome maturation, are related to lipid order changes resulting from the loss of cholesterol and SM in endosomal and lysosomal compartments, as it was previously proposed.15,20
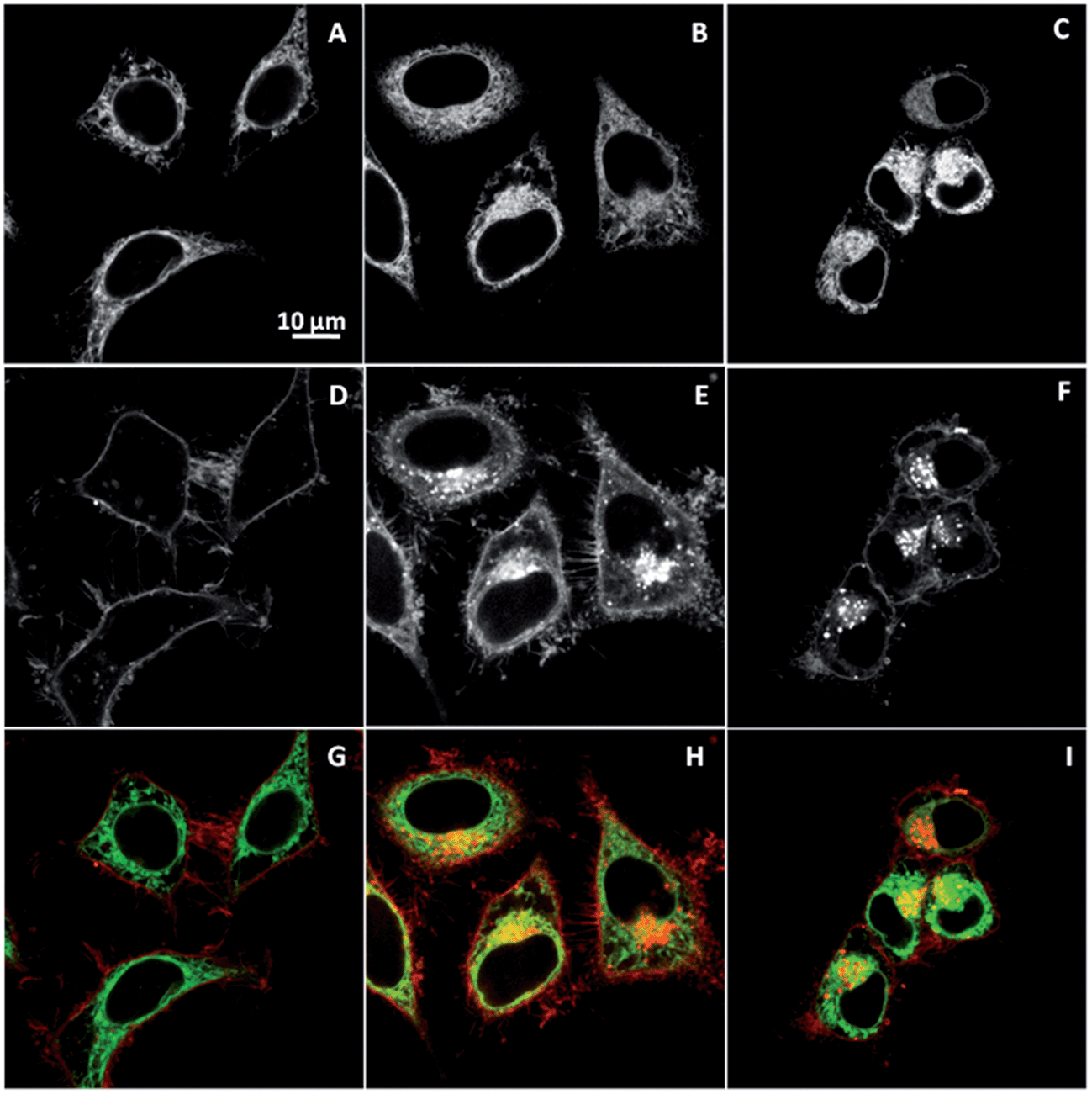 | ||
| Fig. 6 Colocalization of NR12S (0.05 μM) with ER-tracker (0.75 μM) after 5 min (A, D and G), 2 h (B, E and H), and 4 h (C, F and I). Confocal microscopy images correspond to staining with Alexa-488 ER-tracker (upper panels), NR12S (middle panels) and their merge (bottom panels). In the merge figures, NR12S is in red, Alexa-488 ER-tracker is in green, and their colocalization is in yellow. | ||
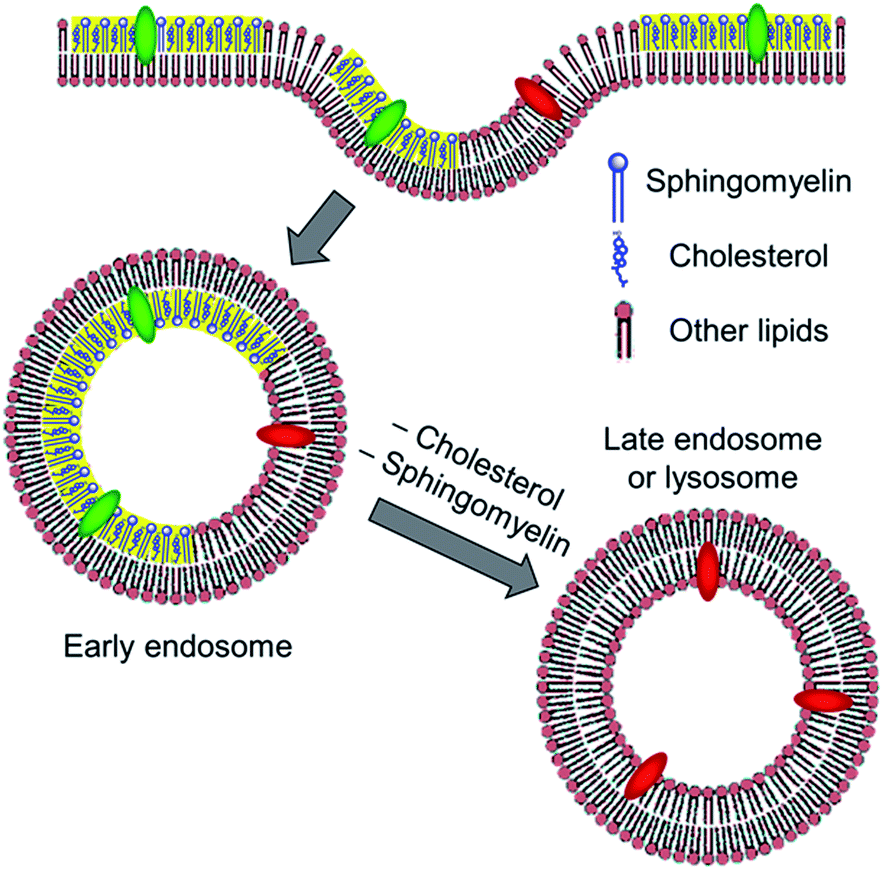 | ||
| Fig. 7 Proposed scheme for the probe colour response during endocytosis. After, binding to the external leaflet of plasma membranes, the probe localizes at the inner leaflet of the endosomes. Change in the colour from green to red represents the probe response to the loss of cholesterol and SM in late endosomes and lysosomes. Yellow parts represent liquid ordered phase formed by SM and cholesterol. | ||
Since NR12S appears as a “smart” marker of endocytic compartments, which could report on the change in their lipid composition with time, we compared it with FM4-64, one of the most common fluorescent membrane probe used for endocytosis studies. Confocal fluorescence imaging shows that NR12S is much brighter than FM4-64 (Fig. 8). Therefore, NR12S could be easily used at a concentration as low as 10 nM for fluorescence imaging,28 while the concentration needed for FM4-64 is 100-fold higher. The second remarkable difference is that FM4-64 internalizes much faster than NR12S. Indeed, while a large fraction of FM4-64 is already internalized after 5 min, showing even some clusters in the perinuclear region (Fig. 8D, arrow), NR12S is mainly observed at the cell plasma membrane for the same incubation time (Fig. 8A). For better comparison of the internalization of both probes, we compared the number of cells with probe clusters inside the cytoplasm (Fig. 8D, arrow). FM4-64 was significantly more internalized after 5 min than NR12S, since 86% of cells show an accumulation of FM4-64 in form of patches in the perinuclear region (42 cells were analysed for each probe) versus only 19% in the case of NR12S. At longer incubation times, FM4-64 shows almost exclusive intracellular fluorescence (Fig. 8E and F), while NR12S shows both plasma membrane and intracellular staining (Fig. 8B and C). This significant difference between the two membrane probes could be related to the differences in their chemical structure. One reason could be the positive charge of FM4-64, which favours faster internalization of the dye as compared to the neutral zwitterionic NR12S probe. Indeed, the positive charge of FM4-64 may favour its binding to lipids and proteins involved in endocytosis, while the neutral NR12S probe may be distributed more homogeneously within the plasma membrane. In addition, the undetermined flip-flop kinetics of FM4-64 may be much faster than the slow flip-flop kinetics evidenced for NR12S,27 so that FM4-64 could rapidly accumulate at the external leaflet of early endosomes, making it available for labelling endosomes in close proximity. In contrast, the possibility that NR12S may inhibit endocytosis could be ruled out, since we observed by fluorescence microscopy that NR12S marginally affects the internalization of labelled transferrin (Fig. S2 in ESI†). Thus, comparison with FM4-64 shows that NR12S is much brighter and its internalization kinetics is slower. These features make it attractive both as a plasma membrane probe and as an endosomal tracker. Moreover, NR12S presents the unique feature of detecting changes in the lipid composition of the membrane, which allows monitoring the loss of lipid order in late endosomes and lysosomes. Finally, we should mention that in all our short and long term experiments, we did not observe any NR12S-related cytotoxicity. However, it should be kept in mind that NR12S can lead to some photo-toxicity, but only at rather high probe concentrations.28
 | ||
| Fig. 8 Fluorescence confocal images of cells incubated with NR12S (A–C) and FM4-64 (D–F) for 5 min (A and D), 30 min (B and E), and 1 h (C and F) at 37 °C. Arrow in (D) shows a patch of FM4-64 already visible after 5 min in contrast to NR12S. Concentration of probes was 1 μM. | ||
4. Conclusions
The internalization of a recently developed membrane probe NR12S, which is sensitive to lipid composition and does not undergo fast flip-flop, was studied in HeLa cells. The probe was found to be internalized through energy-dependent endocytosis pathways and constitute thus, a general marker of endocytosis. Importantly, at long internalization times (≥2 h), the probe localized in late endosomes and lysosomes shows a dramatic change in its emission colour, which could be assigned to a decrease in the membrane lipid order likely connected with cholesterol depletion and sphingomyelin hydrolysis. NR12S constitutes thus, an improved and powerful tool for monitoring endocytosis. Indeed, the probe is simple to use, very bright, compatible with both confocal and two-photon excitation microscopy and can work at concentrations as low as 10 nM. Moreover, its extracellular fluorescence can be turned off, and it allows monitoring changes of lipid composition during endosome maturation.Acknowledgements
This work was supported by ANR JCJC (ANR-11-JS07-014-01). Financial support of ZD from CNRS and Region Alsace is acknowledged.References
- M. Marsh and A. Helenius, Cell, 2006, 124, 729–740 CrossRef CAS PubMed
.
- J. Mercer, M. Schelhaas and A. Helenius, Annu. Rev. Biochem., 2010, 79, 803–833 CrossRef CAS PubMed
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC
- H. Tanno and M. Komada, J. Biochem., 2013, 153, 497–504 CrossRef CAS PubMed
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed
- R. M. Steinman, I. S. Mellman, W. A. Muller and Z. A. Cohn, J. Cell Biol., 1983, 96, 1–27 CrossRef CAS
- M. Jovic, M. Sharma, J. Rahajeng and S. Caplan, Histol. Histopathol., 2010, 25, 99–112 CAS
- W. Stoorvogel, G. J. Strous, H. J. Geuze, V. Oorschot and A. L. Schwartz, Cell, 1991, 65, 417–427 CrossRef CAS
- P. Saftig and J. Klumperman, Nat. Rev. Mol. Cell Biol., 2009, 10, 623–635 CrossRef CAS PubMed
- S. R. Pfeffer, Curr. Opin. Cell Biol., 2013, 25, 414–419 CrossRef CAS PubMed
- C. Lafourcade, K. Sobo, S. Kieffer-Jaquinod, J. Garin and F. G. van der Goot, PLoS One, 2008, 3, e2758 Search PubMed
- C. Bucci, P. Thomsen, P. Nicoziani, J. McCarthy and B. van Deurs, Mol. Biol. Cell, 2000, 11, 467–480 CrossRef CAS
- T. Kobayashi, M.-H. Beuchat, J. Chevallier, A. Makino, N. Mayran, J.-M. Escola, C. Lebrand, P. Cosson, T. Kobayashi and J. Gruenberg, J. Biol. Chem., 2002, 277, 32157–32164 CrossRef CAS PubMed
- X. Du and H. Yang, Acta Biochim. Biophys. Sin., 2013, 45, 11–17 CrossRef CAS PubMed
- B. Karten, K. B. Peake and J. E. Vance, Biochim. Biophys. Acta, 2009, 1791, 659–670 CrossRef CAS PubMed
- E. D. Carstea, J. A. Morris, K. G. Coleman, S. K. Loftus, D. Zhang, C. Cummings, J. Gu, M. A. Rosenfeld, W. J. Pavan, D. B. Krizman, J. Nagle, M. H. Polymeropoulos, S. L. Sturley, Y. A. Ioannou, M. E. Higgins, M. Comly, A. Cooney, A. Brown, C. R. Kaneski, E. J. Blanchette-Mackie, N. K. Dwyer, E. B. Neufeld, T. Y. Chang, L. Liscum, J. F. Strauss Iii, K. Ohno, M. Zeigler, R. Carmi, J. Sokol, D. Markie, R. R. O'Neill, O. P. Van Diggelen, M. Elleder, M. C. Patterson, R. O. Brady, M. T. Vanier, P. G. Pentchev and D. A. Tagle, Science, 1997, 277, 228–231 CrossRef CAS
- E. Lloyd-Evans, A. J. Morgan, X. He, D. A. Smith, E. Elliot-Smith, D. J. Sillence, G. C. Churchill, E. H. Schuchman, A. Galione and F. M. Platt, Nat. Med., 2008, 14, 1247–1255 CrossRef CAS PubMed
- T. Kolter and K. Sandhoff, FEBS Lett., 2010, 584, 1700–1712 CrossRef CAS PubMed
- K. Simons and E. Ikonen, Nature, 1997, 387, 569 CrossRef CAS PubMed
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed
- N. D. Ridgway, Biochim. Biophys. Acta, 2000, 12, 2–3 Search PubMed
- W. Möbius, E. Van Donselaar, Y. Ohno-Iwashita, Y. Shimada, H. F. G. Heijnen, J. W. Slot and H. J. Geuze, Traffic, 2003, 4, 222–231 CrossRef
- S. Bolte, C. Talbot, Y. Boutte, O. Catrice, N. D. Read and B. Satiat-Jeunemaitre, Journal of Microscopy, 2004, 214, 159–173 CrossRef CAS PubMed
- P. C. Hickey, D. J. Jacobson, N. D. Read and N. Louise Glass, Fungal Genet. Biol., 2002, 37, 109–119 CrossRef
- O. A. Kucherak, S. Oncul, Z. Darwich, D. A. Yushchenko, Y. Arntz, P. Didier, Y. Mély and A. S. Klymchenko, J. Am. Chem. Soc., 2010, 132, 4907–4916 CrossRef CAS PubMed
- Z. Darwich, A. S. Klymchenko, O. A. Kucherak, L. Richert and Y. Mely, Biochim. Biophys. Acta, 2012, 1818, 3048–3054 CrossRef CAS PubMed
- J. P. Clamme, J. Azoulay and Y. Mely, Biophys. J., 2003, 84, 1960–1968 CrossRef CAS
- J. M. Bergen, E. J. Kwon, T. W. Shen and S. H. Pun, Bioconjugate Chem., 2007, 19, 377–384 CrossRef PubMed
- O. Faklaris, V. Joshi, T. Irinopoulou, P. Tauc, M. Sennour, H. Girard, C. l. Gesset, J.-C. Arnault, A. Thorel, J.-P. Boudou, P. A. Curmi and F. o. Treussart, ACS Nano, 2009, 3, 3955–3962 CrossRef CAS PubMed
- K. M. Hussain, K. L. J. Leong, M. M.-L. Ng and J. J. H. Chu, J. Biol. Chem., 2010, 286, 309–321 CrossRef PubMed
- H. Parker, K. Chitcholtan, M. B. Hampton and J. I. Keenan, Infect. Immun., 2010, 78, 5054–5061 CrossRef PubMed
- A. Subtil, I. Gaidarov, K. Kobylarz, M. A. Lampson, J. H. Keen and T. E. McGraw, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6775–6780 CrossRef CAS
- S. K. Rodal, G. Skretting, O. Garred, F. Vilhardt, B. van Deurs and K. Sandvig, Mol. Biol. Cell, 1999, 10, 961–974 CrossRef CAS
- R. B. Dickson, J. A. Hanover, M. C. Willingham and I. Pastan, Biochemistry, 1983, 22, 5667–5674 CrossRef CAS
- R. N. Ghosh, D. L. Gelman and F. R. Maxfield, J. Cell Sci., 1994, 107, 2177–2189 CAS
- K. M. Mayle, A. M. Le and D. T. Kamei, Biochim. Biophys. Acta, 2012, 1820, 264–281 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47181k |
| This journal is © The Royal Society of Chemistry 2014 |