Tailoring porosities and electrochemical properties of composites composed of microfibrillated cellulose and polypyrrole
Daniel O.
Carlsson
a,
Albert
Mihranyan
ab,
Maria
Strømme
*a and
Leif
Nyholm
*c
aNanotechnology and Functional Materials, Department of Engineering Sciences, Uppsala University, Box 534, 75121 Uppsala, Sweden. E-mail: Maria.Stromme@angstrom.uu.se
bDivision of Materials Science, Luleå University of Technology, 97187 Luleå, Sweden
cDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 538, 75121, Uppsala, Sweden. E-mail: Leif.Nyholm@kemi.uu.se
First published on 16th January 2014
Abstract
Composites of polypyrrole and nanocellulose (PPy/nanocellulose) have a high potential as electrodes in energy-storage devices and as membranes for electrochemically controlled ion-exchange systems. In the present work, it is demonstrated that such composites with 42–72% porosity can be produced by using microfibrillated cellulose (MFC) prepared through enzymatic pretreatment or carboxymethylation, or by using different amounts of MFC in the composite synthesis. Together with previous work, this shows that the porosity of PPy/nanocellulose composites can be tailored from 30 to 98% with increments of ∼10%. Employing the full porosity range of the composites, it is demonstrated that the electrochemical oxidation rate of the materials depends on their porosity due to limitations in the counter ion diffusion process. By tailoring the porosities of PPy/nanocellulose composites, the electrochemical properties can consequently be controlled. The latter provides new possibilities for the manufacturing of electrochemically controlled ion-extraction and energy storage devices with optimized volumetric energy and power densities.
1. Introduction
The interest in nanocellulose has increased rapidly during the last decade and as a result it constitutes a frequently employed nanomaterial today.1,2 Nanocellulose possesses many interesting properties; it is renewable, inexpensive, non-toxic and has excellent mechanical properties.2–5 In addition, nanocellulose can be extracted from a variety of sources, most commonly from wood, but also from, e.g., filamentous green algae and tunicates, using a range of different preparation methods.2,4,6 The properties of nanocellulose, such as fibre length, width, degree of crystallinity, and surface charge depend on the origin of the nanocellulose and the preparation methods used in its extraction. Microfibrillated cellulose (MFC), was first described in the early 1980's by Turbak et al.7 and Herrick et al.8 MFC comprises nanocellulose fibrils of approximately 3–30 nm width and micrometer lengths and are produced from wood pulp through different homogenization processes. Following the pioneering publications, different pretreatments have been developed to decrease the energy consumption during the homogenization,2,4e.g. involving enzymatic hydrolysis,9,10 carboxymethylation11 and TEMPO-mediated oxidation.12 This development has led to the introduction of a wide range of interesting new nanocellulose materials,2–5 including nanocellulose aerogels,13 magnetic nanopapers and aerogels,14 oil-absorbing aerogels15 and cross-linked aerogels with high charge densities for facile layer-by-layer functionalisation.16During the last few years it has also been demonstrated17,18 that the non-conducting nanocellulose fibrils can be readily coated with a conducting polymer, e.g. polypyrrole (PPy), yielding conductive and electroactive composite fibres. PPy, which is one of the most frequently studied conducting polymers, can be prepared through oxidative electrochemical or chemical polymerization of pyrrole giving rise to a film deposited on a conducting substrate (an electrode) or a powder with poor mechanical properties, respectively.19 As has been described in detail elsewhere,19–21 PPy can be reversibly switched between its reduced, uncharged and non-conducting state and its oxidized, positively charged and conducting state by controlling the applied potential or current. The switching of oxidation states is tightly coupled to the movement of counter ions, required to maintain the charge-neutrality of the PPy. For sufficiently thick PPy films this counter ion mass transport generally limits the rate of the redox reaction whereas very fast oxidation and reduction of the PPy can be found for nanometre-thick, PPy coatings.22–24
The problems associated with the low redox reaction rates for thick PPy films and the poor mechanical properties of PPy are avoided by performing the pyrrole polymerization in the presence of nanocellulose fibrils. The individual nanocellulose fibrils are, in this case, coated by a thin layer of PPy (<50 nm) and after drying, a coherent, free-standing, conductive and electroactive paper-like composite material is obtained. Composites comprising PPy and nanocellulose from wood17,25 or Cladophora sp. algae26 have previously been reported and employed as electrodes in environmentally friendly energy-storage devices22,27 and as an ion-exchange material28 for DNA-extraction29 and hemodialysis.30,31
It has, however, been demonstrated that the redox reaction rates of the PPy/nanocellulose composites are significantly affected by the porosities of the materials.17 The redox rate of PPy/nanocellulose aerogel composites with a porosity of 92–98%, prepared through freeze-drying or supercritical CO2 drying, was shown to be significantly higher than for a composite with identical composition but much lower (i.e. 30–35%) porosity. The importance of high porosity for fast electrochemical processes has also been demonstrated using other, but similar, materials such as aerogels based on the conducting polymer PEDOT (poly(3,4-ethylenedioxythiophene)) and porous carbonaceous foams.32–34
Whereas composites with 30–35% porosity possessed mechanical properties (i.e. 0.51 GPa Young's modulus, 10.93 MPa tensile strength and 2.5% strain to failure) surpassing previously reported values for PPy/cellulose composites when normalized with respect to the cellulose content,35,36 the mechanical properties of the 92–98% porosity aerogel composites were so poor that these could not be measured. These findings, which illustrate the inverse relationship between porosity and mechanical strength for this type of material, imply that, depending on the intended application, a compromise between fast electrochemical processes and mechanical strength generally has to be made.
The composites described above were based on nanocellulose from wood prepared through TEMPO-mediated oxidation, resulting in a porosity of 30–35% when dried in the ambient and 92–98% after specialized drying. It has also been reported that composites based on nanocellulose from Cladophora sp. algae has a porosity of 82% following ambient drying.37 In subsequent work with PPy/nanocellulose composites based on enzymatically pretreated MFC and two forms of Cladophora nanocellulose, porosities ranging from 55% to 75% were obtained after compacting and drying of the composites through pressing.38 Although the electrochemical properties of these composites were not the main focus of the latter work, the results indicated that the porosity affects the redox rates of the composites. Based on these findings it is clear that there is a need for the development of new straightforward methods enabling the porosities of PPy/nanocellulose composites to be readily controlled, as well as for systematic investigations of the impact of the composite porosity on the electrochemical properties covering a large porosity interval.
Due to differences in the fibril properties between different forms of nanocellulose, sheets made thereof have different porosities after drying in air: the porosity of sheets prepared from Cladophora nanocellulose is higher than that for sheets made of MFC prepared through enzymatic pretreatment, which in turn is higher than that for sheets prepared through carboxymethylation.39 In the present work, it is demonstrated that the corresponding PPy/nanocellulose composites follow the same porosity trend. Specifically, it is shown that PPy/nanocellulose composites with porosities between 42 and 72% can be produced in a straightforward way by utilizing two different forms and amounts of MFC from wood, without using any form of specialized drying or compacting procedures. This means that PPy/nanocellulose composites can be tailored to have porosities ranging from 30 to 98% with porosity increments corresponding to ∼10% over the entire range. Utilizing samples covering the full porosity range, the effect of porosity on the electrochemical properties of these composites is clearly demonstrated. By careful selection of the type, or amount, of nanocellulose used in the composite synthesis it is consequently possible to tailor the porosity and the electrochemical properties of PPy/nanocellulose composites.
2. Experimental
2.1 Materials
The MFC, which was produced and provided as 2% hydrogels by Innventia AB (Sweden), was used as received. The MFC generation 1 (i.e. MFC g1) and generation 2 (i.e. MFC g2) were produced from softwood sulphite pulp fibres (Domsjö Dissolving Plus, Domsjö Fabriker AB, Sweden). The preparation of MFC g1 followed the procedure described by Pääkkö et al.,10 encompassing enzymatic hydrolysis as well as mechanical and high-pressure homogenization, yielding micrometer long fibrils, 5–20 nm in width. MFC g2 was produced by carboxymethylation and high-pressure homogenization in conformity with the procedure described by Wågberg et al.,11 yielding micrometer long fibrils with a diameter of 5–15 nm. Typically, MFC g1 is dominated by fibril aggregates of ∼20 nm in width, whereas MFC g2 is dominated by fibril widths closer to 5 nm due to greater fibril repulsion resulting from the introduced surface charges. Pyrrole (Sigma-Aldrich), FeCl3·6H2O (Sigma-Aldrich), HCl (Sigma-Aldrich), NaCl (BDH Prolabo) and Tween-80 (Merck) were used as received and all solutions were prepared using deionised water.2.2 Synthesis
MFC g1 (or MFC g2) hydrogel corresponding to 100 or 300 mg dry cellulose was diluted to a total volume of 50 mL and dispersed employing high-energy ultrasonication (VibraCell 750 W, Sonics, USA). 0.022 mol pyrrole and one drop of Tween-80 were dissolved in 100 mL of 0.5 M HCl and added to the cellulose dispersions followed by mixing for five minutes. Subsequently, 0.048 mol FeCl3 dissolved in 100 mL of 0.5 M HCl was added to initiate the pyrrole polymerization. The polymerization was allowed to proceed for 30 minutes before washing the products in a Büchner funnel at reduced pressure. In order not to degrade the PPy, the composites were washed with 5 L of 0.5 M HCl and 1 L of 0.1 M NaCl.40 The products were subsequently sonicated for two minutes for homogenization purposes before finally collected in a Büchner funnel. The products were ultimately dried in air and stored in the ambient prior to further analysis. The syntheses yielded four composites of roughly the same dimensions (circular shape, diameter 5–6 cm, thickness 0.9–1.3 mm) all of which were rather inflexible and rigid. The composites could be broken into smaller pieces by hand or cut with a scalpel and all characterizations of the composites were carried out using pieces broken off or cut from the synthesized materials. In the text below, the samples are denoted PPy/MFCg1_100, PPy/MFCg1_300, PPy/MFCg2_100 and PPy/MFCg2_300, to reflect the MFC generation and amount used in their synthesis.2.3 PPy content determination
CHN elemental analyses were performed by Analytische Laboratorien (Lindlar, Germany) after drying the samples in a vacuum oven at reduced pressure at 70 °C for at least 24 hours. The amounts of PPy in the composites were calculated from the obtained nitrogen contents, as PPy was the only composite component containing nitrogen.2.4 Scanning electron microscopy (SEM)
Surface and cross-section micrographs were obtained with a LEO1550 field emission SEM instrument (Zeiss, Germany) employing an in-lens secondary electron detector. The samples were mounted on aluminium stubs with double-sided adhesive carbon tape and sample cross-sections were prepared by freezing samples in liquid nitrogen and subsequently breaking the frozen samples.2.5 Density and total porosity determination
Prior to the true density and bulk density measurements the samples were dried in a vacuum oven at reduced pressure at 70 °C for at least 24 hours. The true density (ρt), which corresponds to the actual density of the composite fibres, was measured through He pycnometry (AccuPyc 1340, Micromeritics, USA). The bulk density (ρb) was determined by cutting the samples to an approximately 1 × 2 cm rectangular shape and subsequently measuring the width, length, thickness and weight. The total porosity was calculated using eqn (1).| Total porosity = (1 – ρb/ρt) × 100 | (1) |
2.6 Electrochemical measurements
An Autolab PGSTAT302N potentiostat (Ecochemie, the Netherlands) was employed for all electrochemical measurements using a three-electrode setup comprising a 3 M NaCl Ag/AgCl reference electrode placed in the main compartment of the cell and a coiled platinum wire counter electrode in a separate compartment. All potentials are given with respect to the 3 M NaCl Ag/AgCl reference electrode. Samples (weighing 3.9–4.1 mg) were contacted by means of a paper clip shaped platinum wire and employed as the working electrode positioned in the main compartment. The 2 M NaCl (aq) electrolyte was purged with nitrogen for 15 min prior to the measurements and all measurements were made in a nitrogen atmosphere. Three initial cyclic voltammetry (CV) scans recorded with a scan rate of 5 mV s−1 were made followed by three scans at scan rates of 1, 5, 10, 20, 30, 40, and 50 mV s−1, respectively. The cathodic end potential was set to −0.80 V while the anodic end potential was increased from +0.50 to +0.85 V as the scan rate was increased. The specific charge capacities were calculated by integrating the anodic current from the first point of positive current to the point of minimum current occurring between the PPy oxidation and overoxidation peaks (i.e. at approximately +0.35 to +0.65 V depending on the scan rate and sample). Immediately following the CV-measurements, the samples were subjected to chronoamperometric (CA) measurements involving reduction at a potential of −0.80 V for 600 s followed by oxidation at +0.30 V for 600 s.3. Results and discussion
CHN elemental analyses were carried out to determine the compositions of the composites and the results are shown in Table 1. It can be seen that the composites are composed of roughly 2/3 PPy (by weight), with some variation and that the PPy content is slightly higher when 100 mg rather than 300 mg MFC was used. The differences in the PPy content, especially between PPy/MFCg1_100 and PPy/MFCg1_300, are small and although only one nitrogen content determination was made per sample, the variation in the calculated PPy content can still be expected to be small (i.e. the estimated relative standard deviation is 1.1%) based on previous results obtained with the same method.17 It can also be concluded that values presented in Table 1 are close to those previously obtained for similar composites comprising PPy and nanocellulose derived from wood.17 As the cellulose content of the latter composites was estimated to be about 9% (wt),17 it is reasonable to assume similar cellulose contents also for the present composites.| Sample | Nitrogen content (wt%)a | PPy content (wt%) | PPy content (mmol g−1)b |
|---|---|---|---|
| a The CHN elemental analyses were carried out after drying the composites for ≥24 h at 70 °C and reduced pressure in a vacuum oven. The weight loss, corresponding to water evaporation, was 6.5–7.0 wt%. b mmol per gram composite. | |||
| PPy/MFCg1_100 | 13.9 | 66.4 | 9.89 |
| PPy/MFCg1_300 | 13.0 | 62.2 | 9.27 |
| PPy/MFCg2_100 | 15.2 | 72.8 | 10.8 |
| PPy/MFCg2_300 | 13.1 | 62.7 | 9.35 |
Although no dissimilarities between the structures of the samples could be distinguished by eye, the SEM micrographs revealed significant differences on the micro- and nanoscales as is seen in Fig. 1, depicting representative surface SEM micrographs of the samples at three different magnifications (i.e. 3k×, 50k× and 200k×). At the lowest magnification, all samples appeared non-porous except the PPy/MFCg1_100 sample, for which a few pores of some micrometers width could be observed. The surfaces of the composites based on MFC g1 also appeared rougher than those based on MFC g2. At a magnification of 50k×, both MFC g1 samples displayed fibrous and porous structures, whereas both the MFC g2 samples appeared significantly denser, particularly the PPy/MFCg2_300 sample. The highest magnification micrographs revealed that also the PPy/MFCg2_100 and PPy/MFCg2_300 samples were fibrous although the pores were significantly smaller than for both PPy/MFCg1_100 and PPy/MFCg1_300. More importantly, the micrographs indicated that the composites had different porosities, depending on the amount and type of MFC used, with the porosities decreasing in the following order: PPy/MFCg1_100 > PPy/MFCg1_300 > PPy/MFCg2_100 > PPy/MFCg2_300. Although all samples were fibrous to some extent, a higher degree of composite fibre aggregation was observed when the pore volume decreased.
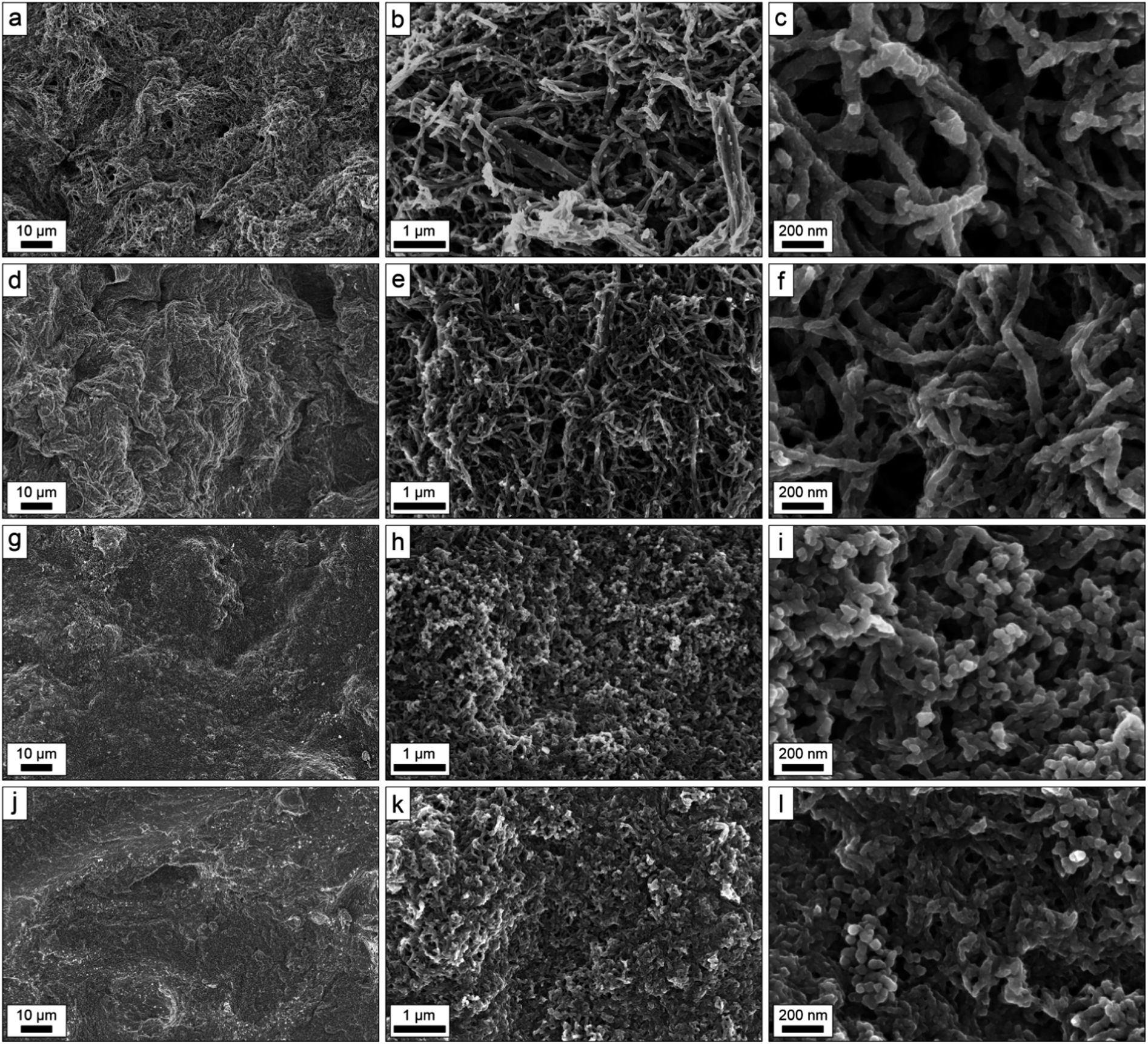 | ||
| Fig. 1 Scanning electron micrographs of the surface of PPy/MFCg1_100 (a–c), PPy/MFCg1_300 (d–f), PPy/MFCg1_100 (g–i) and PPy/MFCg2_300 (j–l), at magnifications of 3k× (left column), 50k× (middle column) and 200k× (right column), respectively. | ||
The widths of the individual composite fibres were estimated using a length measurement tool incorporated in the SEM instrument software. For both the PPy/MFCg1_100 and PPy/MFCg1_300 samples, the fibre widths ranged between ∼60 and 80 nm, while the fibres widths in the PPy/MFCg2_100 and PPy/MFCg2_300 samples ranged between ∼40 and 60 nm. This difference is likely due the differences in fibril widths inherent in these two MFC forms. Based on a fibril width of ∼20 nm for MFC g1 and ∼5 nm for MFC g2, the thickness of the PPy-layer can be estimated to ∼20–30 nm.
To confirm that the observed differences were not confined to the surface only, cross-sections of the samples were prepared to investigate the bulk structures. The micrographs in Fig. 2, which were acquired at magnifications of 200k×, show that the bulk structures of the composites were indeed very similar to the surface structures for all the samples. No uncoated cellulose fibrils could be observed in the bulk of the composite and the bulk porosities, pore volumes and fibre aggregation degrees, followed the same trends as deduced from the surface micrographs (see Fig. 1).
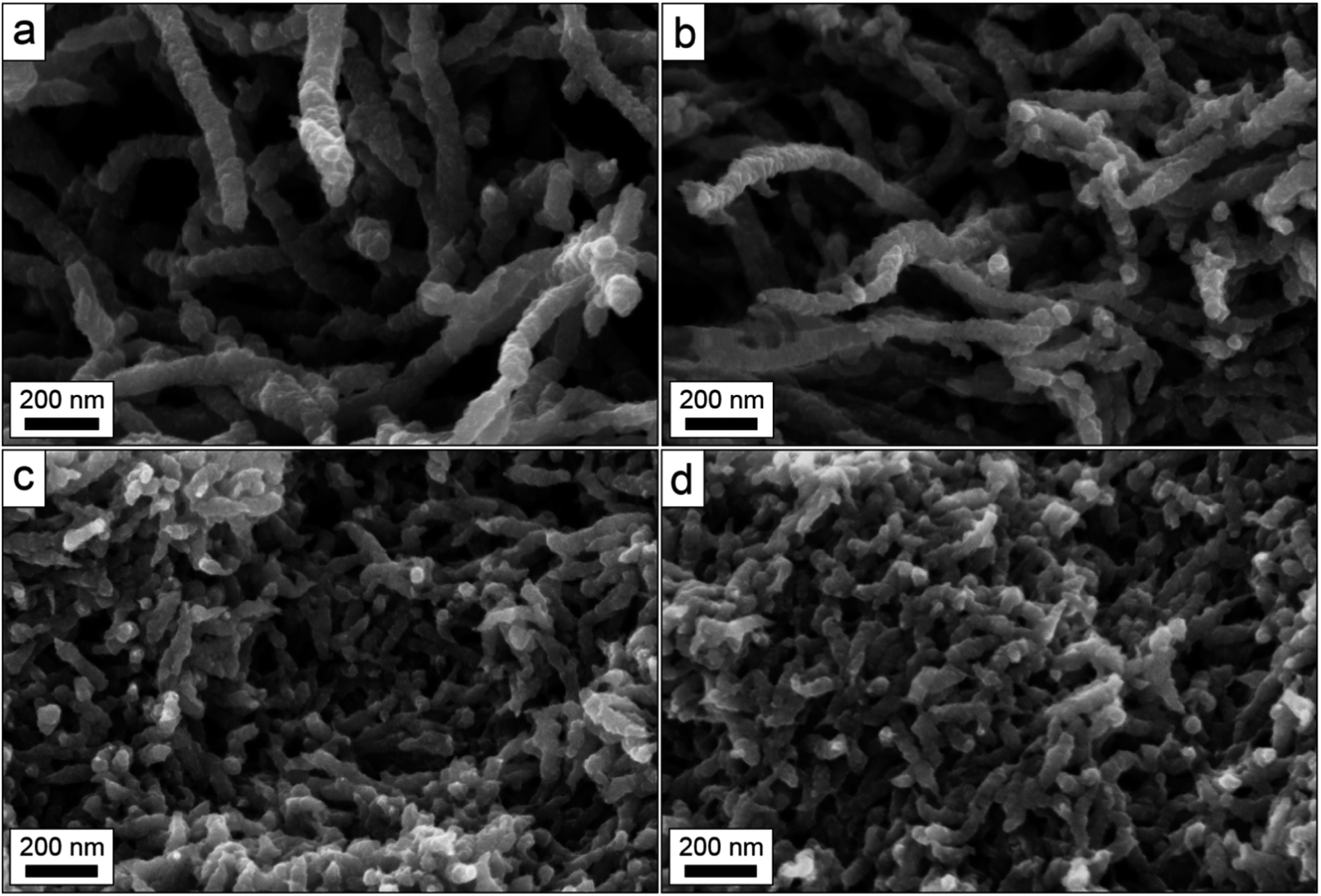 | ||
| Fig. 2 Scanning electron micrographs depicting cross-sections of PPy/MFCg1_100 (a), PPy/MFCg1_300 (b), PPy/MFCg2_100 (c) and PPy/MFCg2_300 (d) samples at 200k× magnification. | ||
In previous work on composites of PPy and Cladophora nanocellulose, it has been shown that the composite fibre structure comprises a nanocellulose fibril core coated with a 30–50 nm thin layer of PPy and that no uncoated fibrils could be observed.26 In the present investigation, no uncoated fibrils or parts thereof could likewise be observed in any of the SEM micrographs (see Fig. 1 and 2), indicating that all fibrils were completely covered with a PPy layer. A closer inspection of the micrographs obtained with 200k× magnification in Fig. 2, however, indicates that the PPy-coating did not form a smooth layer of uniform thickness, probably due to pyrrole polymerization in the solution followed by PPy adsorption on the fibrils, in addition to polymerization directly on the fibrils.
To conclude, the micrographs in Fig. 1 and 2 indicate that the porosity of the composite obtained depends on type and amount of nanocellulose used in the synthesis. The micrographs thus indicate that the porosity decreases in the order PPy/MFCg1_100 > PPy/MFCg1_300 > PPy/MFCg2_100 > PPy/MFCg2_300, and that the porosity decrease is a result of decreased pore volume and increased composite fibre aggregation.
To quantify the porosity differences seen in the micrographs, the total porosities of the samples were determined employing eqn (1), based on the measured true densities and the bulk density values (see Table 2). Whereas only small variations were observed between the true densities for the different samples, the bulk densities differed significantly, resulting in composite porosities ranging from 42% to 72%. In addition, the porosities of the samples decreased in the same order as deduced from the micrographs, i.e. PPy/MFCg1_100 > PPy/MFCg1_300 > PPy/MFCg2_100 > PPy/MFCg2_300. The higher porosity for the MFC g1 samples as compared to that for the MFCg2 samples is in good agreement with the fact that the porosity of pristine MFC g1 sheets has been found to be higher than for pristine MFC g2 sheets.39 It is also clear that the use of a higher amount of cellulose in the synthesis gives rise to less porous samples for both cellulose types used here. The present results regarding porosity are also in good agreement with earlier findings showing that the porosities for pressed PPy/nanocellulose composites followed the same trend as for the corresponding pressed pristine nanocellulose sheets.38 It can hence be concluded that the porosity of PPy/nanocellulose composites can be readily tailored by carefully selecting the type and amount of nanocellulose used in the composite synthesis.
Determinations of the pore size distributions for the present composites are complicated by the fact that pores larger than 100 nm clearly are present in the samples (see Fig. 1 and 2) which means that pore size distributions from nitrogen sorption data do not give a representative picture of the pore size distribution, as was also discussed previously.17
In earlier work17,38 we have shown that the porosities of PPy/nanocellulose composites significantly affect their electrochemical behaviour. It was thus found that highly porous composites with very high porosities composed of individual fibres were oxidized faster than samples with a high degree of fibre aggregation and much lower porosities (i.e. 30–35%).17 In the latter investigation, nanocellulose prepared by TEMPO-mediated oxidation of wood pulp was used together with freeze-drying or supercritical CO2 drying to produce composites with porosities of 92–98%. With air drying (as in the present investigation) composites with porosities of 30–35% were, on the other hand, obtained further demonstrating that the porosity of the composites also can be controlled by the method used to dry the samples.
PPy/nanocellulose composites based on nanocellulose from the Cladophora sp. green algae and dried in air have likewise been found to yield a material with a porosity of approximately 82%.37 The fact that the latter value is significantly higher than the values for the air dried PPy/nanocellulose composite based on TEMPO-oxidized nanocellulose from wood discussed above further emphasizes the influence of the type of the cellulose on the porosity of the composites obtained with PPy.
The composites prepared in the present study were further analyzed with cyclic voltammetry measurements at potential scan rates ranging from 1 to 50 mV s−1. The results of these experiments are shown in Fig. 3, in which the currents have been normalized with respect to the content of electroactive material, i.e. the PPy content. Some voltammograms for the previously reported17 PPy/nanocellulose composites with 30% and 98% porosities have also been included in Fig. 3 for comparison.
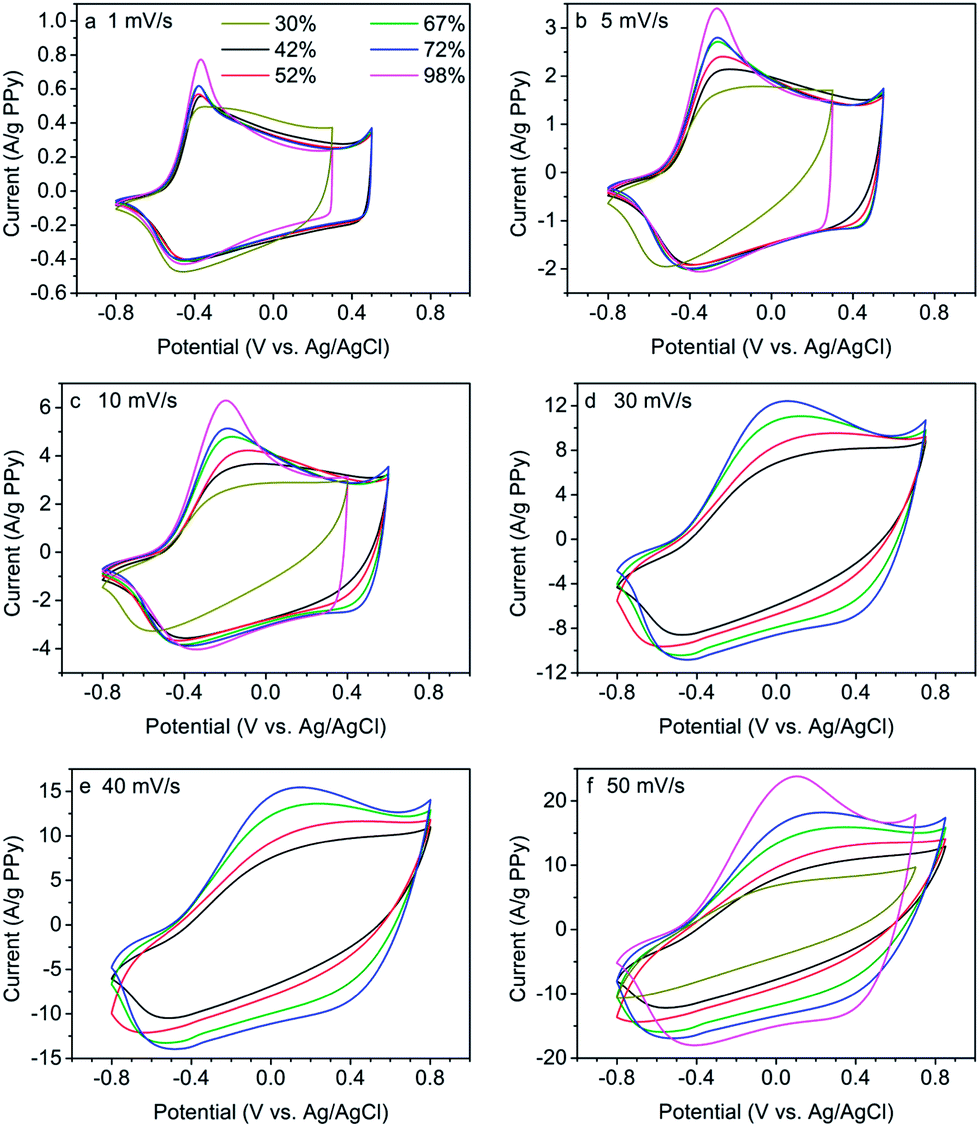 | ||
| Fig. 3 Cyclic voltammograms for PPy/nanocellulose composites with different porosities (porosities indicated in the figure legend in panel a) obtained for a range of potential scan rates. In panels a–c and f, voltammograms for the previously reported17 composites with 30% and 98% porosity have also been included for comparison. The current has been normalized with respect to the PPy contents in the composites. | ||
In Fig. 3a, it is seen that the voltammograms for all composites exhibited a well-defined PPy reduction peak at a potential of about −0.5 V for a potential scan rate of 1 mV s−1 and that the samples with porosities above 42% likewise displayed a well-defined PPy oxidation peak at approximately −0.4 V. For the more compact composite with 30% porosity a less distinctive and broad oxidation peak is instead seen. This electrochemical behaviour can be explained by the dependence of the rate of counter ion diffusion on the porosity of the composites as anions are incorporated and released upon the oxidation and reduction, respectively. The current minima observed at approximately +0.35 V and the subsequent increase in the current for more positive potentials can be explained by the onset of PPy overoxidation.41 In the present study, as well as in the previous study,17 care was taken to reverse the scan direction prior to the onset of the overoxidation since the latter reaction is known to involve PPy degradation.
As is seen in panels b–f in Fig. 3, the PPy oxidation peaks were shifted to more positive potentials with increasing scan rates and the oxidation peaks also became broader and less distinct as the scan rate was increased. It is immediately clear that this dependence of the shape of the voltammograms on the scan rate was most pronounced for the samples with the lowest porosities. For the least porous samples no clear oxidation peaks could therefore be seen which made it difficult to detect the onset of the overoxidation process for scan rates above 5 mV s−1 for the 30% porosity sample. The onset of overoxidation could likewise not be detected in samples with porosities of 42% and 52% at scan rates higher than 30 and 40 mV s−1, respectively. This behaviour thus stems from the fact that charge neutrality during PPy oxidation and reduction requires movement of counter ions. As will be shown in more detail below, the latter process requires longer and longer times for less porous samples due to the decreasing amounts of counter ions present within the samples. For very porous samples, containing a sufficiently large amount of counter ions in the vicinity of the composite fibres, the average counter ion diffusion length is short and the oxidation can proceed rapidly yielding a well-defined oxidation peak typically seen for planar diffusion controlled systems.
For less porous samples, which most likely also have larger tortuosity factors further complicating the mass transport, the counter ions need to diffuse over larger distances within and outside the samples and the diffusion process becomes less and less planar which explains the less distinct oxidation peaks seen for such samples.17,24 It is therefore evident that the voltammetric behaviour of the currently investigated PPy/nanocellulose composites of varying porosity is controlled by the counter ion diffusion process and that this process becomes slower and more complicated as the porosity of the samples decreases.
The effect of the porosity of the composites on the rate of the PPy oxidation can be further illustrated by calculating the specific oxidation charges from the voltammograms for the employed scan rates (see Fig. 4). To minimize the influence of the overoxidation process these charges were calculated from the first point of positive current to the current minimum between the oxidation and overoxidation peaks. When the scan rate was increased it became more difficult to detect the overoxidation onset the lower the porosity was. The maximum scan rate at which the oxidation charge was calculated therefore decreased with decreasing porosity.
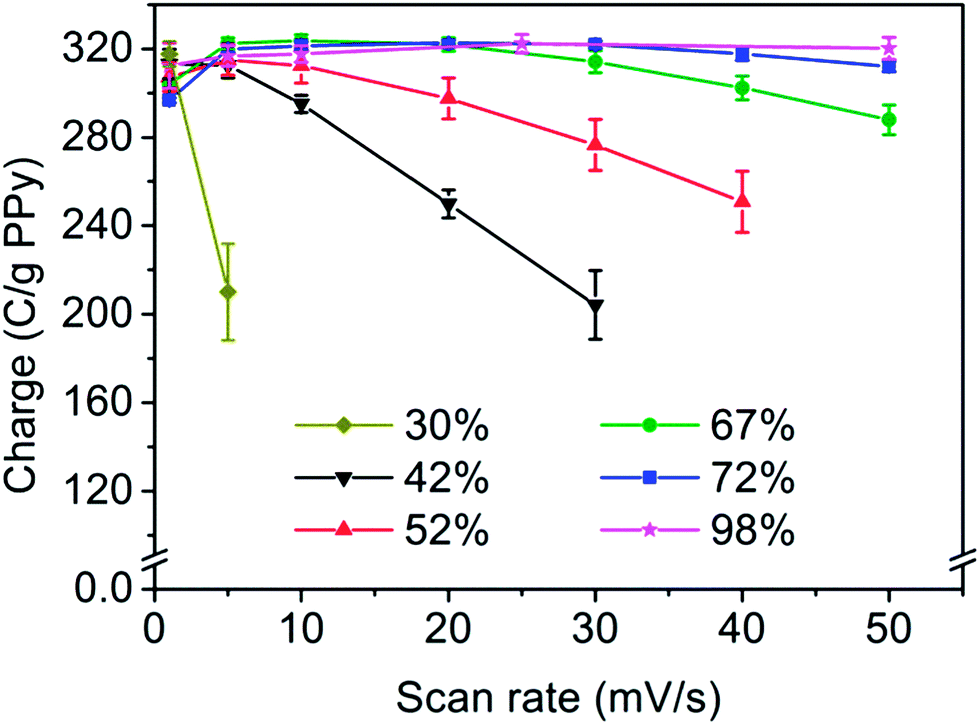 | ||
| Fig. 4 Specific cyclic voltammetric oxidation charges as a function of the scan rate for PPy/nanocellulose composites with different porosities. Previously reported17 composites with 30% and 98% porosity are included for comparison. The charges have been normalized with respect to the PPy amount in the composites. The error bars represent one standard deviation (n = 3) whereas the lines only are intended as guides to the eye. | ||
In Fig. 4, it is clearly seen that all samples exhibited the same specific charge (i.e. about 300 C g−1 PPy, see also Table 3) at the lowest scan rate, i.e. 1 mV s−1. As the same charge, corresponding to a doping degree of about 22% was obtained for all samples, it is reasonable to assume that all the samples could be fully oxidized, irrespective of their porosities, provided that the oxidation proceeded sufficiently slowly. The obtained capacities are also in good agreement with those previously obtained for Cladophora nanocellulose based composites (with a porosity of 82%) for which values of ∼310 C g−1 PPy were obtained at a scan rate of 5 mV s−1.37 As is also seen in Fig. 4, an increase in the scan rate resulted in a drop in the charge capacity for all samples, except for the composite with a porosity of 98%. This decrease was most pronounced for the least porous samples.
| Porosity (%) | Charge capacity at 1 mV s−1 (C g−1 PPy)a | Charge capacity at 1 mV s−1 (C g−1 composite)a | Counter ions in oxidation (mmol g−1 composite)a | Counter ions in composite pores (mmol g−1 composite) |
|---|---|---|---|---|
| a Expressed as the mean ± 1 standard deviation (n = 3). | ||||
| 98 | 312 ± 10 | 224 ± 7 | 2.32 ± 0.08 | 68.14 |
| 72 | 296 ± 1 | 197 ± 1 | 2.04 ± 0.01 | 3.60 |
| 67 | 304 ± 1 | 189 ± 1 | 1.96 ± 0.01 | 2.77 |
| 52 | 307 ± 6 | 224 ± 5 | 2.32 ± 0.05 | 1.50 |
| 42 | 313 ± 7 | 196 ± 4 | 2.03 ± 0.04 | 0.97 |
| 30 | 317 ± 6 | 215 ± 4 | 2.24 ± 0.04 | 0.58 |
To further verify that the oxidation rate depends on the porosity of the samples, chronoamperometric measurements were performed in which the samples were first subjected to reduction at −0.8 V for 600 s and then oxidation using a potential step to +0.3 V. As is evident from the resulting coulograms shown in Fig. 5 (in which analogous data for previously reported17 composites with 30% and 98% porosity also have been included for comparison) the oxidation rate decreases when the porosity is decreased, in agreement with the voltammetric results.
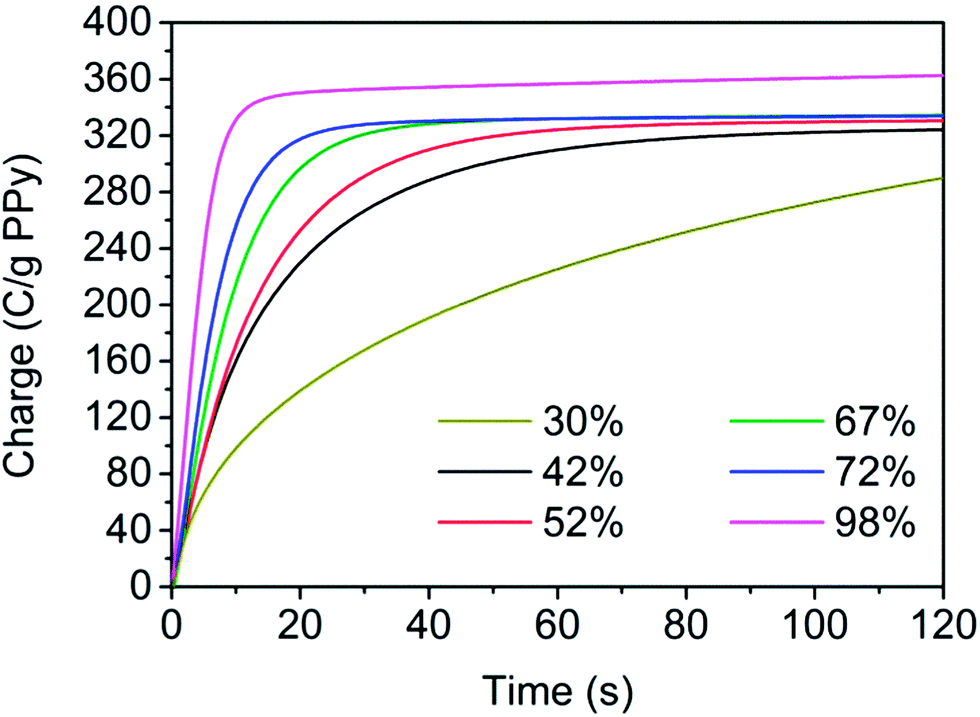 | ||
| Fig. 5 Coulograms for PPy/nanocellulose composites with different porosities obtained for oxidation at +0.3 V after a reductive pre-conditioning step at −0.8 V for 600 s. Previously reported17 composites with 30% and 98% porosity are included for comparison. The charges have been normalized with respect to the PPy amount in the composites. | ||
The results presented above clearly demonstrate that the porous samples are oxidized faster than the less porous ones. This indicates that the amount of counter ions within the pores of the samples was the main limiting factor and that this amount was large enough to yield a rapid oxidation process only for the most porous samples. This hypothesis is in good agreement with the data in Table 3, summarizing the amount of counter ions (i.e. Cl−) required in the PPy oxidation process, based on the average charge capacity obtained at 1 mV s−1 seen in Fig. 4, as well as the amounts of chloride ions estimated to be present in the different composites. The latter were calculated for an electrolyte concentration of 2 M NaCl and the porosities for the different samples. In Table 3, it is observed that the amount of chloride ions present in the least porous samples was insufficient compared to the amount of counter ions required for the charge compensation. This implies that a large number of counter ions would have to diffuse from the bulk electrolyte solution outside the composite and through the composite pore network to reach all PPy coated fibres during the oxidation process. It can also be anticipated that the diffusion would be more and more affected by tortuosity effects as the porosity of the samples decreases. These findings readily explain the long oxidation times obtained for the least porous samples. Conversely, an excess of chloride ions is found to be present in the most porous sample, indicating that the charge compensation process mainly involves diffusion of chloride ions present in the vicinity of the composite fibres. This would give rise to rapid planar diffusion in agreement with the experimental findings. As discussed earlier,17 it is also likely that a higher degree of composite fibre aggregation (which was observed for decreasing porosities) can contribute to the slower oxidation kinetics since this aggregation in essence results in thicker PPy-layers. Based on the present results it can be concluded that the rate of oxidation decreases with decreased porosity due to the increased influence of diffusion caused by the decreasing amount of counter ions within these samples and an increased influence of fibre aggregation and tortuosity effects. It is likewise clear that the present composite preparation approach significantly facilitates studies of the influence of the porosity on the electrochemical properties of PPy/nanocellulose composites. Such studies are very important, for example, in the optimization of electrode materials for charge storage devices as well as ion-extraction membranes.
4. Conclusions
It has been demonstrated that PPy/nanocellulose composites comprising ∼2/3 PPy, and with porosities ranging from 42% to 72%, can be made from MFC obtained by enzymatic hydrolysis or carboxymethylation, employing an ambient drying based approach. The proposed method complements previously described composite manufacturing processes by enabling the porosities to be controlled in a previously inaccessible range. With the present and previously described methods it is possible to prepare PPy/nanocellulose composites with porosities ranging from 30 to 98% with porosity increments of approximately 10%. The porosity of the composite is readily controlled via the type and amount of nanocellulose used in the composite synthesis procedure.It is further shown that the electrochemical properties of the composites are controlled by the porosities of the composites and that very fast oxidations only are seen for porous composites containing a sufficiently large amount of counter ions within the composite itself. For more compact composites, the oxidation requires more time as a result of the larger diffusion lengths and the more complex diffusion process due to the increased influence of tortuosity effects and fibre aggregation. The possibilities of controlling the porosity of PPy/nanocellulose composites presented in this work provide new and powerful means for optimisations of the properties of electrochemically controlled extraction devices and energy storage systems, as well as for fundamental studies of the impact of the porosity on the electrochemical properties of electroactive materials in general. In energy storage devices, such as batteries and supercapacitors, it is important to optimise the performance of the electrodes with respect to their gravimetric and volumetric capacities. The results of this study imply that the composites can be tailored to an optimum porosity, for which the charging rate of the device is sufficiently fast and the volume and thus the weight of the excess electrolyte has been minimized. Another important aspect is the effect that the porosity has on the mechanical properties. This should be investigated in future work employing samples covering the full porosity range.
Acknowledgements
The Swedish Foundation for Strategic Research (SSF), the Swedish Science Council (VR), the Bo Rydin Foundation, the Swedish Energy Agency, The Carl Trygger Foundation, The Knut and Alice Wallenberg Foundation, and the Nordic Innovation Centre (contract number 10014) are gratefully acknowledged for the financial support of this work. Tom Lindström and Eva Ålander at Innventia AB (Stockholm, Sweden) are kindly acknowledged for providing the MFC.Notes and references
- D. H. Milanez, R. M. Do Amaral, L. I. L. De Faria and J. A. R. Gregolin, Mater. Res., 2013, 16, 635–641 CrossRef PubMed.
- N. Lavoine, I. Desloges, A. Dufresne and J. Bras, Carbohydr. Polym., 2012, 90, 735–764 CrossRef PubMed.
- S. Eichhorn, A. Dufresne, M. Aranguren, N. Marcovich, J. Capadona, S. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. Nakagaito, A. Mangalam, J. Simonsen, A. Benight, A. Bismarck, L. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef PubMed.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef PubMed.
- I. Siró and D. Plackett, Cellulose, 2010, 17, 459–494 CrossRef.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- A. F. Turbak, F. W. Snyder and K. R. Sandberg, in Proceedings of the Ninth Cellulose Conference. Applied Polymer Symposia, ed. A. Sarko, Wiley, New York City, 1983, vol. 37, pp. 815–827 Search PubMed.
- F. W. Herrick, R. L. Casebier, J. K. Hamilton and K. R. Sandberg, in Proceedings of the Ninth Cellulose Conference. Applied Polymer Symposia, ed. A. Sarko, Wiley, New York City, 1983, vol. 37, pp. 797–813 Search PubMed.
- M. Henriksson, G. Henriksson, L. A. Berglund and T. Lindström, Eur. Polym. J., 2007, 43, 3434–3441 CrossRef PubMed.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934–1941 CrossRef PubMed.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef PubMed.
- H. Sehaqui, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2011, 71, 1593–1599 CrossRef PubMed.
- R. T. Olsson, M. A. S. Azizi Samir, G. Salazar Alvarez, L. Belova, V. Ström, L. A. Berglund, O. Ikkala, J. Nogues and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584–588 CrossRef PubMed.
- J. T. Korhonen, M. Kettunen, R. H. A. Ras and O. Ikkala, ACS Appl. Mater. Interfaces, 2011, 3, 1813–1816 Search PubMed.
- M. Hamedi, E. Karabulut, A. Marais, A. Herland, G. Nyström and L. Wågberg, Angew. Chem., Int. Ed., 2013, 52, 12038–12042 CrossRef PubMed.
- D. O. Carlsson, G. Nyström, Q. Zhou, L. A. Berglund, L. Nyholm and M. Strømme, J. Mater. Chem., 2012, 22, 19014–19024 RSC.
- M. Pääkkö, J. Vapaavuori, R. Silvennoinen, H. Kosonen, M. Ankerfors, T. Lindström, L. A. Berglund and O. Ikkala, Soft Matter, 2008, 4, 2492–2499 RSC.
- G. Inzelt, Conducting Polymers: A New Era in Electrochemistry, Springer, Berlin, 2008 Search PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef PubMed.
- Conjugated Polymers – Theory, Synthesis, Properties and Characterization, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, Boca Raton, 3rd edn, 2007 Search PubMed.
- L. Nyholm, G. Nyström, A. Mihranyan and M. Strømme, Adv. Mater., 2011, 23, 3751–3769 Search PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef PubMed.
- J.-H. Kim, Y.-S. Lee, A. K. Sharma and C. G. Liu, Electrochim. Acta, 2006, 52, 1727–1732 CrossRef CAS PubMed.
- G. Nyström, A. Mihranyan, A. Razaq, T. Lindström, L. Nyholm and M. Strømme, J. Phys. Chem. B, 2010, 114, 4178–4182 CrossRef PubMed.
- A. Mihranyan, L. Nyholm, A. E. Garcia Bennett and M. Strømme, J. Phys. Chem. B, 2008, 112, 12249–12255 CrossRef CAS PubMed.
- G. Nyström, A. Razaq, M. Strømme, L. Nyholm and A. Mihranyan, Nano Lett., 2009, 9, 3635–3639 CrossRef PubMed.
- K. Gelin, A. Mihranyan, A. Razaq, L. Nyholm and M. Strømme, Electrochim. Acta, 2009, 54, 3394–3401 CrossRef CAS PubMed.
- A. Razaq, G. Nyström, M. Strømme, A. Mihranyan and L. Nyholm, PLoS One, 2011, 6, e29243 CAS.
- N. Ferraz, D. O. Carlsson, J. Hong, R. Larsson, B. Fellström, L. Nyholm, M. Strømme and A. Mihranyan, J. R. Soc., Interface, 2012, 9, 1943–1955 CrossRef CAS PubMed.
- N. Ferraz, A. Leschinskaya, F. Toomadj, B. Fellström, M. Strømme and A. Mihranyan, Cellulose, 2013, 20, 1–12 CrossRef PubMed.
- N. Brun, S. R. S. Prabaharan, C. Surcin, M. Morcrette, H. Deleuze, M. Birot, O. Babot, M. F. Achard and R. Backov, J. Phys. Chem. C, 2012, 116, 1408–1421 CAS.
- Y. Xu, Z. Sui, B. Xu, H. Duan and X. Zhang, J. Mater. Chem., 2012, 22, 8579–8584 RSC.
- X. Zhang, D. Chang, J. Liu and Y. Luo, J. Mater. Chem., 2010, 20, 5080–5085 RSC.
- C. Sasso, D. Beneventi, E. Zeno, M. Petit-Conil, D. Chaussy and M. N. Belgacem, Synth. Met., 2011, 161, 397–403 CrossRef CAS PubMed.
- C. Sasso, E. Zeno, M. Petit-Conil, D. Chaussy, M. N. Belgacem, S. Tapin-Lingua and D. Beneventi, Macromol. Mater. Eng., 2010, 295, 934–941 CrossRef CAS.
- A. Razaq, L. Nyholm, M. Sjödin, M. Strømme and A. Mihranyan, Adv. Energy Mater., 2012, 2, 445–454 CrossRef CAS.
- A. Mihranyan, M. Esmaeili, A. Razaq, D. Alexeichik and T. Lindström, J. Mater. Sci., 2012, 47, 4463–4472 CrossRef CAS.
- K. Hua, D. O. Carlsson, E. Ålander, T. Lindström, M. Strømme, A. Mihranyan and N. Ferraz, RSC Adv., 2014, 4, 2892–2903 RSC.
- D. O. Carlsson, M. Sjödin, L. Nyholm and M. Strømme, J. Phys. Chem. B, 2013, 117, 3900–3910 CrossRef CAS PubMed.
- Y. Li and R. Qian, Electrochim. Acta, 2000, 45, 1727–1731 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |