
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Film-forming polymer nanoparticle strategy for improving the passivation and stability of perovskite solar cells†
Zhenyu
Jia
 *a,
Ran
Wang
a,
Lei
Zhu
b,
Amal
Altujjar
ac,
Polina
Jacoutot
d,
Osama M.
Alkhudhari
ae,
Muhamad Z.
Mokhtar
af,
Ben F.
Spencer
ag,
Nigel W.
Hodson
h,
Xuelian
Wang
a,
Mollie
Osborne-Richards
a,
Andrew G.
Thomas
ag,
Teruo
Hashimoto
a,
Michael
Faulkner
a,
David J.
Lewis
a,
Saif A.
Haque
d,
M. Saiful
Islam
b,
Jennifer M.
Saunders
a and
Brian R.
Saunders
*a
*a,
Ran
Wang
a,
Lei
Zhu
b,
Amal
Altujjar
ac,
Polina
Jacoutot
d,
Osama M.
Alkhudhari
ae,
Muhamad Z.
Mokhtar
af,
Ben F.
Spencer
ag,
Nigel W.
Hodson
h,
Xuelian
Wang
a,
Mollie
Osborne-Richards
a,
Andrew G.
Thomas
ag,
Teruo
Hashimoto
a,
Michael
Faulkner
a,
David J.
Lewis
a,
Saif A.
Haque
d,
M. Saiful
Islam
b,
Jennifer M.
Saunders
a and
Brian R.
Saunders
*a
aDepartment of Materials, University of Manchester, Engineering Building A, Manchester M1 7HL, UK. E-mail: zhenyu.jia@postgrad.manchester.ac.uk; brian.saunders@manchester.ac.uk
bDepartment of Materials, University of Oxford, Oxford OX1 3PH, UK
cBasic Science Department, Deanship of Preparatory Year and Supporting Studies, Imam Abdulrahman Bin Faisal University, Dammam 34221, Kingdom of Saudi Arabia
dDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, Wood Lane W12 0BZ, UK
eDepartment of Chemistry, Taif University, Taif 21974, Saudi Arabia
fDepartment of Water and Environmental Engineering, M46, Faculty of Civil Engineering, Universiti Teknologi Malaysia, 81310 Johor Bahru, Malaysia
gPhoton Science Institute, The Henry Royce Institute, University of Manchester, Manchester M13 9PL, UK
hBioAFM Facility, Faculty of Biology, Medicine and Health, University of Manchester, Stopford Building, Oxford Road, Manchester M13 9PT, UK
First published on 29th August 2024
Abstract
Perovskite solar cells (PSCs) provide excellent potential for large-scale solar energy harvesting because of their high power conversion efficiency (PCE) and facile preparation. However, long-term stability remains the Achilles heel for PSCs and new methods for improving stability are urgently needed. Film-forming polymer nanoparticles are widely used in the surface coatings industry to provide scalable, low-cost protection to surfaces. Inspired by this, here we add three soft, film-forming crosslinked polymer nanoparticle systems (nanogels) with different diameters (60, 100 and 200 nm) to the perovskite (FA0.75MA0.25PbI3) precursor solution and study their effects on PSC performance. The nanogels contain hydrophilic ethylene oxide repeat units and form films when their diameter is less than or equal to 100 nm. Device stability was assessed using shelf-life and light-soaking measurements; in addition, films were subjected to moisture challenge tests. We show that nanogel coverage increases the PCE and stability. Ab initio simulations indicate strong binding of the molecules that comprise the nanogels on the perovskite surface through Pb–O bonds, thereby promoting enhanced surface stability in agreement with the XPS and FTIR signals associated with the binding of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O groups to Pb. Shelf-life stability measurements showed that nanogels with a diameter of 100 nm gave devices that retained >80% of their initial PCE after more than six months compared to 25% for the control. In contrast, the 200 nm nanogels did not form films or increase the PCE or shelf-life stability. The 100 nm nanogels also gave the best stability to light soaking. Our results show that highly deformable hydrophilic film-forming nanogel particles with a size of ∼100 nm provide a scalable alternative approach for improving passivation and stability of PSCs.
O and C–O groups to Pb. Shelf-life stability measurements showed that nanogels with a diameter of 100 nm gave devices that retained >80% of their initial PCE after more than six months compared to 25% for the control. In contrast, the 200 nm nanogels did not form films or increase the PCE or shelf-life stability. The 100 nm nanogels also gave the best stability to light soaking. Our results show that highly deformable hydrophilic film-forming nanogel particles with a size of ∼100 nm provide a scalable alternative approach for improving passivation and stability of PSCs.
Broader contextWhilst silicon solar cells continue to dominate deployment of solar cell technology, the rise of perovskite solar cells (PSCs) is beginning to gain commercial momentum. Nevertheless, stability improvements for PSCs are required in order to successfully compete with the 25-year warranties offered for silicon solar cells. A major source of instability for PSCs is surface defects and new approaches for decreasing trap density via passivation are urgently needed. Nanogels (swellable crosslinked polymer nanoparticles) that are highly deformable and expose previously hidden binding groups upon deposition are a new macro-passivating additive for PSCs. Here, we investigate the effect of nanogel size on the passivation and stability of PSCs and report that the optimum size is 100 nm. We demonstrate that our film-forming nanogels decrease trap density and increase PSC stability. Using a combination of ab initio simulation and experimental techniques, we discuss the mechanisms and relations that underpin these findings. |
Introduction
Perovskite solar cells (PSCs) have received tremendous attention owing to their outstanding properties such as high absorption coefficient, large carrier diffusion length, tuneable bandgap, as well as facile and potentially low cost construction.1–9 Since the seminal study of Miyasaka et al. in 2009,10 the power conversion efficiency (PCE) for PSCs has increased dramatically from 3.8% to 26.1%.11 Accordingly, PSCs have considerable potential for commercial exploitation. However, one of the main obstacles to such exploitation is their relatively poor stability,12 which originates from mobile ions,13,14 defects,15–17 sensitivity to oxygen,18 water19,20 and light21,22 as well as poor mechanical toughness.23 Enhanced ion migration and non-radiative recombination can occur at defect sites, leading to degradation and PCE reduction.24,25 Defects at interfaces and grain boundaries can become conduits where moisture invades.26,27 To address these issues, additives have been used to engineer the perovskite layer which has resulted in increases in PCE and stability.1,28–31 However, even the highest PCE systems still degrade over relatively short periods of time compared to silicon solar cells which have warranties of 25 years.32 Our motivation for the present study is to address the urgent need to explore new types of additives for improving PSC stability.Self-assembled monolayers (SAMs) have shown promise as an effective approach to improve PSC stability.33 An iodine-terminated SAM was used to increase the adhesion toughness at the interface between the electron transfer layer (ETL) and perovskite as well as stability.34 The approach has been extended to include flexible PSCs.35 In addition, a ruthenium dye-based SAM has been used as an interlayer between perovskite and the hole transfer layer (HTL) to increase PSC stability.36
Additive engineering aims to use species added to the perovskite precursor solution or interfaces to decrease the defect density and thereby increase PCE and stability.17,37 Additives have principally involved small molecules1,28,38,39 and conventional polymers.40,41 Small molecules that have been successful in improving the properties include phenethylammonium iodide,42 methylammonium chloride1 and diammonium spacer ligands.37 Examples of polymers used include polyacrylic acid,40,43,44 polyethyleneimine,40,43 polyethylene glycol,40,45,46 polyacrylonitrile47 and poly(ethylene dimethacrylate-polyhedral oligomeric silsesquioxane).48
Polymer additives can increase the nucleation rate and retard crystal growth49,50 to enlarge crystal grain size and effectively reduce the defect density at the grain boundaries. In addition, polymers can also improve the adhesion strengths and reinforce the interfaces of PSCs.51 However, the use of polymers as additives in PSCs poses a dilemma. The strong multiple interactions that would benefit the mechanical and device stability of the perovskite films also prevent formation of compact perovskite films due to interference with perovskite crystallization.40 This problem could be overcome by using deformable polymer nanoparticles such as nanogels that flatten and form films after perovskite crystallization to present previously hidden functional groups to the perovskite surface; such an approach is the focus of the present study. Nanogels are nanosized crosslinked polymer colloid particles that swell in a good solvent.52 Comparing dispersions of polymer nanogels to polymer solutions, the former have much lower viscosities which enables facile delivery of the polymer to surfaces.
We have used poly(N-isopropylacrylamide) (PNP) microgels to improve the performance of semi-transparent PSCs.53,54 (Microgels are nanogels at a larger length scale.) Here, a new alternative family of nanogels is investigated (Fig. 1a) in non-semitransparent PSCs that deliver much higher PCEs. The nanogels used in the present study are comprised of a different polymer to PNP and uniquely display film-forming behavior in the perovskite layer. The advantage of using nanogels or microgels is that, provided the particles swell in the precursor solvent (which acts as a good solvent), they do not agglomerate during perovskite film formation because of strong steric stabilization. The latter originates from the entropy decrease from a loss of configurational freedom if polymer chains of (swollen) neighboring nanogels interpenetrate. We have also found that the size of these particles determines the morphology of the perovskite films with large microgels dictating the morphology53 whilst smaller nanogels can be dispersed throughout the film.55 The present study exploits the excellent colloidal stability of film-forming nanogels to passivate the perovskite and improve stability.
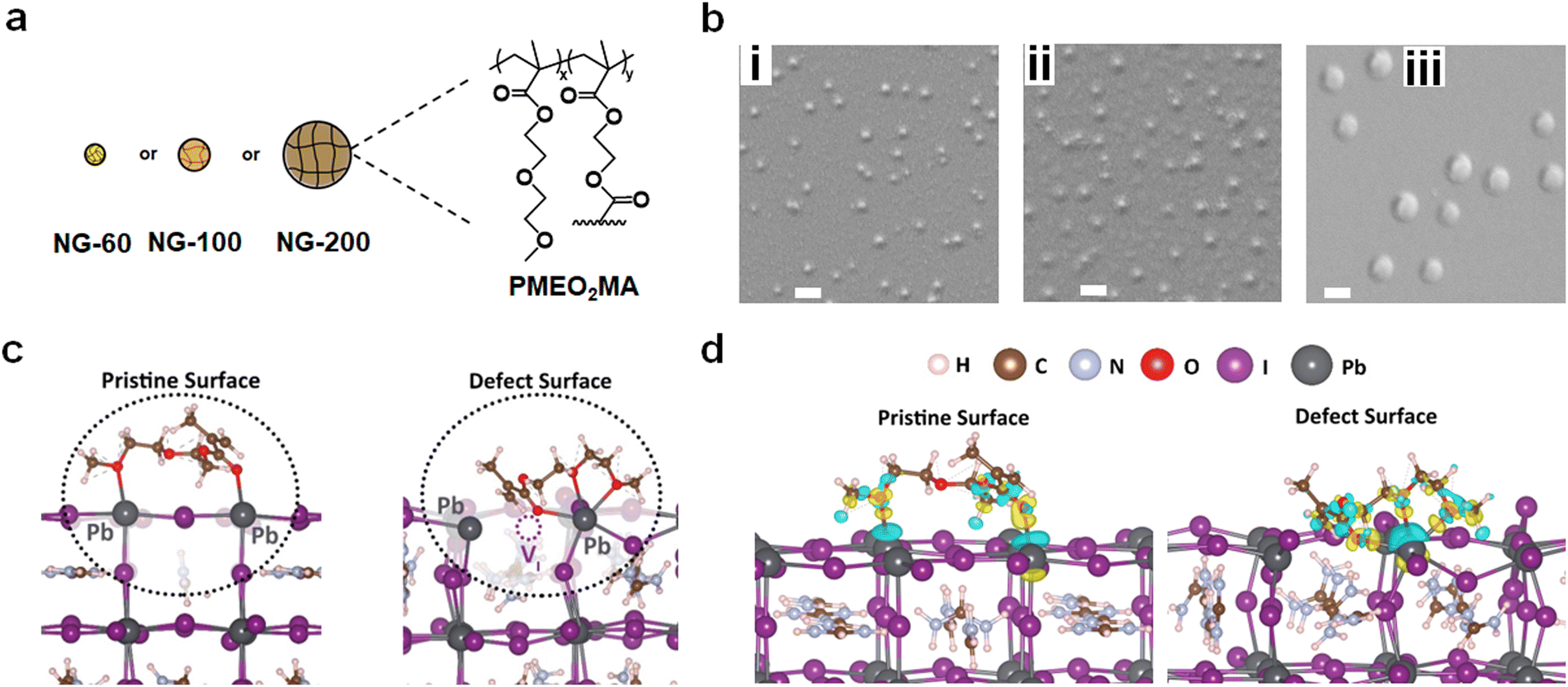 | ||
| Fig. 1 (a) Three dispersions with different PMEO2MA nanogel diameters are used in this study: small (NG-60), relatively large (NG-100) and very large (NG-200). (b) SEM images for (i) NG-60, (ii) NG-100 and (iii) NG-200 particles. Scale bars 200 nm. (c) Depiction of the nature of MEO2MA bonding on the pristine (left) and the defect (right) (001) PbI2-terminated FAPbI3 surface. (d) Side view of the lowest-energy configuration and charge transfer density difference of MEO2MA bonding on the pristine (left) and the defect (right) surface. Charge density gain and loss are colored in yellow and blue, respectively. | ||
Here, we employ poly(2-(2-methoxyethoxy)ethylmethacrylate) (PMEO2MA) nanogels (Fig. 1a) as film-forming nanoparticles. (Note that x ≫ y for the structure in Fig. 1a.) PMEO2MA has a low glass transition temperature56 and a strong tendency to deform when deposited.57 By flattening on surfaces nanogels increase their surface area and expose previously buried functional groups. Furthermore, nanogels can partially coalesce on surfaces when the solvent evaporates.58 Such a process has been the basis for surface coatings for decades59,60 and is investigated here in PSCs for the first time. The nanogel additive approach used in this study is inherently simple because the nanogels are added to the perovskite precursor solution prior to deposition. Our approach has similarities to the use of polymer binders in the preparation of organic field-effect transistors,61–63 in which linear polymers are employed to provide property tuning. The approach used in this study is distinct in that we use polymer colloid nanogel particles and apply them to the challenge of PSC stability improvement.
We apply a facile nanogel film-forming strategy for delivering PMEO2MA to perovskite grain surfaces. Three nanogel systems with different diameters are used (Fig. 1a). We report that the film-forming nanogels provide much improved performance compared to the non-film-forming system. We explain these results in terms of increased contact and passivation of the PMEO2MA chains with the perovskite. FTIR and ab initio simulation results show that PMEO2MA binds to Pb2+ and transient absorption spectroscopy studies reveal that the thin nanogel film does not obstruct hole transport. The film-forming nanogels are also shown to improve the mechanical properties of the perovskite films as assessed by adhesion tests. The deformable film-forming nanogel approach introduced in this study is scalable and should be applicable to other PSCs.
Results and discussion
Nanogel characterization and insights into interactions between PMEO2MA and the perovskite surface
Three PMEO2MA nanogel systems were prepared (see Experimental details and Table S1, ESI†) and are denoted as NG-60, NG-100 and NG-200 based on their diameters in nm (Fig. 1a). Scanning electron microscopy (SEM) images obtained by drying diluted nanogel dispersions gave average diameters of 60, 103 and 202 nm, respectively, for NG-60, NG-100 and NG-200 (Fig. 1b and Table S2, ESI†), with their size distributions shown in Fig. S1 (ESI†). Atomic force microscopy (AFM) data measured for the NG-200 particles deposited from water are shown in Fig. S2 (ESI†). The average particle diameter and height are 237 and 69 nm, respectively, giving an aspect ratio (diameter/height) of 3.4. Hence, the PMEO2MA nanogel particles tend to flatten when deposited from the swollen state64 as can be seen from the line profile of a NG-200 particle (Fig. S2, ESI†). Additional discussion and characterization data for the nanogels are given in the ESI† (Note 1 and Fig. S3, ESI†).To gain preliminary atomic-scale insights into the interactions of these nanogels at the perovskite surface, density functional theory (DFT) simulation techniques were employed. An optimized monomeric representation of the PMEO2MA molecule, namely, MEO2MA (Fig. S4, ESI†), was placed on top of formamidinium lead tri-iodide (FAPbI3), which constitutes about 75% of the perovskite composition in our experiments. It is known that iodide vacancy defects play a role in non-radiative recombination and ion migration, which can decrease the PSC device efficiency.65,66 Therefore, our computational investigation focuses on the interactions with the pristine surface as well as the surface where an iodine vacancy defect is present. The (001) PbI2-terminated surface was constructed, as this surface configuration has been reported to be one of the most thermodynamically stable and highly studied halide perovskite surfaces.67 (Computational details are provided in the Experimental details in the ESI.†)
Fig. 1c (left) illustrates the optimized configuration of MEO2MA on the perovskite surface, which shows the molecule reshaped into a less planar configuration with two O atoms interacting with two nearest Pb2+ ions at the perovskite surface. An increased band gap of about 30 meV was found compared to that for the pure FAPbI3 surface, suggesting that a blue shift may be found spectroscopically, which is shown to be in agreement with our measurements below.
The binding energy at the perovskite surface is found to be −1.42 eV, indicating favourable bonding affinity between the MEO2MA molecule and surface. The Pb–O distances between the O atom of the methacrylate moiety carbonyl (CO) group and the O atom of the 2-(2-methoxyethoxy)ethanol moiety C–O group with their bonded Pb2+ ions are 2.55 Å and 2.70 Å respectively. Notably, both of these values are significantly shorter than the Pb–I bond length (3.14 Å) in bulk FAPbI3, indicating the stabilization of the undercoordinated surface Pb2+ ions. The chemical bonding was further confirmed by the high charge density around the oxygen atoms and the charge transfer profile at both Pb–O bonds from the charge density difference (Fig. 1d, left). Such O–Pb bonding is shown to be in agreement with our XPS and FTIR data below.
To further investigate the interactions, ab initio molecular dynamics (AIMD) simulations were carried out at 300 K, starting at the lowest-energy monomer-surface configuration. On the pristine and defective surfaces, the dynamics of the molecule and surface relaxations (shown in ESI,† Videos S1 and S2) indicate that the additive molecule remains close to the perovskite surface, regardless of the presence of the facile vacancy defect.
The interactions on the defective surface indicate the two O atoms from the 2-(2-methoxyethoxy)ethanol moiety (C–O) form bonds with the same nearest Pb2+ ions, while the O atom of the carbonyl group (CO) stabilizes near the iodide vacancy site, effectively reducing the surface vacancy density. This oxygen is likely to bond with either of the nearest Pb2+ ions adjacent to the vacancy site, but the lowest-energy configuration was found in which three oxygen atoms bond to the Pb2+ ions (Fig. 1c, right). The binding energy is −2.23 eV, which is about 0.8 eV more favourable than that for the pristine surface indicating strong defect passivation. It should be noted that the bonding scenario where one O atom bonds to the other adjacent Pb2+ ion was found slightly less favourable (−1.97 eV) and shown in Fig. S5 (ESI†). The bonding between O atoms and the Pb2+ ion is further confirmed by the O–Pb bond lengths (2.65–2.92 Å) and the charge density transfer at the O–Pb bonds from the DFT simulations (Fig. 1d, right). Such strong molecule–perovskite interactions increase the anchoring strength on the surface, stabilize the undercoordinated Pb, and passivate the surface iodine vacancy, all of which are expected to improve the perovskite performance and stability.
We next investigate experimental data for nanogel film formation, perovskite films and devices prepared using precursor solutions containing the nanogels. The precursor compositions used for all of the perovskite films are given in Table S3 (ESI†).
Nanogel film formation
The nanogel concentrations employed in this work are given in terms of the mass concentration of nanogel in the precursor solution (mg g−1). A preliminary study using the NG-100 system established that a nanogel concentration of 3.0 mg g−1 was optimum for device PCE (see Note 2 and Fig. S6 and S7, as well as Table S4, ESI†). (The effect of nanogel concentration for the NG-60 and NG-200 systems is studied later.) A nanogel concentration of 3.0 mg g−1 is used unless otherwise stated.We investigated the effect of spin-coating for the three nanogel dispersions onto bl-TiO2/ITO using SEM (see Fig. 2). Interestingly, individual NG-60 and NG-100 particles are not readily apparent. The NG-60 system showed nanosized films of ∼1000 nm in diameter (Fig. 2a). Such features are due to localized nanogel coalescence and nanofilm formation. NG-100 also showed such nanofilms; however, they are smaller with a size of ∼200 nm, which is indicative of localized coalescence with fewer nanogels coalesced per nanofilm (Fig. 2b). In contrast, NG-200 showed almost exclusively isolated particles (Fig. 2c) with only occasional limited inter-nanogel coalescence.
 | ||
| Fig. 2 SEM images ITO/bl-TiO2 substrates after spin-coating DMF/DMSO dispersions (3.0 mg g−1) of (a) NG-60, (b) NG-100 and (c) NG-200 particles. An SEM of the bare ITO/bl-TiO2 substrate is shown in (d) for comparison. The yellow circles highlight coalesced nanogels. Scale bars: 200 nm. | ||
These trends arise because the average separation of the nanogels reduces with decreasing nanogel size at constant mass surface concentration (i.e., nanogel mass per unit area). Furthermore, the likelihood of nanogel coalescence (and film formation) on a surface increases as the average interparticle separation decreases. Hence, nanofilm formation is more favorable at a concentration of 3.0 mg g−1 for NG-60 and NG-100, but less so for NG-200.
Morphology of perovskite films prepared using nanogels with different sizes
SEM images are shown for the perovskite films prepared with different nanogels (Fig. 3). Nanocrystals that grow vertically are apparent for some of the systems and we will discuss their origin below. The average grain size (Fig. S8, ESI†) decreased in the order NG-60 (1820 nm) > NG-100 (1625 nm) > CTRL (1375 nm) > NG-200 (1270 nm). As shown in the ESI† (Note 3), the estimated specific surface areas (As) of the NG-60, NG-100 and NG-200 particles are 165, 103 and 34 m2 g−1, respectively. Consequently, the nanogels have high surface areas available to bind to perovskite colloids that are present during crystallization. We conducted a crystallization rate experiment by following the change in the color of the films visually as a function of time during annealing (Fig. S9, ESI†). We used a nanogel concentration of 10 mg g−1 for this experiment to amplify the differences to enable clear visualization of the effect of nanogels on crystallization rate. (This produced some macroscopic particles which adversely affected compact perovskite structure formation and contributed to the decreased PCE for the 10 mg g−1 NG-100 system in Table S4 (ESI†).) The results show a significantly decreased crystallization rate in the presence of the nanogels. Such a decrease in the perovskite growth rate would increase the average grain sizes as shown by McMeekin et al.68 Indeed, it is likely that nanogels binding with PbI2 contributed to the decreased crystallization rate. In support of this proposal, DFT calculations of the charge transfer density (Fig. S10a, ESI†) and binding energies (Fig. S10b, ESI†) showed that PbI2 interacts favorably with MEO2MA monomer via Pb–O bonding.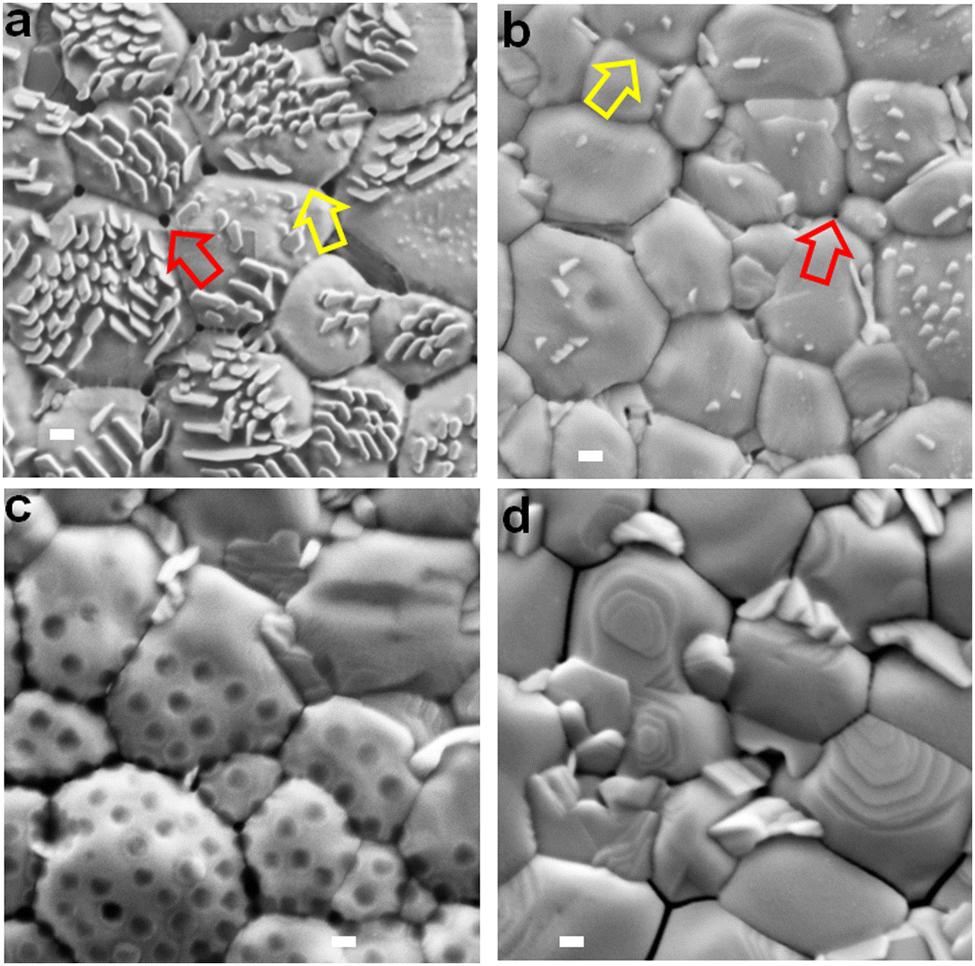 | ||
| Fig. 3 Top view SEM images of the perovskite films prepared using (a) NG-60, (b) NG-100, (c) NG-200 and (d) the control without nanogels. The yellow arrows highlight nanofilm bridges between perovskite grains. The red arrows highlight small pinholes in the bridging nanofilms at the junction of three perovskite grains. Scale bars: 200 nm. See also Fig. S11 (ESI†). | ||
Close examination of the SEM images of the perovskite/nanogel systems reveals that perovskite grains for the NG-60 system (Fig. 3a) are more rounded and have a less crystalline morphology compared to the control (Fig. 3d) as well as to those prepared with NG-100 (Fig. 3b) and NG-200 (Fig. 3c). Whilst individual NG-60 and NG-100 particles are absent, nanofilm segments can be seen bridging grains (yellow arrows). In contrast, the NG-200 system shows features due to NG-200 particles (Fig. 3c) with no evidence of nanofilms. NG-200 particles that bridge grains can also be seen. The control film shows large gaps between the grains (Fig. 3d). (These features are also shown in expanded SEM images in Fig. S11, ESI.†) Pinholes in the nanofilms are evident at the junctions of three grains (red arrows) in Fig. 3a and b, which are smallest for the NG-100 system. We conclude that nanofilms are present for the perovskites prepared with NG-60 and NG-100. In contrast, only individual nanogels (and not nanofilms) are present for perovskite/NG-200.
We were intrigued by the vertical nanocrystals and found that the NG-200 particles when used at 10 mg g−1 favor the growth of vertical nanocrystals adjacent to them (see Fig. S12, ESI†). It is likely that such a process also occurred for the smaller nanogels. Indeed, increasing the NG-100 concentration to greater than 3.0 mg g−1 increases the vertical nanocrystal surface concentration (Fig. S13, ESI†). Consequently, the surface concentration of vertical nanocrystals is an indirect measure of nanogel coverage. The link between vertical nanocrystals and nanogel coverage is discussed in more detail in the ESI† (Note 4). It follows from Fig. 3 that the nanogel film coverage was greatest for the NG-60 system and least for the NG-200 system. This suggestion was tested by conducting water contact angle measurements on the films (Fig. S14, ESI†). The nanogels are hydrophilic as discussed in the ESI† (Note 1). The contact angle for the perovskite films decreases in the order: CTRL > NG-200 > NG-100 > NG-60. Hence the surface becomes increasingly hydrophilic as the nanogel size decreases, which is attributed to greater coverage by nanogel nanofilms. This conclusion is congruent with the As calculations (Note 3, ESI†) wherein, the area of surface covered by a fixed nanogel mass concentration will increase in the order NG-200 < NG-100 < NG-60.
An interesting question concerns why a higher nanogel concentration is required to trigger vertical nanocrystal growth for NG-100 and NG-200 systems compared to NG-60. The concentration required to obtain a given coverage of the perovskite by the nanogels will increase as As decreases. A concentration of 3 mg g−1 for NG-60 (As = 165 m2 g−1) was required to produce a high surface concentration of vertical nanocrystals (Fig. 3a). However, perovskites prepared using 3 mg g−1 NG-100 (As = 103 m2 g−1) and NG-200 (As = 34 m2 g−1) had fewer vertical nanocrystals (Fig. 3b and c) due to less surface coverage by these nanogels. Accordingly, a higher nanogel concentration (e.g., 10 mg g−1) was required for NG-100 and NG-200 to give sufficient surface coverages to trigger growth of high vertical nanocrystal concentrations (Fig. S12 and S13, ESI†).
Energy-dispersive X-ray (EDX) analysis was employed to analyze the composition of the vertical nanocrystals on the surface of perovskite films (Fig. S15, ESI†). A perovskite/NG-60 film (3 mg g−1) was examined as well as the control film. Three distinct positions were scanned in each film. The corresponding elemental weight percentages at these positions are detailed in Table S5 (ESI†) as well as the Pb/I ratios. In the regions where only planar grains were analyzed (positions 1–3 of the control and positions 2 and 3 of the NG-60 sample), the Pb/I ratios were in the range of 0.64–0.86. This can be compared to the value expected of 0.56 based on the masses of PbI2, FAI and MAI used for FA0.75MA0.25PbI3 synthesis. Hence, the planar grain surfaces are iodine-deficient. Iodide ions can easily diffuse due to their relatively low migration energy.14,69 Also, I2 liberation at surfaces is well known for perovskites.70,71 Interestingly, a probe of the vertical nanocrystals (position 1 of Fig. S15(b), ESI†) gave a Pb/I ratio of 0.52, which is very close to that expected. This may indicate that the vertical nanocrystals are less susceptible to loss of I.
SEM cross-sectional images were also obtained for the films (Fig. S16, ESI†). The cross-sections for the NG-60 (Fig. S16a, ESI†) and NG-100 (Fig. S16b, ESI†) systems show many aligned amorphous grooves and undulations. In contrast, the NG-200 (Fig. S16c, ESI†) and control (Fig. S16d, ESI†) cross-sections show well-defined crystalline grains. Accordingly, the morphologies of the NG-60 and NG-100 perovskite fracture surfaces are indicative of plastic flow; whereas, those for the NG-200 and control films are typical of brittle fracture. To confirm this suggestion, we prepared a perovskite/NG-100 film using a nanogel concentration of 20 mg g−1 and obtained SEM cross-section images. A representative image (Fig. S17, ESI†) shows extensive plastic flow, due to a thicker NG-100 film. It is important to note that the inset from the cross-section (Fig. S17, ESI†) captures individual and coalesced NG-100 particles deep within the perovskite film between two grains. Hence, not only do these nanogels form nanofilms at the perovskites surface, nanogels reside at grain boundaries in the perovskite interior.
Solar cell performance and stability
PSCs were constructed using a planar structure: glass/ITO/TiO2/perovskite (nanogels)/Spiro-OMeTAD/Au (Fig. 4a). Box plots showing the measured device data appear in Fig. 4b. Photocurrent density–voltage (J–V) curves for the best performing devices of each class are shown in Fig. 4c and the data are listed in Table S4 (ESI†). The overall best performing device is NG-100 with a champion PCE of 20.08%. This is almost 1.7% higher than the champion control device (18.40%). The best PCEs for the NG-60 (19.12%) and NG-200 (17.84%) systems are higher and lower, respectively, than the control device. Hence, both NG-100 and NG-60 improve the PCE of the PSCs; whereas, NG-200 decreases the PCE. Results from a literature survey shown in Table S6 (ESI†) reveal that the best PCE measured in this study is above average for comparable systems reported in the literature. Our devices were not designed for maximizing the PCE. We stress that the focus of this study is on passivation and stability in the model FA/MA system. Comprehensive studies on FA/MA/Cs or FA/Cs perovskites are planned for future detailed investigations. The data shown in Fig. 4b demonstrate that variations of the open-circuit voltage (Voc) and fill factor (FF) are primarily responsible for the PCE changes. Moreover, the data shown in Fig. S18 (ESI†) reveal that the average PCE measured during the reverse sweeps has a linear relationship with the product of the Voc and (FF/100). Both of these quantities are increased by NG-60 and NG-100.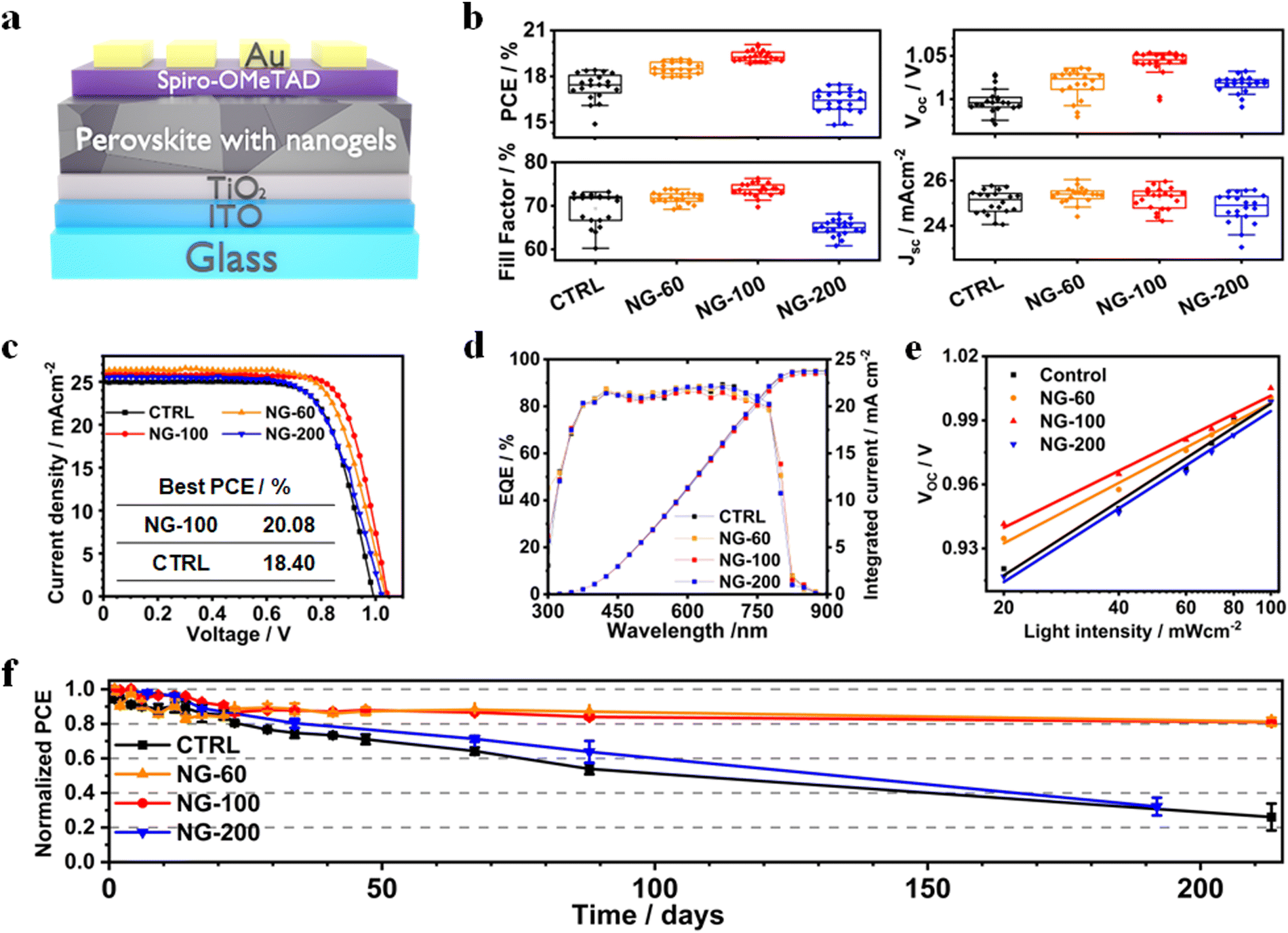 | ||
| Fig. 4 (a) Device architecture used. (b) Measured PCEs, fill factors, open-circuit voltages and short-circuit current densities for the devices. (c) J–V curves for selected devices. (d) EQE data for the devices. (e) The variation of the open-circuit voltage with incident light intensity. (f) Normalized PCE shelf-life storage data for the PSCs. The unencapsulated devices were stored at room temperature in the dark under ambient conditions, RH = 35%. | ||
The EQE spectra (Fig. 4d) show that the integrated short-circuit current density (Jsc) values agree with those from the J–V curves to within 8%. The hysteresis index (Fig. S19 and Table S4, ESI†) is a minimum of 7.6% for the NG-100 system compared to 9.1% for the control. The average values for the NG-60 and NG-200 devices are 12.7% and 9.8%, respectively. Hence, these results suggest that NG-100 is better able to oppose the build-up of mobile ions that are the major contributor to hysteresis in PSCs.72
Light intensity variation against Voc data were measured (Fig. 4e) and analyzed to investigate charge recombination behavior. The ideality factor (n) is calculated using eqn (1),73
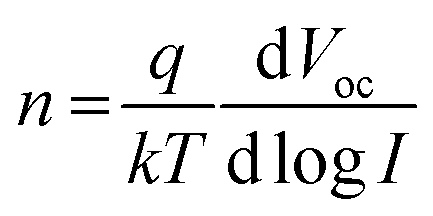 | (1) |
Space-charge-limited current (SCLC) tests were performed to analyse the trap density in the perovskite films using electron-only (ITO/TiO2/perovskite/PC61BM/Au) devices. Fig. S21 (ESI†) shows the dark current–voltage characteristics of various devices and the trap-filled limit voltage (VTFL) values. The calculation of trap density used77
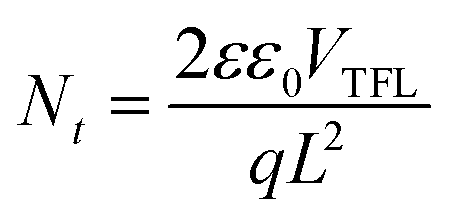 | (2) |
To explore the stability of the PSCs, we monitored the PCE variation of unencapsulated devices for more than 210 days (Fig. 4f). The devices were stored in the dark at 25 °C. The time to reach 80% of the initial PCE (t80) values for the control and NG-200 systems are 23 and 34 days, respectively. In contrast, the t80 values for the NG-60 and NG-100 devices are greater than 212 days, which is a record shelf-life stability for perovskite solar cells containing microgels or nanogels. Light soaking tests were also performed for non-encapsulated devices in ambient air (Fig. S22(a), ESI†). We used the linear portion of the normalized PCE versus time data to calculate the PCE decay rates (Fig. S22(b), ESI†). These data show that the rate of PCE decay increased in the order NG-100 < NG-60 < NG-200 < control. These device-based stability results demonstrate that NG-60 and NG-100 significantly improve PSC stability. UV-visible spectra and film photographs for the control, NG-60, NG-100 and NG-200 perovskite/nanogel systems over 21 days exposure to high moisture content air (85% RH, Fig. S23, ESI†) demonstrate that the NG-60 and NG-100 particles greatly improve the film stability against degradation by water vapour. In contrast, the NG-200 system did not exhibit good protection for the perovskite. These results provide experimental confirmation of the stability improvement predicted by the DFT simulations.
While protecting the perovskite from degradation at the interface, the influence of nanogels on the yield of photoinduced hole transfer across the perovskite-NG/spiro-OMeTAD heterojunction is of particular importance. For example, it is critically important that the presence of the NG material does not impede this hole transfer process. To study this, we employed transient absorption spectroscopy (TAS) using the NG-100 particles as they gave the best device performance. The TA kinetics for thin films with and without NG-100 nanofilm interlayer are shown in Fig. S24 in the ESI.† Thin films of TiO2/perovskite plus NG-100/Spiro on glass were optically excited using nanosecond laser pulses at 510 nm, ensuring selective charge photogeneration in the perovskite layer.80 Following the charge transfer from the perovskite conduction band to the hole transporting layer, a near-infrared probe (at 1600 nm) was used to track the polaron absorption kinetics in the spiro-OMeTAD layer. As such, early-time kinetics are attributed to hole injection yield, while signal decay is assigned to bimolecular recombination between the perovskite conduction band and hole carriers in Spiro81–83 (see the schematic in Fig. S24c, ESI†). Given the similar signal amplitude at early times ∼10 μs for both samples (Fig. S24a and b, ESI†) at different excitation densities, the charge injection yield from the perovskite into the hole transporting layer appears unaffected by the presence of the NG-100 nanofilm. This, in turn, is consistent with the earlier observation of similar Jsc values for both NG-100 and control PSCs (Fig. 4b and Table S4, ESI†). Moreover, we observed no significant variation in the hole recombination dynamics (i.e. TA signal decay) at the interface between the NG-100-modified and control samples as can be seen in the lifetimes (τ) from the monoexponential fittings of the TA signals (Table S7) in the ESI.† This further indicates that the NG-100 nanofilm does not disrupt the interfacial charge dynamics.
Effects of the nanogels on perovskite structure and optoelectronic properties
X-ray diffraction patterns were measured for the systems (Fig. S25a, ESI†) and are dominated by the (100) and (200) peaks. This suggests that the (100) plane is parallel to the substrate.1 The intensity of the (100) peak was strongest for the NG-60 and NG-100 systems and weakest for the NG-200 system (Fig. S25b, ESI†). Furthermore, the full-width at half maximum height (FWHM) values determined for the (100) peak are smallest for the NG-60 and NG-100 systems (Fig. S25c, ESI†). These data indicate an increase in average crystal size for the NG-60 and NG-100 systems, which qualitatively agrees with the trend of the average grain size values measured from SEM (Fig. S8b, ESI†). The intensity ratio of the (100)/(210) peaks (Fig. S25d, ESI†) gives a measure of the grain orientation uniformity.84 This is highest for NG-60 and NG-100 and lowest for the NG-200 and the control. These results imply that NG-60 and NG-100 have a constructive effect on grain alignment; whereas, the larger NG-200 particles disrupt alignment. We attribute this effect to the relative large size of the NG-200 particles which prevents good registry of neighboring grains being achieved.UV-visible spectra were measured for the films (Fig. S26, ESI†) and the spectra are similar. Steady-state photoluminescence (PL) measurements were obtained for the films (Fig. 5a). A blue shift in PL maxima was observed (Fig. S27, ESI†) for the NG-60 (812 nm) and NG-100 (807 nm) films compared to the control film (817 nm); whereas, this is absent for the NG-200 system (819 nm). The PL intensity increases for the NG-60 and NG-100 films relative to the control, as seen in Fig. 5a, while it decreases for the NG-200 system. These trends are indicative of passivation85 for the NG-60 and NG-100 systems. To confirm this proposal, time-resolved PL (TRPL) measurements were performed (Fig. 5c) and fitted using86,87
I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2) | (3) |
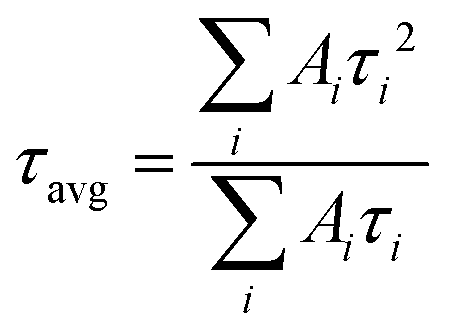 | (4) |
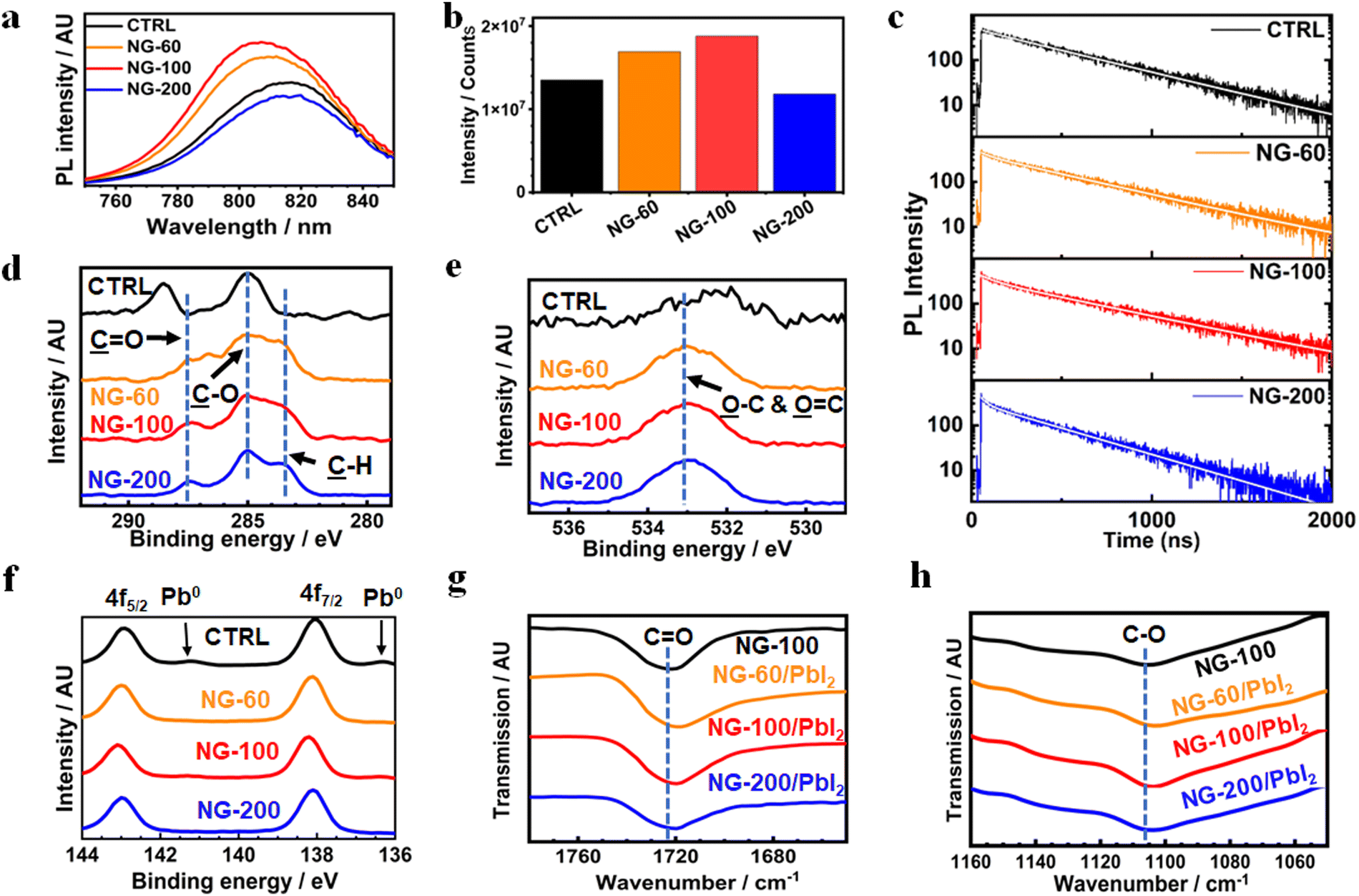 | ||
| Fig. 5 (a) PL spectra for various perovskite/nanogel systems. (b) The maximum PL intensity from the spectra in (a). (c) TRPL data for various perovskite films. (d) C 1s, (e) O 1s and (f) Pb 4f core level spectra from XPS measurements for the control perovskite and films prepared with nanogels (3 mg g−1). (g) CO and (h) C–O stretching regions of the FTIR spectra measured for NG-100 and the various nanogels mixed with PbI2. | ||
The average lifetimes in this study are greater than 300 ns (see Fig. S28 and Table S8, ESI†) and follow the same trend as the PL intensities (Fig. 5c). The large lifetimes are consequences of the large grain sizes for the films.90 The τavg values increase with inclusion of NG-60 (466 ns) and NG-100 (500 ns), whereas the value for NG-200 (358 ns) is less than that for the control (446 ns). Hence, the TRPL and steady-state PL data show evidence for passivation of the perovskite by the NG-60 and NG-100 particles, but not the NG-200 particles. We attribute the passivation for the NG-60 and NG-100 systems to the improved coverage of the grains by the nanogels as discussed above. These results experimentally confirm the DFT prediction of a strong interaction between the PMEO2MA units and perovskite.
Passivation of perovskite shown from PL measurements is often reflected in improved device performance. Here, we investigated the relationship between VocFF/100 for the devices and the intensity from the steady-state PL measurements (see Fig. S29(a), ESI†). A very strong linear relationship is evident with an R2 value of 0.98. The relationship between VocFF/100 and carrier lifetime from TRPL was also tested (Fig. S29(b), ESI†). In the latter case, there was some evidence of linearity with an R2 value of 0.84. These data establish useful correlations between device performance and PL which we exploit later.
To investigate the surface of the films, an XPS study was conducted. XPS C 1s and O 1s spectra show that the C signal (Fig. 5d) due to perovskite and O signal (Fig. 5e) due to adsorbed oxygen species37 are replaced by signals from PMEO2MA for the nanogel systems. The assignments for the control perovskite signals and those from the nanogel systems are based on related perovskites37 and polymers reported elsewhere.91–93 These data further confirm that nanogels are present at the perovskite film surface. XPS spectra for the Pb 4f region (Fig. 5f) show that all three nanogel systems decreased the presence of Pb0. Such metallic Pb on the perovskite surface may originate from the oxidation of I− into I2 and is an indication of instability. This can increase the nonradiative recombination in the perovskite film due to the high level of iodine vacancies after oxidation.94 The films containing nanogels have very weak or negligible Pb0 peaks which is attributed to stabilization of the perovskite by the nanogels.
FTIR spectra were measured for PbI2/nanogel mixtures after solvent removal to probe possible interactions between these species (Fig. S30, ESI†). The band due to CO (Fig. 5g) for the pure NG-100 (1722 cm−1) moved to 1719 cm−1 for NG-60/PbI2 and 1720 cm−1 for NG-100/PbI2 and NG-200/PbI2. Analysis of the C–O region (Fig. 5h) shows the band for the nanogel at 1106 cm−1 moved to 1104 cm−1 for all three nanogel/PbI2 mixtures. These data provide support for binding of the CO groups95 and C–O groups96 to Pb. Such binding was predicted from the DFT simulation data discussed above and involves Pb2+.
Mechanical property enhancement and moisture trapping
To probe the effect of the nanogels on the mechanical properties of the films, we conducted adhesion tests (Fig. S31, ESI†). These tests involve applying adhesive to the surface and then peeling it off and examining the film and tape afterwards.55 The photographs of the films before and after tape adhesion show that a relatively high amount of perovskite was removed for the control. Less perovskite was removed for the nanogel films. The % detachment was measured and decreased in the order CTRL > NG-200 > NG-100–NG-60 (see Fig. S31c, ESI†). It follows that NG-60 and NG-100 most favored trans-granular adhesion23 and perovskite grain-to-substrate adhesion. These effects likely contributed to the improved stability of the NG-60 and NG-100 films noted above because mechanical stability and device performance stability are closely linked.23,34 We attribute the greater mechanical stability for the NG-100 and NG-60 systems to their greater nanogel coverage of the perovskite grains. The NG-200 coverage is lower due to a lower specific surface area.A contributing factor for the stability enhancements may be that the nanogels trap moisture. We tested the ability of the dried NG-200 films to absorb moisture using a gravimetric experiment. The dried NG-200 particle films absorbed moisture rapidly from humid air (Fig. S32, ESI†) and gained about 10% of their dry weight within 1 day and the uptake was slower after that. It follows that the hydrophilic nanogels in our perovskite films would also absorb water as moisture moved through the film and, in so doing, protect the perovskite from water-triggered degradation.
Proposed mechanism for the nanogel size effects on solar cell performance
The coverage of the perovskite by the nanogels increases as the nanogel size decreases as judged by the contact angles (Fig. S14b, ESI†) and specific surface areas (Note 3, ESI†). Furthermore, the NG-60 and NG-100 particles form nanofilms due to localized inter-nanogel coalescence. In contrast, the NG-200 particles have greater separation between the particles and little tendency to form films. Our SEM data show that NG-200 and NG-100 particles are located at grain boundaries, at the top surface (Fig. S11c, ESI†), deep inside the film (Fig. S17, ESI†) and also at the buried surface (Fig. S33, ESI†). Hence, the nanogels are distributed throughout the perovskite films.It is likely that all parts of the perovskite surface covered by nanogels are electrically isolated from the adjacent material (perovskite grain, HTL, ETL) because the nanogels are insulating. By partially covering the perovskite surface, the nanogels prevent access of photogenerated charges to underlying perovskite surface defects as indicated by the ab initio simulations. From the device performance perspective, as the nanogel size decreases at constant nanogel concentration the coverage of the perovskite surface increases resulting in a trade-off between removing access of photocarriers to underlying defects and charge transport to the electrodes. The NG-100 particle system gives the best compromise for surface/interface passivation and charge transport to the electrodes whilst also providing compact grain structure and perovskite structural ordering. This gave the best PCE as well as mechanical stability as measured by adhesion. Additionally, the vertical crystals (Fig. 3a and b) may provide a conductive contact for charge transport between the underlying perovskite grains and the Spiro layer for all systems. In contrast, the large NG-200 particles had the lowest coverage and did not form films. Those nanogels left large areas of the surface vulnerable to moisture attack because of their lower coverage and also interfere with perovskite grain orientation alignment due to their relatively large size. Hence, there is an optimum size for these nanogels (100 nm) in terms of boosting the performance of the PSCs studied here. Their ability to increase performance benefited from exposure of previously hidden functional groups upon the particle deformation (flattening) that occurred when the nanogels are deposited.
Given that 3 mg g−1 was the optimum concentration for the NG-100 system, an interesting question concerns whether the same concentration is optimum for NG-60 and NG-200. We address this question using PL measurements for each system using concentrations of 1, 3 and 5 mg g−1 (Fig. S34, ESI†). For the NG-60 system, the maximum PL intensity was greatest for 3 mg g−1; whilst, for the NG-200 system the PL intensity decreased with increasing NG concentration. The same trends were found for τavg from the TRPL measurements (see Fig. S35, ESI†). We then applied the two linear relationships established in Fig. S29 (ESI†) between VocFF/100 and these PL parameters. As a check of accuracy and validation of this method, Fig. S36(a) (ESI†) shows that the measured device values for VocFF/100 and those calculated using our PL method agree to within 4%. The calculated VocFF/100 values for the NG-60 systems (Fig. S36(b), ESI†) show that the maximum value occurred at 3 mg g−1. In contrast, the calculated VocFF/100 values for the NG-200 systems (Fig. S36(c), ESI†) show decreasing values at all NG-200 concentrations. We therefore conclude that 3 mg g−1 is the optimum concentration for NG-60 and that all concentrations of NG-200 are detrimental for that system as judged by the calculated VocFF/100 values.
Conclusions
In summary, new methods for increasing the stability of PSCs are urgently needed if they are to be commercialized, and this study demonstrates improved PSC passivation and stability using deformable film-forming polymer nanoparticles. Here, we included three soft, film-forming crosslinked nanogels with different diameters (60, 100 and 200 nm) in the perovskite precursor solution. The nanogels form nanofilms when their diameter is less than or equal to 100 nm and increased the PCE and device stability. Ab initio simulations indicate strong binding of the nanogel macromolecules on the perovskite surface through Pb–O bonds, thereby promoting enhanced defect passivation and surface stability; such interactions were confirmed by the observed XPS and FTIR signals associated with the binding of the CO and C–O groups to Pb2+. Nanogels with a diameter of 100 nm gave devices with a best PCE of 20.08% compared to 18.40% for the control. Moreover, devices from these film-forming nanogels retained more than 80% of their initial PCEs after more than 6 months compared to only 25% for the control. In contrast, the 200 nm nanogels did not form films or provide a PCE or stability improvement. The film-forming nanogel strategy reported in this work is scalable, as nanogels can be prepared on a multi-tonne scale and simply added to the perovskite precursor solution. Our study therefore demonstrates that highly deformable hydrophilic film-forming polymer nanoparticles may provide a scalable and widely-applicable approach for enhancing the PCE and stability of PSCs without introducing extra processing steps.
Author contributions
Z. Jia was responsible for film and device construction, measurements, data analysis, and contributed to the writing. R. Wang conducted the contact angle test, and helped with the FTIR, EQE, SCLC, light soaking test, and PL studies. L. Zhu conducted the DFT studies and assisted in editing the manuscript. M. S. Islam assisted with DFT interpretation and the writing of the manuscript. A. Altujjar helped with the measurements for EQE, SCLC and PL studies. P. Jacoutot conducted the TAS experiments, analyzed and interpreted the data, and assisted in writing the manuscript. S. A. Haque supervised the TAS experiments, assisted with data interpretation, and helped write the manuscript. O. M. Alkhudhari and M. Z. Mokhtar helped with solar cell construction and measurement. B. F. Spencer and A. G. Thomas obtained and assisted in interpreting the XPS data. T. Hashimoto and M. Faulkner obtained the EDX data and helped with data analysis. N. W. Hodson conducted the AFM test and provided help with data analysis. X. Wang and M. Osborne-Richards conducted the adhesive tape test. J. M. Saunders assisted with polymer synthesis as well as interpreted DLS data. B. R. Saunders conceived the study, assisted with data interpretation and contributed to the writing of the manuscript.Data availability
Data will be made available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. R. S., M. S. I., L. Z., S. A. H. and P. J. gratefully acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC, EP/X012263/1; EP/X012484/1; EP/X012344/1). M. S. I. and L. Z. acknowledge the UK's HEC Materials Chemistry Consortium (EP/R029431) for the use of the ARCHER2 high-performance computing facilities. We are grateful for access to the University of Manchester BioAFM facility.References
- X. Shen, B. M. Gallant, P. Holzhey, J. A. Smith, K. A. Elmestekawy, Z. Yuan, P. V. G. M. Rathnayake, S. Bernardi, A. Dasgupta, E. Kasparavicius, T. Malinauskas, P. Caprioglio, O. Shargaieva, Y.-H. Lin, M. M. McCarthy, E. Unger, V. Getautis, A. Widmer-Cooper, L. M. Herz and H. J. Snaith, Adv. Mater., 2023, 35, 2211742 CrossRef CAS PubMed.
- E. Ruggeri, M. Anaya, K. Gałkowski, A. Abfalterer, Y.-H. Chiang, K. Ji, Z. Andaji-Garmaroudi and S. D. Stranks, Adv. Mater., 2022, 34, 2202163 CrossRef CAS PubMed.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- L. Tan, J. Zhou, X. Zhao, S. Wang, M. Li, C. Jiang, H. Li, Y. Zhang, Y. Ye, W. Tress, L. Ding, M. Grätzel and C. Yi, Adv. Mater., 2023, 35, 2205027 CrossRef CAS PubMed.
- C. Li, X. Wang, E. Bi, F. Jiang, S. M. Park, Y. Li, L. Chen, Z. Wang, L. Zeng, H. Chen, Y. Liu, C. R. Grice, A. Abudulimu, J. Chung, Y. Xian, T. Zhu, H. Lai, B. Chen, R. J. Ellingson, F. Fu, D. S. Ginger, Z. Song, E. H. Sargent and Y. Yan, Science, 2023, 379, 690–694 CrossRef CAS PubMed.
- Z. Ni, C. Bao, Y. Liu, Q. Jiang, W.-Q. Wu, S. Chen, X. Dai, B. Chen, B. Hartweg, Z. Yu, Z. Holman and J. Huang, Science, 2020, 367, 1352–1358 CrossRef CAS PubMed.
- W. Hui, L. Chao, H. Lu, F. Xia, Q. Wei, Z. Su, T. Niu, L. Tao, B. Du, D. Li, Y. Wang, H. Dong, S. Zuo, B. Li, W. Shi, X. Ran, P. Li, H. Zhang, Z. Wu, C. Ran, L. Song, G. Xing, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Science, 2021, 371, 1359–1364 CrossRef CAS PubMed.
- Y. Hassan, J. H. Park, M. L. Crawford, A. Sadhanala, J. Lee, J. C. Sadighian, E. Mosconi, R. Shivanna, E. Radicchi, M. Jeong, C. Yang, H. Choi, S. H. Park, M. H. Song, F. De Angelis, C. Y. Wong, R. H. Friend, B. R. Lee and H. J. Snaith, Nature, 2021, 591, 72–77 CrossRef CAS PubMed.
- J. Chen, X. Zhao, S.-G. Kim and N.-G. Park, Adv. Mater., 2019, 31, 1902902 CrossRef PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- National Renewable Energy Laboratory, Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html (accessed November, 2023) Search PubMed.
- G. Nazir, S.-Y. Lee, J.-H. Lee, A. Rehman, J.-K. Lee, S. I. Seok and S.-J. Park, Adv. Mater., 2022, 34, 2204380 CrossRef CAS PubMed.
- H. Zai, Y. Ma, Q. Chen and H. Zhou, J. Energy Chem., 2021, 63, 528–549 CrossRef CAS.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, L. Shi, Z. Liu, J. Huang, J. S. Yun, Y. Zeng, A. Pu, K. Sun, Z. Hameiri, J. A. Stride, J. Seidel, M. A. Green and X. Hao, Adv. Energy Mater., 2018, 8, 1800138 CrossRef.
- L. Zhu, X. Zhang, M. Li, X. Shang, K. Lei, B. Zhang, C. Chen, S. Zheng, H. Song and J. Chen, Adv. Energy Mater., 2021, 11, 2100529 CrossRef CAS.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- C. Caddeo, M. I. Saba, S. Meloni, A. Filippetti and A. Mattoni, ACS Nano, 2017, 11, 9183–9190 CrossRef CAS PubMed.
- N. Aristidou, C. Eames, M. S. Islam and S. A. Haque, J. Mater. Chem. A, 2017, 5, 25469–25475 RSC.
- B. Chen, J. Song, X. Dai, Y. Liu, P. N. Rudd, X. Hong and J. Huang, Adv. Mater., 2019, 31, 1902413 CrossRef PubMed.
- S. Gharibzadeh, P. Fassl, I. M. Hossain, P. Rohrbeck, M. Frericks, M. Schmidt, T. Duong, M. R. Khan, T. Abzieher, B. A. Nejand, F. Schackmar, O. Almora, T. Feeney, R. Singh, D. Fuchs, U. Lemmer, J. P. Hofmann, S. A. L. Weber and U. W. Paetzold, Energy Environ. Sci., 2021, 14, 5875–5893 RSC.
- N. Rolston, A. D. Printz, J. M. Tracy, H. C. Weerasinghe, D. Vak, L. J. Haur, A. Priyadarshi, N. Mathews, D. J. Slotcavage, M. D. McGehee, R. E. Kalan, K. Zielinski, R. L. Grimm, H. Tsai, W. Nie, A. D. Mohite, S. Gholipour, M. Saliba, M. Grätzel and R. H. Dauskardt, Adv. Energy Mater., 2018, 8, 1702116 CrossRef.
- D. Meggiolaro, E. Mosconi and F. De Angelis, ACS Energy Lett., 2018, 3, 447–451 CrossRef CAS.
- J. Warby, F. Zu, S. Zeiske, E. Gutierrez-Partida, L. Frohloff, S. Kahmann, K. Frohna, E. Mosconi, E. Radicchi, F. Lang, S. Shah, F. Peña-Camargo, H. Hempel, T. Unold, N. Koch, A. Armin, F. De Angelis, S. D. Stranks, D. Neher and M. Stolterfoht, Adv. Energy Mater., 2022, 12, 2103567 CrossRef CAS.
- C. Park, H. Ko, D. H. Sin, K. C. Song and K. Cho, Adv. Funct. Mater., 2017, 27, 1703546 CrossRef.
- T. Zhang, X. Meng, Y. Bai, S. Xiao, C. Hu, Y. Yang, H. Chen and S. Yang, J. Mater. Chem. A, 2017, 5, 1103–1111 RSC.
- C. Zhi, S. Wang, S. Sun, C. Li, Z. Li, Z. Wan, H. Wang, Z. Li and Z. Liu, ACS Energy Lett., 2023, 8, 1424–1433 CrossRef CAS.
- R. Lu, Y. Liu, D. Zhao, X. Guo and C. Li, J. Mater. Chem. A, 2022, 10, 21422–21429 RSC.
- X. Yuan, R. Li, Z. Xiong, P. Li, G. O. Odunmbaku, K. Sun, Y. Deng and S. Chen, Adv. Funct. Mater., 2023, 33, 2215096 CrossRef CAS.
- Y. Kong, W. Shen, H. Cai, W. Dong, C. Bai, J. Zhao, F. Huang, Y.-B. Cheng and J. Zhong, Adv. Funct. Mater., 2023, 33, 2300932 CrossRef CAS.
- Y. Rong, Y. Hu, A. Mei, H. Tan, M. I. Saidaminov, S. I. Seok, M. D. McGehee, E. H. Sargent and H. Han, Science, 2018, 361, eaat8235 CrossRef PubMed.
- F. H. Isikgor, S. Zhumagali, L. V. T. Merino, M. De Bastiani, I. McCulloch and S. De Wolf, Nat. Rev. Mater., 2023, 8, 89–108 CrossRef CAS.
- Z. Dai, S. K. Yadavalli, M. Chen, A. Abbaspourtamijani, Y. Qi and N. P. Padture, Science, 2021, 372, 618–622 CrossRef CAS PubMed.
- Z. Dai, S. Li, X. Liu, M. Chen, C. E. Athanasiou, B. W. Sheldon, H. Gao, P. Guo and N. P. Padture, Adv. Mater., 2022, 34, 2205301 CrossRef CAS PubMed.
- L. Liu, Y. Yang, M. Du, Y. Cao, X. Ren, L. Zhang, H. Wang, S. Zhao, K. Wang and S. Liu, Adv. Energy Mater., 2023, 13, 2202802 CrossRef CAS.
- T. Niu, Y.-M. Xie, Q. Xue, S. Xun, Q. Yao, F. Zhen, W. Yan, H. Li, J.-L. Brédas, H.-L. Yip and Y. Cao, Adv. Energy Mater., 2022, 12, 2102973 CrossRef CAS.
- J. Xiong, P. N. Samanta, Y. Qi, T. Demeritte, K. Williams, J. Leszczynski and Q. Dai, ACS Appl. Mater. Interfaces, 2022, 14, 5414–5424 CrossRef CAS PubMed.
- H. Zhu, Y. Liu, F. T. Eickemeyer, L. Pan, D. Ren, M. A. Ruiz-Preciado, B. Carlsen, B. Yang, X. Dong, Z. Wang, H. Liu, S. Wang, S. M. Zakeeruddin, A. Hagfeldt, M. I. Dar, X. Li and M. Grätzel, Adv. Mater., 2020, 32, 1907757 CrossRef CAS PubMed.
- D. J. Fairfield, H. Sai, A. Narayanan, J. V. Passarelli, M. Chen, J. Palasz, L. C. Palmer, M. R. Wasielewski and S. I. Stupp, J. Mater. Chem. A, 2019, 7, 1687–1699 RSC.
- S. Wang, Z. Zhang, Z. Tang, C. Su, W. Huang, Y. Li and G. Xing, Nano Energy, 2021, 82, 105712 CrossRef CAS.
- J. Zhuang, P. Mao, Y. Luan, X. Yi, Z. Tu, Y. Zhang, Y. Yi, Y. Wei, N. Chen, T. Lin, F. Wang, C. Li and J. Wang, ACS Energy Lett., 2019, 4, 2913–2921 CrossRef CAS.
- L. Zuo, H. Guo, D. W. deQuilettes, S. Jariwala, N. De Marco, S. Dong, R. DeBlock, D. S. Ginger, B. Dunn, M. Wang and Y. Yang, Sci. Adv., 2017, 3, e1700106 CrossRef PubMed.
- C. Zhao, H. Zhang, M. Almalki, J. Xu, A. Krishna, F. T. Eickemeyer, J. Gao, Y. M. Wu, S. M. Zakeeruddin, J. Chu, J. Yao and M. Grätzel, Adv. Mater., 2023, 35, 2211619 CrossRef CAS PubMed.
- M. Wang, Y. Zhao, X. Jiang, Y. Yin, I. Yavuz, P. Zhu, A. Zhang, G. S. Han, H. S. Jung, Y. Zhou, W. Yang, J. Bian, S. Jin, J.-W. Lee and Y. Yang, Joule, 2022, 6, 1032–1048 CrossRef CAS.
- Y. Zhao, J. Wei, H. Li, Y. Yan, W. Zhou, D. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- D. Liu, X. Wang, X. Wang, B. Zhang, X. Sun, Z. Li, Z. Shao, S. Mao, L. Wang, G. Cui and S. Pang, Angew. Chem., Int. Ed., 2023, 62, e202301574 CrossRef CAS PubMed.
- L. Luo, H. Zeng, Z. Wang, M. Li, S. You, B. Chen, A. Maxwell, Q. An, L. Cui, D. Luo, J. Hu, S. Li, X. Cai, W. Li, L. Li, R. Guo, R. Huang, W. Liang, Z.-H. Lu, L. Mai, Y. Rong, E. H. Sargent and X. Li, Nat. Energy, 2023, 8, 294–303 CAS.
- D. Bi, C. Yi, J. Luo, J.-D. Décoppet, F. Zhang, S. M. Zakeeruddin, X. Li, A. Hagfeldt and M. Grätzel, Nat. Energy, 2016, 1, 16142 CrossRef CAS.
- Z. Huang, X. Hu, C. Liu, L. Tan and Y. Chen, Adv. Funct. Mater., 2017, 27, 1703061 CrossRef.
- Z. Li, C. Jia, Z. Wan, J. Xue, J. Cao, M. Zhang, C. Li, J. Shen, C. Zhang and Z. Li, Nat. Commun., 2023, 14, 6451 CrossRef CAS PubMed.
- A. H. Milani, J. M. Saunders, N. T. Nguyen, L. P. D. Ratcliffe, D. J. Adlam, A. J. Freemont, J. A. Hoyland, S. P. Armes and B. R. Saunders, Soft Matter, 2017, 13, 1554–1560 RSC.
- O. M. Alkhudhari, A. Altujjar, M. Z. Mokhtar, B. F. Spencer, Q. Chen, A. G. Thomas, N. W. Hodson, X. Wang, P. Hill, J. Jacobs, R. J. Curry and B. R. Saunders, J. Mater. Chem. A, 2022, 10, 10227–10241 RSC.
- O. M. Alkhudhari, R. Wang, Z. Jia, N. W. Hodson, A. Alruwaili, A. Altujjar, E. Picheo and B. R. Saunders, RSC Adv., 2024, 14, 6190–6198 RSC.
- A. Altujjar, M. Z. Mokhtar, Q. Chen, J. Neilson, B. F. Spencer, A. Thomas, J. M. Saunders, R. Wang, O. Alkhudhari, A. Mironov and B. R. Saunders, ACS Appl. Mater. Interfaces, 2021, 13, 58640–58651 CrossRef CAS PubMed.
- D. Portoghese and A. F. Routh, Chem. Eng. Res. Des., 2005, 83, 139–142 CrossRef CAS.
- A. Toussaint and M. De Wilde, Prog. Org. Coat., 1997, 30, 113–126 CrossRef CAS.
- E. Buratti, I. Sanzari, F. Dinelli, T. Prodromakis and M. Bertoldo, Polymers, 2020, 12, 2638 CrossRef CAS PubMed.
- D. Saatweber and B. Vogt-Birnbrich, Prog. Org. Coat., 1996, 28, 33–41 CrossRef CAS.
- J. Engström, C. J. Brett, V. Körstgens, P. Müller-Buschbaum, W. Ohm, E. Malmström and S. V. Roth, Adv. Funct. Mater., 2020, 30, 1907720 CrossRef.
- S. Galindo, A. Tamayo, F. Leonardi and M. Mas-Torrent, Adv. Funct. Mater., 2017, 27, 1700526 CrossRef.
- M.-B. Madec, J. J. Morrison, V. Sanchez-Romaguera, M. L. Turner and S. G. Yeates, J. Mater. Chem., 2009, 19, 6750–6755 RSC.
- A. Campos, S. Riera-Galindo, J. Puigdollers and M. Mas-Torrent, ACS Appl. Mater. Interfaces, 2018, 10, 15952–15961 CrossRef CAS PubMed.
- H. Bachman, A. C. Brown, K. C. Clarke, K. S. Dhada, A. Douglas, C. E. Hansen, E. Herman, J. S. Hyatt, P. Kodlekere, Z. Meng, S. Saxena, M. W. Spears Jr, N. Welsch and L. A. Lyon, Soft Matter, 2015, 11, 2018–2028 RSC.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS PubMed.
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS.
- D. P. McMeekin, Z. Wang, W. Rehman, F. Pulvirenti, J. B. Patel, N. K. Noel, M. B. Johnston, S. R. Marder, L. M. Herz and H. J. Snaith, Adv. Mater., 2017, 29, 1607039 CrossRef PubMed.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. S. Islam and P. J. Cameron, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Nat. Energy, 2016, 2, 16195 CrossRef.
- T. Webb and S. A. Haque, Energy Environ. Sci., 2024, 17, 3244–3269 RSC.
- G. Tumen-Ulzii, T. Matsushima, D. Klotz, M. R. Leyden, P. Wang, C. Qin, J.-W. Lee, S.-J. Lee, Y. Yang and C. Adachi, Commun. Mater., 2020, 1, 31 CrossRef.
- A. Mahapatra, N. Parikh, P. Kumar, M. Kumar, D. Prochowicz, A. Kalam, M. M. Tavakoli and P. Yadav, Molecules, 2020, 25, 2299 CrossRef CAS PubMed.
- L. Xie, S. Du, J. Li, C. Liu, Z. Pu, X. Tong, J. Liu, Y. Wang, Y. Meng, M. Yang, W. Li and Z. Ge, Energy Environ. Sci., 2023, 16, 5423–5433 RSC.
- V. D. Mihailetchi, H. X. Xie, B. deBoer, L. J. A. Koster and P. W. M. Blom, Adv. Funct. Mater., 2006, 16, 699–708 CrossRef CAS.
- L. Liu, C. Zheng, Z. Xu, Y. Li, Y. Cao, T. Yang, H. Zhang, Q. Wang, Z. Liu, N. Yuan, J. Ding, D. Wang and S. Liu, Adv. Energy Mater., 2023, 13, 2300610 CrossRef CAS.
- Y. Huang, L. Li, Z. Liu, H. Jiao, Y. He, X. Wang, R. Zhu, D. Wang, J. Sun, Q. Chen and H. Zhou, J. Mater. Chem. A, 2017, 5, 8537–8544 RSC.
- Q. Han, S.-H. Bae, P. Sun, Y.-T. Hsieh, Y. Yang, Y. S. Rim, H. Zhao, Q. Chen, W. Shi, G. Li and Y. Yang, Adv. Mater., 2016, 28, 2253–2258 CrossRef CAS PubMed.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Science, 2019, 366, 749–753 CrossRef CAS PubMed.
- A. Marchioro, J. Teuscher, D. Friedrich, M. Kunst, R. van de Krol, T. Moehl, M. Grätzel and J.-E. Moser, Nat. Photonics, 2014, 8, 250–255 CrossRef CAS.
- R. J. E. Westbrook, T. J. Macdonald, W. Xu, L. Lanzetta, J. M. Marin-Beloqui, T. M. Clarke and S. A. Haque, J. Am. Chem. Soc., 2021, 143, 12230–12243 CrossRef CAS PubMed.
- T. Webb, X. Liu, R. J. E. Westbrook, S. Kern, M. T. Sajjad, S. Jenatsch, K. D. G. I. Jayawardena, W. H. K. Perera, I. P. Marko, S. Sathasivam, B. Li, M. Yavari, D. J. Scurr, M. R. Alexander, T. J. Macdonald, S. A. Haque, S. J. Sweeney and W. Zhang, Adv. Energy Mater., 2022, 12, 2200666 CrossRef CAS.
- F. T. F. O'Mahony, Y. H. Lee, C. Jellett, S. Dmitrov, D. T. J. Bryant, J. R. Durrant, B. C. O'Regan, M. Graetzel, M. K. Nazeeruddin and S. A. Haque, J. Mater. Chem. A, 2015, 3, 7219–7223 RSC.
- Z. Xu, Z. Liu, N. Li, G. Tang, G. Zheng, C. Zhu, Y. Chen, L. Wang, Y. Huang, L. Li, N. Zhou, J. Hong, Q. Chen and H. Zhou, Adv. Mater., 2019, 31, 1900390 CrossRef PubMed.
- M. Salado, A. D. Jodlowski, C. Roldan-Carmona, G. de Miguel, S. Kazim, M. K. Nazeeruddin and S. Ahmad, Nano Energy, 2018, 50, 220–228 CrossRef CAS.
- Z. Wu, M. Jiang, Z. Liu, A. Jamshaid, L. K. Ono and Y. Qi, Adv. Energy Mater., 2020, 10, 1903696 CrossRef CAS.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006, pp. 27–61 Search PubMed.
- W. Zhao, H. Lin, Y. Li, D. Wang, J. Wang, Z. Liu, N. Yuan, J. Ding, Q. Wang and S. Liu, Adv. Funct. Mater., 2022, 32, 2112032 CrossRef CAS.
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S.-H. Jang, J. Zhang, X. Lu, D. Lei, C.-S. Lee, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134–20142 CrossRef CAS PubMed.
- Q. An, F. Paulus, D. Becker-Koch, C. Cho, Q. Sun, A. Weu, S. Bitton, N. Tessler and Y. Vaynzof, Matter, 2021, 4, 1683–1701 CrossRef CAS.
- Y. Chang, C.-Y. Ko, Y.-J. Shih, D. Quémener, A. Deratani, T.-C. Wei, D.-M. Wang and J.-Y. Lai, J. Membr. Sci., 2009, 345, 160–169 CrossRef CAS.
- L. Islas, J.-C. Ruiz, F. Muñoz-Muñoz, T. Isoshima and G. Burillo, Appl. Surf. Sci., 2016, 384, 135–142 CrossRef CAS.
- F. Zhang, E. T. Kang, K. G. Neoh, P. Wang and K. L. Tan, Biomaterials, 2001, 22, 1541–1548 CrossRef CAS PubMed.
- J. Guo, B. Wang, D. Lu, T. Wang, T. Liu, R. Wang, X. Dong, T. Zhou, N. Zheng, Q. Fu, Z. Xie, X. Wan, G. Xing, Y. Chen and Y. Liu, Adv. Mater., 2023, 35, 2212126 CrossRef CAS PubMed.
- T. Zhu, L. Shen, S. Xun, J. S. Sarmiento, Y. Yang, L. Zheng, H. Li, H. Wang, J.-L. Bredas and X. Gong, Adv. Mater., 2022, 34, 2109348 CrossRef CAS PubMed.
- C. Ma and N.-G. Park, ACS Energy Lett., 2020, 5, 3268–3275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee01073f |
| This journal is © The Royal Society of Chemistry 2024 |