
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lithium–silver alloys in anode-less batteries: comparison in liquid- and solid-electrolytes†
Ju-Hyeon
Lee
a,
Jeong Yeon
Heo
a,
Ji Young
Kim
b,
Ki Yoon
Bae
b,
Samick
Son
b and
Ji Hoon
Lee
 *a
*a
aSchool of Materials Science and Engineering and KNU Advanced Material Research Institute, Kyungpook National University, Daegu, 41566, Republic of Korea. E-mail: jihoonlee@knu.ac.kr
bAdvanced Battery Development Group, Hyundai Motor Company, Hwaseong-si, Gyeongi-do 16082, Republic of Korea
First published on 10th July 2024
Abstract
This study comprehensively investigates the phase evolution of silver–carbon composite (Ag/C) layers in anode-less batteries with both liquid and solid electrolytes. The results of in situ X-ray diffraction and cross-sectional electron microscopy analyses reveal that the alloying reaction of Ag and Li is more homogeneous in solid–electrolyte-based cells compared to liquid-electrolyte-based cells. This homogeneity is attributed to diffusional Coble creep across the heterogeneous interfaces of Ag/C layers and solid electrolytes.
From a battery cell design perspective, an anode-less system has the potential to enhance energy densities to their theoretical limits.1,2 Additionally, it can considerably reduce the cell volume required for anode stacks, thus providing a cost-effective and simplified fabrication process.3 Consequently, this approach has been widely implemented in next-generation lithium-ion battery (LIB) systems, such as all-solid-state batteries (ASSBs) and lithium–metal batteries.4–6 However, anode-less LIB systems suffer from significant capacity degradation during cycling, primarily due to the formation of Li dendrites caused by the absence of layers to host metallic Li deposits.7,8 To address this challenge, a thin layer (≈10 μm) of silver (Ag)/porous carbon (C) composites has been applied to the current collector (CC).9–11 This layer allows Ag nanoparticles to act as nucleation sites for Li deposits through the electrochemical alloying reaction (yAg + x(Li+ + e−) → LixAgy), thereby preventing the growth of metallic Li dendrites on the anode.6,12,13 Despite its broad applications in various LIB systems, there has been limited research directly comparing the alloying reactions of Ag with Li in solid-state versus liquid-state electrolyte systems.
This study investigates the distinct alloying reactions of Ag/C layers in anode-less battery systems using solid- and liquid-electrolytes (Fig. 1). During the charging process (i.e., Li plating), homogeneous Li deposits formed between the Ag/C layer and the CC in the cells based on a solid electrolyte (SE, Li6PS5Cl). Conversely, in cells using a liquid electrolyte (LE, 1.15 M lithium hexafluorophosphate dissolved in a carbonate solution), preferential Li electrodeposition was observed on the top surface of the Ag/C layers (referred to as top-plating), with unreacted Ag/C layers situated between the Li deposits and the CC. This distinct behaviour stems from the interfacial properties between the electrolytes and the Ag/C layers. Under cathodic polarization conditions, Li deposits resulting from the alloying reactions of Li and Ag diffuse across a well-defined heterogeneous interface between the SE and Ag/C layers (SE|Ag/C), promoting the formation of homogeneous and uniform Li–Ag layers as Li deposits between Ag/C and CC (refer to Fig. S1 for a detailed illustration of the Li nucleation, diffusion, and Li–Ag alloying processes, ESI†). However, in the LE-based cells, the relatively homogeneous interface between the LE and Ag/C layers (LE|Ag/C), due to the electrolyte infiltration and good Li ion diffusivity, facilitates sufficient Li flux near individual Ag particles. Consequently, Li–Ag alloy particles formed in the initial Li plating step are more effective in further alloying reactions than unreacted Ag particles, accelerating the heterogeneous formation of Li–Ag alloying layers on top of the remaining Ag/C layers.
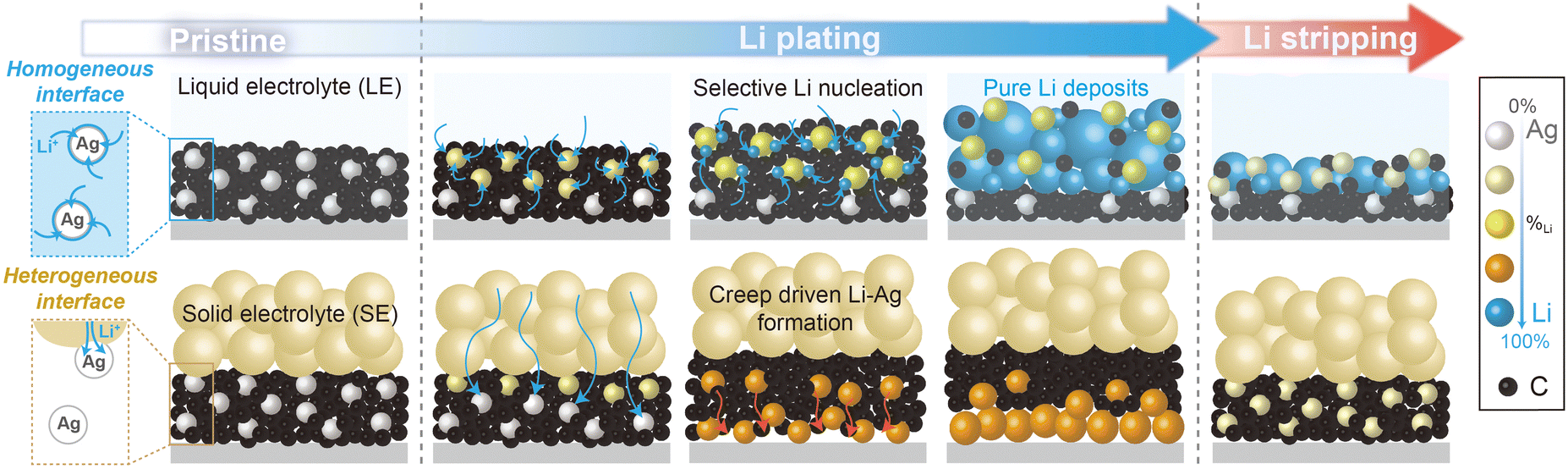 | ||
| Fig. 1 Schematic illustration showing the comparative alloying reactions of Ag/C with Li in LE and SE. | ||
Fig. 2A and B and Fig. S2 (ESI†) present the typical galvanostatic Li plating/stripping profiles of Ag/C layers in LE- and SE-based cells at a current density of 0.5 mA cm−2, with a cutoff capacity of 2 mA h cm−2 (refer to the ESI† for experimental details). Both cells exhibited plateau overpotentials relative to 0 V vs. Li+/Li (referred to as “V” unless otherwise stated in this work), indicating the formation of Li deposits during the plating process and their dissolution during the stripping process. Building on previous studies,14,15 we evaluated the overpotential for Li nucleation (ηn) and growth (ηg, including Li–Ag alloying reactions), as shown in Fig. S2C and D (ESI†). The Ag/C layers in the SE-based cell exhibited a higher ηg value (32 mV) but a lower ηn value (2 mV) than those in the LE-based cell (ηg = 18 mV, ηn = 7 mV). The higher ηg in the SE-based cell is attributed to sluggish Li diffusion in the SE, across the Ag/C|SE interface, and within the Ag/C layers. Conversely, the lower ηn value can be attributed to the Li–Ag alloying reactions, which significantly reduces the ηn value. In contrast, in the LE-based cell, the lower ηg can be explained by the rapid access of Li ions onto the surface of the Ag/C layer, while the higher ηn value reflects the electrochemical reduction of Li ions to metallic Li in the absence of a suitable host structure. These observations indicate that the kinetics of Li diffusion dominate over the thermodynamics of Li electrodeposition in SE-based cells. The overpotential was ≈14 mV higher in the SE-based cell compared to the LE-based cell, likely due to the slower Li ion diffusion in the SE matrix and across the SE|Ag/C interfaces. A sharp voltage dip during the plating process of the LE-based cell corresponded to the nucleation overpotential required for the formation of Li–Ag alloys with sufficiently large nuclei for the growth of Li deposits.14,15 Conversely, it is noteworthy that the SE-based cell did not exhibit such a sharp voltage dip, suggesting a different Li electrodeposition mechanism16–19 in LE- and SE-based cells. Considering the smaller voltage dips but larger Li growth plateaus for the SE-based cell compared to the LE-based cell, the kinetics of Li diffusion appear to outweigh the thermodynamics of Li electrodeposition in the SE-based cell.
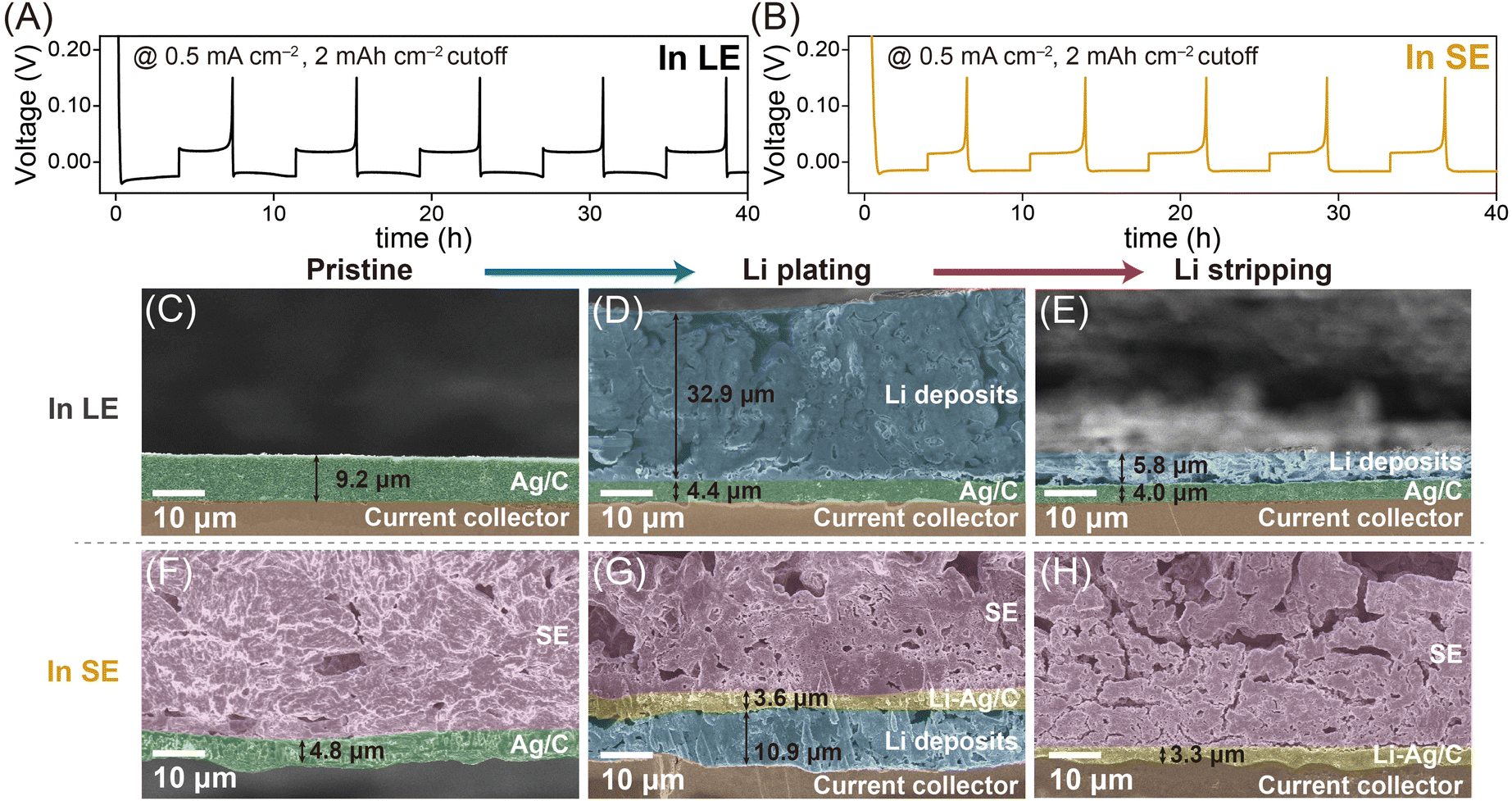 | ||
| Fig. 2 Comparative galvanostatic Li plating/stripping profiles of the Ag/C layers in (A) LE- and (B) SE-based cells at 60 °C. Cross-sectional SEM images of Ag/C-containing electrodes in (C)–(E) LE- and (F)–(H) SE-based cells at different charging states (Left: Pristine, middle: 2 mA h cm−2 of Li plating. Right: Li stripping at 0.15 V). The holes and pitches in the SE layers are formed during sampling of the SE|Ag/C|CC pellet from the cell housing. | ||
This difference in the primary mechanism of Li electrodeposition in the LE- and SE-based cells resulted in distinct locations of Li deposits. As shown in Fig. 2C–E and Fig. S3 (ESI†), Li deposit layers (≈32.9 μm) grew on the Ag/C layers in the LE-based cell. The significant amount of fluorine detected in the Li deposits in the energy dispersive spectroscopy (EDS) mapping results (Fig. S3, ESI†) implies the presence of solid electrolyte interface (SEI) layers resulting from the decomposition of the LE. To address the safety concerns caused by accelerated LE decomposition on top-plated Li deposits, researchers have explored various strategies, including Li-confinable hosts, mesh-type hosts, and modified separators.20 Notably, even after the Li plating process, unreacted Ag/C layers (≈4.4 μm) remained between the Li deposits and the CC, likely because Li preferentially alloys with the Li–Ag alloys21 initially formed during the plating step, rather than with unreacted Ag under the LE system, where Li diffusion onto the Ag/C layers is relatively rapid. Upon the stripping process, the thickness of the Li deposits significantly decreased to approximately 5.8 μm, but clear interfaces remained between the Li deposits and unreacted Ag/C layers. For reference, the Ag/C layers were also evaluated in the cell using an ether-based LE (Fig. S4, ESI†). Notably, the galvanostatic plating/stripping profile in the ether-based LE exhibited no significant difference compared with that in the carbonate-based LE (Fig. 2A).
In contrast, in the SE-based cell, Li deposits diffused across the SE|Ag/C interfaces and were plated between the Ag/C layers and the CC (Fig. 2F–H and Fig. S5, ESI†),9 attributed to the diffusional Coble creep-controlled Li electrodeposition mechanism.16–19 Li deposits formed by filling the pores in the Ag/C layers with Li nucleates, followed by an alloying reaction with the nearby Ag particles to form Li–Ag alloys. Notably, as discussed in the subsequent section, the alloying reactions in the SE-based cell occurred uniformly, leaving no unreacted metallic Ag phase, unlike in the LE-based cell (Fig. 2D). After the stripping process, the Li deposit layers vanished, leaving Li–Ag/C layers between the SE and CC. The distinct locations of Li deposits in the LE- and SE-based cells remained consistent when paired with a high-voltage cathode (LiNbO3-coated LiNi0.8Co0.1Mn0.1O2, NCM), indicating that Li-containing electrodes (i.e., NCM or Li foil) do not affect the Li deposition behaviour on the Ag/C layers (Fig. S6 and S7, ESI†).
To understand the different locations of Li deposits and their correlation with the phase evolution behaviours of Ag/C layers observed through SEM analysis during LE- and SE-based cell operations, we conducted in situ X-ray diffraction (XRD) analysis (Fig. 3 and Fig. S8–S10, ESI†). Initially, we confirmed that the Ag/C layers exhibited similar plating/stripping behaviours in the SE-based coin cell configuration, ensuring a fair comparison of their phase evolution behaviours in LE- and SE-based cells during in situ XRD characterizations (Fig. S11, ESI†). According to the well-defined Li–Ag binary phase diagram,15,22–25 the nucleation of Li on the Ag matrix leads to sequential phase transitions of Li–Ag alloys, resulting in various intermetallic compounds (e.g., β (Li1Ag1), γ3 (Li9Ag4), γ2 (Li4Ag1), and γ1 (Li9Ag1)). During the plating process below 0 V in the LE-based cell (Fig. 3A and B), the β phase ((110), ∼17.8°) initially formed by consuming the Ag phase, followed by the gradual appearance of the γ3 phase ((33![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) and (333), ∼17.6°). The (33) and (333) peaks associated with the γ3 phase shifted toward lower 2θ values due to unit cell expansion, possibly related to the significant solubility of Li in the γ3 phase. Further plating resulted in the formation of the γ2 phase ((220), ∼17.2°) from the γ3 matrix, but the transition from γ2 to γ1 phases was not observed. Additionally, the Ag(111) peak did not fully disappear, aligning with cross-sectional SEM images that showed clear interfaces between the Li deposits (i.e., Li–Ag alloys) and the unreacted Ag/C layers (Fig. 2D). During the stripping process up to 0.15 V, the phase transitions of the Li–Ag alloy occurred in the reverse order of the plating process, with the coexistence of the γ3 phase and the unreacted Ag phase at 0.15 V.
) and (333), ∼17.6°). The (33) and (333) peaks associated with the γ3 phase shifted toward lower 2θ values due to unit cell expansion, possibly related to the significant solubility of Li in the γ3 phase. Further plating resulted in the formation of the γ2 phase ((220), ∼17.2°) from the γ3 matrix, but the transition from γ2 to γ1 phases was not observed. Additionally, the Ag(111) peak did not fully disappear, aligning with cross-sectional SEM images that showed clear interfaces between the Li deposits (i.e., Li–Ag alloys) and the unreacted Ag/C layers (Fig. 2D). During the stripping process up to 0.15 V, the phase transitions of the Li–Ag alloy occurred in the reverse order of the plating process, with the coexistence of the γ3 phase and the unreacted Ag phase at 0.15 V.
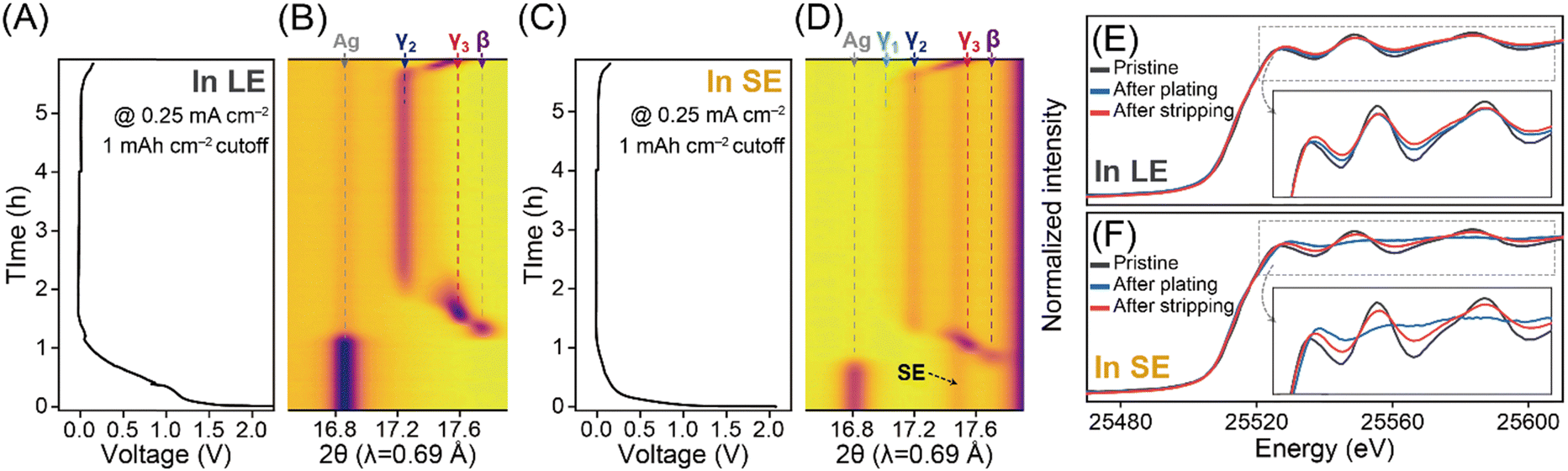 | ||
| Fig. 3 (A)–(D) In situ XRD analysis for the Ag/C layers. (A) Galvanostatic Li plating/stripping profiles at a current density of 0.25 mA cm−2 in the LE-based cell and (B) its corresponding contour map displaying the phase evolution of Ag and Li–Ag alloys (β, γ3, γ2, and γ1). (C) and (D) The same considerations in the SE-based cell. Ex-situ XAFS analysis at the Ag K-edge for the Ag/C layers prepared from the (E) LE- and (F) SE-based cells at different charging states. | ||
In the SE-based cell (Fig. 3C and D), the Ag/C layers demonstrated a phase transition trend similar to that observed in the LE-based cell during the plating process. However, under identical plating conditions, the alloying reaction advanced further into the two-phase region of the γ1 and γ2 phases in the SE-based cell, as evidenced by the appearance of the XRD peak for the γ1 phase at ∼17.0°, which was not observed in the LE-based cell. This finding indicates that the movement of Li along the SE, Ag, and C interfaces, governed by diffusional Coble creep,16–19 produces Li–Ag alloys while promoting their deposition between Ag/C and CC by penetrating through the Ag/C layers in the SE-based cell. Conversely, in the LE-based cell, after the formation of the γ2 phase, it is more likely that pure Li plating and the formation of the SEI layer (Fig. S3J, ESI†) through further electrolyte decomposition are favoured over the Li–Ag alloying reaction. This could also explain the thicker Li deposit layers observed in the LE-based cell (Fig. 2D and Fig. S12, ESI†), which may contain pure Li deposits and SEI layers, compared to the SE-based cell. Upon the completion of the stripping process, only the γ3 phase was present at 0.15 V, without a full recovery of the Ag phase.
We also performed X-ray absorption fine structure (XAFS) analysis at the Ag K-edge for Ag/C layers prepared in the LE- and SE-based cells at different states of charge (Fig. 3E and F). The X-ray absorption near edge structure (XANES) profiles revealed three key observations: (1) upon Li plating of 2.0 mA h cm−2, the XANES profiles in both cells shifted toward higher energy values, resulting from the higher Li content in the Li–Ag alloy matrix and more homogeneous alloying in the SE-based cell. (2) The oscillations in the XANES profile were significantly weaker in the SE-based cell compared to the LE-based cell, suggesting that the crystallinity of the Ag matrix decreased due to the high Li content in the alloying matrix. The weak extended XAFS (EXAFS) intensity for the Ag–Ag path in the Ag/C layers at the plating of 2.0 mA h cm−2 in the SE-based cell also supported the crystallinity weakening in the Li–Ag alloys with high Li contents (Fig. S13–S17 and Tables S1–S7, ESI†). (3) Even after the stripping process at 0.15 V, the XANES and EXAFS profiles did not fully return to those of the pure Ag phase, which is consistent with the in situ XRD results.
This study investigates the correlation between the locations of Li deposits and the physicochemical property evolution of Ag/C layers in anode-less battery systems using XRD, XAFS, and cross-sectional SEM characterizations. In the LE-based cell, Li deposits were plated on the surface of the Ag/C layers, leaving unreacted Ag/C layers between the Li deposits and the current collector (CC). This phenomenon is attributed to the faster electron transfer on the Ag/C layers compared to Li conduction within the Ag/C layers. In contrast, in the SE-based cell, Li movement across the SE|Ag/C interfaces, controlled by diffusional Coble creep, resulted in homogeneous Li deposits (extending to the γ1 phase) plated between the Ag/C layers and the CC. This Coble creep-mediated Li movement promotes homogeneous Li–Ag alloying behaviours, not only in terms of the location of Li deposits but also in terms of the phase evolution of Li–Ag alloys. If the electrochemical environment did not facilitate the effective operation of Coble creep, Li deposits would have formed between the Ag/C and SE.16 This study aims to enhance the fundamental understanding of Li electrodeposition in anode-less battery systems that use lithiophilic element/porous carbon composite layers.
This research was supported by the National Research Foundation of Korea (NRF-2022R1C1C1004171). The authors also acknowledge technical support with 5A (MS-XRS) and 10C (Wide XAFS) beamlines of the Pohang Light Source-II in the Pohang Accelerating Laboratory.
Data availability
The data supporting this article have been included as part of the ESI.† And, data are available from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- J. Lee, S. H. Choi, G. Im, K.-J. Lee, T. Lee, J. Oh, N. Lee, H. Kim, Y. Kim, S. Lee and J. W. Choi, Adv. Mater., 2022, 34, 2203580 CrossRef CAS PubMed.
- N. Lee, J. Oh and J. W. Choi, Mater. Futures, 2023, 2, 013502 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- H. J. Choi, D. W. Kang, J.-W. Park, J.-H. Park, Y.-J. Lee, Y.-C. Ha, S.-M. Lee, S. Y. Yoon and B. G. Kim, Adv. Sci., 2022, 9, 2103826 CrossRef CAS PubMed.
- J. A. Lewis, C. Lee, Y. Liu, S. Y. Han, D. Prakash, E. J. Klein, H.-W. Lee and M. T. McDowell, ACS Appl. Mater. Interfaces, 2022, 14, 4051–4060 CrossRef CAS PubMed.
- Z. Liang, Y. Xiang, K. Wang, J. Zhu, Y. Jin, H. Wang, B. Zheng, Z. Chen, M. Tao, X. Liu, Y. Wu, R. Fu, C. Wang, M. Winter and Y. Yang, Nat. Commun., 2023, 14, 259 CrossRef CAS PubMed.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- J.-S. Kim, G. Yoon, S. Kim, S. Sugata, N. Yashiro, S. Suzuki, M.-J. Lee, R. Kim, M. Badding, Z. Song, J. Chang and D. Im, Nat. Commun., 2023, 14, 782 CrossRef CAS PubMed.
- C. Wu, B. Emley, L. Zhao, Y. Liang, Q. Ai, Z. Chen, F. C. Robles Hernández, F. Wang, S. Risal, H. Guo, J. Lou, Y. Yao and Z. Fan, Nano Lett., 2023, 23, 4415–4422 CrossRef CAS PubMed.
- B. Li, Z. Sun, N. Lv, Y. Hu, L. Jiang, Z. Zhang and F. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 37738–37746 CrossRef CAS PubMed.
- D. Spencer-Jolly, V. Agarwal, C. Doerrer, B. Hu, S. Zhang, D. L. R. Melvin, H. Gao, X. Gao, P. Adamson, O. V. Magdysyuk, P. S. Grant, R. A. House and P. G. Bruce, Joule, 2023, 7, 503–514 CrossRef CAS.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS.
- S. H. Park, D. Jun, J. E. Jung, S. G. Lee, G. H. Lee and Y. J. Lee, J. Mater. Chem. A, 2022, 10, 21995–22006 RSC.
- S. H. Park, D. Jun, G. H. Lee, S. G. Lee, J. E. Jung, K. Y. Bae, S. Son and Y. J. Lee, Adv. Sci., 2022, 9, 2203130 CrossRef CAS PubMed.
- Y. Chen, Z. Wang, X. Li, X. Yao, C. Wang, Y. Li, W. Xue, D. Yu, S. Y. Kim, F. Yang, A. Kushima, G. Zhang, H. Huang, N. Wu, Y.-W. Mai, J. B. Goodenough and J. Li, Nature, 2020, 578, 251–255 CrossRef CAS PubMed.
- Z. Wang, X. Li, Y. Chen, K. Pei, Y.-W. Mai, S. Zhang and J. Li, Chem, 2020, 6, 2878–2892 CAS.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- S. Li, T. Zhao, K. Wang, C. Sun, W. Jia, M. Zhang, H. Wang, A. Shao and Y. Ma, Adv. Funct. Mater., 2022, 32, 2203010 CrossRef CAS.
- S. Zhang, G. Yang, Z. Liu, S. Weng, X. Li, X. Wang, Y. Gao, Z. Wang and L. Chen, ACS Energy Lett., 2021, 6, 4118–4126 CrossRef CAS.
- S. Jin, Y. Ye, Y. Niu, Y. Xu, H. Jin, J. Wang, Z. Sun, A. Cao, X. Wu, Y. Luo, H. Ji and L.-J. Wan, J. Am. Chem. Soc., 2020, 142, 8818–8826 CrossRef PubMed.
- A. D. Pelton, Bull. Alloy Phase Diagrams, 1986, 7, 223–228 Search PubMed.
- C.-M. Park, H. Jung and H.-J. Sohn, Electrochem. Solid-State Lett., 2009, 12, A171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical data, electron microscopy images, XRD patterns, EXAFS results, and digital images of Ag/C layers. See DOI: https://doi.org/10.1039/d4cc02704c |
| This journal is © The Royal Society of Chemistry 2024 |