
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of vioprolide B and its dehydrobutyrine-glycine analogue†
Noé
Osorio Reineke
a,
Franziska A. V.
Elsen
 b,
Hanusch A.
Grab
a,
Dietrich
Mostert
b,
Stephan A.
Sieber
b and
Thorsten
Bach
*a
b,
Hanusch A.
Grab
a,
Dietrich
Mostert
b,
Stephan A.
Sieber
b and
Thorsten
Bach
*a
aTechnische Universität München, School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Lichtenbergstraße 4, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de
bTechnische Universität München, School of Natural Sciences, Department of Bioscience and Center for Functional Protein Assemblies, Ernst-Otto-Fischer-Straße 8, 85747 Garching, Germany
First published on 15th July 2024
Abstract
Herein, we describe the total synthesis of the depsipeptide vioprolide B and of an analogue, in which the (E)-dehydrobutyrine amino acid was replaced by glycine. The compounds were studied in biological assays which revealed cytotoxicity solely for vioprolide B presumably by covalent binding to cysteine residues of elongation factor eEF1A1 and of chromatin assembly factor CHAF1A.
Natural products and other biologically active compounds frequently display an α,β-unsaturated carboxylic ester or amide group as a potential Michael acceptor. The functional group invites conjugate addition reactions which can lead to irreversible binding to a target protein. If selectivity is achieved towards a specific protein and if binding leads to regulation of the protein function, this mode of action can be a useful starting point for drug discovery.1 However, the presence of a Michael acceptor in a molecule does not necessarily imply it to be active by conjugate addition. Our groups have for some time been interested in the biological chemistry of a class of depsipeptides,2 called vioprolides.3 The compounds were first isolated by Reichenbach, Höfle and co-workers from the myxo-bacterium Cystobacter violaceus Cb vi35.4 Detailed biosynthetic studies were performed in the group of R. Müller which revealed the individual steps of the nonribosomal peptide synthesis including the formation of less common structural elements.5 A key feature of the compound class is the presence of E-dehydrobutyrine which evolves biosynthetically from threonine by elimination. The configuration of the double bond imposes notable strain on the molecule and prohibits peptide bond formation to the adjacent amino acid.6 In the so far only total synthesis7 of a vioprolide, vioprolide D,3b the double bond configuration was established in the final stages by Z → E isomerization. In the present study, we have interrogated the role of the double bond for the biological activity of the vioprolides. To this end, we prepared vioprolide B (1) which displayed in previous work a higher anticancer activity than vioprolide D.3a The compound was compared with a synthetic analogue 2, in which the dehydrobutyrine (Dhb) entity was replaced by a glycine (Gly) fragment (Fig. 1). It was found that compound 2 was completely inactive in assays against HeLa and Jurkat cells. Possible biological targets of vioprolide B were identified by competitive activity-based protein profiling (ABPP) with cysteine-reactive iodoacetamide alkyne probes.8
 | ||
| Fig. 1 Structures of vioprolide B (1) and an analogue 2, in which the potential Michael acceptor (E)-dehydrobutyrine (Dhb) is replaced by a glycine (Gly). | ||
The synthesis of vioprolide B commenced with the known7c southern fragment 3, to the N-terminal site of which N-tert-butyloxycarbonyl(Boc)-protected pipecolic acid (N-Boc-Pip) was attached by peptide coupling.9,10 After releasing the Boc-protecting group from piperidine 4, a peptide coupling10 of compound 5 with the northern fragment3b of vioprolide B was probed. Various attempts with the free C-terminal carboxylic acid failed which is why the corresponding pentafluorophenyl ester116 was prepared (see the ESI† for further details). Stirring of secondary amine 5 with the activated ester 6 at 50 °C led to a smooth bond formation to product Z-7 which comprises the complete skeleton of the natural product. As for vioprolide D, the adjustment of the relative configuration from Z- to E-Dhb was postponed to the final step. Hence, the deprotection of the ester at the glycerate12 was followed by removal of the N-Boc group at the N-terminal amino acid alanine. Macrolactamization13 and global silyl deproctection resulted in depsipeptide Z-8. Thiazoline ring formation14 was the final bond forming step before the adjustment of the double bond configuration was completed. Iodination and elimination15 delivered a Z-configured alkenyl iodide the iodine–carbon bond of which was hydrogenolytically cleaved16 under retention of configuration. After purification by preparative HPLC, vioprolide B (1) was obtained as a diastereomerically pure compound, the analytical data of which were in full agreement with the natural product (Scheme 1).
 | ||
| Scheme 1 Total synthesis of vioprolide B (1) by linkage of a northern (6) and southern (5) fragment followed by macrolacamization, thiazoline formation, and adjustment of the double configuration (Z → E) at the dihydrobutyrine fragment. Abbreviations: TFA = trifluoroacetic acid; HOBt = hydroxybenzotriazole; EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; DCE = dichloroethane; HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; DIAD = Di-iso-propyl azodicarboxylate; NIS = N-iodosuccinimide; DABCO = 1,4-diazabicyclo[2.2.2]octane. | ||
The introduction of the glycine fragment as required for analogue 9 commenced at an initial phase of the total syn-thesis. Glycine-serine dipeptide 9 was reacted with the known17 activated thioproline-substituted benzotriazole 10. Deprotection of the proline led to tripeptide 11 which was coupled with the D-leucine–alanine dipeptide 12 thus completing the assembly of the northern half of the target molecule. Since we relied again on an activated ester for combining the northern and the southern fragment, methyl ester 13 was converted into the pentafluorophenyl ester 14. The coupling with fragment 5 was successfully performed by stirring at ambient temperature and delivered product 15. The final steps of the synthesis followed the protocol employed for vioprolide B (Scheme 2). Macrolactamization, thiazoline formation and deprotection enabled the conversion to thioamide 16. Thiazoline formation was accomplished with the Burgess reagent18 since the impurities of the Mitsunobu protocol were impossible to separate from the final product. A full comparison of the NMR data of 1 and 2 are found in the ESI.†
 | ||
| Scheme 2 Synthesis of vioprolide B analogue 2 by replacing the Dhb unit with a glycine. Burgess reagent = (methoxycarbonylsulfamoyl)triethylammonium hydroxide. | ||
To evaluate the cytotoxicity of both molecules, we determined the metabolic activity of Jurkat cells upon addition of 1 and 2via the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay (Fig. 2). Interestingly, while synthetic vioprolide B (1) displayed potent cytotoxicity with an IC50 value of 123 nM [94–148 nM 95% confidence interval (CI)], the Dhb analogue 2 lost its bioactivity (IC50 > 10 μM) highlighting the relevance of the Michael acceptor. The activity of the synthetic material matched well the previously studied cytotoxicity determined for vioprolide B isolated from the natural producer. The IC50 value for Jurkat cells had been determined as 187 ± 24 nM for the natural product.3a
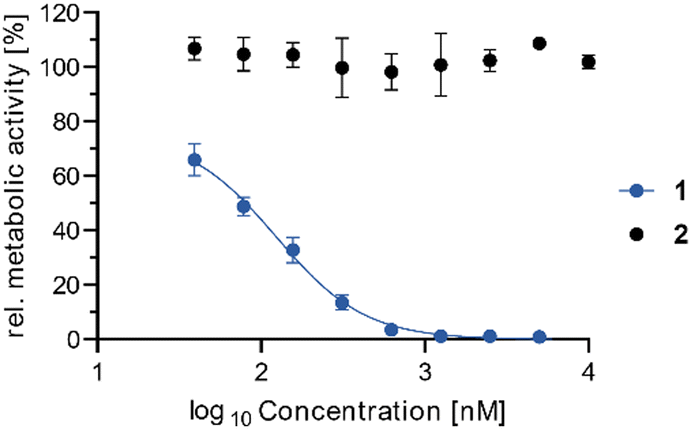 | ||
| Fig. 2 Dose-dependent inhibition of Jurkat cell proliferation by compounds 1 (vioprolide B) and 2 (Dhb-Gly analogue). IC50 is determined by MTT assay with 48 h treatment and calculated as 123 nM (94–148 nM 95% confidence intervall) for 1 and >10 μM for 2. Data points result from four biologically independent experiments performed in three technical replicates. | ||
In earlier work, we had seen that the nature of the exocyclic double bond in vioprolide D has a strong influence on its activity,3b and the present result further supported the key role of the E-Dhb entity. We, thus, hypothesized that the electrophilic Michael acceptor is crucial for the covalent interaction with nucleophilic cysteines on cellular proteins. To unravel these cellular protein targets, we performed activity-based protein profiling (ABPP) in a competitive mode utilizing the cysteine reactive iodoacetamide alkyne probe (IAA) (Scheme 3).8 Jurkat cells were pre-incubated with 10 μM of 1 or 2 as control for 1 h, followed by the addition of IAA to label residual free cysteines in the proteome. The cells were subsequently lysed and modified with isotopically labelled light or heavy desthiobiotin azide tags (iso-DTB) via click chemistry.8c Enrichment of probe-bound proteins via streptavidin beads followed by tryptic digest revealed light and heavy isotopically labelled peptides which were analysed via LC-MS/MS. The corresponding volcano plot depicts most significantly enriched proteins which bind IAA treatment and disappear upon pre-incubation with 1. Among the most significant hits, we identified the elongation factor 1-alpha 1 (eEF1A1) as well as the chromatin assembly factor 1 subunit a (CHAF1A), both with essential roles for cell viability. Moreover, using the MS-Fragger software, we identified residue Cys 31 in eEF1A1 and Cys 79 in CHAF1A as the modified sites.19 As part of the ribosomal elongation complex, eEF1A1 catalyses the transfer of the aminoacyl-tRNA to the ribosome during protein biosynthesis.20 CHAF1A forms the largest subunit of CAF-1, an essential chromatin assembly factor involved in the replication fork progression in DNA replication.21
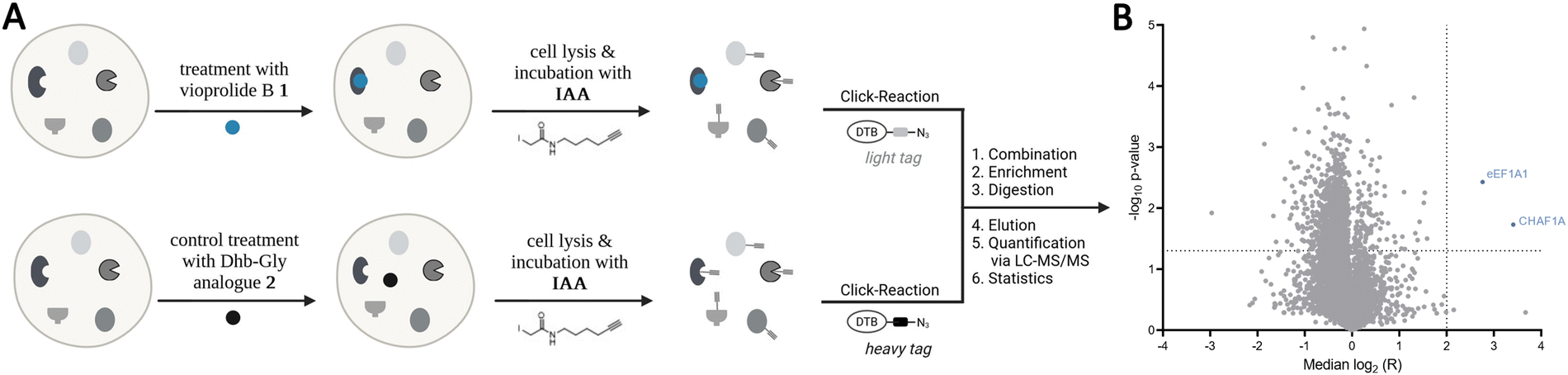 | ||
| Scheme 3 (A) Schematic overview of the competitive isotopically labelled desthiobiotin azide (isoDTB)-activity-based protein profiling (ABPP) workflow. Two identical sample sets of Jurkat cells were treated in situ with compound 1 (upper) or 2 as control (lower), lysed, incubated with cysteine-reactive iodoacetamide alkyne (IAA) and clicked to isotopically labelled light respective heavy tags. Samples were combined, enriched, digested with trypsin, eluted from streptavidin beads and quantified by LC-MS/MS. The difference in MS1 signal intensity between heavy (2-treated, control) and light (1-treated) labelled peptides is represented by the competition ratio R. (B) The results of (A) are shown in the volcano plot and represent the median log2(R) and the statistical −log10(p) by a one-sample t-test of for all quantified cysteines. All data result from at least three biologically independent replicates. (A) was created with https://BioRender.com. | ||
Overall, our study provides for the first time evidence for the importance of the exocyclic double bond present in the vioprolides. We here demonstrate its essential role for cancer cell toxicity and its binding to cysteine residues within proteins regulating translation and chromatin assembly. These results highlight that the concept of targeted covalent modification is highly relevant for natural product cytotoxicity and that the structural complexity of vioprolides by itself is not sufficient for cellular target engagement.
Financial support by the Deutsche Forschungsgemeinschaft (Ba 1372/23) is gratefully acknowledged. We thank O. Ackermann (TU München) for support with the HPLC measurements. NOR warmly thanks J. Domack and T. Ziegelmeier for synthetic support. We thank M. Zollo and S. M. Hacker for the useful discussion on proteomics. F. A. V. Elsen is supported by a Kekulé-Stipendium of the Fonds der Chemischen Industrie (FCI).
Data availability
The data supporting this article have been included as part of the ESI.† Primary data are available at the ProteomeXchange Consortium (PRIDE partner repository with the dataset identifier PXD053104) and at Zenodo (https://doi.org/10.5281/zenodo.11576894).Conflicts of interest
There are no conflicts to declare.Notes and references
- Reviews: (a) M. Gersch, J. Kreuzer and S. A. Sieber, Nat. Prod. Rep., 2012, 29, 659–682 RSC; (b) P. Gehrtz and N. London, Trends Pharmacol. Sci., 2021, 42, 434–447 CrossRef CAS PubMed; (c) S.-T. Liang, C. Chen, R.-X. Chen, R. Li, W.-L. Chen, G.-H. Jiang and L.-L. Du, Front. Pharmacol., 2022, 13, 1033003 CrossRef CAS PubMed.
- Reviews: (a) H. I. Farah, U. Supratman, A. T. Hidayat and R. Maharani, ChemistrySelect, 2022, 7, e202103470 CrossRef CAS; (b) X. Wang, X. Gong, P. Li, D. Lai and L. Zhou, Molecules, 2018, 23, 169 CrossRef PubMed; (c) S. C. Stolze and M. Kaiser, Molecules, 2013, 18, 1337–1367 CrossRef CAS PubMed; (d) S. C. Stolze and M. Kaiser, Synthesis, 2012, 1755–1777 CAS.
- (a) V. C. Kirsch, C. Orgler, S. Braig, I. Jeremias, D. Auerbach, R. Müller, A. M. Vollmar and S. A. Sieber, Angew. Chem., Int. Ed., 2020, 59, 1595–1600 CrossRef CAS PubMed; (b) H. A. Grab, V. C. Kirsch, S. A. Sieber and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 12357–12361 CrossRef CAS PubMed.
- D. Schummer, E. Forche, V. Wray, T. Domke, H. Reichenbach and G. Höfle, Liebigs Ann., 1996, 971–978 CrossRef CAS.
- (a) F. Yan, D. Auerbach, Y. Chai, L. Keller, Q. Tu, S. Hüttel, A. Glemser, H. A. Grab, T. Bach, Y. Zhang and R. Müller, Angew. Chem., Int. Ed., 2018, 57, 8754–8759 CrossRef CAS PubMed; (b) F. Yan and R. Müller, ACS Chem. Biol., 2019, 14, 99–105 CrossRef CAS PubMed.
- E. Butler, L. Florentino, D. Cornut, G. Gomez-Campillos, H. Liu, A. C. Regan and E. J. Thomas, Org. Biomol. Chem., 2018, 16, 6935–6960 RSC.
- For other synthetic efforts towards the vioprolides, see: (a) N. Chopin, F. Couty and G. Evano, Lett. Org. Chem., 2010, 7, 353–359 CrossRef CAS; (b) H. Liu and E. J. Thomas, Tetrahedron Lett., 2013, 54, 3150–3153 CrossRef CAS; (c) N. Osorio Reineke, H. A. Grab and T. Bach, Synthesis DOI:10.1055/s-0043-1763750 , in press.
- (a) K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. Gonzalez-Paez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed; (b) E. Weerapana, et al. , Nature, 2010, 468, 790–795 CrossRef CAS PubMed; (c) P. R. A. Zanon, L. Lewald and S. M. Hacker, Angew. Chem., Int. Ed., 2020, 59, 2829–2836 CrossRef CAS PubMed.
- (a) Y. Chen, M. Bilban, C. A. Foster and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 5431–5440 CrossRef CAS PubMed; (b) J. Yao, H. Liu, T. Zhou, H. Chen, Z. Miao, G. Dong, S. Wang, C. Sheng and W. Zhang, Tetrahedron, 2012, 68, 3074–3085 CrossRef CAS; (c) P. Barbie and U. Kazmaier, Org. Lett., 2016, 18, 204–207 CrossRef CAS PubMed.
- Reviews: (a) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (b) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS; (c) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed.
- (a) L. Kisfaludy, J. E. Roberts, R. H. Johnson, G. L. Mayers and J. Kovacs, J. Org. Chem., 1970, 35, 3563–3565 CrossRef CAS PubMed; (b) J. Yang, H. Huang and J. Zhao, Org. Chem. Front., 2023, 10, 1817–1846 RSC.
- K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee and B. S. Safina, Angew. Chem., Int. Ed., 2005, 44, 1378–1382 CrossRef CAS PubMed.
- (a) L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS; (b) L. A. Carpino and A. El-Faham, J. Org. Chem., 1994, 59, 695–698 CrossRef CAS; (c) L. A. Carpino, A. El-Faham and F. Albericio, J. Org. Chem., 1995, 60, 3561–3564 CrossRef CAS.
- (a) O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS; (b) N. Galéotti, C. Montagne, J. Ponce and P. Jouin, Tetrahedron Lett., 1992, 33, 2807–2810 CrossRef.
- (a) R. S. Hoerrner, D. Askin, R. P. Volante and P. J. Reider, Tetrahedron Lett., 1998, 39, 3455–3458 CrossRef CAS; (b) G. J. Roff, R. C. Lloyd and N. J. Turner, J. Am. Chem. Soc., 2004, 126, 4098–4099 CrossRef CAS PubMed; (c) P. M. T. Ferreira, L. S. Monteiro and G. Pereira, Eur. J. Org. Chem., 2008, 4676–4683 CrossRef CAS; (d) Y. Yasuno, A. Nishimura, Y. Yasukawa, Y. Karita, Y. Ohfune and T. Shinada, Chem. Commun., 2016, 52, 1478–1481 RSC.
- (a) S. M. Kupchan and A. Afonso, J. Org. Chem., 1960, 25, 2217–2218 CrossRef CAS; (b) A. P. Kozikowski and K. Sugiyama, Tetrahedron Lett., 1980, 21, 4597–4600 CrossRef CAS; (c) D. R. Boyd, N. D. Sharma, J. F. Malone and C. C. R. Allen, Chem. Commun., 2009, 3633–3635 RSC; (d) S. Hanessian, G. Huang, C. Chenel, R. Machaalani and O. Loiseleur, J. Org. Chem., 2005, 70, 6721–6734 CrossRef CAS PubMed.
- (a) M. A. Shalaby, C. W. Grote and H. Rapoport, J. Org. Chem., 1996, 61, 9045–9048 CrossRef CAS PubMed; (b) B. McKeever and G. Pattenden, Tetrahedron, 2003, 59, 2701–2712 CrossRef CAS.
- (a) G. M. Atkins, Jr. and E. M. Burgess, J. Am. Chem. Soc., 1968, 90, 4744–4745 CrossRef; (b) P. Wipf and P. C. Fritch, Tetrahedron Lett., 1994, 35, 5397–5400 CrossRef CAS; (c) C. D. J. Boden and G. Pattenden, Tetrahedron Lett., 1995, 36, 6153–6156 CrossRef CAS.
- (a) A. T. Kong, F. da Veiga Leprevost, D. M. Avtonomov, D. Mellacheruvu and A. I. Nesvizhskii, Nat. Methods, 2017, 14, 513–520 CrossRef CAS PubMed; (b) F. Yu, et al. , Nat. Commun., 2020, 11, 4065 CrossRef CAS PubMed.
- T. E. Dever, J. D. Dinman and R. Green, CSH Perspect. Biol., 2018, vol. 10, p. a032649 Search PubMed.
- M. Hoek and B. Stillman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12183–12188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02946a |
| This journal is © The Royal Society of Chemistry 2024 |