
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignin-derivable alternatives to petroleum-derived non-isocyanate polyurethane thermosets with enhanced toughness†
Sampanna V.
Mhatre
 a,
Jignesh S.
Mahajan
a,
Thomas H.
Epps
III
*abc and
LaShanda T. J.
Korley
*abc
a,
Jignesh S.
Mahajan
a,
Thomas H.
Epps
III
*abc and
LaShanda T. J.
Korley
*abc
aDepartment of Materials Science and Engineering, University of Delaware, Newark, Delaware 19716, USA. E-mail: thepps@udel.edu; lkorley@udel.edu
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA
cCenter for Research in Soft matter and Polymers (CRiSP), University of Delaware, Newark, Delaware 19716, USA
First published on 30th November 2022
Abstract
The structural similarities between lignin-derivable bisguaiacols and petroleum-derived bisphenol A/F (BPA/BPF) suggest that bisguaiacols could be ideal biobased alternatives to BPA/BPF in non-isocyanate polyurethane (NIPU) thermosets. Herein, bisguaiacol/bisphenol-derived cyclic carbonates with variations in methoxy content and bridging-carbon substitution were cured with two triamines of different chain lengths, and the impact of these differences on the thermomechanical properties of NIPU networks was examined. The methoxy groups present in the lignin-derivable cyclic carbonates led to thermosets with significantly improved toughness (∼49–59 MJ m−3) and elongation at break (εb ∼195–278%) vs. the BPA/BPF-based benchmarks (toughness ∼ 26–35 MJ m−3, εb ∼ 86–166%). Furthermore, the addition of dimethyl substitution on the bridging carbon resulted in increased yield strength (σy) – from ∼28 MPa for networks with unsubstituted bridging carbons to ∼45 MPa for the dimethyl-substituted materials. These enhancements to mechanical properties were achieved while retaining essential thermoset properties, such as application-relevant moduli and thermal stabilities. Finally, the triamine crosslinkers provided substantial tunability of thermomechanical properties and produced NIPUs that ranged from rigid materials with a high yield strength (σy ∼ 65–88 MPa) to flexible and tough networks. Overall, the structure-property relationships presented highlight a promising framework for the design of versatile, bio-derivable, NIPU thermosets.
Introduction
Polyurethanes (PUs) are a class of thermoplastic (linear chain) or thermosetting (crosslinked) polymers with numerous uses including in building and industrial insulation, cars, furniture, clothing, and shoes – and as coatings, sealants, and adhesives.1–6 Given these various applications, PUs rank 6th in global polymer production, with more than 70 wt% of PUs employed as thermosetting materials.4,5 Yet, PUs are primarily manufactured from petroleum feedstocks, and only ∼1% of the total global PU production was biobased as of 2020.7,8 Additionally, the synthesis of PUs typically involves the use of hazardous isocyanates.2,3 Two of the most common thermosetting PU precursors, methylene diphenyl diisocyanate (MDI) and toluene diisocyanate (TDI), are considered carcinogenic, mutagenic, and reprotoxic,4,6 and extended exposure to isocyanates can lead to other severe health concerns, such as asthma, dermatitis, conjunctivitis, and acute poisoning.3,6,9 Moreover, most isocyanates are manufactured from toxic and hazardous phosgene gas.10 Compounding these toxicity and potential non-renewability considerations at their end of life, waste thermoset PUs are either incinerated or land-filled, releasing toxins and depleting land resources6 with a few exceptions, such as the utilization of PU foam mattresses for soil-free food production.11To address the challenges associated with isocyanates, non-isocyanate polyurethanes (NIPUs), e.g., poly(hydroxyurethanes), have emerged as more benign alternatives to traditional PUs.1,6,12 Typical NIPU thermosets are synthesized by reacting multifunctional cyclic carbonates and amines (as opposed to multifunctional isocyanates and diols in traditional PUs), which leads to pendant hydroxyl groups adjacent to the urethane linkages in the NIPU polymer backbone.3,13,14 These additional hydroxyl units can enhance the thermomechanical properties and also impart reprocessability to NIPU thermosets through associative transcarbomylation.13–16 The thermomechanical properties of these polymeric systems are governed by the nature of the building blocks. For example, aromatic building blocks provide the required rigidity to networks for high-performance applications, while aliphatic building blocks offer weather and ultraviolet light resistance.4,5,15–17 Although NIPU thermosets derived from biobased feedstocks have been reported in the literature,1,18,19 many of these approaches have been primarily limited to aliphatic/cycloaliphatic, bifunctional or multifunctional cyclic carbonates derived from soybean oil,20–22 linseed oil,23 glycerol, pentaerythritol, trimethylolpropane,24 or limonene.25 The aliphatic or cycloaliphatic backbones present in most biobased NIPUs result in thermosets with relatively low moduli and tensile strengths.1,18–20,22–25 In contrast, petroleum-based bisphenol A (BPA)-derived cyclic carbonate (BPACC), prepared from the dicarbonation of BPA diglycidyl ether (BADGE), is often employed to make higher-moduli NIPU networks.15,16,26 Unfortunately, BPA is considered non-sustainable27,28 and is a suspected endocrine disruptor, as it can potentially bind to estrogen receptors.17,29 Thus, there is a significant need to find and utilize non-toxic, biobased, aromatic building blocks that structurally resemble BPA (or MDI) and can be useful as precursors in NIPU thermosets.
Lignin is a high-volume renewable source of aromatic building blocks,30–34 and it can be deconstructed to yield a broad range of substituted phenols that then can be converted into bisphenols through traditional chemical approaches.15,17,30,35–42 Moreover, these lignin-derivable bisphenols (particularly bisguaiacols) possess lower estrogenic activity in comparison to petroleum-derived BPA and bisphenol F (BPF) due to the methoxy groups that restrict access to estrogen receptors.43,44 These bisguaiacols have been studied as replacements for BPA/BPF in epoxy-amine networks, wherein the relative number of methoxy substituents, and their regioisomers on the aromatic ring impacted the glass transition temperatures (Tgs) and storage moduli of the thermosets.30,45,46 Lignin-derivable syringaresinol dicyclic carbonate crosslinked with tris(2-aminoethyl) amine (TREN) also has been investigated as an alternative to a BPA-based NIPU network.16 The bio-derivable NIPU had a Tg of ∼62 °C that was comparable to a TREN-cured BPACC control.16 However, in these studies application-significant tensile properties (e.g., toughness, elongation at break [εb], and tensile strength at yield [σy]) were not explored. Overall, there is a need for a systematic design and evaluation of lignin-derivable NIPU networks that are structurally similar to petroleum-based bisaromatic NIPUs. Accordingly, this work describes a significant expansion of the library of biobased cyclic carbonates by utilizing lignin-derivable cyclic carbonates for NIPU thermoset formation with robust thermomechanical performance that can be evaluated against aromatic, petroleum-derived analogues.
Herein, the thermomechanical properties of a series of lignin-derivable NIPU thermosets are explored. Specifically, the impact of structural differences (e.g., the number of methoxy groups on the aromatic rings and bridging-carbon substitutions in the lignin-derivable monomers) on Tg, thermal stability, modulus, σy, toughness, and εb are probed. For this study, bio-derivable bisguaiacols with methoxy groups on the aromatic rings were functionalized to form diglycidyl ethers and then dicyclic carbonates (Scheme 1). The behavior of these lignin-inspired materials was benchmarked against petroleum-derived analogues generated from BPA and BPF. The various cyclic carbonates were cured by two different amines, TREN (short-chain triamine) and JEFFAMINE® T-403 (T403, long-chain, flexible, polyether triamine), to assess structure–property relationships of both tightly and loosely crosslinked NIPU networks (Scheme 2). Using this approach, the thermomechanical versatility of the lignin-derivable NIPU thermosets became apparent, in which the mechanical properties, in particular the toughness, were significantly enhanced in comparison to the petroleum-based benchmarks, while the advantageous thermal properties were retained.
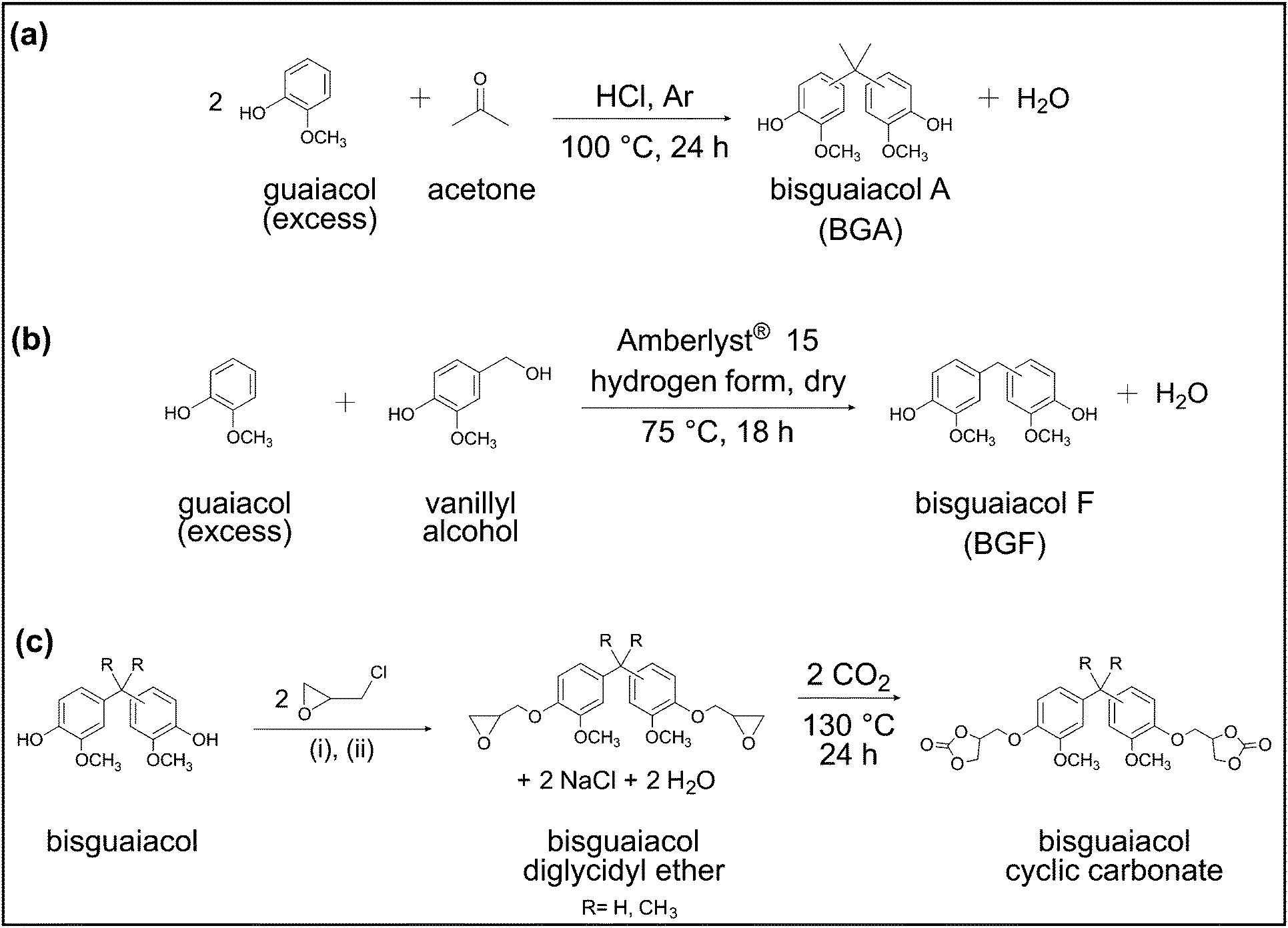 | ||
| Scheme 1 Synthetic scheme of lignin-derivable bisguaiacols, diglycidyl ethers, and cyclic carbonates. (a) Condensation of guaiacol and acetone to give bisguaiacol A (BGA). (b) Reaction between vanillyl alcohol and guaiacol to give bisguaiacol F (BGF). (c) Glycidylation of bisguaiacols to form diglycidyl ethers, followed by carbonation to give cyclic carbonates. (i) 0.1 mol tetrabutylammonium bromide (TBAB), argon (Ar), 50 °C for 2 h, (ii) 4 mol sodium hydroxide (NaOH, aq, 40 wt%), 50 °C for 18 h. | ||
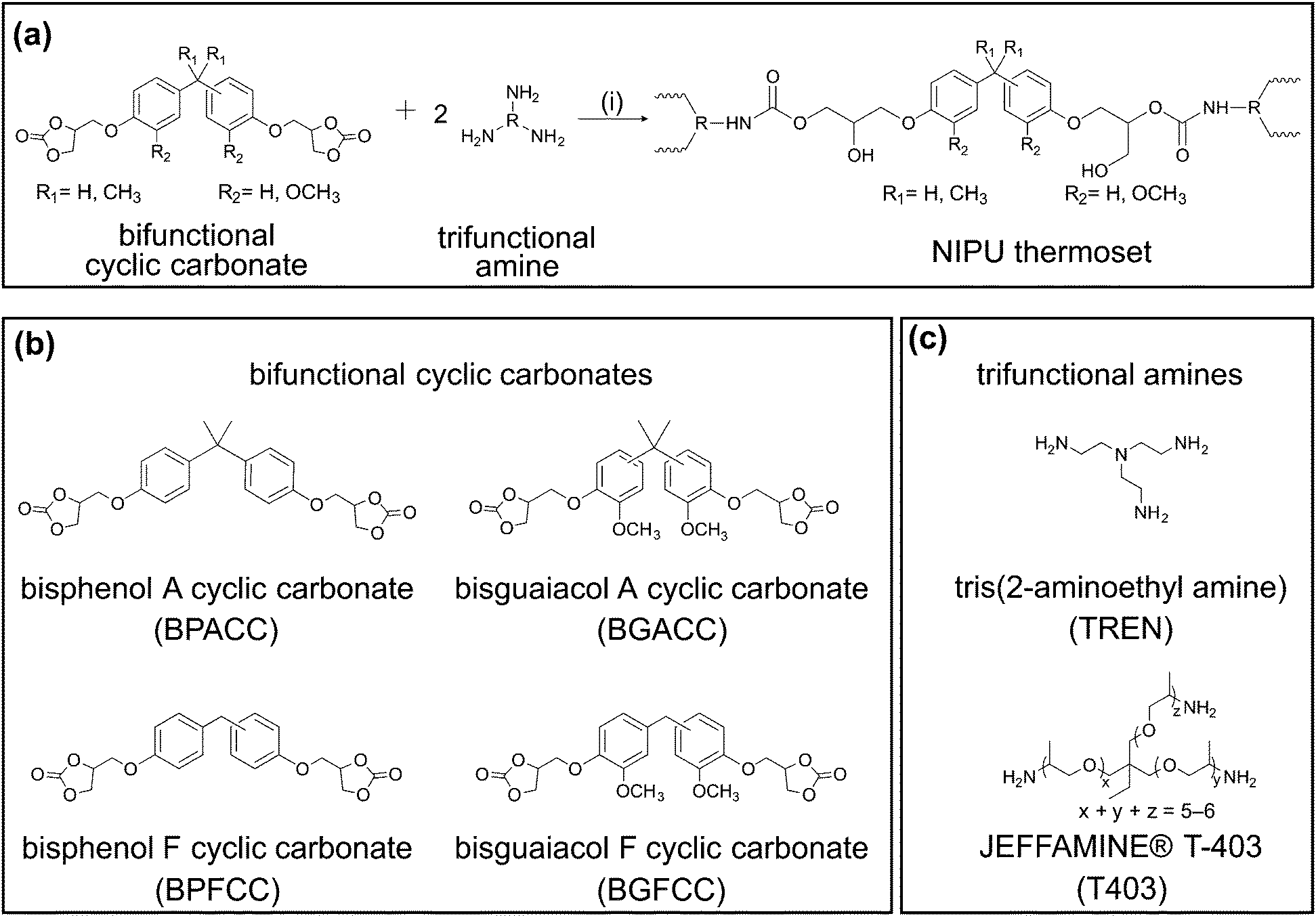 | ||
| Scheme 2 NIPU thermosets: synthetic scheme and molecular structures of cyclic carbonates and triamines. (a) Reaction of bifunctional cyclic carbonates with trifunctional amines to form NIPU thermosets. (i) N,N-dimethylformamide (DMF), 100 °C for 72 h. (b) Molecular structures of cyclic carbonates, wherein the lignin-derivable compounds (on the right) have one methoxy group both on the right-most and the left-most aromatic ring (1,1), and the petroleum-derived analogues on the left have no methoxy group on either ring (0,0). The compounds on top have a dimethyl-substituted bridging carbon (Me), and the bottom compounds have an unsubstituted bridging carbon (Un). (c) Molecular structures of trifunctional amines used as curing agents. | ||
Experimental
Materials
BADGE (molecular weight = 340.41 g mol−1, extent of oligomerization = ∼0.02, physical form = solid), guaiacol (≥98% food grade), vanillyl alcohol (≥98%), thioglycolic acid (98%), and Amberlyst® 15 hydrogen form (dry) were purchased from Sigma-Aldrich. Bisphenol F diglycidyl ether (BFDGE, EPON™ 862, epoxy equivalent weight = 170 g mol−1), a product of Hexion was purchased through Chemical Marketing Concepts Inc. T403 (molecular weight = ∼440 g mol−1), a product of The Huntsman Corporation, was supplied by Azelis Americas CASE. Tetrabutylammonium bromide (TBAB, 99+%), epichlorohydrin (99%), and deuterated dimethyl sulfoxide (DMSO-d6) were purchased from Acros Organics. TREN (97%), acetone (≥99.5%), hydrochloric acid (HCl, 36.5 to 38.0% [w/w]), sodium hydroxide (≥97%), N,N-dimethylformamide (DMF, ≥99.8%), magnesium sulfate (anhydrous), sodium bicarbonate (≥99.7%), sodium chloride (≥99.5%), dichloromethane (DCM, ≥99.5%), hexanes (99%), ethyl acetate (≥99.5%), acetonitrile (≥99.9%) and chloroform (>99.8%) were purchased from Thermo Fisher Scientific. All chemicals were used as received. Carbon dioxide (CO2, grade 5), argon (Ar, grade 5), and nitrogen (N2, grade 5) were purchased from Keen Compressed Gas.Synthesis of bisguaiacol A (BGA)
BGA was prepared by the acid-catalyzed condensation of guaiacol and acetone (Scheme 1a) in a manner similar to the industrial approach for the synthesis of BPA.38 In a typical BGA synthesis, 40 g (0.32 mol) of guaiacol, and 3.4 mL (0.046 mol) of acetone (dried over magnesium sulfate) were weighed in a 250 mL, single-neck, round-bottom flask equipped with a condenser and magnetic stir bar. Next, 4 mL of concentrated HCl (4 g, ∼15% w/w relative to guaiacol) was slowly added to the above reactants, followed by the addition of 0.17 mL of thioglycolic acid. The reaction mixture was purged with Ar for 10 min and subsequently stirred at 100 °C under reflux for 24 h. Later, the solution was allowed to cool to ∼20 °C and then dissolved in 15 mL of DCM. The mixture was washed three times with saturated sodium bicarbonate solution in a separatory funnel. The organic phase was washed three times with deionized (DI) water followed by three washes with brine solution. The residue was purified by automated flash column chromatography (Biotage® Selekt, Sfär Silica column—60 μm particle size, 100 Å pore size, 100 g silica gel) with a step gradient of ethyl acetate (25% v/v) and hexanes (75% v/v) for elution. The solvents were removed from the product by rotary evaporation, after which, the concentrated product was dried in an oven under vacuum at 50 °C for 24 h. The desired product obtained was a white solid (purity: >99%, yield: ∼40 mol%). Proton (1H) Nuclear Magnetic Resonance (NMR) (600 MHz, DMSO-d6) δ 8.70 (s, 2H), 6.79 (dd, J = 8.5, 2.7 Hz, 0H), 6.71 (t, J = 3.5 Hz, 2H), 6.69–6.49 (m, 4H), 3.72 (s, 1H), 3.68 (d, J = 2.4 Hz, 4H), 2.51 (s, 5H), 1.55 (d, J = 18.2 Hz, 5H). Fourier Transform Mass Spectrometry (FTMS) (Electrospray Ionization [ESI], m/z): calcd for C17H20O4 288.1362; found 288.1354. The 1H NMR spectrum with peak assignments is in Fig. S1 (ESI†).Synthesis of bisguaiacol F (BGF)
BGF was synthesized by the electrophilic aromatic substitution reaction between vanillyl alcohol and guaiacol (Scheme 1b) as described in the literature.30,37 In a typical synthesis of BGF, guaiacol (0.32 mol, 40 g) and vanillyl alcohol (0.08 mol, 12 g) were weighed into a 250 mL, single-neck, round-bottom flask equipped with a magnetic stir bar.37 The mixture was heated at 75 °C, stirred, and sparged with Ar for 40 min. Subsequently, a solid catalyst, dry Amberlyst® 15 hydrogen form (3.6 g, 30% w/w relative to vanillyl alcohol) was added to the reaction mixture. The reaction was allowed to proceed for 16 h. The reaction mixture was then cooled to ∼20 °C, dissolved in ∼40 mL of DCM, and the catalyst was removed using a Büchner funnel equipped with Whatman filter paper (grade 1). The filtrate was washed three times with ∼40 mL of DI water in a separatory funnel. Excess DCM from the organic phase was removed by rotary evaporation, and the residue was further purified by automated flash column chromatography (Biotage® Selekt, Sfär Silica column—60 μm particle size, 100 Å pore size, 100 g silica gel) with a step gradient of ethyl acetate (25% v/v) and hexanes (75% v/v) for elution. Solvents were removed from the product by rotary evaporation, after which, the concentrated product was dried in an oven under vacuum at 50 °C for 24 h to give BGF as a white solid (purity: >99%, yield: ∼40 mol%).1H NMR (600 MHz, DMSO-d6) δ 8.67 (s, 2H), 6.87–6.50 (m, 6H), 3.71 (d, J = 7.0 Hz, 8H). FTMS (ESI, m/z): calcd for C15H16O4 260.1049; found 260.1041. The 1H NMR spectrum with peak assignments is in Fig. S2 (ESI†).General procedure for the synthesis of diglycidyl ethers
Bisguaiacol diglycidyl ethers were synthesized as described in Nicastro et al.30 In a typical synthesis of a diglycidyl ether, epichlorohydrin (0.20 mol), bisguaiacol (0.02 mol), and TBAB (0.002 mol) were weighed into a 50 mL, single-neck, round-bottom flask equipped with a magnetic stir bar. The mixture was stirred, sparged with Ar for 30 min at ∼20 °C, and then heated at 50 °C for 2 h in an oil bath. The reaction flask was immediately cooled in an ice bath (∼0 °C) for 10 min. The flask was then transferred to a water bath (maintained at ∼20 °C), and 40 wt% of aqueous NaOH (0.08 mol) was added dropwise to the reaction mixture. The reaction was continued for 16 h at ∼20 °C. After completion, the reaction mixture was dissolved in ∼10 mL of DCM, washed with DI water in a separation funnel until the aqueous layer reached neutral pH, and then washed three times with brine. The organic phase was purified by automated flash column chromatography (Biotage® Selekt, Sfär Silica column—60 μm particle size, 100 Å pore size, 100 g silica gel) with a step gradient of ethyl acetate (60% v/v) and hexanes (40% v/v) for elution. The solvents were removed from the product using a rotary evaporator, after which, the concentrated product was dried in an oven under vacuum at 45 °C for 24 h. The 1H NMR spectra of all the diglycidyl ethers with peak assignments are in Fig. S3–S6 (ESI†).General procedure for the synthesis of cyclic carbonates
Cyclic carbonates were synthesized by the catalytic carbonation of diglycidyl ethers (Scheme 1c). In a typical synthesis of bisphenolic cyclic carbonate (using BPACC as an example), BADGE (0.03 mol, 10 g), and TBAB (0.12 g, 1.2% w/w relative to diglycidyl ether) were weighed in a 25 mL Teflon™ cylindrical liner equipped with a magnetic stir bar. The liner was sealed in a 25 mL Parr reactor and purged with CO2 three times at ∼20 °C. The reaction mixture was then pressurized with ∼20 bar CO2, heated at 130 °C using a ceramic band heater connected to a proportional-integral-derivative controller, and stirred for 24 h. After the reaction time, the reactor was allowed to cool to ∼20 °C and was then depressurized. The resulting product was dissolved in acetone (∼2 mL) and precipitated in DI water (∼20 mL) two times. The precipitated solid was dried in an oven under vacuum at 65 °C for 24 h to afford BPACC. The other cyclic carbonates were synthesized similarly. The 1H NMR spectra of the cyclic carbonates with peak assignments are in Fig. S7–S10 (ESI†).NMR spectroscopy
All NMR samples were prepared in DMSO-d6, and spectra were recorded on a Bruker AVANCE III 600 MHz spectrometer. 1H NMR spectra were processed and evaluated using the MestreNova software package.Mass spectrometry
The masses of bisguaiacols, diglycidyl ethers, and cyclic carbonates were determined by ESI-FTMS using a Q-Exactive Orbitrap (Thermo Fisher Scientific) mass spectrometer. Samples were dissolved in acetonitrile at a concentration of 0.1 mg mL−1, and the ESI-FTMS was conducted in a positive ion mode by direct syringe injection of samples in the mass spectrometer.Preparation of NIPU thermosets
All NIPU networks were prepared by reacting equivalent functional groups of cyclic carbonates and amines (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.67 [mol:mol] on a molecule basis). Herein, the cyclic carbonate functionality was determined based on the molecular structures (confirmed by 1H NMR spectroscopy and mass spectrometry). The high purity (>99%) of cyclic carbonates indicated that the cyclic carbonates are bifunctional. In a typical thermoset preparation as illustrated through the formation of BGACC–TREN networks, 0.6 mmol of BGACC and 0.4 mmol of TREN were dissolved in 1.0 mL and 0.5 mL of DMF, respectively. After solubilization, the solutions of BGACC and TREN were added to a 20 mL scintillation vial and mixed by shaking on a shaker plate at 800 rpm for 20 min. The solution was then poured into a rectangular silicone mold having dimensions of 33 mm × 25 mm × 16 mm (length × width × height). For uniform and defect-free film formation, the reaction mixture was first cured in air at 60 °C for 2 h, then at 80 °C for 2 h, followed by 100 °C for 20 h. To ensure complete solvent removal and maximum conversion, the sample was postcured at 100 °C for 48 h under vacuum. The other 7 networks with either TREN or T403 were prepared similarly using stoichiometric equivalents (1:0.67 [mol:mol]) of the cyclic carbonates to amines to provide 0.1–0.2 mm-thick NIPU network films (Fig. S11, ESI†).
:0.67 [mol:mol] on a molecule basis). Herein, the cyclic carbonate functionality was determined based on the molecular structures (confirmed by 1H NMR spectroscopy and mass spectrometry). The high purity (>99%) of cyclic carbonates indicated that the cyclic carbonates are bifunctional. In a typical thermoset preparation as illustrated through the formation of BGACC–TREN networks, 0.6 mmol of BGACC and 0.4 mmol of TREN were dissolved in 1.0 mL and 0.5 mL of DMF, respectively. After solubilization, the solutions of BGACC and TREN were added to a 20 mL scintillation vial and mixed by shaking on a shaker plate at 800 rpm for 20 min. The solution was then poured into a rectangular silicone mold having dimensions of 33 mm × 25 mm × 16 mm (length × width × height). For uniform and defect-free film formation, the reaction mixture was first cured in air at 60 °C for 2 h, then at 80 °C for 2 h, followed by 100 °C for 20 h. To ensure complete solvent removal and maximum conversion, the sample was postcured at 100 °C for 48 h under vacuum. The other 7 networks with either TREN or T403 were prepared similarly using stoichiometric equivalents (1:0.67 [mol:mol]) of the cyclic carbonates to amines to provide 0.1–0.2 mm-thick NIPU network films (Fig. S11, ESI†).
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy
ATR-FTIR spectroscopy was performed using a Thermo Nicolet NEXUS 870 FTIR with deuterated triglycine sulfate/potassium bromide (DTGS/KBr) detector. All cyclic carbonates and NIPU network films were scanned at a resolution of 4 cm−1, and 64 scans were collected in the range of 4000–500 cm−1. NIPU formation was assessed by examination of the cyclic carbonate carbonyl stretching band at ∼1790 cm−1, the urethane carbonyl stretching band at ∼1700 cm−1, and the broad hydroxyl stretching band at ∼3500–3300 cm−1 (Fig. S12 and S13, ESI†).Soxhlet extraction
The gel content of crosslinked NIPU networks was determined through Soxhlet extraction. A representative sample of each NIPU network film (∼60 mg) was weighed and placed in a cellulose extraction thimble (obtained from Whatman). The sample-containing thimble was then placed in a Soxhlet extractor. Chloroform (∼100 mL) was used as the extraction solvent, and one cycle of thimble filling and emptying took about 10–12 min. The Soxhlet extraction was done for 24 h at 80 °C with ∼140 cycles of filling and emptying of the thimble. The sample was then dried in a vacuum oven at 80 °C until a constant dried weight was obtained (∼48 h). The gel content was estimated by mdry/m0, for which mdry and m0 are the weights of the dried sample and the sample before Soxhlet extraction, respectively. Three measurements were performed for each sample, and the reported values are the average gel content with errors representing the standard deviations.Differential scanning calorimetry (DSC)
A TA Instruments Discovery Series DSC instrument was employed to characterize the Tgs of the NIPU networks. Each sample was encapsulated in a Tzero aluminum pan and sealed using a Tzero lid. Two heating/cooling cycles were performed between −20 and 200 °C under a continuous N2 flow (50 mL min−1) at a heating/cooling rate of 10 °C min−1. Midpoints of the inflections in the second heating traces were used to determine the Tg. The reported Tg values are an average of three repeats with errors representing the standard deviations. DSC cooling curves are presented in Fig. S14 (ESI†).Thermogravimetric analysis (TGA)
A Discovery Series Thermogravimetric Analyzer TA Q500 (TA Instruments) was used to assess the thermal stability of the NIPU networks. Approximately 4–5 mg of each sample was placed in a 100 μL platinum pan, and a continuous flow of N2 (50 mL min−1) was passed over the sample and balance pans. The samples were first heated to 160 °C at a heating rate of 10 °C min−1 and held at this temperature to facilitate residual solvent/moisture removal. The samples were cooled to 25 °C and then heated to 700 °C at a rate of 10 °C min−1. For the reported TGA traces (Fig. S15, ESI†), the weight loss of each sample was normalized to its weight after the 160 °C isothermal hold. The temperature at 5 wt% loss (Td,5%) was used to gauge the thermal stability of crosslinked NIPU films. The reported values are an average of three repeats with errors representing the standard deviations.Dynamic mechanical analysis (DMA)
A TA Instruments RSA-G2 instrument was used to determine the storage modulus (E′), loss modulus (E′′), and tan δ (E′′/E′) of various specimens. Rectangular samples with dimensions of 30 mm × 10 mm × 0.15–0.2 mm (length × width × thickness) were first heated from −10 °C to 150 °C, then cooled back to −10 °C, followed by a second heating cycle to 150 °C, all at a heating/cooling rate of 3 °C min−1. Data from the second heating trace were used for analysis and are reported herein. The tests were conducted under oscillatory tension mode with a strain amplitude of 0.3%, grip-to-grip separation of ∼10 mm, frequency of 1 Hz, and an axial force of 0.4 N. The reported moduli and tan δ values are an average of three independent repeats with errors representing the standard deviations.Tensile tests
The tensile properties of NIPU networks were measured on a Zwick/Roell tensile tester with a load cell of 100 N and a clamp force of 200 N at ∼20 °C. Dog-bone-shaped samples were cut from the NIPU network films using a Zwick/Roell bent-lever manual cutting press (ZCP 020). The specimens with dimensions (Fig. S16, ESI†) of 9.5 mm × 2.5 mm × 0.15–0.2 mm (grip-to-grip separation × width × thickness) were subjected to an extension rate of 10 mm min−1 until film breakage. The tensile strength at yield (σy), elongation at break (εb), Young's modulus (E0), and toughness (area under the tensile stress–strain curve) results are reported as the average values of five specimens with errors representing the standard deviations.Results and discussion
The structural similarities between lignin-derivable bisguaiacols and petroleum-derived BPA/BPF suggested that the bio-derivable NIPUs could be alternatives to petroleum-based NIPU thermosets. We exploited variations in methoxy content and bridging-carbon substitution as dictated through building-block choice to tune thermomechanical properties. Towards these objectives, lignin-derivable BGA was synthesized through the condensation of guaiacol and acetone (Scheme 1a),38 and lignin-derivable BGF was synthesized via the acid-catalyzed electrophilic aromatic substitution reaction between guaiacol and vanillyl alcohol (Scheme 1b).30,37 Then, the bisguaiacol diglycidyl ethers were synthesized by glycidylation of BGA and BGF (Scheme 1c).30 Carbonation of the diglycidyl ethers generated BGACC and BGFCC, respectively (Scheme 1c). For comparative purposes, BPACC and BPFCC were prepared using a similar approach. The chemical structure of the respective compounds was confirmed using 1H NMR spectroscopy, and the molecular weight was determined using mass spectrometry. Thus, the four cyclic carbonates (Scheme 2b) differed in the number of methoxy groups, with either one methoxy group on each of the aromatic rings (1,1) or no methoxy groups on each ring (0,0). The cyclic carbonates also varied in bridging-carbon substitution (i.e., unsubstituted bridging carbon [Un] or dimethyl-substituted bridging carbon [Me]). The cyclic carbonates were cured by reaction with either a short-chain triamine (TREN) or a long-chain triamine (T403) (Scheme 2c) to investigate the impact of bisphenol structural features in densely vs. lightly crosslinked networks.Preparation of NIPU thermosets
All cyclic carbonates cured with TREN exhibited complete conversion of cyclic carbonate functionalities, within error, as indicated by FTIR data (Fig. S12, ESI†), in which there is a disappearance of the cyclic carbonate carbonyl stretching band at ∼1790 cm−1. Additionally, the appearance of urethane carbonyl and hydroxyl stretching bands at ∼1700 cm−1 and ∼3300–3400 cm−1, respectively, is noted. The gel contents are provided in Table 1 and also suggest conversions >99%. In contrast, the T403-based networks had a urethane carbonyl stretching band (∼1700 cm−1) and hydroxyl stretching band (∼3300–3400 cm−1), but they also had a weak absorbance of the cyclic carbonate carbonyl stretching band (∼1790 cm−1) (Fig. S13, ESI†). The gel contents of the T403-cured NIPUs were ∼87% in all cases, as shown in Table 1. This lower gel content and incomplete conversion of cyclic carbonates in T403-NIPUs could be a result of the low reactivity of T403, wherein the primary amine group is attached to a secondary carbon in contrast to TREN with the amine group attached to a primary carbon.6| Sample | Gel content (%)ab |
|---|---|
| a Determined by Soxhlet extraction of NIPU networks at 80 °C with chloroform as an extraction solvent. b The reported values are the average of three repeats with errors representing the standard deviation. | |
| BPACC–TREN | 99.4 ± 0.5 |
| BGACC–TREN | 99.2 ± 1.0 |
| BPFCC–TREN | 99.4 ± 0.7 |
| BGFCC–TREN | 99.0 ± 0.5 |
| BPACC–T403 | 87.2 ± 2.8 |
| BGACC–T403 | 86.0 ± 1.3 |
| BPFCC–T403 | 87.5 ± 2.3 |
| BGFCC–T403 | 87.1 ± 1.5 |
Thermal properties of NIPU thermosets
The presence of methoxy groups in lignin-derivable NIPU networks slightly lowered the Tgs of the materials as shown in Fig. 1 and 2. For example, BGACC–TREN had a Tg of ∼67 °C, and BPACC–TREN had a Tg of ∼79 °C, while BGFCC–TREN had a Tg of ∼64 °C, and BPFCC–TREN had a Tg of ∼69 °C (Fig. 1a and 2a). A similar trend was noted for the T403-based NIPUs as shown in Fig. 1b and 2b, though the magnitude of changes was somewhat smaller. This trend in Tgs is consistent with the previous literature for lignin-derivable epoxy-amine thermosets,30,45,47 aliphatic/aromatic polyesters,48 and cyanate ester networks.49 Interestingly, unlike many NIPUs reported in the literature, there was physical aging noted in both TREN and T403-cured NIPU thermosets, as indicated by the dips in the DSC heating traces near the Tg. However, lignin-derivable and petroleum-derived NIPU networks had comparable physical aging (excess enthalpy values) as shown in Fig. 1 and Table S1 (ESI†). Furthermore, the Tg trend in Fig. 1 and 2 was consistent with the DSC cooling curves (Fig. S14 and Table S1, ESI†), suggesting that physical aging may not significantly impact the reported Tg behavior.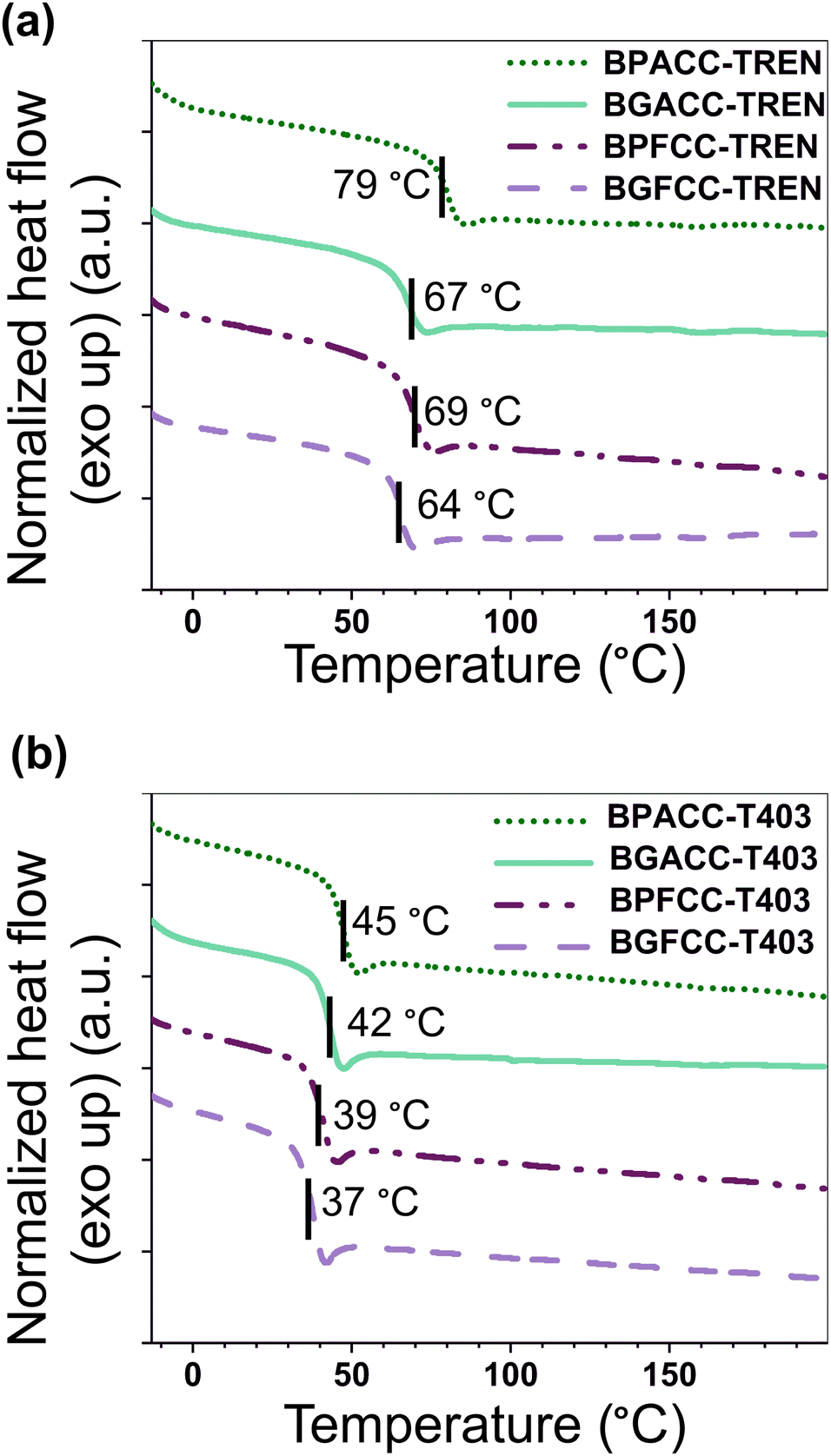 | ||
| Fig. 1 Representative DSC second heating traces of (a) TREN-based and (b) T403-based NIPU networks (10 °C min−1, N2). Curves are shifted vertically for clarity with Tg values marked on the respective curves. Each tick mark on the y-axis represents a 0.1 W g−1 increment. | ||
 | ||
| Fig. 2 T g as a function of the number of methoxy groups on the rings with the type of bridging-carbon substitution as bar labels for (a) TREN-based NIPU networks and (b) T403-based NIPU networks. Error bars indicate standard deviations of the measurements. | ||
The nature of bridging-carbon substitution also affected the Tgs of NIPU networks. Dimethyl substitution on bridging carbon resulted in higher Tgs vs. unsubstitution (Fig. 2). For example, the Tgs of BPACC–TREN (∼79 °C) and BGACC–TREN (∼67 °C) were higher than the Tgs of BPFCC–TREN (∼69 °C) and BGFCC–TREN (∼64 °C), Fig. 2a. This increase in Tg is likely related to the reduced rotational freedom caused by the dimethyl substituents.45 A similar trend existed for the T403-based networks as shown in Fig. 2b. In general, the Tgs of T403-based networks (∼37–45 °C) were lower than the TREN-based networks (∼64–79 °C). The Tgs obtained from DMA experiments (the temperature at maximum tan δ, Table S2, ESI†) showed a comparable trend as above.
The thermal stabilities and degradation behavior (Fig. S15, ESI†) of the lignin-derivable NIPU networks were similar to those of the BPA/BPF-based NIPU thermosets. Here, the Td,5% obtained from TGA was used as the comparative metric. The TREN-based networks had Td,5%s between 265 °C and 272 °C, while the T403-based thermosets had Td,5%s between 286 °C and 298 °C (Table 2). These Td,5% values were comparable with, or even higher than, similar traditional PU systems (Td,5% ∼ 250 °C).17 Interestingly, the methoxy groups in the bio-derivable networks did not appear to have a detrimental impact on thermal stability, which is notable because it has been reported that thermal degradation is accelerated by the presence of methoxy groups.30,47,49 Additionally, the T403 networks had increased Td,5%s relative to the TREN networks, which is possibly influenced by the longer distance between urethane bonds and the smaller number of such thermally labile urethane linkages per volume in the TREN-based thermoset.9,50,51
| Sample | T g (°C) | T d,5% (°C) |
|
|
σ y (MPa) | Toughnesse (MJ m−3) | ε b (%) |
|---|---|---|---|---|---|---|---|
| a Determined from the second heating trace of DSC at a heating rate of 10 °C min−1 in a N2 atmosphere. b The reported values are the average of three repeats with errors representing the standard deviation. c Determined by TGA at a heating rate of 10 °C min−1 in an N2 atmosphere. d Determined from the second heating trace of DMA experiments conducted in oscillatory film tension mode at a heating rate of 3 °C min−1, strain amplitude of 0.3%, frequency of 0.1 Hz, and an axial force of 0.4 N. e All data are based on tensile stress–strain curves obtained from the uniaxial tensile testing using a load cell of 100 N and an extension rate of 10 mm min−1 at ∼20 °C. Toughness is calculated from the area under stress-strain curves. The reported values are the average of five repeats with errors representing the standard deviation. | |||||||
| BPACC–TREN | 79 ± 0.9 | 265 ± 2 | 1.7 ± 0.2 | 4.4 ± 0.2 | 88 ± 8 | 3 ± 1 | 5 ± 1 |
| BGACC–TREN | 67 ± 0.3 | 272 ± 5 | 1.2 ± 0.2 | 2.5 ± 0.1 | 76 ± 9 | 2 ± 1 | 4 ± 1 |
| BPFCC–TREN | 69 ± 0.7 | 267 ± 5 | 2.1 ± 0.2 | 3.4 ± 0.5 | 72 ± 10 | 3 ± 1 | 7 ± 1 |
| BGFCC–TREN | 64 ± 0.6 | 265 ± 8 | 1.5 ± 0.5 | 4.3 ± 0.5 | 65 ± 8 | 2 ± 1 | 5 ± 1 |
| BPACC–T403 | 45 ± 1.5 | 286 ± 5 | 1.0 ± 0.1 | 1.1 ± 0.2 | 45 ± 2 | 26 ± 8 | 86 ± 26 |
| BGACC–T403 | 42 ± 0.2 | 298 ± 4 | 1.1 ± 0.1 | 1.3 ± 0.2 | 42 ± 3 | 49 ± 6 | 195 ± 8 |
| BPFCC–T403 | 39 ± 0.3 | 287 ± 4 | 1.5 ± 0.3 | 2.0 ± 0.2 | 28 ± 2 | 35 ± 3 | 166 ± 7 |
| BGFCC–T403 | 37 ± 0.7 | 290 ± 10 | 1.2 ± 0.1 | 1.1 ± 0.3 | 28 ± 4 | 59 ± 9 | 278 ± 34 |
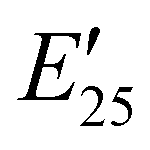
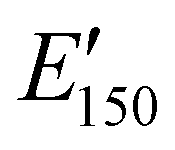
Mechanical properties of NIPU thermosets
The viscoelastic properties of NIPU thermosets were investigated through DMA and are summarized in Table 2 and Table S2 (ESI†). As illustrated in Fig. 3, the E′ data show a pattern found in a typical thermosetting polymer,52 with a sharp drop in E′ appearing near the Tg followed by a rubbery plateau with no terminal relaxation. The storage moduli at 25 °C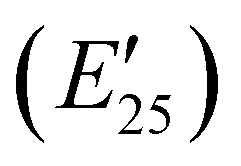 for the NIPU networks ranged from ∼1.0 GPa to 2.1 GPa. These
for the NIPU networks ranged from ∼1.0 GPa to 2.1 GPa. These 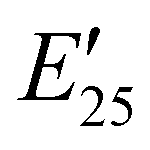 values are consistent with E0 values (∼1.2 GPa to 2.2 GPa) obtained from the tensile testing of NIPU networks (Table S3, ESI†), and they are also comparable to similar traditional PU thermosets (∼1 GPa).17 The storage moduli at 150 °C (
values are consistent with E0 values (∼1.2 GPa to 2.2 GPa) obtained from the tensile testing of NIPU networks (Table S3, ESI†), and they are also comparable to similar traditional PU thermosets (∼1 GPa).17 The storage moduli at 150 °C (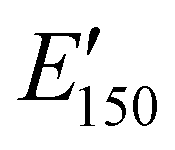 ; i.e., the rubbery plateau moduli) ranged from 2.5 MPa to 4.4 MPa for TREN NIPUs, while the
; i.e., the rubbery plateau moduli) ranged from 2.5 MPa to 4.4 MPa for TREN NIPUs, while the 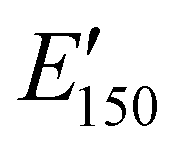 s of T403 NIPUs were between 1.1 MPa and 2 MPa. The lower
s of T403 NIPUs were between 1.1 MPa and 2 MPa. The lower 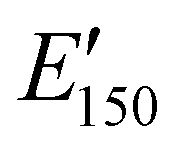 values of T403-based networks vs. those of the TREN-based counterparts was likely the result of the higher molecular weight between crosslinks of T403 NIPUs as influenced by T403 chain length.
values of T403-based networks vs. those of the TREN-based counterparts was likely the result of the higher molecular weight between crosslinks of T403 NIPUs as influenced by T403 chain length.
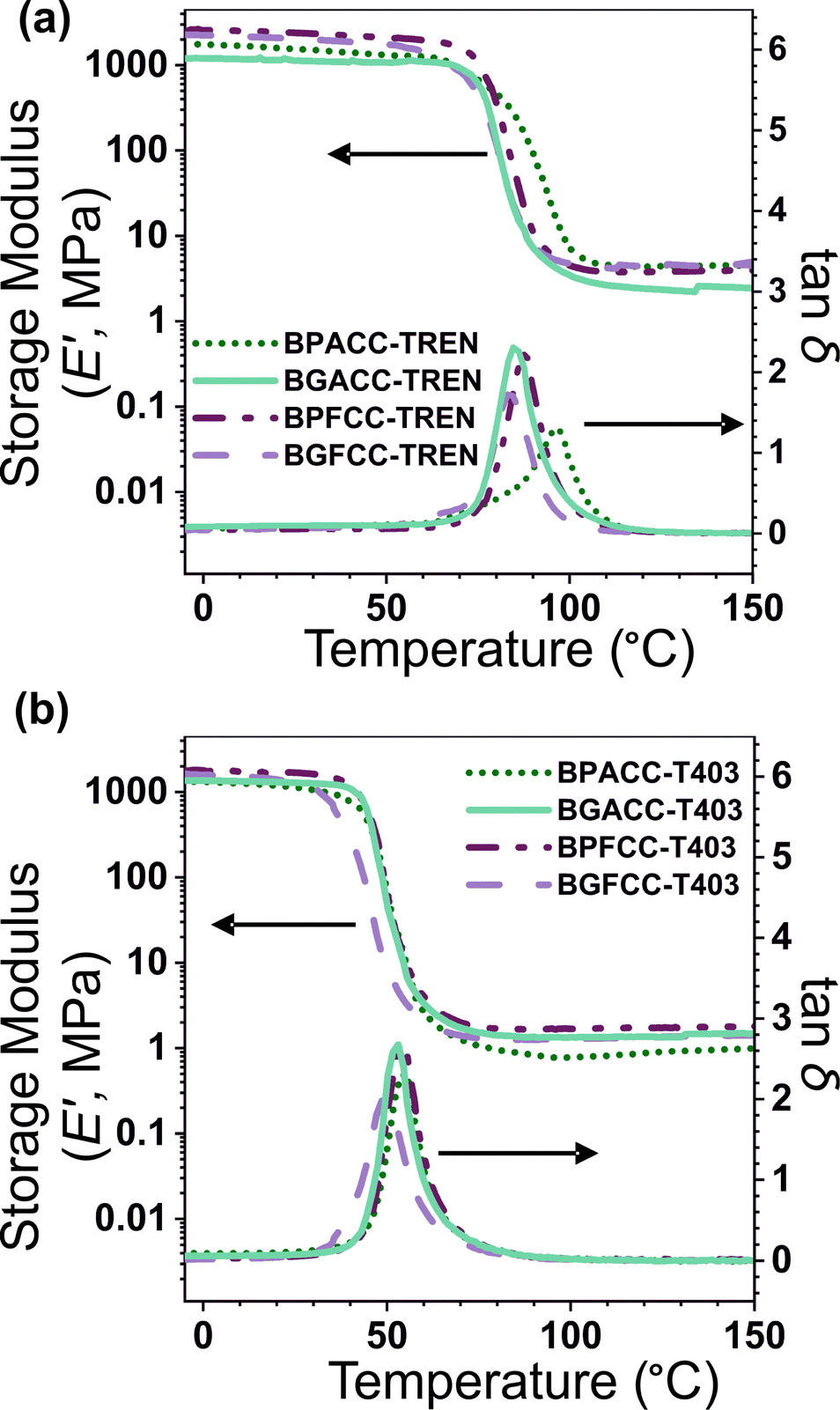 | ||
| Fig. 3 Storage modulus and tanδ as a function of temperature for (a) TREN-based and (b) T403-based NIPU networks. Data shown are from the second heating cycle of DMA experiments, in oscillatory film tension mode at a heating rate of 3 °C min−1, strain amplitude of 0.3%, frequency of 0.1 Hz, and an axial force of 0.4 N. | ||
The effect of structural differences in cyclic carbonates and triamines on the tensile properties of NIPU thermosets were examined through uniaxial tensile testing. The TREN-based networks showed elastic deformation and immediate fracture after the yield point, as expected for highly crosslinked thermosets (Fig. 4a).52 The σy of all the TREN-based NIPU networks were in the range of 65–88 MPa (Table 2). Though all values are generally within experimental error, Fig. 4a strongly suggests that the dimethyl-substituted networks have substantially increased yield strengths vs. their unsubstituted counterparts across both petroleum-based and lignin-derivable constructs.
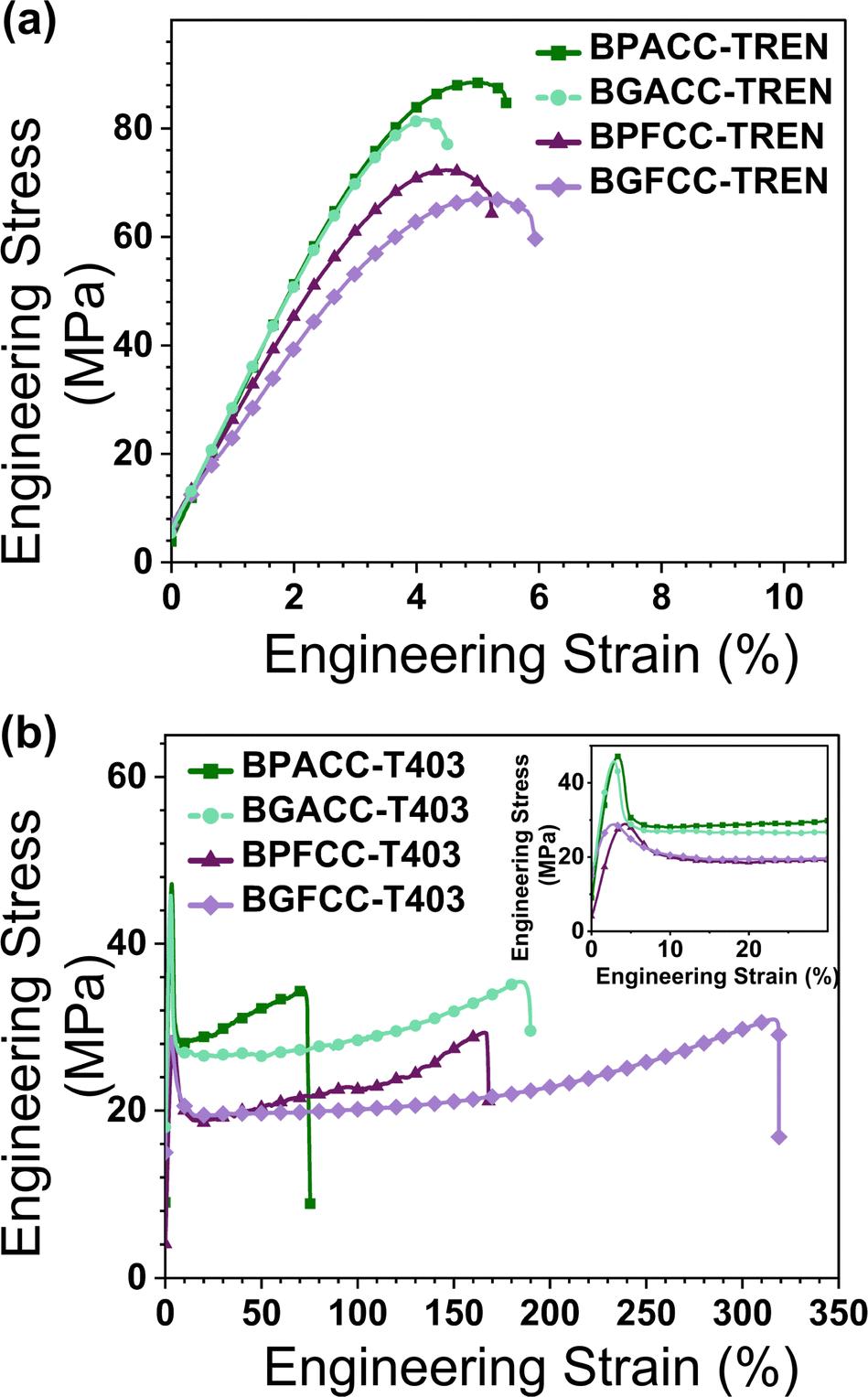 | ||
| Fig. 4 Representative engineering tensile stress versus engineering strain of (a) TREN-based NIPU networks and (b) T403-based NIPU networks obtained from uniaxial tensile testing using a load cell of 100 N and an extension rate of 10 mm min−1 at ∼20 °C. | ||
The brittle nature of the TREN-based thermosets provided limited insights into the impact of methoxy groups and bridging-carbon substitutions on the toughness, σy, and εb. In contrast, the lightly crosslinked T403 NIPUs allowed more detailed exploration of the role of these structural variations on mechanical behavior. Fig. 4b shows representative tensile stress–strain curves for T403 NIPUs. All T403-NIPU thermosets have similar stress–strain curves with initial elastic behavior followed by plastic deformation after the yield point. Also, T403-cured NIPUs exhibited necking during tensile testing that may have led to a sharp yield point. The plastic deformation and high toughness of T403-based networks are strongly impacted by the flexible polyether aliphatic chain of triamine.53 Additionally, distinct differences were noted in the tensile properties as a function of methoxy group content and the nature of bridging-carbon substitution (Fig. 5a–c). Surprisingly, the toughness and εb of lignin-derivable, T403-based NIPU networks were significantly higher than those of the thermosets derived from BPA and BPF analogues. For example, the toughness of lignin-derivable BGFCC–T403 (59 ± 9 MJ m−3) was ∼66% higher than that of BPFCC–T403 (35 ± 3 MJ m−3). Also, BGFCC–T403 (εb = 278 ± 34%) was more extensible than the BPFCC–T403 (εb = 166 ± 7%) network. Yet, the σy values of the two networks were quite comparable (∼28 MPa). Moreover, the water uptake of all the thermoset films (Table S4, ESI†) were generally comparable, which suggests that moisture uptake may not be the major driver impacting the difference in tensile results. A similar trend occurred in the toughness, εb, and σy of BGACC–T403 and BPACC–T403 networks as shown in Fig. 5, suggesting that the bio-derivable BGA and BGF platforms increase toughness and extensibility without sacrificing tensile strength.
 | ||
| Fig. 5 (a) Toughness, (b) εb, and (c) σy as a function of the number of methoxy groups on the rings with the bridging-carbon substitution as bar labels. | ||
As described above, the methoxy groups in lignin-derivable NIPUs led to networks with higher toughness and εb than the BPA/BPF-analogues without any appreciable reduction in σy or E0. These methoxy moieties on the aromatic backbone can hinder close chain packing,30 which could result in network plasticization and enhanced deformability (εb). However, such plasticizing effects usually reduce the E0 and σy,52 which was not the case herein. NIPUs inherently have associating hydrogen bonds between the urethane linkages and the pendant hydroxyl groups,1,22 and the lignin-derivable versions provide additional inter- and intra-molecular hydrogen-bonding sites via their methoxy substituents, which could increase the hydrogen-bonding potential.45,54 These amplified associations may be responsible for the enhanced tensile strength of the polymers.53 Thus, a combined effect of less efficient chain packing and increased hydrogen bonding possibly resulted in lignin-derivable NIPUs that were tougher than the petroleum-derived systems. This improved toughness is consistent with a recent study on non-isocyanate thermoplastic polythiourethanes, wherein a bisphenol-S cyclic thiocarbonate was cured by amino-terminated dimer-acid polyamides.53
Finally, the bridging-carbon substitution also influenced the tensile properties, wherein the dimethyl substituted materials had enhanced σyvs. their unsubstituted counterparts (Fig. 5c). For instance, the σy of BGACC/BPACC–T403 (∼42–45 MPa) was ∼17 MPa higher than BGFCC/BPFCC–T403 (σy ∼ 28 MPa). However, the dimethyl substituents reduced the toughness and εb (Fig. 5b and c), possibly due to more limited chain mobility in comparison to the unsubstituted versions.45,53,55 This trend also held across networks, and it is further apparent that TREN-based NIPUs had higher σy but lower extensibility than T403-based NIPU thermosets, most likely due to their densely crosslinked architecture obtained from the short-chain triamine (Table 2).56
Conclusions
This work examined the effect of structural features in lignin-derivable cyclic carbonates, such as methoxy content and bridging-carbon substitutions, on the thermomechanical properties of NIPU thermosets. The methoxy groups in lignin-derivable cyclic carbonates led to NIPU networks with substantially higher toughness (∼49–59 MJ m−3) and εb (∼195–278%) in comparison to those of BPA/BPF-based benchmarks (toughness ∼26–35 MJ m−3, εb ∼86–166%). Moreover, the dimethyl substitution on bridging carbons generated networks with higher σy (∼45 MPa) than those with unsubstituted bridging carbons (σy ∼28 MPa). This improved mechanical performance of lignin-derivable NIPU thermosets was achieved without any detrimental effects on other crucial thermoset properties such as moduli and thermal stabilities. Furthermore, the Tgs of the networks were tunable; the introduction of methoxy groups lowered the Tgs, while the dimethyl bridging carbon substituents raised the Tgs, allowing for performance-based optimization. Overall, the structural characteristics, such as methoxy moieties on the rings, the nature of bridging carbon substituents, and/or chain-length of crosslinker, afford thermomechanical versatility to lignin-derivable NIPU networks, highlighting their potential as building blocks in NIPU thermosets.Abbreviations
| ATR-FTIR | Attenuated total reflectance-Fourier transform infrared |
| BADGE | Bisphenol A diglycidyl ether |
| BFDGE | Bisphenol F diglycidyl ether |
| BGA | Bisguaiacol A |
| BGACC | Bisguaiacol A cyclic carbonate |
| BGADGE | Bisguaiacol A diglycidyl ether |
| BGF | Bisguaiacol F |
| BGFCC | Bisguaiacol F cyclic carbonate |
| BGFDGE | Bisguaiacol F diglycidyl ether |
| BPA | Bisphenol A |
| BPACC | Bisphenol A cyclic carbonate |
| BPF | Bisphenol F |
| BPFCC | Bisphenol F cyclic carbonate |
| DCM | Dichloromethane |
| DI | Deionized |
| DMA | Dynamic mechanical analysis |
| DMF | N,N-dimethylformamide |
| DMSO-d6 | Deuterated dimethyl sulfoxide |
| DSC | Differential scanning calorimetry |
| DTGS | Deuterated triglycine sulfate |
| E′ | Storage modulus |
| E′′ | Loss modulus |
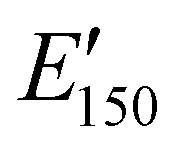
| Storage modulus at 150 °C |
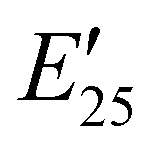
| Storage modulus at 25 °C |
| E 0 | Young's modulus |
| ESI | Electrospray ionization |
| FTMS | Fourier transform mass spectrometry |
| MDI | Methylene diphenyl diisocyanate |
| Me | Dimethyl-substituted bridging carbon |
| NIPU | Non-isocyanate polyurethane |
| NMR | Nuclear Magnetic Resonance |
| PU | Polyurethane |
| T403 | Jeffamine® T-403 |
| TBAB | Tetrabutylammonium bromide |
| T d,5% | Temperature of 5% weight loss |
| TDI | Toluene diisocyanate |
| T g | Glass transition temperature |
| TGA | Thermogravimetric analysis |
| TREN | Tris(2-aminoethyl) amine |
| Un | Unsubstituted bridging carbon |
| ε b | Elongation at break |
| σ y | Tensile strength at yield |
Author contributions
Conceptualization: S. V. M., T. H. E., and L. T. J. K. Investigation and formal analysis: S. V. M. and J. S. M. Writing (original draft): S. V. M. and J. S. M. Writing (review and editing): S. V. M., J. S. M., T. H. E., and L. T. J. K. Funding acquisition, supervision, and project administration: T. H. E. and L. T. J. K.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a National Science Foundation grant (NSF DMR POL 2004682) awarded to L. T. J. K. and T. H. E. The authors thank the University of Delaware (UD) Advanced Materials Characterization Laboratory for the use of the ATR-FTIR, DSC, and TGA instruments, and the UD NMR facility and UD Mass Spectrometry facility for the use of NMR and Mass Spectrometers. The authors also thank the Center for Plastics Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under award DE-SC0021166, for the use of a Parr reactor and the RSA-G2 instrument.References
- C. Carre, Y. Ecochard, S. Caillol and L. Averous, ChemSusChem, 2019, 12, 3410–3430 CrossRef CAS PubMed.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- C. Liang, U. R. Gracida-Alvarez, E. T. Gallant, P. A. Gillis, Y. A. Marques, G. P. Abramo, T. R. Hawkins and J. B. Dunn, Environ. Sci. Technol., 2021, 55, 14215–14224 CrossRef CAS PubMed.
- M. Szycher, Szycher's handbook of polyurethanes, CRC Press, Boca Raton, FL, 2nd edn, 2013 Search PubMed.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- Market volume of polyurethane worldwide from 2015 to 2025, with a forecast for 2022 to 2029 (in million metric tons), https://www.chemintel360.com/reportdetails/Global-Polyurethane-Market/25, accessed September 3, 2022.
- Bio-based Building Blocks and Polymers – Global Capacities, Production and Trends 2020 – 2025, https://renewable-carbon.eu/publications/product/bio-based-building-blocks-and-polymers-global-capacities-production-and-trends-2020-2025-graphics-figure-6/, accessed September 3, 2022.
- V. Besse, R. Auvergne, S. Carlotti, G. Boutevin, B. Otazaghine, S. Caillol, J.-P. Pascault and B. Boutevin, React. Funct. Polym., 2013, 73, 588–594 CrossRef CAS.
- X. Meng, S. Zhang, B. Scheidemantle, Y.-y Wang, Y. Pu, C. E. Wyman, C. M. Cai and A. J. Ragauskas, Ind. Crops Prod., 2022, 178, 114579 CrossRef CAS.
- M. A. Al Meselmani, H. C. Wright, D. D. Cameron and A. J. Ryan, Nat. Rev. Earth Environ., 2020, 1, 439 CrossRef.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- Q. Chen, K. Gao, C. Peng, H. Xie, Z. K. Zhao and M. Bao, Green Chem., 2015, 17, 4546–4551 RSC.
- M. Janvier, P.-H. Ducrot and F. Allais, ACS Sustainable Chem. Eng., 2017, 5, 8648–8656 CrossRef CAS.
- J. S. Mahajan, R. M. O’Dea, J. B. Norris, L. T. J. Korley and T. H. Epps, III, ACS Sustainable Chem. Eng., 2020, 8, 15072–15096 CrossRef CAS.
- M. A. C. Mhd Haniffa, K. Munawar, Y. C. Ching, H. A. Illias and C. H. Chuah, Chem. – Asian J., 2021, 16, 1281–1297 CrossRef CAS PubMed.
- M. Ghasemlou, F. Daver, E. P. Ivanova and B. Adhikari, Eur. Polym. J., 2019, 118, 668–684 CrossRef CAS.
- A. Salanti, L. Zoia, M. Mauri and M. Orlandi, RSC Adv., 2017, 7, 25054–25065 RSC.
- S. Hu, X. Chen and J. M. Torkelson, ACS Sustainable Chem. Eng., 2019, 7, 10025–10034 CrossRef CAS.
- J. Dong, B. Liu, H. Ding, J. Shi, N. Liu, B. Dai and I. Kim, Polym. Chem., 2020, 11, 7524–7532 RSC.
- M. Bähr and R. Mülhaupt, Green Chem., 2012, 14, 483–489 RSC.
- M. Fleischer, H. Blattmann and R. Mülhaupt, Green Chem., 2013, 15, 934–942 RSC.
- B. Tamami, S. Sohn and G. L. Wilkes, J. Appl. Polym. Sci., 2004, 92, 883–891 CrossRef CAS.
- D. C. Webster, Prog. Org. Coat., 2003, 47, 77–86 CrossRef CAS.
- S. F. Koelewijn, S. Van den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2017, 19, 2561–2570 RSC.
- G. Yang, B. J. Rohde, H. Tesefay and M. L. Robertson, ACS Sustainable Chem. Eng., 2016, 4, 6524–6533 CrossRef CAS.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- K. H. Nicastro, C. J. Kloxin and T. H. Epps, III, ACS Sustainable Chem. Eng., 2018, 6, 14812–14819 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2016, 4, 6082–6089 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- M. Jawerth, M. Lawoko, S. Lundmark, C. Perez-Berumen and M. Johansson, RSC Adv., 2016, 6, 96281–96288 RSC.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- G. F. Bass and T. H. Epps, III, Polym. Chem., 2021, 12, 4130–4158 RSC.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- K. H. S. Reno, J. F. Stanzione III, R. P. Wool, J. M. Sadler, J. J. LaScala and E. D. Hernandez, US Pat., 15313656, 2017 Search PubMed.
- T. H. Epps, III, L. T. J. Korley, M. D. Green, J. S. Mahajan and H. Shokrollahzadeh, US Pat. application, 17616245, 2022 Search PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- R. M. O’Dea, J. A. Willie and T. H. Epps, III, ACS Macro Lett., 2020, 9, 476–493 CrossRef PubMed.
- Y. Peng, K. H. Nicastro, T. H. Epps, III and C. Wu, Food Chem., 2021, 338, 127656 CrossRef CAS PubMed.
- A. Amitrano, J. S. Mahajan, L. T. J. Korley and T. H. Epps, III, RSC Adv., 2021, 11, 22149–22158 RSC.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, III, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed.
- S. Zhao, X. Huang, A. J. Whelton and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2018, 6, 7600–7608 CrossRef CAS.
- A. Yamamoto, K. Nemoto, M. Yoshida, Y. Tominaga, Y. Imai, S. Ata, Y. Takenaka, H. Abe and K. Sato, RSC Adv., 2020, 10, 36562–36570 RSC.
- B. G. Harvey, A. J. Guenthner, W. W. Lai, H. A. Meylemans, M. C. Davis, L. R. Cambrea, J. T. Reams and K. R. Lamison, Macromolecules, 2015, 48, 3173–3179 CrossRef CAS.
- A. Farhadian, A. Ahmadi, I. Omrani, A. B. Miyardan, M. A. Varfolomeev and M. R. Nabid, Polym. Degrad. Stab., 2018, 155, 111–121 CrossRef CAS.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- A. J. Kinloch and R. J. Young, in Fracture Behaviour of Polymers, ed. A. J. Kinloch and R. J. Young, Springer Netherlands, Dordrecht, 1995 Search PubMed.
- B. Xu, Q. Yin, C. Su, J. Cheng, J. Zhang and J. Zhao, ACS Macro Lett., 2022, 11, 517–524 CrossRef CAS PubMed.
- B. Wang, S. Ma, Q. Li, H. Zhang, J. Liu, R. Wang, Z. Chen, X. Xu, S. Wang, N. Lu, Y. Liu, S. Yan and J. Zhu, Green Chem., 2020, 22, 1275–1290 RSC.
- G. Pan, Z. Du, C. Zhang, C. Li, X. Yang and H. Li, Polym. J., 2007, 39, 478–487 CrossRef CAS.
- J. Huang, P. Fu, W. Li, L. Xiao, J. Chen and X. Nie, RSC Adv., 2022, 12, 23048–23056 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00895e |
| This journal is © The Royal Society of Chemistry 2023 |