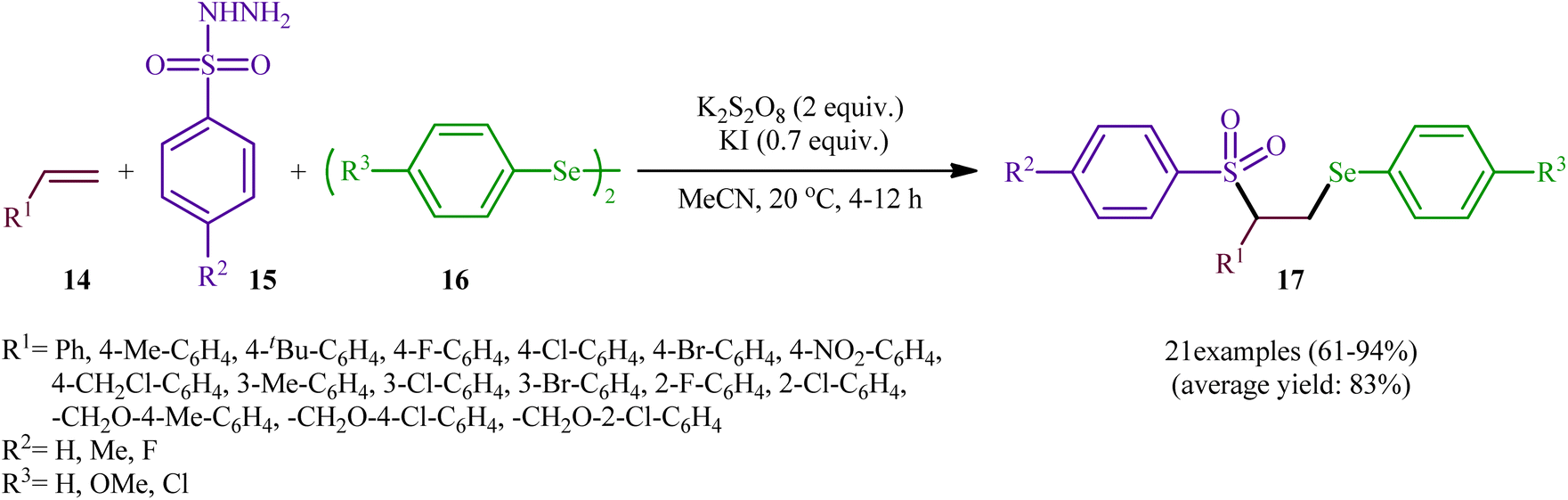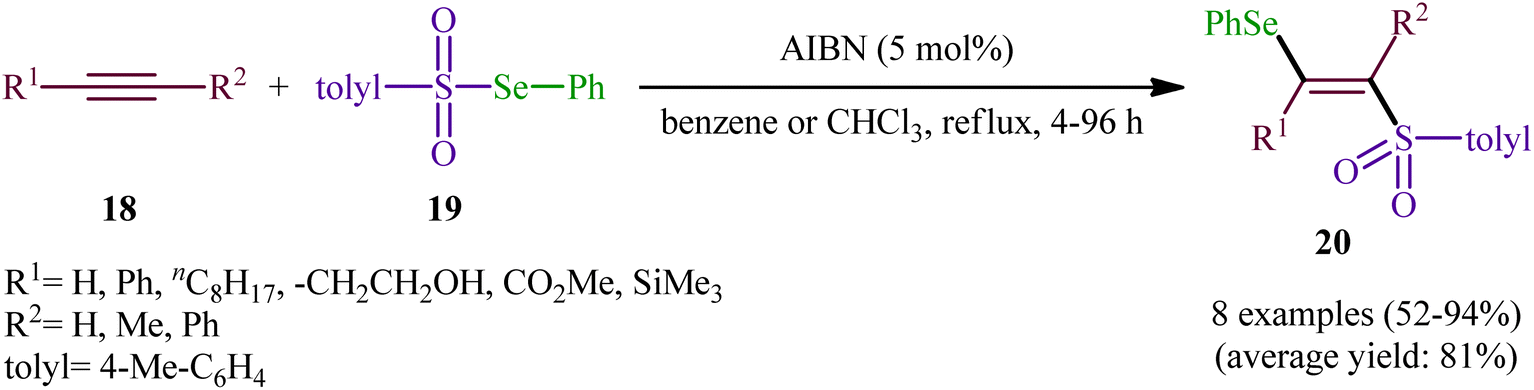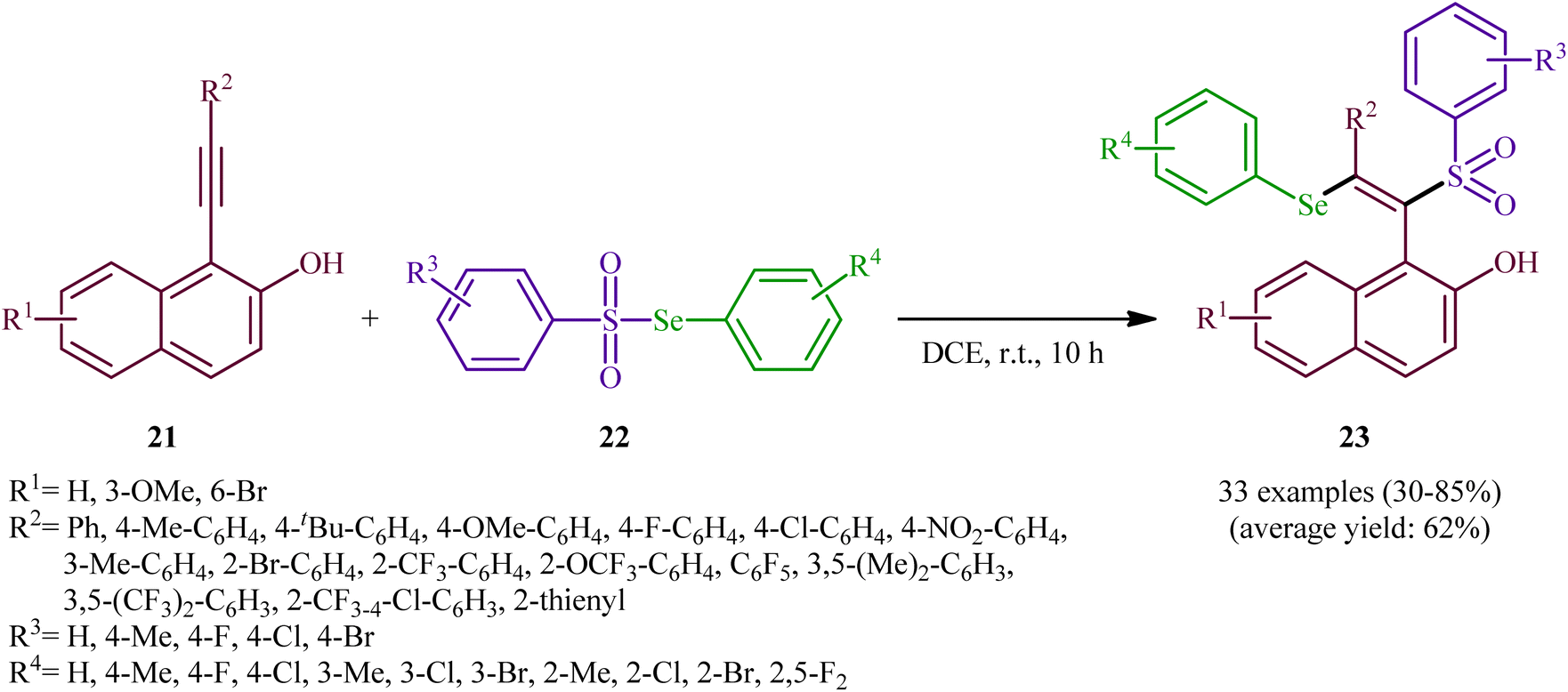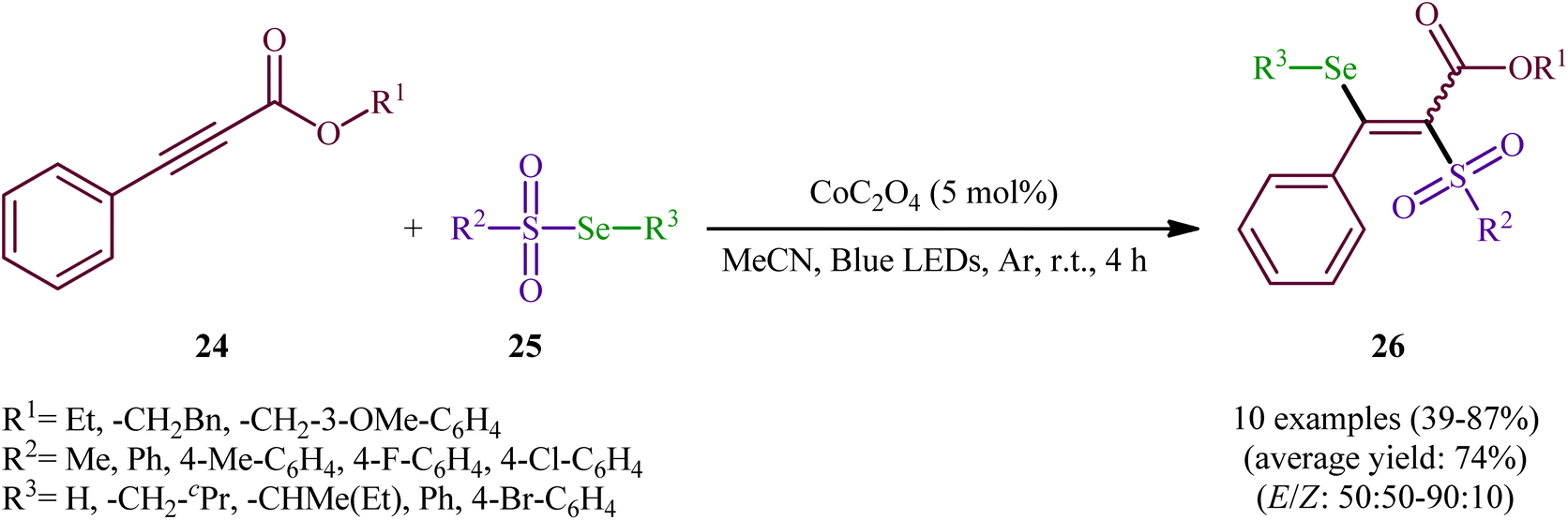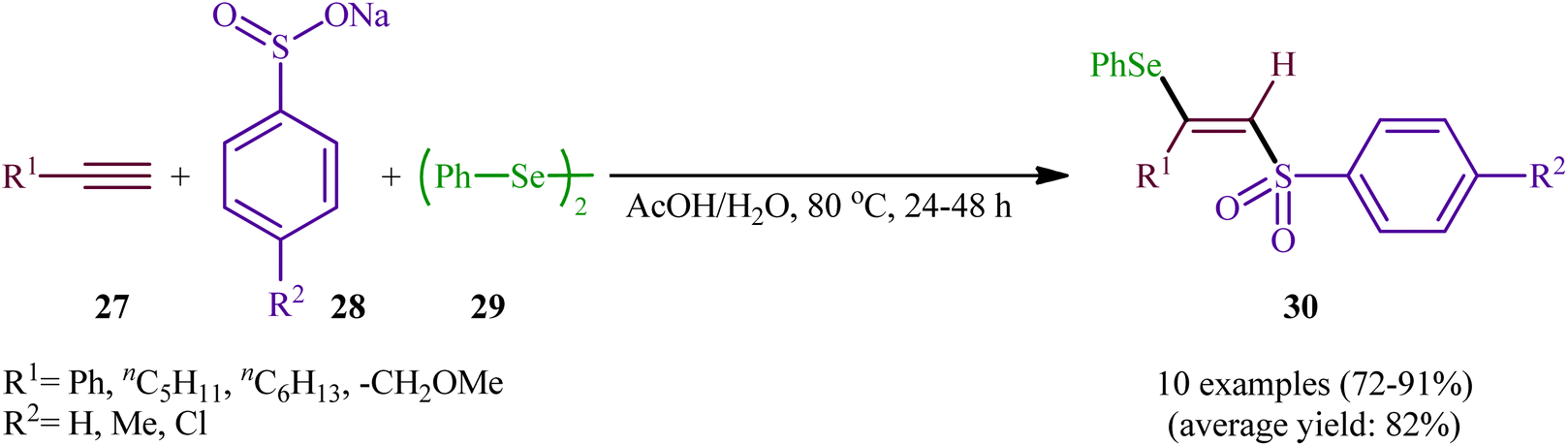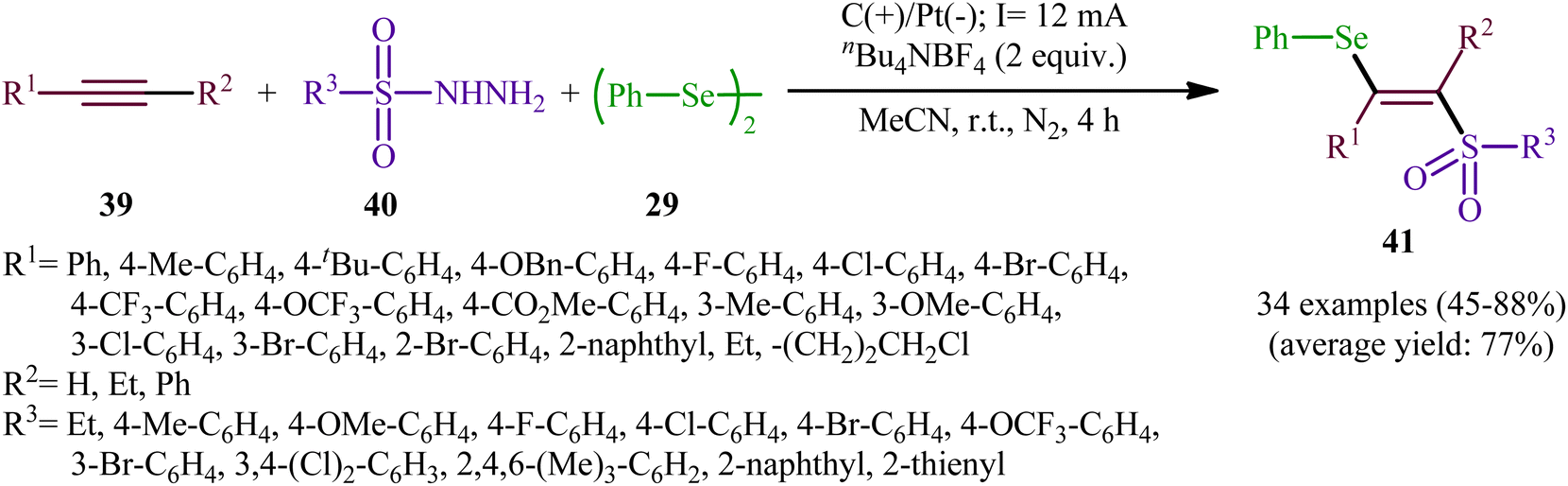DOI:
10.1039/D2RA04128F
(Review Article)
RSC Adv., 2022,
12, 30564-30576
Direct selenosulfonylation of unsaturated compounds: a review
Received
4th July 2022
, Accepted 21st September 2022
First published on 26th October 2022
Abstract
In this review, we have discussed recent developments on the direct selenosulfonylation of unsaturated compounds which lead to the formation of two new carbon-sulfur and carbon-selenium bonds in a single operation. The reactions were classified based on the type of starting unsaturated compound and product. Thus, the review is divided into three major sections. The first describes the current literature on selenosulfonylation of alkenes. The second section covers the available literature on selenosulfonylation of alkynes. The third focuses exclusively on selenosulfonylation of allenes.
1. Introduction
Double functionalization of unsaturated compounds (e.g., alkenes, alkynes), as an engrossing and powerful strategy to promptly increase molecular complexity within a single process, has emerged as a hot topic during the past years for the selective construction of two new carbon–carbon and/or carbon–heteroatom bonds in a step-economic fashion.1 This methodology does not require laborious isolation or purification intermediates, minimizes the formation of waste and makes synthetic schemes simpler and cleaner.2
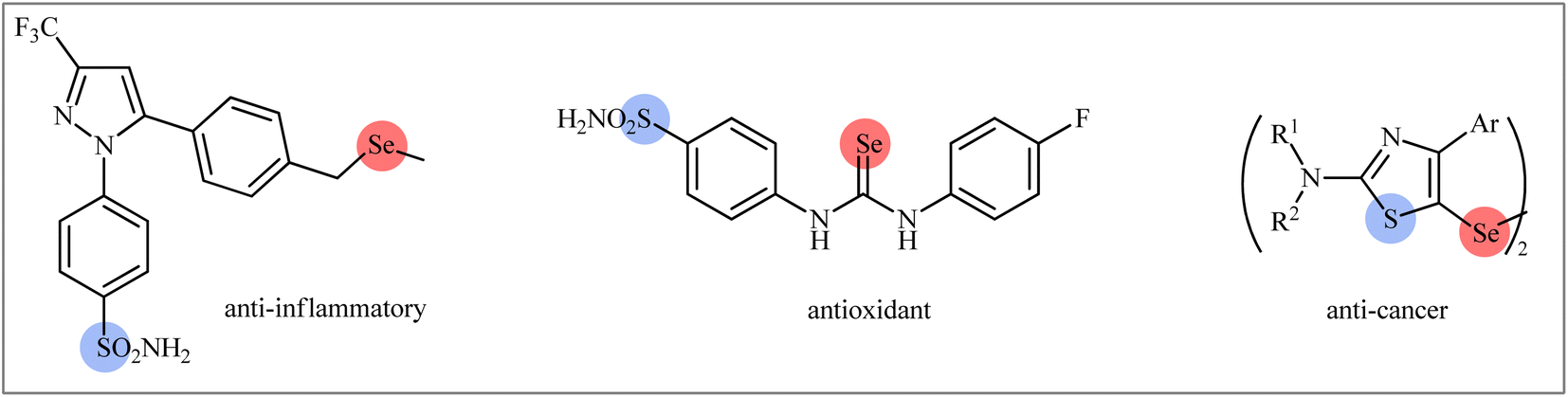
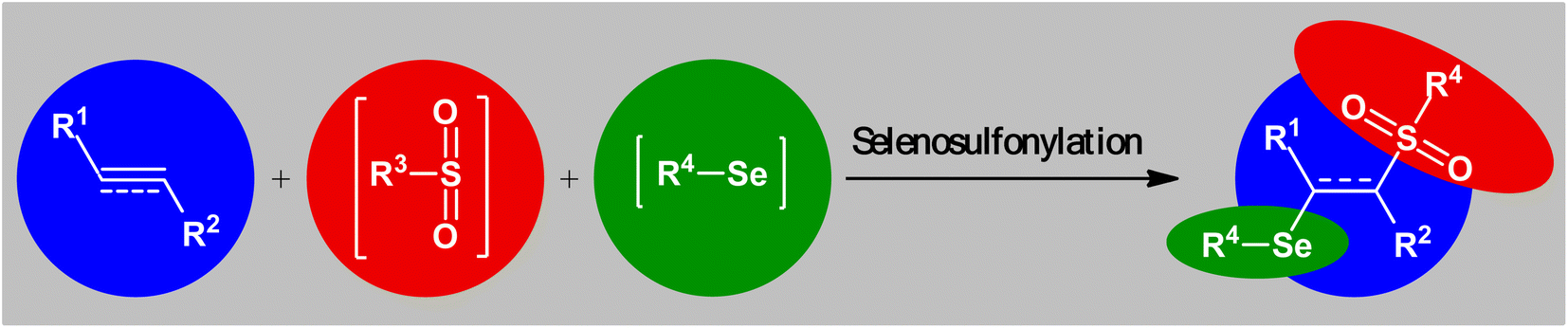
Sulfur-containing organic compounds have found a huge number of applications in many different fields such as medical, pharmaceuticals, agrochemicals, and polymers.3 Interestingly, more than 280 FDA-approved drugs contain at least one sulfur atom in their structures and many scientists around the world are working on the development of new sulfur-based medicines.4 Similarly, selenium-containing organic compounds have a broad range of applications in a wide variety of research areas,5,6 particularly in pharmaceutical science.7 Consequently, organic molecules possessing both sulfur and selenium atoms have recently attracted the attention of medicinal chemists because of their unique biological activities (Scheme 1).8 In this family of compounds, β-selenosulfone derivatives have drawn a lot of interest from the synthetic community not only because of their potential biological properties but also widespread synthetic applications.9–19 The most straightforward and commonly used strategy for the preparation of this special class of organosulfur-selenium compounds is the direct selenosulfonylation of unsaturated compounds (Fig. 1) and has been the subject of a number of papers in recent years. In 1999, TG Back highlighted this strategy of β-selenosulfones synthesis in an interesting book entitled “organoselenium chemistry: a practical approach”.20 Since several important and striking advantages on this difunctionalization reaction have occurred during the past several years, it seems it is an appropriate time to summarize those developments. In connection with our interest on difunctionlization reactions,21 organosulfur chemistry22 and modern organic synthesis,23 herein, we intend to summarize the literature reports on the selenosulfonylation of unsaturated compounds from 1980 till date. For clarity, the topic is divided into three major sections. The first describes the current literature on selenosulfonylation of alkenes. The second section covers selenosulfonylation of alkynes. The third focuses exclusively on selenosulfonylation of allenes.
 |
| | Scheme 1 Selected examples of biologically active compounds bearing both sulfur and selenium atoms in their structures. | |
 |
| | Fig. 1 Direct selenosulfonylation of unsaturated C–C bonds. | |
2. Selenosulfonylation of alkenes
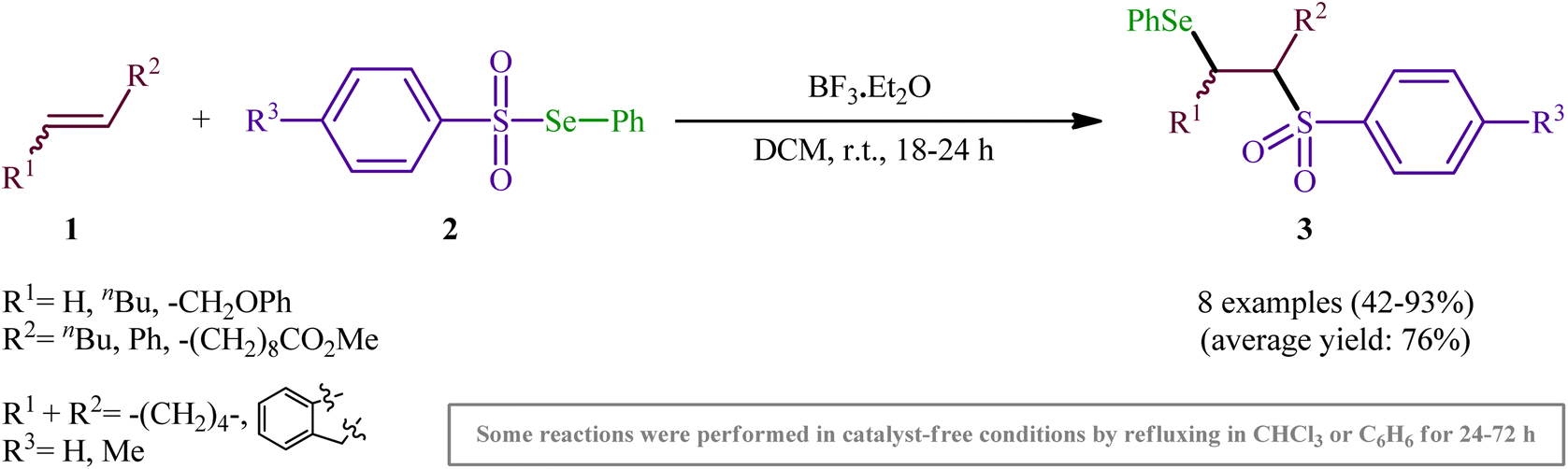
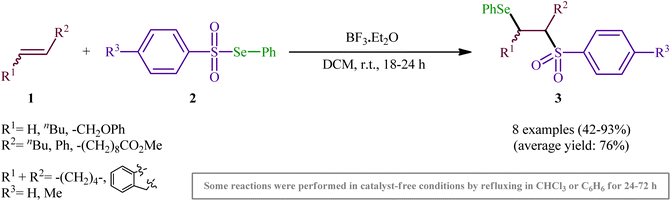
One of the earliest reports of the direct selenosulfonylation of alkenes was published by Back and Collins in 1980,24 who showed that the treatment of various unhindered alkenes 1 with the pre-prepared Se-phenyl areneselenosulfonates 2 in the presence of a catalytic amount of BF3·Et2O at room temperature resulted in the formation β-phenylselenosulfones 3 in moderate to excellent yields (Scheme 2). The target bifunctional adducts was also achieved by refluxing the reactants in chloroform or benzene in the absence of a Lewis acid, or by heating them neat in a sealed glass tube. Both terminal and internal alkenes were applicable to these protocols and in all cases the corresponding β-phenylselenosulfones were selectively obtained. Some important information of this synthetic procedure is: (i) Lewis acid-catalyzed additions of Se-phenyl areneselenosulfonates to aromatic alkenes (i.e., styrene) proceeded with predominantly Markovnikoff orientation. In contrast, the uncatalyzed thermally initiated additions provided adducts of opposite regiochemistry; (ii) catalytic reaction of aliphatic alkenes (i.e., methyl 10-undecenoate) with Se-phenyl areneselenosulfonates afforded mixtures of the two regioisomers while uncatalyzed selenosulfonatlon of aliphatic alkenes (i.e., allyl phenyl ether) selectively provided the anti-Markovnikoff adducts; and (iii) the addition of Se-phenyl areneselenosulfonates to cyclic alkenes (i.e., cyclohexene and to indene) under both catalytic and non-catalytic conditions afforded solely the trans adducts. As for the mechanism, the authors speculated that the catalyzed reaction most likely proceeds through an electrophilic addition pathway involving bridged seleniranium ions while a free radical addition mechanism has been implicated for uncatalyzed reaction.25 Subsequently, Kice's research group provided further examples of β-phenylselenosulfones synthesis by the same methodology and demonstrated that they can be readily converted to the synthetically important vinylic sulfones via oxidation with hydrogen peroxide.26,27 This research group also indicated the photodecomposition of selenosulfonates and demonstrated their addition to alkenes to form β-phenylselenosulfones in good yields.28
 |
| | Scheme 2 Back's synthesis of β-phenylselenosulfones 3. | |
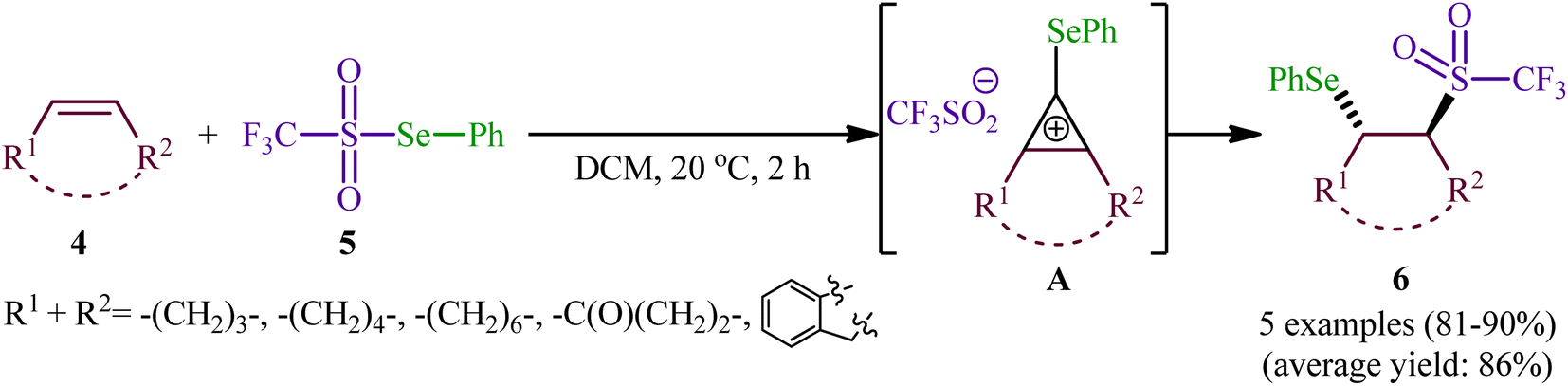
In 1999, a similar difunctionalization strategy was applied by Billard and Langlois for the synthesis of β-selenyl-trifluoromethylsulfone derivatives.29 In this report they demonstrated that the reaction between cyclic alkenes 4 and strongly electrophilic phenyl trifluoromethane-selenosulfonate (CF3SO2SePh; 5) under catalyst-free conditions in DCM at ambient temperature afforded the corresponding β-selenyl-trifluoromethylsulfones 6 in high yields and outstanding regioselectivity, in which trans-Markovnikov adducts were sole products obtained from unsymmetrical cyclic alkenes (Scheme 3). The exclusive formation of trans-products indicates that the reaction proceeds via an anti-addition involving seleniranium ions as intermediates A. However, when a terminal alkene (i.e., 1-undecene) was subjected to this reaction, a mixture of regioisomers was obtained. Concerning the addition to dienes, it was observed that the selenosulfonylation of 1,3-cyclohexadiene was completely regioselective (1,2-adduct) and stereoselective (trans-adduct) whereas 1,4-addition product was exclusively obtained from 2,3-dimethyl-l,3-butadiene, albeit without any stereoselectivity (Z/E = 60/40).
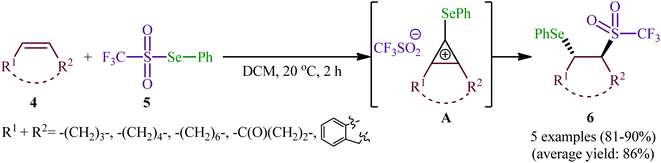 |
| | Scheme 3 Synthesis of β-selenyl-trifluoromethylsulfones 6 developed by Billard. | |
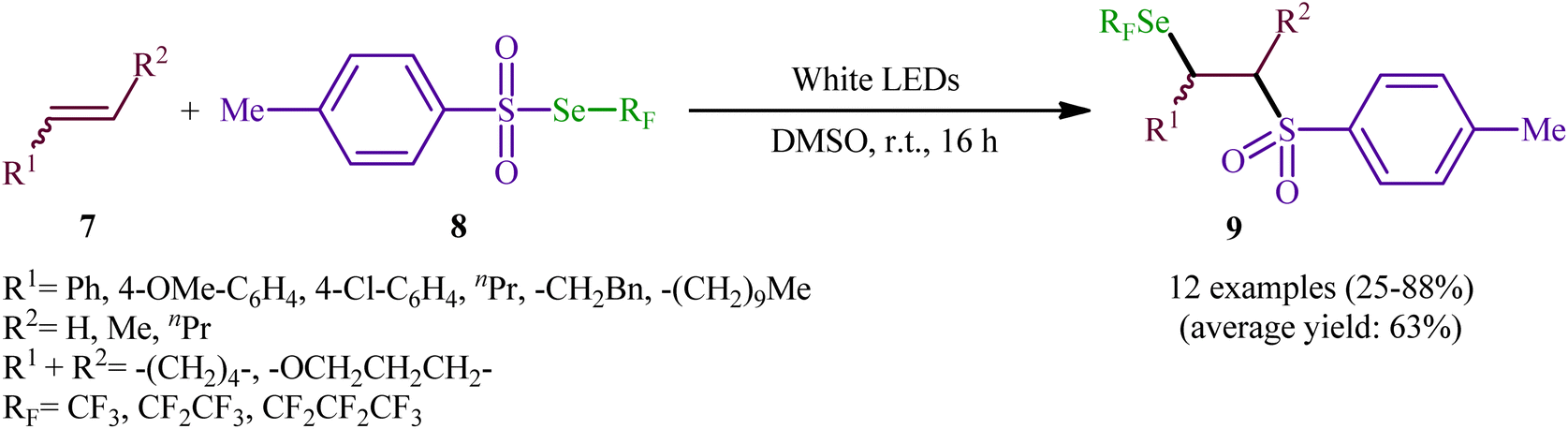
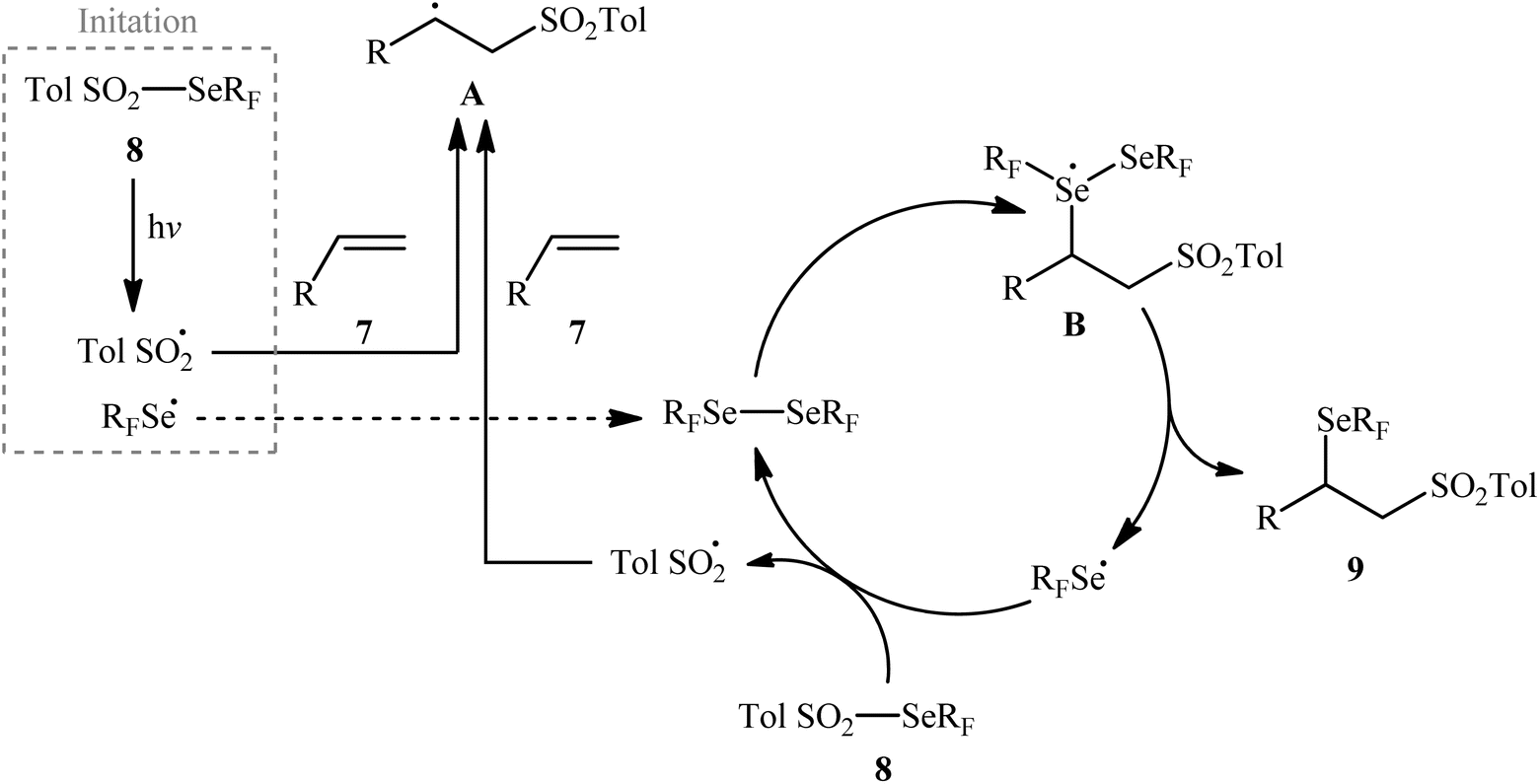
Near two decades later, the same authors reported their results on the visible-light promoted fluoroalkylselenolation of various terminal and internal alkenes 7 with fluoroalkylselenotoluenesulfonates 8 (Scheme 4).30 Optimal conditions for this photoreaction were the use of white LEDs as the light source, and DMSO as the solvent. The reaction proceeded cleanly at room temperature and the desired β-fluoroalkylselenolated sulfones 9 were obtained in fair to high yields within 16 h. In the case of both terminal and internal aromatic alkenes, the reaction exhibited a high degree of regioselectivity, in which the selenyl groups are predominantly placed on the carbon atom adjacent to the aryl group and sulfonyl group added to the carbon atom near the alkyl group. Terminal aliphatic and internal cyclic alkenes also provided the anti-Markovnikoff adducts exclusively, whereas internal acyclic alkenes provided a mixture of regioisomers. Noteworthy, replacing alkene substrates with alkynes gave the corresponding β-(seleno)-vinyl sulfones in moderate to excellent yields. Regarding the reaction mechanism, the authors proposed the following reaction pathways (Scheme 5): (i) homolytic cleavage of the S–Se bond of fluoroalkylselenotoluenesulfonates 8 upon white LED irradiation to form the selenyl radical and tosyl radical; (ii) addition of the tosyl radical to the double bond of alkene 7 to produce the radical intermediate A; (iii) reaction of radical species A with the diselenide (formed via the dimerization of selenyl radicals) to afford radical intermediate B; and (iv) release of a selenium radical from intermediate B to give the target product 9.
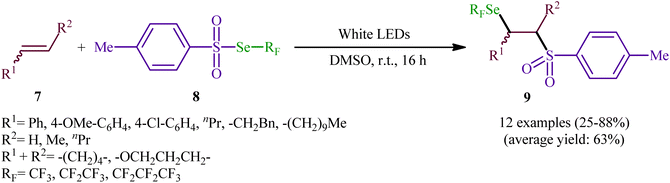 |
| | Scheme 4 Visible-light promoted fluoroalkylselenolation of alkenes 7 with fluoroalkylselenotoluenesulfonates 8. | |
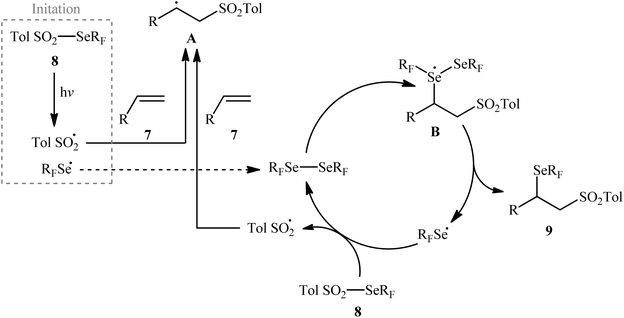 |
| | Scheme 5 Proposed mechanism for the reaction in Scheme 4. | |
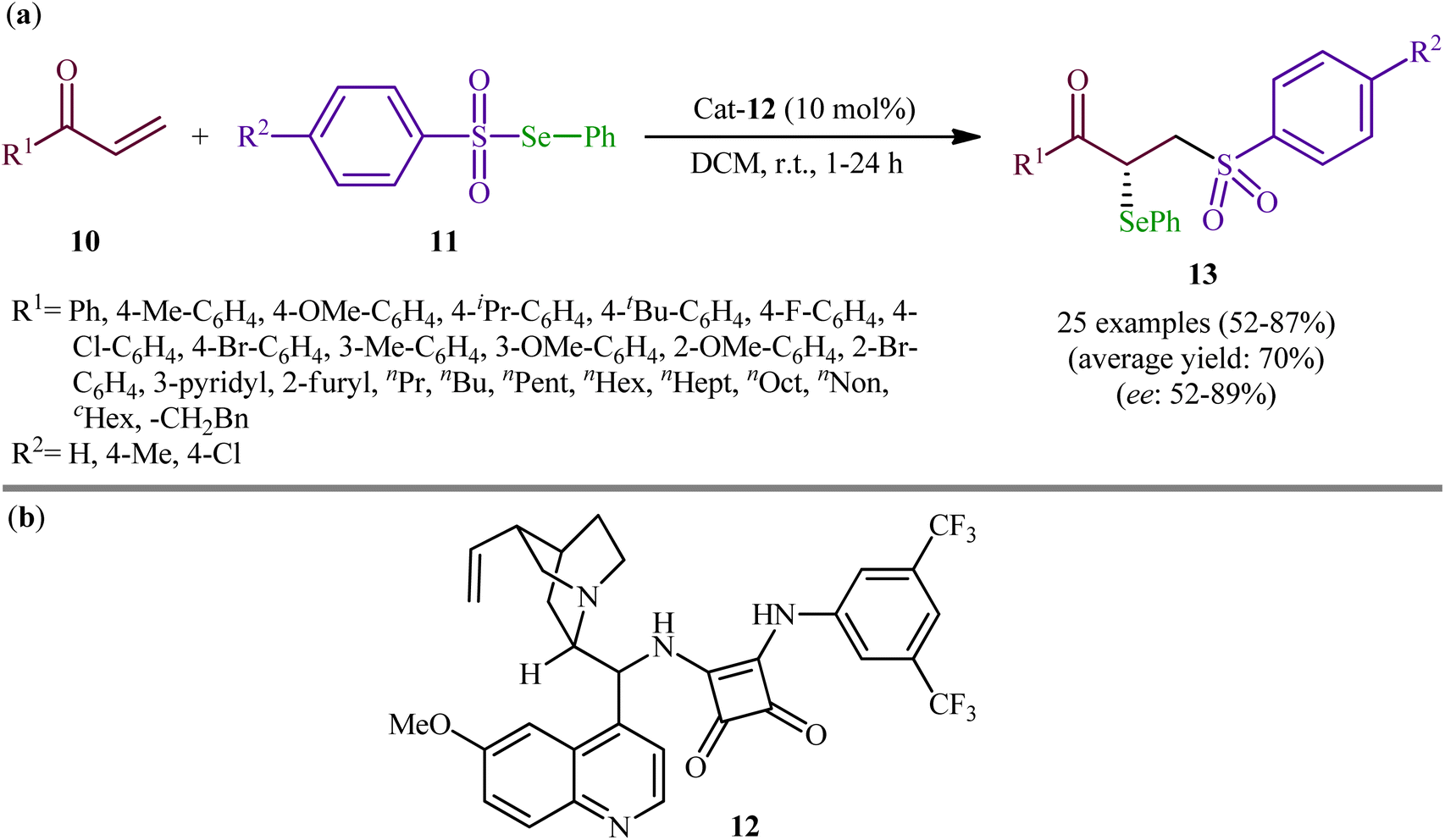
Following these works, the group of Yan recently demonstrated that a diverse range of chiral α-selenylated β-sulfonyl ketones 13 could be obtained in good yields (up to 87%) and high enantioselectivity (up to 89% ee) from the reaction of α,β-unsaturated ketones 10 with Se-phenyl areneselenosulfonates 11 employing a chiral bifunctional squaramide 12 as efficient organo-catalyst in DCM at room temperature (Scheme 6).31 The examples showed that various aliphatic, aromatic, and heteroaromatic α,β-unsaturated ketones were compatible with this methodology. A series of useful functional groups such as methoxy, fluoro, chloro, and bromo were tolerated under these reaction conditions, thus promising further manipulation of products. The authors also pointed out the synthetic utility of their strategy by high yielding gram-scale synthesis of 1-(4-(tert-butyl)phenyl)-2-(phenylselanyl)-3-tosylpropan-1-one (75% yield on 10 mmol scale). However, beta-substituted enones were incompatible in this reaction. Subsequently, Mao and Qin elegantly solved this limitation by performing the process in saturated NaCl solution, which afforded the expected α-selenyl β-sulfonyl ketone derivatives with two contiguous stereogenic centers in decent yields.32
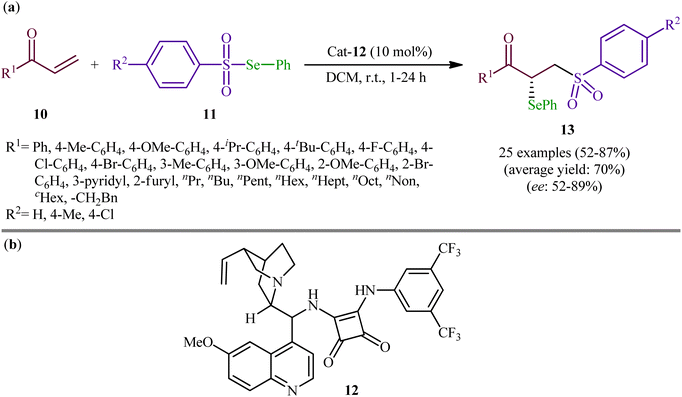 |
| | Scheme 6 (a) Organo-catalyzed enantioselective addition of Se-phenyl areneselenosulfonates 11 to α,β-unsaturated ketones 10; (b) chemical structure of squaramide catalyst 12. | |
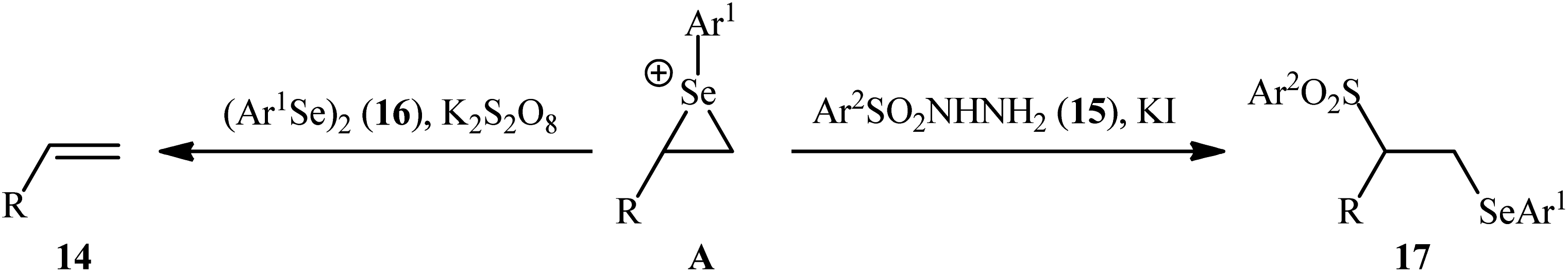
In a significant contribution in this research arena, Sun and Zhang along with their co-workers developed an efficient three-component synthesis of β-phenylselenosulfones 17 from terminal alkenes 14, arylsulfonyl hydrazides 15 and diaryl diselenides 16 under catalyst-free conditions (Scheme 7).33 By employing styrene, 4-methyl benzene sulfonyl hydrazine, and diphenyl diselenide as the model reactants, several oxidants such as (NH4)2S2O8, K2S2O8, ozone, and tert-butyl hydroperoxide (TBHP) were carefully screened. Among them, K2S2O8 displayed the excellent result for this reaction, whereas KI was found to be the most effective additive among the others tested (e.g., Na2CO3). MeCN was found to be the best solvent for this transformation and, among several common solvents tested, DCE was found to be less effective. Evaluation of the substrate scope showed that the reaction was tolerant to both electron-rich and electron-poor arylsulfonyl hydrazides and diaryl diselenides. However, the scope of alkenes was restricted to the use of styrene and (allyloxy)benzene derivatives. Based on literature report and some control experiments, the authors postulated a plausible pathway for this reaction as shown in Scheme 8.
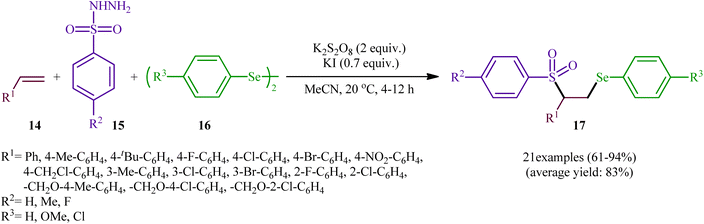 |
| | Scheme 7 Synthesis of β-phenylselenosulfones 17 via three-component reaction between terminal alkenes 14, arylsulfonyl hydrazides 15 and diaryl diselenides 16. | |
 |
| | Scheme 8 Mechanistic proposal for the formation of β-phenylselenosulfones 17. | |
3. Selenosulfonylation of alkynes
In the present section, we describe the most important developments on the direct selenosulfonylation of alkynes to give the corresponding β-selenovinyl sulfones. For clarity, the section is divided into three major subsections according to the type of reactions. Two-component reactions are covered first. This is followed by three-component reactions. Finally, the only reported example of four-component reactions will be discussed at the end of the section.
3.1. Two-component reactions
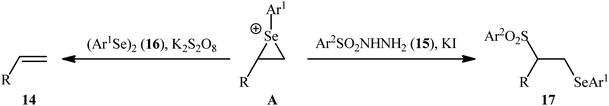
After their seminal work on the synthesis of a small library of β-selenovinyl sulfones though the simple heating of the corresponding alkynes and Se-phenyl p-tolueneselenosulfonate in common organic solvents (i.e., DCM, C6H6, and CHCl3) under catalyst- and additive-free conditions,34 the first general and practical methodology for the direct selenosulfonylation of alkynes was published by Back and co-workers in 1983.35 In this report they demonstrated that the radical-type reaction between alkynes 18 and Se-phenyl p-tolueneselenosulfonate 19 in the presence of azodiisobutyronitrile (AIBN) as a radical initiator afforded the respective β-selenovinyl sulfones 20 in moderate to excellent yields and outstanding regio- and stereo-selectivity in which in all examples anti-Markovnikov (E)-isomers were obtained as the sole products (Scheme 9). Mechanistically, this selenosulfonylation reaction was speculated to be followed analogous pathway to the one depicted for alkenes in Scheme 5. Subsequently, the authors demonstrated that the prepared β-selenovinyl sulfones were easily converted into various useful organic chemicals such as selenoxides, acetylenic sulfones, vinylic sulfones, allenic sulfones, enamines sulfones among others.9–19 As early as in the beginning of the 2000s, Qian and Huang utilized a conceptually similar strategy to fabrication of a library of β-selenovinyl sulfone resins utilizing polystyrene-supported selenosulfonates as selenosulfonylating reagents and successfully converted them to the corresponding acetylenic sulfones through stereospecific selenoxide syn-elimination using hydrogen peroxide as the oxidant.36
 |
| | Scheme 9 AIBN-catalyzed addition of Se-phenyl p-tolueneselenosulfonate 19 to alkynes 18. | |
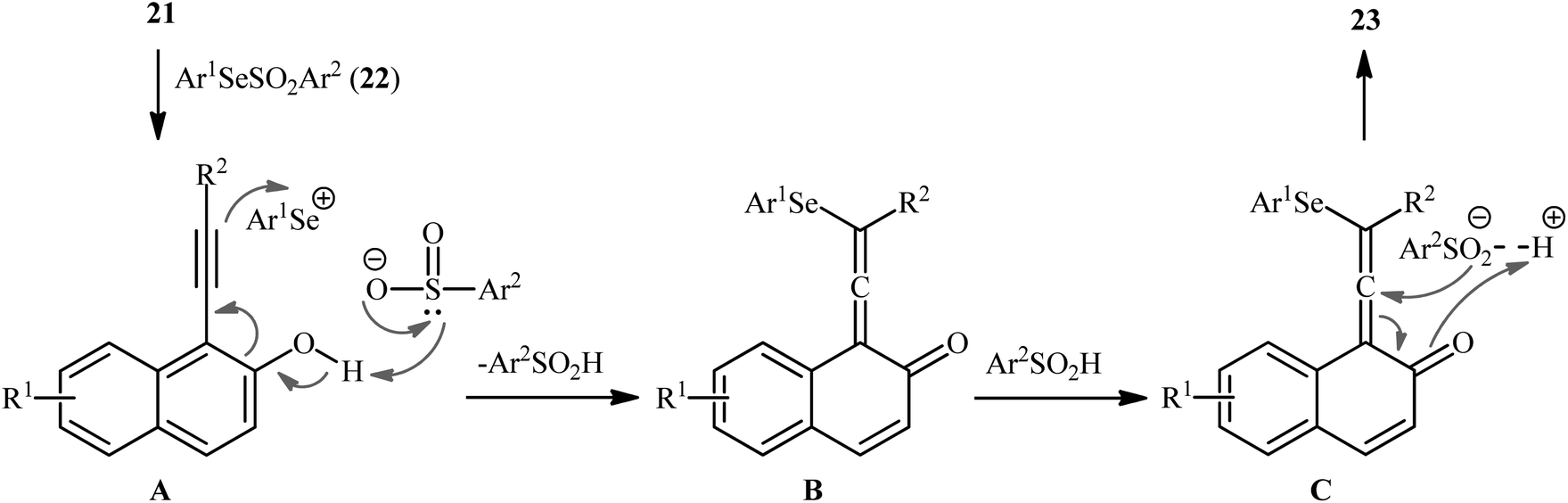
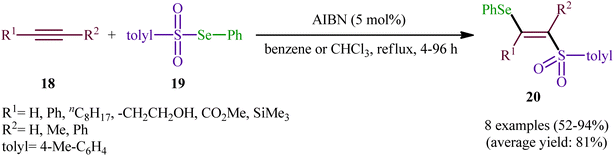
In 2019, Qin's research group disclosed a related selenosulfonylation of 1-ethynylnaphthalen-2-ol derivatives 21 with Se-aryl areneselenosulfonates 22 under mild conditions which exhibited considerably better substrate scope for selenosulfonylating agents when compared to the previous works.37 The transformation was performed under 1,2-dichloroethane (DCE) in the absence of any catalysts or additives under an inert atmosphere, which afforded the 1-(2-(arylselanyl)-1-(arylsulfonyl)vinyl)naphthalen-2-ol products 23 in moderate to high yields with excellent E-selectivity (Scheme 10). However, 2-ethynylphenol was inert under the standard conditions. The authors solved this limitation by performing the process in the presence of triethyl amine in refluxing toluene. Furthermore, they elegantly applied the prepared tetrasubstituted olefins as starting materials to the fabrication of useful naphtho[2,1-b]furan and benzofuran scaffolds through oxidation with 3-chloroperoxybenzoic acid (m-CPBA) and then treatment with aq. KOH. Although the detailed mechanistic picture remains unclear, the authors suggest that vinylidene ortho-quinone methide (VQM) intermediate might be the key intermediate for this selenosulfonylation reaction (Scheme 11).
 |
| | Scheme 10 Catalyst-free regioselective selenosulfonylation of 1-ethynylnaphthalen-2-ols 21 with Se-aryl areneselenosulfonates 22. | |
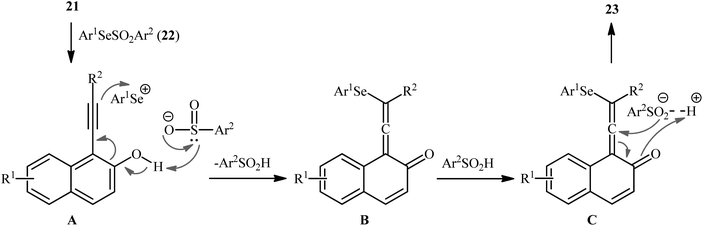 |
| | Scheme 11 Proposed mechanism for the formation of 1-(2-(arylselanyl)-1-(arylsulfonyl)vinyl)naphthalen-2-ols 23. | |
In the same year, Wang-Ji and co-workers extended the substrate scope of this chemistry to 2-alkynoates.38 Thus, a library of alkyl/benzyl 2-alkynoates 24 were reacted with alkyl/aryl selenosulfonates 25 in the presence of a catalytic amount of CoC2O4 under an inert atmosphere and blue light irradiation at room temperature leading to the respective β-selenovinyl sulfones 26 in moderate to high yields and good stereoselectivities in favor of the (E)-products (Scheme 12). The results showed that alkyl 2-alkynoates afforded significantly better yields compared to the benzyl-substituted ones and sterically less hindered alkyl-substituted 2-alkynoates (e.g., ethyl 2-alkynoates) provided higher stereoselectivities than more sterically hindered alkyl 2-alkynoates (e.g., phenethyl 2-alkynoates). It is worthy of note that the presence of both catalyst and light were crucial for the success of this transformation. No product was obtained in the absence of either of them. According to the authors, the presence of light source was crucial for cleavage of Se–S bond and the metal catalyst had a fundamental influence on the addition of free radicals to alkyne. Under the slightly modified conditions [Cu(0) (2 mol%), MeCN, Ar, blue LEDs, r. t., 4 h], they also executed the direct selenosulfonylation of a panel of alkynamides, offering a decent yield of the corresponding β-selenovinyl sulfones.
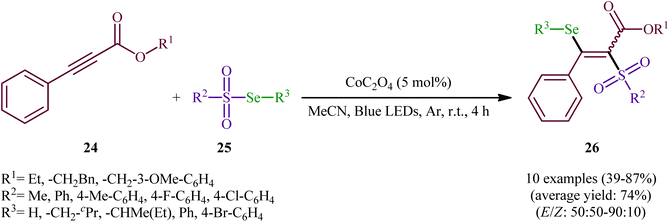 |
| | Scheme 12 Visible light-induced Co-catalyzed selenosulfonylation of alkynes 24 with alkyl/aryl selenosulfonates 25. | |
3.2. Three-component reactions
In 2002, the Huang group developed an interesting three-component method for the preparation of β-selenovinyl sulfones 30 through the reaction between alkynes 27, sodium arenesulfinates 28, and diphenyl diselenide 29 in the absence of any catalyst or additive in aqueous acetic acid at 80 °C (Scheme 13).39 Although the reactions of terminal alkynes are regio- (anti-Markovnikov) and stereoselective, (anti), internal alkynes were unsuitable substrates under the optimal reaction. Furthermore, the applicability of aliphatic sodium sulfonates or aliphatic diselenides were not investigated under the conditions employed.
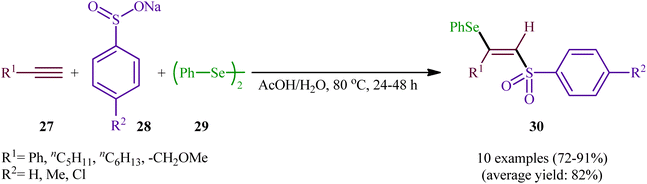 |
| | Scheme 13 Catalyst-free three-component reaction between alkynes 27, sodium arenesulfinates 28, and diphenyl diselenide 29. | |
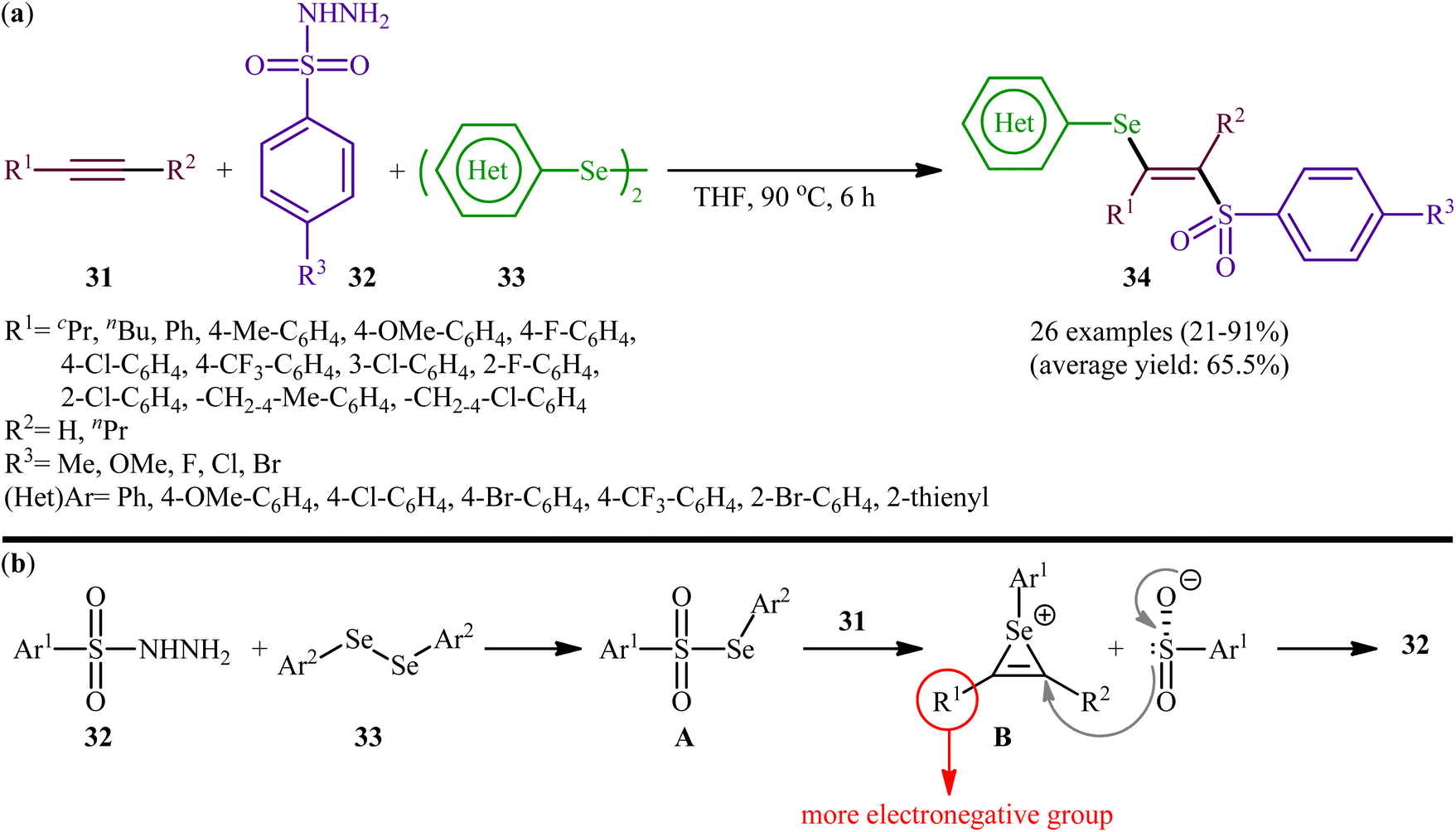
Fifteen years later, Liu and colleagues developed a similar selenosulfonylation of alkynes 31 using arylesulfonyl hydrazides 32 and diaryl diselenides 33 as the sources of sulfonyl and selenyl groups, respectively, under catalyst-free conditions by simply mixing of the substrates in refluxing THF (Scheme 14a).40 The procedure was shown to be general and a diverse range of terminal and internal alkynes participated in the reaction. Moreover, either electron-rich or electron-poor derivatives of both arylesulfonyl hydrazides and diaryl diselenides exhibited good applications under standard conditions and provided the expected β-selenovinyl sulfones 34 in reasonable yields. In addition, the reaction could be performed efficiently on gram scale (e.g., (E)-(2-((4-methoxyphenyl)sulfonyl)-1-phenylvinyl)(phenyl))selane was obtained in 1.0 g scale in high yield of 80%). To gain mechanistic insights, several control experiments were performed. The possibility of a radical pathway was neglected since the reaction with radical scavengers such as TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl)) and BHT (butylated hydroxytoluene) did not prevent the product formation. Based on these results, it was confirmed that this reaction most likely proceeds via a cationic-species-induced pathway. Initially formed Se-aryl areneselenosulfonate intermediate A (via the reaction of arylesulfonyl hydrazides 32 and diaryl diselenides 33) undergoes heterolysis on alkynes 31 to generate seleniranium cation B that after SN2-type reaction with benzenesulfonyl anion leads to the desired product 34 (Scheme 14b).
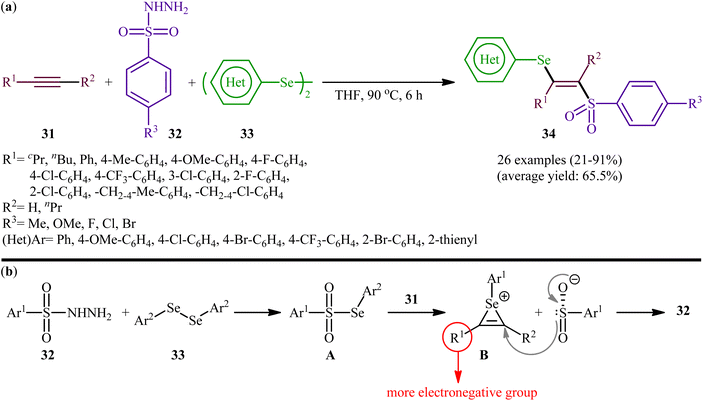 |
| | Scheme 14 (a) Catalyst-free three-component synthesis of β-selenovinyl sulfones 34 from alkynes 31, arylesulfonyl hydrazides 32 and diaryl diselenides 33; (b) possible mechanism for the generation of β-selenovinyl sulfones 34. | |
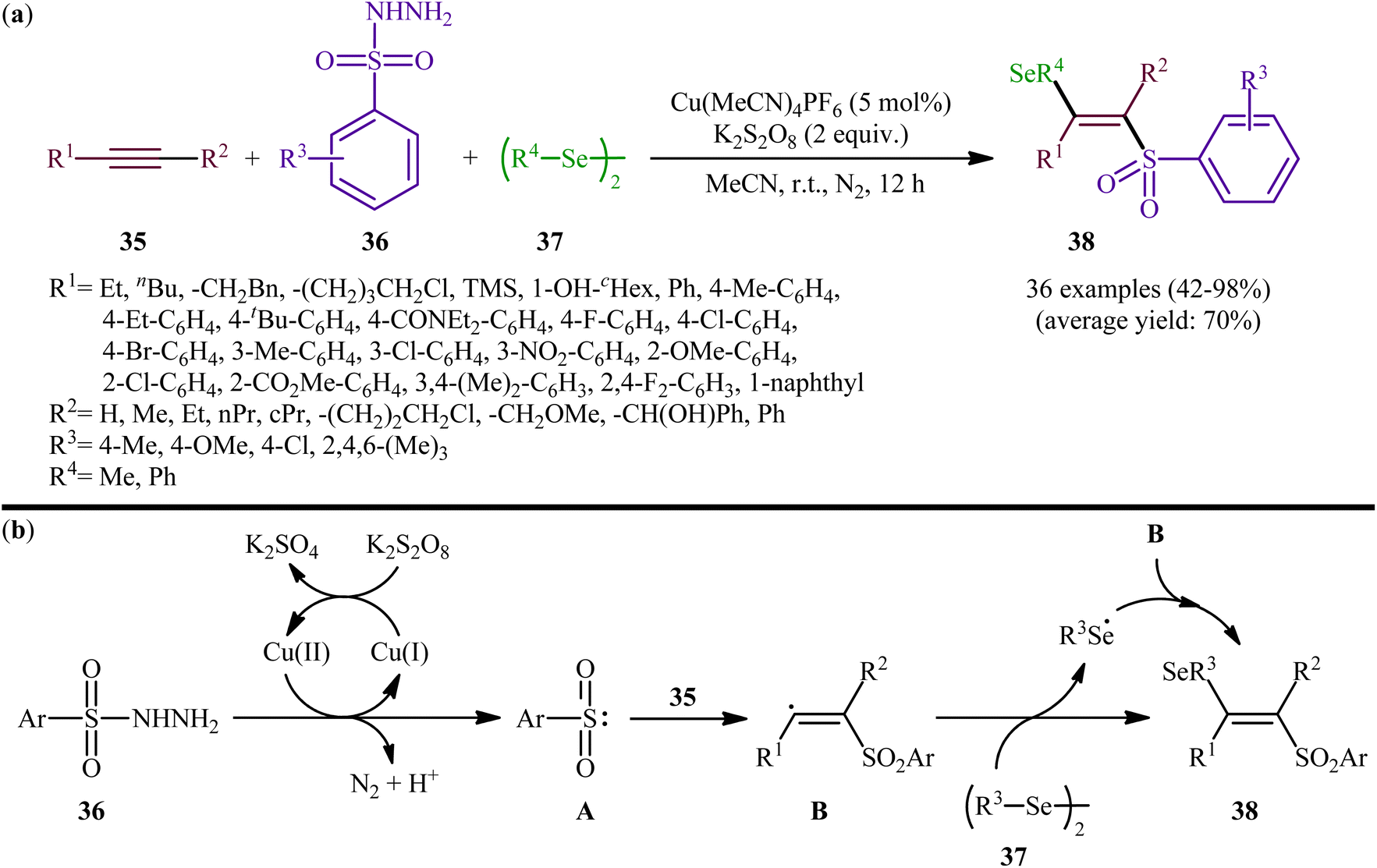
Concurrently, Li and Zhang along with their co-workers investigated the catalytic version of this transformation employing Cu(MeCN)4PF6 as the catalyst and K2S2O8 as the oxidant.41 The reactions were carried out in MeCN at room temperature, tolerated various terminal and internal alkynes 35, both aromatic and aliphatic diselenides 37, as well as several arylesulfonyl hydrazides 36 with either electron-donating or electron-withdrawing substituents, and provided moderate to almost quantitative yields of the desired products 38 within 12 h (Scheme 15a). The results indicated that this protocol selectively gave higher yields of (E)-β-selenovinyl sulfone products compared to Liu's methodology. To gain mechanistic insights, some preliminary control experiments were performed. No products were obtained by treatment of prop-1-yn-1-ylbenzene with Se-phenyl p-tolueneselenosulfonate under the optimized conditions. Therefore, the possibility of existence of selenosulfonate intermediate in the mechanism of this reaction was neglected. In parallel, the radical trapping experiments with TEMPO and BHT pointed toward a radical reaction mechanism. Based on these results and literature, it was suggested that this transformation most likely proceeds via a pathway similar to the one depicted in Scheme 15b.
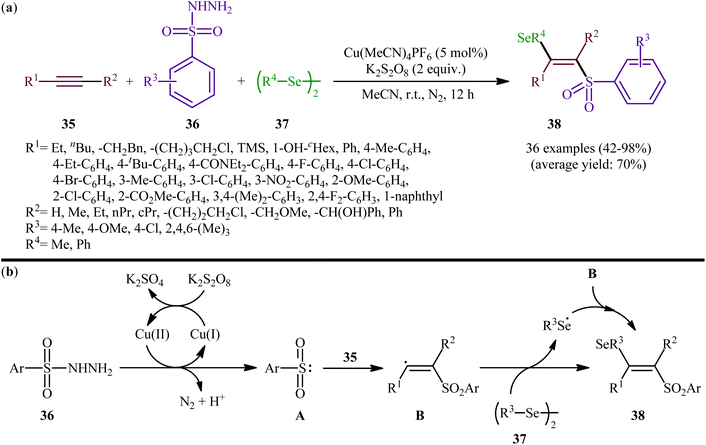 |
| | Scheme 15 (a) Cu-catalyzed selenosulfonation of alkynes 35 with arylesulfonyl hydrazides 36 and di-aryl/alkyl diselenides 37; (b) mechanism proposed to explain the formation of β-selenovinyl sulfones 38. | |
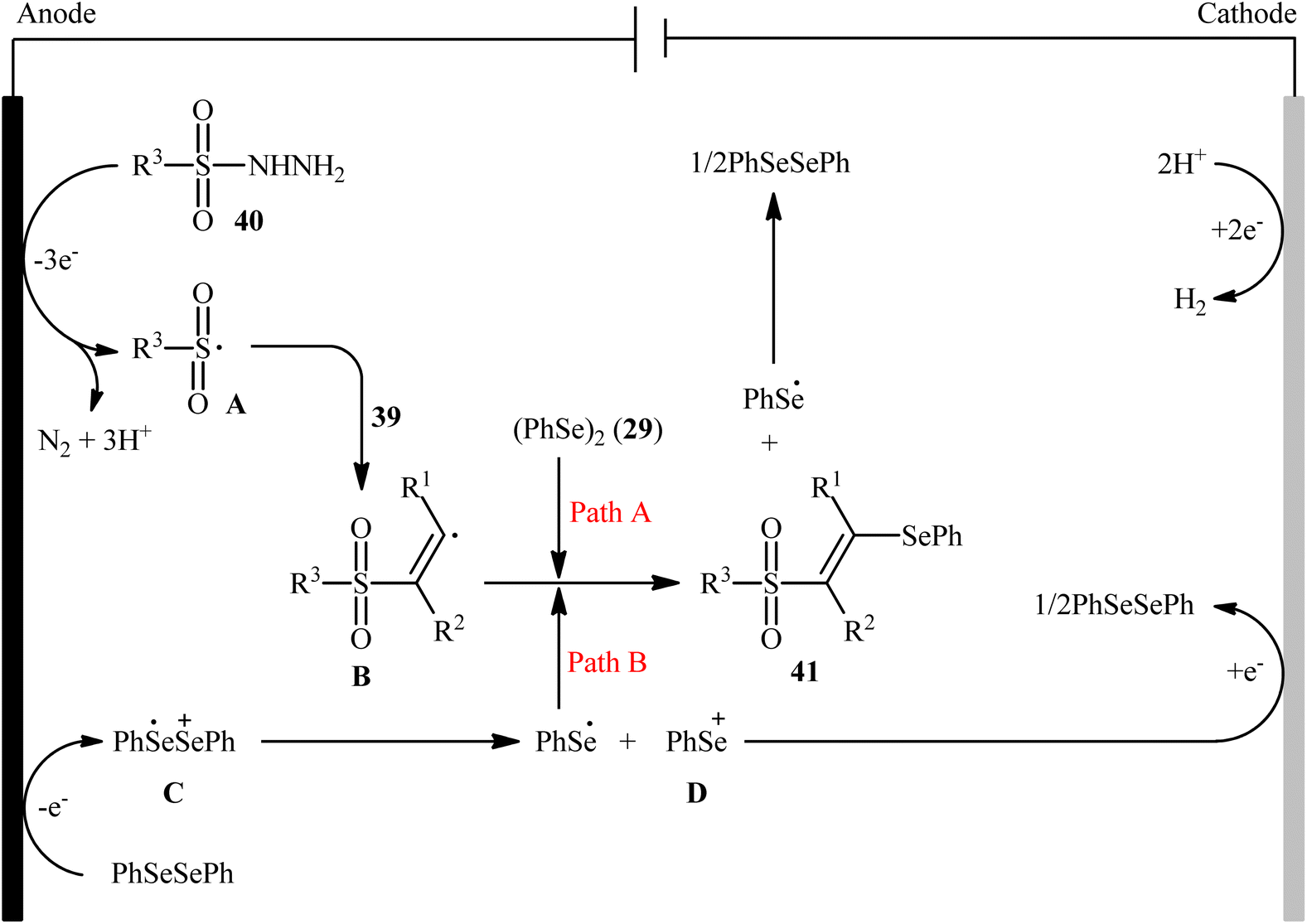
Recently, Chen and Xu along with their colleagues developed an efficient electrochemical approach for regioslective selenosulfonylation of both terminal and internal alkynes 39 employing sulfonyl hydrazines 40 and diphenyl diselenide 29.42 The optimal system was identified using nBu4NBF4 (tetrabutylammonium tetrafluoroborate) as a supporting electrolyte with an undivided graphite/Pt-cell in MeCN under an inert atmosphere and constant current of 12 mA at room temperature. This electrochemical protocol efficiently provided the expected β-selenovinyl sulfones 41 in good yields under catalyst-free conditions and without assistance of any oxidizing agent or additive (Scheme 16). Although the reported examples in this study are generally limited to the use of only diphenyl diselenide as selenenylating agent, the scope of sulfonyl hydrazines that underwent reaction was broad enough to include alkyl, aryl, heteroaryl and naphthyl derivatives. Importantly, the process can also be scaled up to provide multi-gram quantities of β-selenovinyl sulfone products without further optimization and without loss in the yield or outcome. To gain mechanistic insights, several control experiments were performed. No products were obtained by treatment of phenylacetylene with PhSeSO2Ph under standard conditions. Therefore, selenosulfonates might not be the real reactive intermediates in this transformation. The TEMPO experiment confirmed the possibility of radical species since the reaction with TEMPO completely prevent the product formation. Furthermore, cyclic voltammetric measurements revealed a reversal reduction peak at 0.13 V assigned to phenyl selenol radical to phenyl selenol cation. Based on the above results, the authors speculated that the plausible mechanism for the formation of β-selenovinyl sulfones 41 includes the generation of sulfonyl radical A through the electrooxidation of sulfonyl hydrazine 40 along with deprotonation and release of a molecular nitrogen, its reaction with the alkyne 39 to give relatively stable β-sulfonyl vinyl radical B, and the atom transfer reaction of B with diphenyl diselenide 29 (Scheme 17, path (a)). In another possibility, anodic oxidation of diphenyl diselenide 29 affords cationic radical intermediate C, which dissociates into phenylselenium radical and phenyl selenium cation D. Finally, the phenyl selenol radical couples with β-sulfonyl vinyl radical B to form the target product 41. At the same time, cathodic reduction of the radical cation of diphenyl diselenide C regenerates diphenyl diselenide 29 and completes the catalytic cycle (Scheme 17, path (b)).
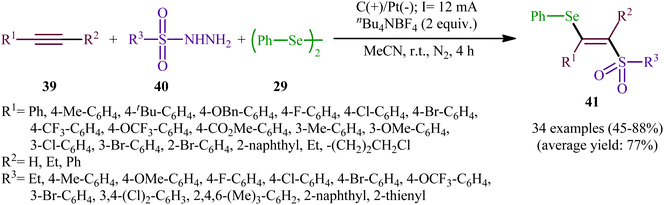 |
| | Scheme 16 Electrochemical selenosulfonylation of alkynes 39 employing sulfonyl hydrazines 40 and diphenyl diselenide 29. | |
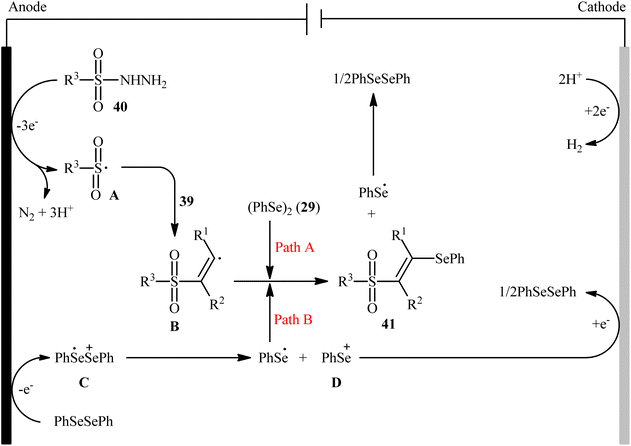 |
| | Scheme 17 Plausible mechanism for electrosynthesis of β-selenovinyl sulfones 41. | |
3.3. Four-component reactions
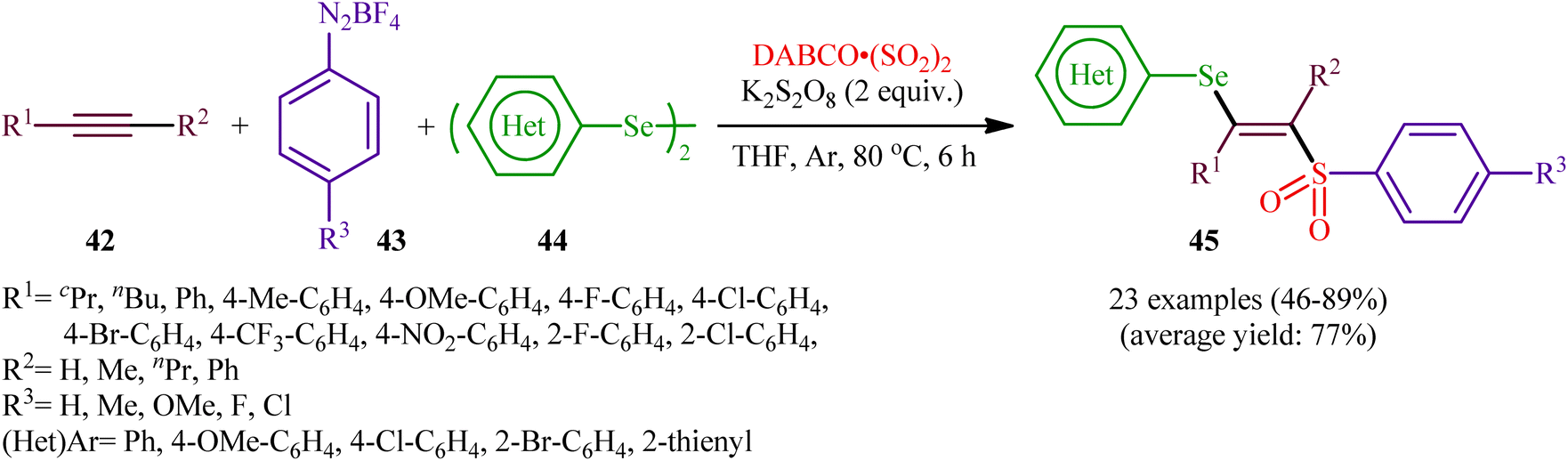
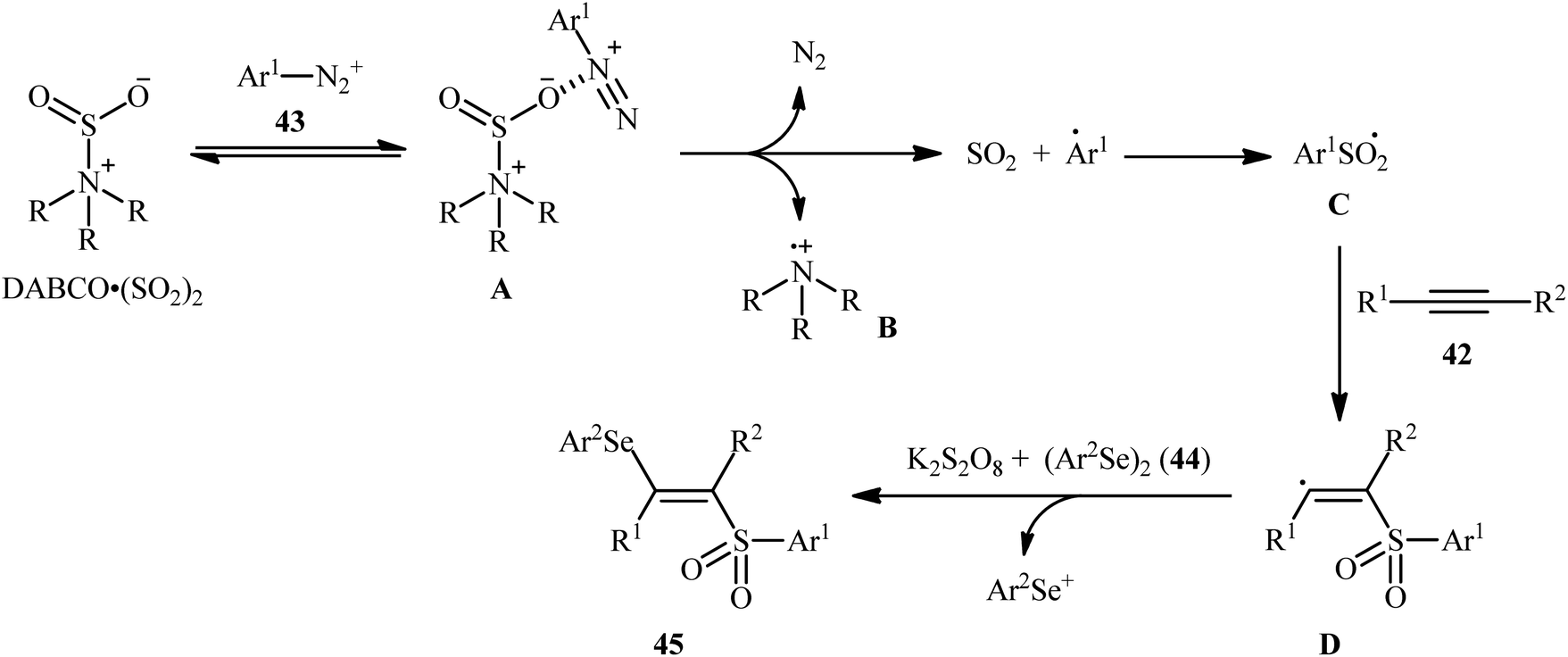
In 2018, Sun's research group studied the possibility of regio- and stereospecific synthesizing (E)-β-selenovinyl sulfones 45 through the catalyst-free four-component reaction between alkynes 42, aryldiazonium tetrafluoroborates 43, di(hetero)aryl diselenides 44, and 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABCO·(SO2)2) via the insertion of sulfur dioxide.43 Thus, the scrupulous analysis of the optimized reactions revealed that performing the process in the presence of over-stoichiometric amounts of K2S2O8 as an oxidant in refluxing THF under an inert atmosphere was the optimum condition, giving the desired (E)-β-selenovinyl sulfones 45 in moderate to high yield, ranging from 46% to 89% (Scheme 18). Interestingly, the authors further demonstrated that β-sulfonyl-vinyl tellanes could also be achieved in good yields when diaryl diselenides were replaced with diarylditellanes. The results showed that the outcome of reaction was independent of the electronic nature of aryldiazonium tetrafluoroborates and di(hetero)aryl diselenides. Therefore, either electron-donating or electron-deficient functionalities on both partners were well tolerated by this protocol. Concerning the substrate scope of alkynes, the decreasing order of reactivity is terminal aromatic alkynes ≥ internal aryl-alkyl alkynes ≈ internal aryl–aryl alkynes > terminal aliphatic alkynes. The mechanistic studies suggested that this transformation presumably proceeds through a radical pathway as shown in Scheme 19. It is noteworthy to mention that during a model reaction between 4-methylphenyldiazonium tetrafluoroborate, DABSO, and diphenyl diselenide, and in the absence of alkyne, the adduct PhSeSO2Ph was detected and isolated. Then, the reaction of this adduct with phenylacetylene was performed under optimized conditions. Interestingly, no reaction was occurred and the PhSeSO2Ph was recovered completely. This result indicated that the selenosulfonate intermediates were formed in situ but were not involved in the selenosulfonation process. Recently, He and Wu along with their co-workers utilized a conceptually similar strategy to fabrication of cyanoalkylsulfonylated selenoallenes44 and 3-((arylselanyl)methylene)pyrrolidines45 in acceptable yields through selenosulfonylation of 1,3-enynes and 1,6-enynes, respectively, employing cycloketone oxime esters as the cyanoalkyl sources.
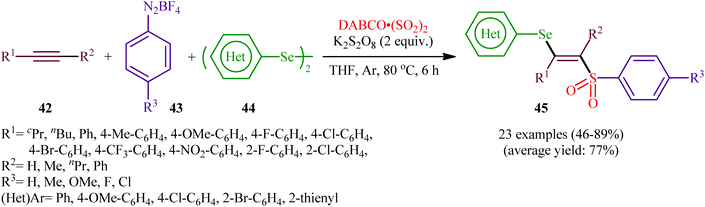 |
| | Scheme 18 Sun's synthesis of (E)-β-selenovinyl sulfones 45. | |
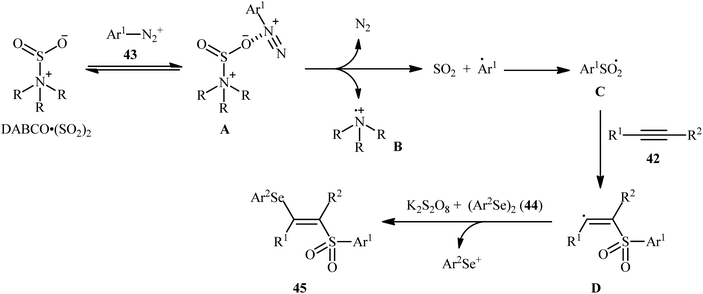 |
| | Scheme 19 Mechanistic proposal for the reaction in Scheme 18. | |
4. Selenosulfonylation of allenes
Compared with the relatively well-developed selenosulfonylation of alkenes and alkynes, the direct selenosulfonylation of allenes is still uncommon. In fact, only one preliminary report of such a reaction was published in the literature till date.
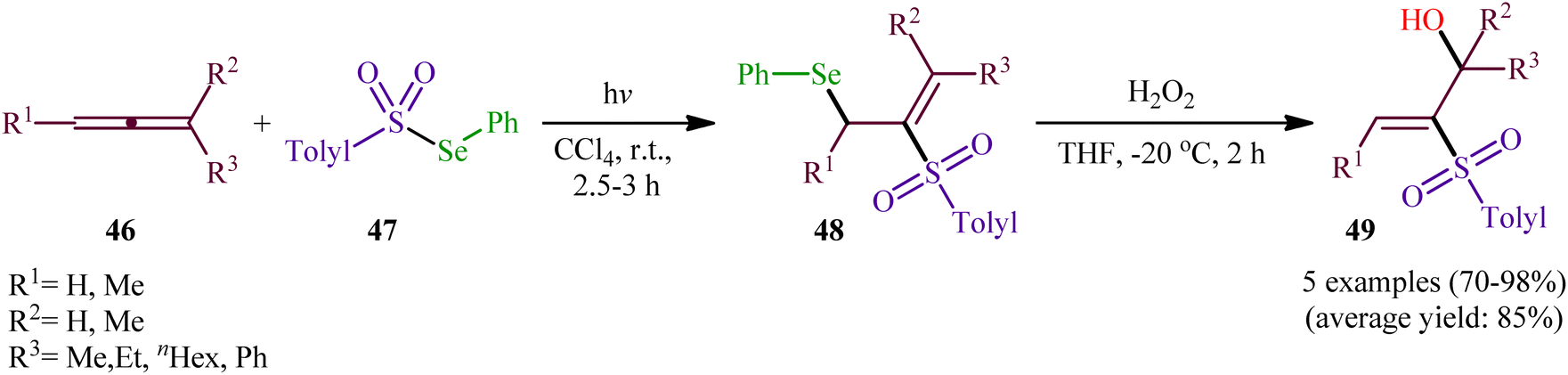
In 1982, Kang and Kice published the first and only example of the direct conversion of allenes 46 into the corresponding β-(sulfonyl)allylselanes 48 using Se-phenyl 4-methylbenzenesulfonoselenoate 47 as selenosulfonylating agent in Rayonet photoreactor.46,47 Five phenyl(2-(tolylsulfonyl)allyl)selanes 48 were synthesized and then converted in good to excellent yields to the corresponding 2-(tolylsulfonyl)prop-2-en-1-ols 49 by oxidative elimination of the phenylselanyl group using H2O2 as the oxidant (Scheme 20). Regarding the regioselectivity of this transformation, in all examples the sulfonyl group selectively attacked the central carbon of the allenic system and selanyl group was attached to the less highly substituted of the two terminal carbons.
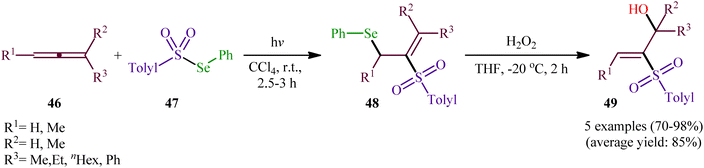 |
| | Scheme 20 Kice's synthesis of phenyl(2-(tolylsulfonyl)allyl)selanes 48. | |
5. Conclusion
Difunctionalization reactions of unsaturated compounds, the incorporation of two functional groups onto a carbon–carbon π-double bond in a single operation, have emerged as a powerful methodology to generate complex molecules from simple starting materials that would otherwise be difficult to prepare. In this regard, 1,2-selenosulfonylation reactions which provide a robust approach for the synthesis of β-selenosulfones derivatives have gained a great deal of attention. As illustrated, various unsaturated compounds such as alkenes, alkynes, and allenes were successfully applied in this procedure. Most of the reactions covered in this review have been carried out under catalyst-free and mild conditions and can be easily scaled up to gram levels without further optimization. However, there are still a number of challenges to be addressed in this methodology. Some of these are listed below: (i) majority of methods for selenosulfonylation of alkenes rely on the use of pre-prepared Se-aryl selenosulfonates. Thus, development of new and efficient approaches that use simple and commercially available reagents is highly desirable; (ii) the stereoselectivity of selenosulfonylation reactions of alkynes is dominated by the (E)-products. Therefore, additional studies are needed to develop efficient processes that also allow these reactions to occur in (Z)-selective manner; and (iii) the number of reported examples in selenosulfonylation of allenes are too narrow and certainly much more work must be done to study the scope and limitations of this double functionalization reaction.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) R. M. Romero, T. H. Woeste and K. Muniz, Asian J. Chem., 2014, 9, 972–983 CrossRef CAS PubMed;
(b) X. W. Lan, N. X. Wang and Y. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef CAS;
(c) Z. L. Li, G. C. Fang, Q. S. Gu and X. Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC;
(d) W. Liu and W. Kong, Org. Chem. Front., 2020, 7, 3941–3955 RSC;
(e) Y. A. Elshaier, M. T. Nemr, M. Al Refaey, W. A. Fadaly and A. Barakat, New J. Chem., 2022, 46, 13383–13400 RSC;
(f) K. Kikushima, E. E. Elboray, J. O. C. Jiménez-Halla, C. R. Solorio-Alvarado and T. Dohi, Org. Biomol. Chem., 2022, 20, 3231–3248 RSC.
- Z. Hossaini, E. A. Mahmood, M. R. P. Heravi, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 21651–21665 RSC.
-
(a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed;
(b) P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed;
(c) A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2014, 54, 185–267 CrossRef CAS.
-
(a) K. A. Scott and T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef;
(b) M. Abdoli, A. Angeli, M. Bozdag, F. Carta, A. Kakanejadifard, H. Saeidian and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2017, 32, 1071–1078 CrossRef CAS PubMed;
(c) M. Abdoli, M. Bozdag, A. Angeli and C. T. Supuran, Metabolites, 2018, 8, 37 CrossRef PubMed;
(d) M. Abdoli, S. Giovannuzzi, C. T. Supuran and R. Žalubovskis, J. Enzyme Inhib. Med. Chem., 2022, 37, 1568–1576 CrossRef CAS PubMed;
(e) A. H. M. Hussein, A. A. Khames, A. B. A. El-Adasy, A. A. Atalla, M. Abdel-Rady, M. I. Hassan, M. T. Nemr and Y. A. Elshaier, RSC Adv., 2020, 10, 29723–29736 RSC;
(f) M. T. Nemr and A. M. AboulMagd, Bioorg. Chem., 2020, 103, 104134 CrossRef CAS PubMed;
(g) M. T. Nemr, A. Sonousi and A. A. Marzouk, Bioorg. Chem., 2020, 105, 104446 CrossRef CAS PubMed.
-
(a) S. Otocka, M. Kwiatkowska, L. Madalinska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed;
(b) E. E. Alberto, V. D. Nascimento and A. L. Braga, J. Braz. Chem. Soc., 2010, 21, 2032–2041 CrossRef CAS;
(c) A. Arora, S. Singh, P. Oswal, D. Nautiyal, G. K. Rao, S. Kumar and A. Kumar, Coord. Chem. Rev., 2021, 438, 213885 CrossRef CAS;
(d) Q. Li, Y. Zhang, Z. Chen, X. Pan, Z. Zhang, J. Zhu and X. Zhu, Org. Chem. Front., 2020, 7, 2815–2841 RSC;
(e) F. Chen, J. Ma, Y. Zhu, X. Li, H. Yu and Y. Sun, J. Hazard. Mater., 2022, 426, 128064 CrossRef CAS PubMed;
(f) W. Yang, H. Zhang, Y. Liu, C. Tang, X. Xu and J. Liu, RSC Adv., 2022, 12, 14435–14438 RSC;
(g) J. Zhang, J. Lv and J. Wang, Z. Kristallogr. - New Cryst. Struct., 2022, 237, 385–387 CrossRef CAS;
(h) Y. Yang, H. Zhu, X. Xu, L. Bao, Y. Wang, H. Lin and C. Zheng, Microporous Mesoporous Mater., 2021, 324, 111289 CrossRef CAS.
-
(a) P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef;
(b) S. Panda, A. Panda and S. S. Zade, Coord. Chem. Rev., 2015, 300, 86–100 CrossRef CAS.
-
(a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed;
(b) C. W. Nogueira, G. Zeni and J. B. Rocha, Chem. Rev., 2004, 104, 6255–6286 CrossRef CAS PubMed;
(c) C. W. Nogueira, N. V. Barbosa and J. B. Rocha, Arch. Toxicol., 2021, 95, 1179–1226 CrossRef CAS PubMed;
(d) P. Collery, J. Trace Elem. Med. Biol., 2018, 50, 498–507 CrossRef CAS;
(e) D. Radomska, R. Czarnomysy, D. Radomski and K. Bielawski, Int. J. Mol. Sci., 2021, 22, 1009 CrossRef CAS PubMed.
-
(a) D. Desai, N. Kaushal, U. H. Gandhi, R. J. Arner, C. D'Souza, G. Chen, H. Vunta, K. El-Bayoumy, S. Amin and K. S. Prabhu, Chem.-Biol. Interact., 2010, 188, 446–456 CrossRef CAS PubMed;
(b) A. Angeli, D. Tanini, T. S. Peat, L. Di Cesare Mannelli, G. Bartolucci, A. Capperucci, C. Ghelardini, C. T. Supuran and F. Carta, ACS Med. Chem. Lett., 2017, 8, 963–968 CrossRef CAS PubMed;
(c) S. Shaaban, H. E. Gaffer, M. Alshahd and S. S. Elmorsy, Int. J. Res. Dev. Pharm. Life Sci., 2015, 4, 1654–1668 CAS.
- T. G. Back, S. Collins, U. Gokhale and K. W. Law, J. Org. Chem., 1983, 48, 4776–4779 CrossRef CAS.
- T. G. Back, S. Collins and K. W. Law, Tetrahedron Lett., 1984, 25, 1689–1692 CrossRef CAS.
- T. G. Back, S. Collins and K. W. Law, Can. J. Chem., 1985, 63, 2313–2318 CrossRef CAS.
- T. G. Back, J. R. Proudfoot and C. Djerassi, Tetrahedron Lett., 1986, 27, 2187–2190 CrossRef CAS.
- T. G. Back, M. V. Krishna and K. R. Muralidharan, Tetrahedron Lett., 1987, 28, 1737–1740 CrossRef CAS.
- T. G. Back, S. Collins, M. V. Krishna and K. W. Law, J. Org. Chem., 1987, 52, 4258–4264 CrossRef CAS.
- T. G. Back and K. R. Muralidharan, J. Org. Chem., 1989, 54, 121–125 CrossRef CAS.
- T. G. Back, M. V. Krishna and K. R. Muralidharan, J. Org. Chem., 1989, 54, 4146–4153 CrossRef CAS.
- T. G. Back, K. Brunner, M. Vijaya Krishna and E. K. Lai, Can. J. Chem., 1989, 67, 1032–1037 CrossRef CAS.
- T. G. Back, E. K. Lai and K. R. Muralidharan, J. Org. Chem., 1990, 55, 4595–4602 CrossRef CAS.
- T. G. Back and M. V. Krishna, J. Org. Chem., 1991, 56, 454–457 CrossRef CAS.
- Organoselenium chemistry: a practical approach, ed. T.G.
Back, 1999, OUP Oxford Search PubMed.
-
(a) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2021, 11, 470–483 RSC;
(b) B. Azizi, M. R. P. Heravi, Z. Hossaini, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 13138–13151 RSC;
(c) L. Yan-mei, F. Jin-feng, H. Long-qiang, L. Wei-na and E. Vessally, RSC Adv., 2021, 11, 24474–24486 RSC;
(d) Y. Zhang and E. Vessally, RSC Adv., 2021, 11, 33447–33460 RSC;
(e) Y. Cao, S. Soleimani-Amiri, R. Ahmadi, A. Issakhov, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 32513–32525 RSC.
-
(a) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef CAS;
(b) E. Vessally, R. Mohammadi, A. Hosseinian, K. Didehban and L. Edjlali, J. Sulfur Chem., 2018, 39, 443–463 CrossRef CAS;
(c) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(d) A. Hosseinian, L. Zare Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur Chem., 2018, 39, 674–698 CrossRef CAS;
(e) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef PubMed;
(f) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS;
(g) A. Hosseinian, Y. J. Sadeghi, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 565–585 CrossRef CAS;
(h) M. Hamzehloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef CAS;
(i) N. H. Jabarullah, K. Jermsittiparsert, P. A. Melnikov, A. Maseleno, A. Hosseinian and E. Vessally, J. Sulfur Chem., 2020, 41, 96–115 CrossRef CAS;
(j) Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC;
(k) Y. Cao, S. Abdolmohammadi, R. Ahmadi, A. Issakhov, A. G. Ebadi and E. Vessally, RSC Adv., 2021, 11, 32394–32407 RSC;
(l) M. R. J. Sarvestani, N. Mert, P. Charehjou and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed;
(m) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(n) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed;
(o) E. A. Mahmood, B. Azizi and S. Majedi, Chem. Rev. Lett., 2020, 3, 2–8 CAS;
(p) N. Salehi and B. Azizi, et. al, J. Chem. Lett., 2021, 2, 2–8 Search PubMed;
(q) D. Tayde and M. Lande, Chem. Rev. Lett., 2021, 4, 30–36 CAS.
-
(a) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(b) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 1–19 CrossRef PubMed;
(c) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(d) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS;
(e) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS PubMed;
(f) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC;
(g) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 1–32 CrossRef PubMed;
(h) Z. He, D. Wu and E. Vessally, Top. Curr. Chem., 2020, 378, 1–30 CrossRef PubMed;
(i) W. Xu, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2021, 43, 101358 CrossRef CAS;
(j) A. Hassanpour, M. R. P. Heravi, A. Ebadi, A. Hosseinian and E. Vessally, J. Fluorine Chem., 2021, 245, 109762 CrossRef CAS;
(k) A. Karbakhshzadeh, M. R. P. Heravi, Z. Rahmani, A. G. Ebadi and E. Vessally, J. Fluorine Chem., 2021, 248, 109806 CrossRef CAS.
- T. G. Back and S. Collins, Tetrahedron Lett., 1980, 21, 2215–2218 CrossRef CAS.
- T. G. Back and S. Collins, J. Org. Chem., 1981, 46, 3249–3256 CrossRef CAS.
- Y. H. Kang and J. L. Kice, Tetrahedron Lett., 1982, 23, 5373–5374 CrossRef CAS.
- Y. H. Kang and J. L. Kice, J. Org. Chem., 1984, 49, 1507–1511 CrossRef CAS.
- R. A. Gancarz and J. L. Kice, J. Org. Chem., 1981, 46, 4899–4906 CrossRef CAS.
- T. Billard and B. R. Langlois, Tetrahedron, 1999, 55, 8065–8074 CrossRef CAS.
- C. Ghiazza, L. Khrouz, C. Monnereau, T. Billard and A. Tlili, ChemComm, 2018, 54, 9909–9912 RSC.
- S. Luo, N. Zhang, Z. Wang and H. Yan, Org. Biomol. Chem., 2018, 16, 2893–2901 RSC.
- Z. Chen, F. Hu, S. Huang, Z. Zhao, H. Mao and W. Qin, J. Org. Chem., 2019, 84, 8100–8111 CrossRef CAS PubMed.
- K. Sun, K. Lv, Z. Shi, F. Fu, C. Zhang and Z. Zhang, Sci. China: Chem., 2017, 60, 730–733 CrossRef CAS.
- T. G. Back and S. Collins, Tetrahedron Lett., 1981, 22, 5111–5114 CrossRef CAS.
- T. G. Back, S. Collins and R. G. Kerr, J. Org. Chem., 1983, 48, 3077–3084 CrossRef CAS.
- H. Qian and X. Huang, Tetrahedron Lett., 2002, 43, 1059–1061 CrossRef CAS.
- S. Huang, Z. Chen, H. Mao, F. Hu, D. Li, Y. Tan, F. Yang and W. Qin, Org. Biomol. Chem., 2019, 17, 1121–1129 RSC.
- R. Zhang, P. Xu, S. Y. Wang and S. J. Ji, J. Org. Chem., 2019, 84, 12324–12333 CrossRef CAS PubMed.
- X. Huang, Q. Xu, C. G. Liang and Q. W. He, Synth. Commun., 2002, 32, 1243–1249 CrossRef CAS.
- K. Sun, X. Wang, F. Fu, C. Zhang, Y. Chen and L. Liu, Green Chem., 2017, 19, 1490–1493 RSC.
- Y. Liu, G. Zheng, Q. Zhang, Y. Li and Q. Zhang, J. Org. Chem., 2017, 82, 2269–2275 CrossRef CAS PubMed.
- X. Kong, K. Yu, Q. Chen and B. Xu, Asian J. Org. Chem., 2020, 9, 1760–1764 CrossRef CAS.
- K. Sun, Z. Shi, Z. Liu, B. Luan, J. Zhu and Y. Xue, Org. Lett., 2018, 20, 6687–6690 CrossRef CAS PubMed.
- F. S. He, P. Bao, F. Yu, L. H. Zeng, W. P. Deng and J. Wu, Org. Lett., 2021, 23, 7472–7476 CrossRef CAS PubMed.
- F. S. He, Y. Yao, Z. Tang, W. Xie and J. Wu, Org. Chem. Front., 2021, 8, 6119–6124 RSC.
- K. H. Kang and J. L. Kice, Tetrahedron Lett., 1982, 23, 5373–5374 CrossRef.
- J. L. Kice and Y. H. Kang, Tetrahedron, 1985, 41, 4739–4746 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence h
h