
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed anti-Markovnikov hydroarylation of alkenes†
Julia
Nguyen
,
Andrea
Chong
and
Gojko
Lalic
 *
*
Department of Chemistry, University of Washington, Seattle, Washington 98195, USA. E-mail: lalic@chem.washington.edu
First published on 13th February 2019
Abstract
We have developed a nickel-catalyzed hydroarylation of alkenes using aryl halides as coupling partners. Excellent anti-Markovnikov selectivity is achieved with aryl-substituted alkenes and enol ethers. We also show that hydroarylation occurs with alkyl substituted alkenes to yield linear products. Preliminary examination of the reaction mechanism suggests irreversible hydrometallation as the selectivity determining step of the hydroarylation.
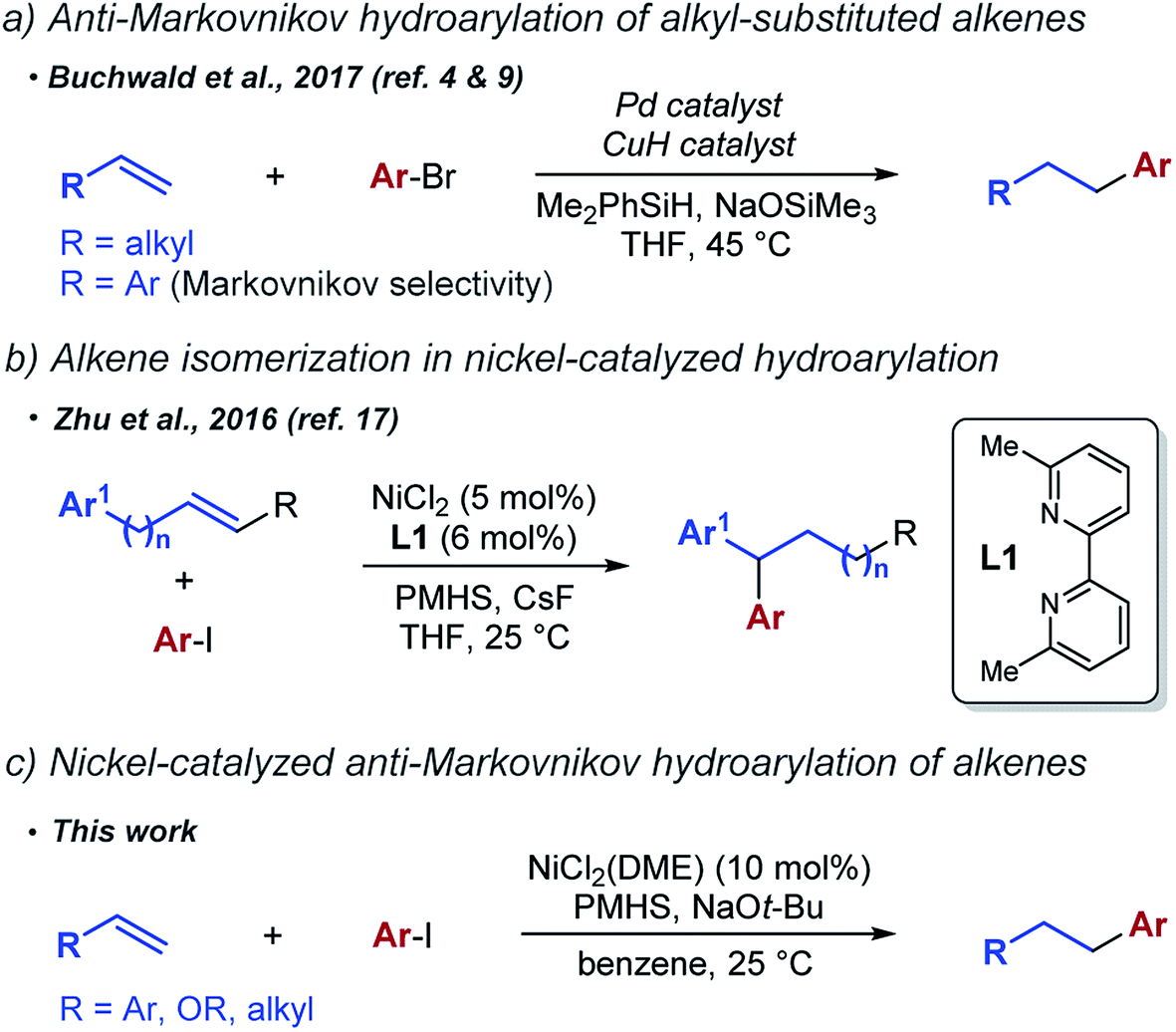
Catalytic hydroarylation of alkenes has been targeted as an attractive synthetic route to alkylarenes, a common structural motif in organic chemistry. Traditionally, hydroarylation is accomplished either via classical Friedel–Crafts reaction1 or through methods based on metal catalyzed C–H activation of arenes.2 More recently, a third complementary strategy has been developed based on the reductive coupling of aryl halides and alkenes in the presence of a hydride donor. The key advance offered by reductive cross coupling relative to Friedel–Crafts and C–H activation methods is excellent site selectivity of the arene alkylation regardless of the substitution pattern of the starting arene.
The reductive cross coupling approach has been effectively used in several methods for the Markovnikov hydroarylation of alkenes. In 2016, Nakao et al. reported a Markovnikov hydroarylation of aryl alkenes using a dual palladium/copper catalyst system.3 The same year, Buchwald et al. reported an enantioselective version of the reaction using a similar approach.4 Markovnikov hydroarylation of alkenes has also been achieved by Herzon et al.5,6 using metal hydride hydrogen atom transfer (HAT) to initiate radical alkylation of arenes. Concurrently, Shenvi et al.7,8 developed a distinct approach based on metal hydride HAT coupled with nickel-catalyzed cross-coupling.
Anti-Markovnikov selectivity in the reductive cross coupling of aryl halides and alkenes is relatively rare. At the time we were developing this project, it had been achieved only in reactions of simple terminal alkenes (Scheme 1a). Buchwald et al. have shown that a dual palladium/copper catalyst system promotes coupling of unactivated alkenes with aryl halides to generate linear hydroarylation products.9,10 Notably, the same catalyst system was previously shown to give Markovnikov selectivity in the hydroarylation of aryl alkenes.4 Recently, an alternative approach based on reductive Heck reaction has been successfully used by Engle et al.11 and later Hu et al.12 to access the same products. Relative to the excellent scope of the Engle's reaction of terminal alkyl alkenes, the scope of the Hu's reaction with aryl alkenes was limited.
 | ||
| Scheme 1 Reductive cross coupling of alkenes with aryl halides. | ||
We were interested in developing a hydroarylation reaction that would complement existing methods and allow anti-Markovnikov hydroarylation of aryl alkenes and possibly other new substrate classes. Furthermore, we were hoping to achieve these transformations using a single metal complex as a catalyst. This catalyst would have to promote both the hydrometallation of alkenes and the subsequent cross coupling with aryl halides.
We focused on nickel complexes as particularly well suited for the task. Insertion of alkenes into nickel hydride complexes is well documented13,14 and alkyl nickel complexes have been implicated as intermediates in cross-coupling reactions with organohalides.15 Following an important precedent established by Liu et al.,16 the general feasibility of nickel-catalyzed hydroarylation was recently demonstrated by a reductive arylation reaction reported by Zhu et al. (Scheme 1b).17,18 This transformation involved extensive isomerization of the alkene and the exclusive formation of 1,1-diaryl products regardless of the original position of the alkene relative to the arene.19
In this work, we report the anti-Markovnikov hydroarylation of alkenes using a simple nickel catalyst. We are able to achieve this linear selectivity with a number of different substrate classes, including aryl alkenes, enol ethers and simple unactivated alkenes (Scheme 1c).
While exploring the reactivity of various nickel complexes in the catalytic hydroarylation of aryl alkenes, we discovered that NiCl2(DME) provides the hydroarylation product with excellent anti-Markovnikov selectivity (Table 1, entry 1). Essential for the anti-Markovnikov selectivity and the overall success of the reaction was the absence of a ligand. A variety of ligands, including L1 used by Zhu et al., resulted in no formation of the desired product (entries 2 and 3) (see ESI for additional data†). For those ligands that did generate product, such as N-heterocyclic carbine ligand IPr, regioselectivity and yield were low (entry 4). Using different nickel sources, such as NiCl2 (entry 5), resulted in lower yields. Interestingly, Ni(COD)2, a Ni(0) source, proved to be competent in this reaction (entry 6). Aromatic solvents, such as benzene and toluene (entry 7), were essential for high yield of the hydroarylation product, while most other solvents were completely ineffective (see ESI for details†). Interestingly, ethereal solvents, such as 1,4-dioxane (entry 8) and THF (entry 9), provided significantly lower regioselectivity.
|
|
|||
|---|---|---|---|
| Entry | Variations from above | Yieldb (%) | L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Bc :Bc |
|
a Conditions: 1 (2 equiv.), 2 (1 equiv.), NaOt-Bu (4 equiv.), PMHS (4 equiv.).
b Yields were determined by GC analysis of the crude reaction mixture using 1,3,5-trimethylbenzene as the internal standard. Number in parentheses are yields of isolated products.
c L:B is the ratio of the linear product to the branched product as determined by GC analysis of the crude reaction mixture.
|
|||
| 1 | None | 89 (81) | 55:1 |
| 2 | L1 as a ligand | 0 | — |
| 3 | L2 as a ligand | 0 | — |
| 4 | IPr as a ligand | 24 | 1.3:1 |
| 5 | NiCl2 instead of NiCl2(dme) | 44 | 15:1 |
| 6 | Ni(COD)2 instead of NiCl2(dme) | 39 | 12:1 |
| 7 | Toluene instead of benzene | 80 | 48:1 |
| 8 | 1,4-Dioxane | 35 | 3:1 |
| 9 | THF instead of benzene | 8 | 2.5:1 |
| 10 | (Me2HSi)2O instead of PMHS | 57 | 29:1 |
| 11 | LiOt-Bu instead of NaOt-Bu | 30 | 7:1 |
| 12 | KOt-Bu instead of NaOt-Bu | 0 | — |

|
|||

Polymethylhydrosiloxane (PMHS) was the best hydride source. Other silanes, including closely related (Me2HSi)2O (entry 10), provided lower yields and worse regioselectivity (see ESI† for details). Similarly, NaOt-Bu was essential for the reaction and even the closely related Li and K analogs (entries 11 and 12) provided significantly inferior results. Exclusion of any component of this reaction resulted in no formation of product 3.
Hydroarylation could be achieved with a range of aryl- and alkyl-substituted alkenes (Table 2). With aryl alkenes, ortho, meta and para substitution is tolerated (compounds 7, 8, 4). Aryl alkenes containing both electron-donating and electron-withdrawing substituents are viable substrates (5 and 6). Good selectivities and yields were also obtained in hydroarylation of aryl alkenes containing a wide range of heteroarenes, including benzofuran (13), indole (14, 15), pyrrole (16), benzopyrazole (17), and pyrrolopyridine (18). Finally, substitution on the α position of the alkene is also tolerated, as indicated by the formation of product 19 in the reaction with α-methylstyrene.
| a Conditions: aryl halide (1 equiv.), alkene (2 equiv.), PMHS (4 equiv.), NaOt-Bu (4 equiv.). Yields of isolated products are reported. The ratios of linear to branched products were determined by GC analysis of the crude reaction mixture and are given in parenthesis. |
|---|

|
Nickel-catalyzed hydroarylation of various other classes of alkenes was also examined. Unactivated terminal alkenes were suitable coupling partners (20–23, 27–29), although the regioselectivity was somewhat lower. Surprisingly, a 1,3-diene substrate gave product 24 with high yield and good regioselectivity. Another unusual class of alkenes that were compatible with our reaction conditions are enol ethers. We were able to isolate the product of the reaction with ethyl vinyl ether (25) in good yield (78%) while the benzyl protected enol ether provided the expected product 26 in 63% yield. To our knowledge, these are the only examples of anti-Markovnikov reductive cross coupling reactions with 1,3-dienes or enol ethers.
We have also explored the performance of various arene coupling partners, and both aryl iodides and aryl bromides are viable substrates (Table 2). The electronic properties of aryl electrophiles seem to play a significant role as electron-neutral and electron-rich aryl halides generally gave high yields (30, 37, 46), while electron-poor aryl halides, though still competent, provided lower yields of the hydroarylation products (31 and 32). Again, we found that various heteroarenes are tolerated, including pyrrolopyridine (41), quinoline (42, 43), indole (44), benzofuran (45), pyrimidine (47), and benzopyrazole (48).
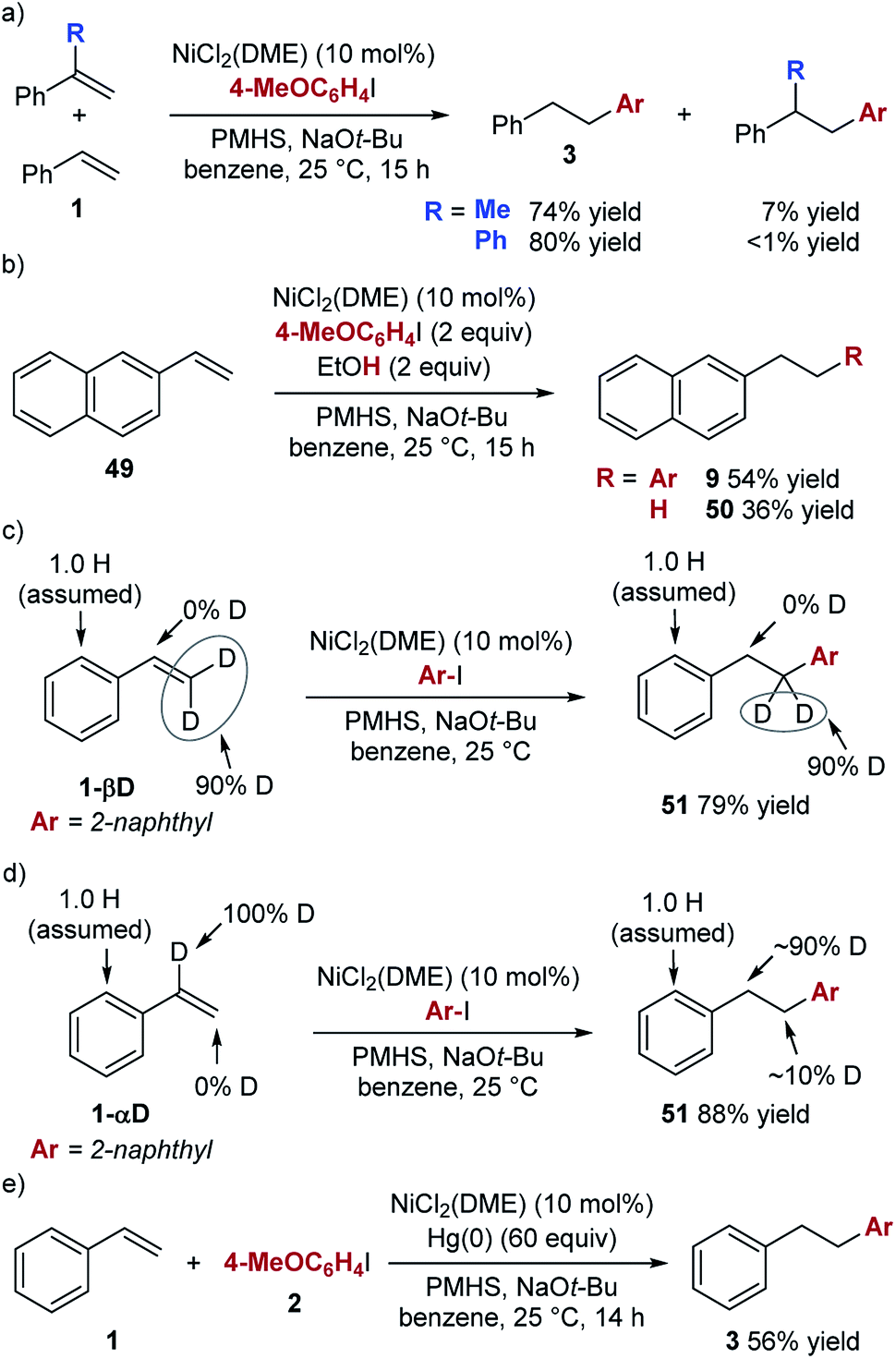
The unique aspect of this new hydroarylation reaction is the anti-Markovnikov selectivity obtained with a range of alkenes, including aryl alkenes and enol ethers. The observed selectivity is consistent with an addition of an aryl radical to the alkene, which would favour addition to the alkene terminus and formation of the more stable secondary alkyl radical. To test this mechanistic hypothesis, we performed experiments in the presence of a radical trap. The presence of 10 mol% of TEMPO did lower the yield of the desired product and a full equivalent completely blocked its formation (see ESI† for details). However, in all cases, we observed only trace amounts (<5% yield) of the aryl–TEMPO adduct, suggesting that TEMPO likely interacts with another intermediate20 and not with an aryl radical.21 In competition experiments (Scheme 2a), we found that simple styrene reacts faster than α-methylstyrene or α-phenylstyrene, both of which should undergo radical addition faster than styrene. The results of these experiments are not consistent with a radical addition mechanism, leading us to explore other options.
 | ||
| Scheme 2 Probing the reaction mechanism. | ||
A mechanism involving hydrometallation followed by arylation of the alkyl nickel intermediate was proposed by Zhu et al. for the related hydroarylation reaction (see Scheme 1b). We used the experiment shown in Scheme 2b to explore the intermediacy of the alkyl nickel complex in our hydroarylation reaction.
In the presence of 2 equiv. of EtOH, the hydroarylation product is formed in a lower yield than in the absence of EtOH (54% vs. 82%), while the alkane product (50) is formed in 36% yield. This result is consistent with competitive protonation of the alkyl nickel intermediate by EtOH. Based on these considerations, we propose that the hydroarylation reaction proceeds according to the mechanism shown in Scheme 3.
 | ||
| Scheme 3 Proposed mechanism of hydroarylation. | ||
In the context of this mechanism, we were interested in the step that determines the regioselectivity of the reaction. In hydrofunctionalization reactions that proceed through hydrometallation, the product determining step varies depending on the reversibility of the hydrometallation step. When irreversible, hydrometallation is the regio-determining step. If hydrometallation is fast and reversible, there is a Curtin–Hammett scenario, and regioselectivity is determined by the relative rates of the irreversible reductive elimination at different sites and not by hydrometallation. Alkene insertion into nickel hydride complexes is often fast and reversible,22 as documented by Zhu et al. in their reductive arylation of alkenes.17 Because the C–C bond-forming reductive elimination from nickel complexes tends to be exothermic,23 the rates of reductive elimination are often based on the ground state stability of the alkyl metal intermediates. The result is that the reductive elimination from the most stable alkyl metal complex is favoured.
To explore the reversibility of the hydrometallation step in the hydroarylation reaction, we used deuterium-labelled styrene substrates (Scheme 2c and d). In the reaction with substrate 1-βD we observed no deuterium scrambling, while the reaction with 1-αD, resulted in minimal scrambling of the deuterium label.24 In the context of the reaction mechanism shown in Scheme 3, the two deuterium scrambling experiments point to hydrometallation being irreversible and product determining step of the reaction. These results indicate that a strong kinetic preference for the formation of a homobenzylic alkyl nickel intermediate over the benzylic regioisomer is responsible for the regioselectivity of the hydroarylation.
The unusual selectivity in the hydrometallation of styrenes raises questions about the nature of the active catalyst. The absence of ligand in our hydroarylation reaction makes the formation of nanoparticles likely, and we observe an immediate formation of particles upon the addition of the alkoxide to the reaction mixture. Mixtures of alkoxides and reducing reagents have previously been used to generate highly reactive nickel nanoparticle catalysts, and sodium alkoxide has been proposed to stabilize “ligandless” nickel catalysts.25,26 Furthermore, in the hydroarylation reaction, we observed a burst of initial activity (64% yield after 10 minutes), followed by a very slow reaction over the next 14 h when the maximum yield is achieved. Finally, we found that the addition of mercury significantly lowers the yield of the hydroarylation (56% vs. 89% GC yields) (Scheme 2e).27 These observations are consistent with a heterogeneous active catalyst.
Conclusions
We have developed a new method for the anti-Markovnikov hydroarylation of alkenes. The reaction involves reductive cross coupling of alkenes and aryl halides in the presence of a silane as a hydride donor. Anti-Markovnikov selectivity is obtained with a wide range of alkenes, including alkyl- and aryl-substituted alkenes, and enol ethers. Preliminary investigation of the reaction mechanism provides evidence for a mechanism that involves hydrometallation of the alkene, followed by coupling of the alkyl nickel intermediate with an aryl halide. The evidence points to highly selective hydrometallation of the alkene as the regioselectivity determining step of the reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by NSF (NSF CAREER Award 1254636) and NIH (1R01GM125791-01A1) is acknowledged.Notes and references
-
G. A. Olah, R. Krishnamurti and G. K. Surya Prakash, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamom Press, Oxford, 1991, vol. 3, pp. 293–339 Search PubMed
.
- For a comprehensive review of this large research area, see: Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed
- K. Semba, K. Ariyama, H. Zheng, R. Kameyama, S. Sakaki and Y. Nakao, Angew. Chem., Int. Ed., 2016, 55, 6275–6279 CrossRef CAS PubMed
- S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372 CrossRef CAS PubMed
- X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 8718 CrossRef CAS PubMed
- X. Ma, H. Dang, J. A. Rose, P. Rablen and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 5998 CrossRef CAS PubMed
- S. A. Green, J. L. M. Matos, A. Yagi and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 12779 CrossRef CAS PubMed
- S. L. Shevick, C. Obradors and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 12056–12068 CrossRef CAS PubMed
- S. D. Friis, M. T. Pirnot, L. N. Dupuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 7242 CrossRef CAS
- For a recent example of anti-Markovnikov hydropyridylation of alkenes using photocatalysis, see: A. J. Boyington, M. L. Y. Riu and N. T. Jui, J. Am. Chem. Soc., 2017, 139, 6582 CrossRef CAS
- J. A. Gurak Jr and K. M. Engle, ACS Catal., 2018, 8, 8987–8992 CrossRef PubMed
- L. Jin, J. Qian, N. Sun, B. Hu, Z. Shen and X. Hu, Chem. Commun., 2018, 54, 5752 RSC
- N. A. Eberhardt and H. Guan, Chem. Rev., 2016, 116, 8373 CrossRef CAS
- R. Cariou, T. W. Graham and D. W. Stephan, Dalton Trans., 2013, 42, 4237 RSC
- T. J. Anderson, G. D. Jones and D. A. Vicic, J. Am. Chem. Soc., 2004, 126, 8100 CrossRef CAS
- Liu et al. have reported two examples of nickel-catalyzed hydroarylation reaction: X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef
- F. Chen, K. Chen, Y. Zhang, Y. He, Y. M. Wang and S. Zhu, J. Am. Chem. Soc., 2017, 139, 13929 CrossRef CAS
- For examples of related reductive cross-coupling reactions, see: P. Shukla, A. Sharma, B. Pallavi and C. H. Cheng, Tetrahedron, 2015, 71, 2260 CrossRef CAS
- Interestingly, Zhao et al. have also reported Markovnikov selectivity in a redox neutral nickel-catalysed hydroarylation of styrenes, see: L. J. Xiao, L. Cheng, W. M. Feng, M. L. Li, J. H. Xie and Q. L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461 CrossRef CAS PubMed
- TEMPO has been shown to react rapidly with metal hydrides. See: A. C. Albéniz, P. Espinet, R. López-Fernández and A. J. Sen, J. Am. Chem. Soc., 2002, 124, 11278 CrossRef CAS
- We cannot exclude the possibility that the Ar–TEMPO adduct reacts further under the reaction conditions.
- C. A. Tolman, J. Am. Chem. Soc., 1972, 94, 2994 CrossRef CAS
- V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS
- The deuterium scrambling is also observed in the recovered 1-αD and is higher at higher conversions. Furthermore, if the reaction with 1-αD is stopped after 10 minutes, product 51 is isolated in 49% yield and no deuterium scrambling is observed in either the product or the recovered starting material (see page S31 of the ESI†). These results suggest that the scrambling may be independent of the hydroarylation reaction.
- I. Buslov, F. Song and X. Hu, Angew. Chem., Int. Ed., 2016, 55, 12295 CrossRef CAS PubMed
- J. J. Brunet, D. Besozzi, A. Courtois and P. Caubere, J. Am. Chem. Soc., 1982, 104, 7130 CrossRef CAS
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05445b |
| This journal is © The Royal Society of Chemistry 2019 |