
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 18F-difluoromethylarenes using aryl boronic acids, ethyl bromofluoroacetate and [18F]fluoride†
Jeroen B. I.
Sap
a,
Thomas C.
Wilson
a,
Choon Wee
Kee
 a,
Natan J. W.
Straathof
a,
Christopher W. am
Ende
b,
Paramita
Mukherjee
b,
Lei
Zhang
b,
Christophe
Genicot
c and
Véronique
Gouverneur
*a
a,
Natan J. W.
Straathof
a,
Christopher W. am
Ende
b,
Paramita
Mukherjee
b,
Lei
Zhang
b,
Christophe
Genicot
c and
Véronique
Gouverneur
*a
aChemistry Research Laboratory, Department of Chemistry, Oxford University, OX1 3TA Oxford, UK. E-mail: veronique.gouverneur@chem.ox.ac.uk; Tel: +44 (0)1865 285002
bPfizer Inc., Medicine Design, Eastern Point Road, Groton, Connecticut 06340, and 1 Portland Street, Cambridge, Massachusetts 02139, USA
cGlobal Chemistry, UCB New Medicines, UCB Biopharma Sprl, 1420 Braine-L'Alleud, Belgium
First published on 29th January 2019
Abstract
Herein, we report the radiosynthesis of 18F-difluoromethylarenes via the assembly of three components, a boron reagent, ethyl bromofluoroacetate, and cyclotron-produced non-carrier added [18F]fluoride. The two key steps are a copper-catalysed cross-coupling reaction, and a Mn-mediated 18F-fluorodecarboxylation.
Introduction
Positron emission tomography (PET) is a molecular imaging technique that requires molecules labelled with a positron-emitting radionuclide. Fluorine-18 is a widely used positron emitting radionuclide in part due to its favourable decay properties, and the numerous clinical applications of 2-deoxy-2-[18F]fluoro-D-glucose, a radiopharmaceutical prepared from [18F]fluoride.1 While radiochemists have in recent years focused their efforts on methods enabling 18F-fluorination2 and 18F-trifluoromethylation of (hetero)arenes,2,318F-difluoromethylation reactions have been less studied despite the importance of the CF2H motif4 in radioligand design for drug discovery programmes. In 2013, we reported a Ag(I)-mediated 18F-fluorodecarboxylation of 2-fluoro-2-arylacetic acids with [18F]Selectfluor (bis)triflate leading to [18F]ArCF2H.5 Subsequently, we disclosed a Ag(I)-mediated halogen exchange reaction using [18F]fluoride.6 In 2016, a multi-step method to label [18F]ArCF2H from aryl (pseudo)halides was disclosed by Ritter and co-workers.7 Later, Liang and co-workers demonstrated that halogen exchange of benzyl (pseudo)halides with [18F]fluoride followed by oxidative benzylic C–H fluorination with Selectfluor afforded [18F]ArCF2H with improved molar activity.8 Despite these advances, 18F-difluoromethylation remains a challenging problem, especially for structurally complex targets. We initially considered adapting difluoromethylation reactions operating via C–H functionalisation.9 Whilst this strategy is ideal for (hetero)arenes with innate reactivity leading to site-selective 18F-difluoro-methylation, substrates that are not reactive or too reactive would be unsuitable, thereby limiting applicability for radioligand synthesis. We therefore opted to develop a method using pre-functionalised aryl boron reagents; these are amenable to 18F-fluorination and 18F-trifluoromethylation,10 so extension to 18F-difluoromethylation was viewed as a valuable development. Building on our Ag(I)-mediated 18F-fluorodecarboxylation towards [18F]ArCF2H,5 a reaction requiring [18F]Selectfluor (bis)triflate (Scheme 1A),11 and on the Mn-mediated fluorodecarboxylation reported by Groves and co-workers, a reaction using [18F]fluoride (Scheme 1B),12,13 we envisaged that the 18F-fluorodecarboxylation of 2-fluoro-2-arylacetic acids with [18F]fluoride could afford [18F]ArCF2H. The beneficial effect of fluorine substitution on radical stabilisation would be favorable for this process.5,14 This approach would require a robust method to cross-couple the aryl boron reagent with ethyl bromofluoroacetate followed by hydrolysis to access the carboxylic acid precursor; we gave preference to a coupling methodology applying Cu-catalysis instead of Pd or Ni, a decision driven by guidelines for residual metals in (radio)pharmaceuticals.15 The proposed strategy therefore relies on three readily available components, the boron reagent, ethyl bromofluoroacetate, and [18F]fluoride (Scheme 1C).16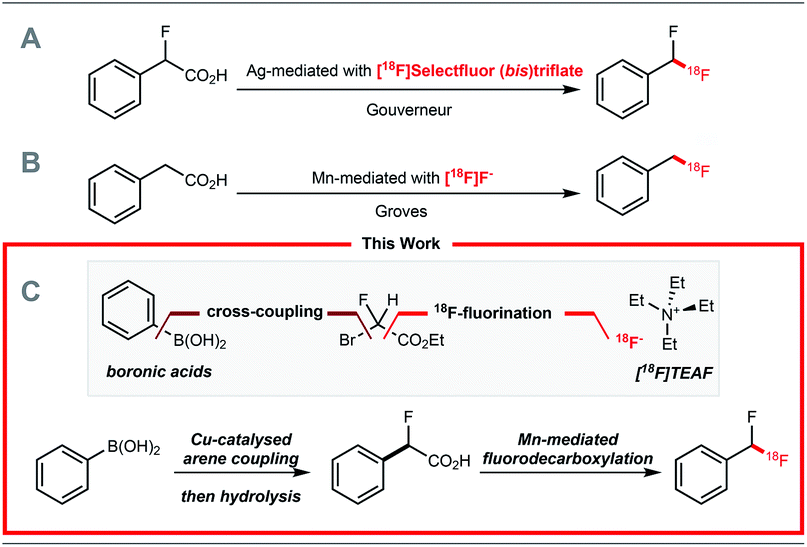 | ||
| Scheme 1 (A) Ag(I)-mediated 18F-fluorodecarboxylation with [18F]Selectfluor (bis)triflate. (B) Mn(III)-mediated 18F-fluorodecarboxylation with [18F]fluoride towards [18F]ArCH2F. (C) Synthetic plan towards [18F]ArCF2H from boron reagents and [18F]fluoride. | ||
Results and discussion
Preliminary experiments demonstrated that the model fluoro-substituted carboxylic acid 1a is amenable to fluorodecarboxylation with fluoride. When an equimolar mixture of 1a and 2a was treated with Mn(tmp)Cl (2.5 mol%), Et3N·3HF (1.2 equiv.) and PhIO (3.3 equiv.) in MeCN at 50 °C, 3a and 4a were obtained in 44% and 20% yield, respectively. This result indicates that the fluorine-substituted precursor 1a is more reactive than non-fluorinated 2a towards fluorodecarboxylation (Scheme 2A). We verified that product 4a did not undergo fluorination via C–H functionalisation under these conditions.17 When an excess of 1a (1 equiv.) was treated with TBAF (0.1 equiv.), PhIO (0.5 equiv.) and Mn(tmp)Cl (0.2 equiv.) in MeCN, 3a was obtained in 50% yield (determined by 19F NMR based on TBAF consumption) (Scheme 2B). Notably, quantitative fluoride incorporation was observed applying similar reaction conditions to the preformed hypervalent iodine complex 5a (Scheme 2C). These preliminary data boded well for 18F-labeling with [18F]fluoride as the limiting reagent, and prompted the development of a robust protocol to convert aryl boron reagents into 2-fluoro-2-arylacetic acids. | ||
| Scheme 2 (A) Competition studies evaluating the effect of fluorine substitution on fluorodecarboxylation. (B) Reaction with sub-stoichiometric fluoride. (C) Reaction of iodine(III) complex 5a with sub-stoichiometric fluoride. Yields of isolated products. Mn(tmp)Cl = Mn(III) meso-tetra(2,4,6-trimethylphenyl)porphyrin chloride. aYield determined by 19F NMR using α,α,α-trifluorotoluene as internal standard. | ||
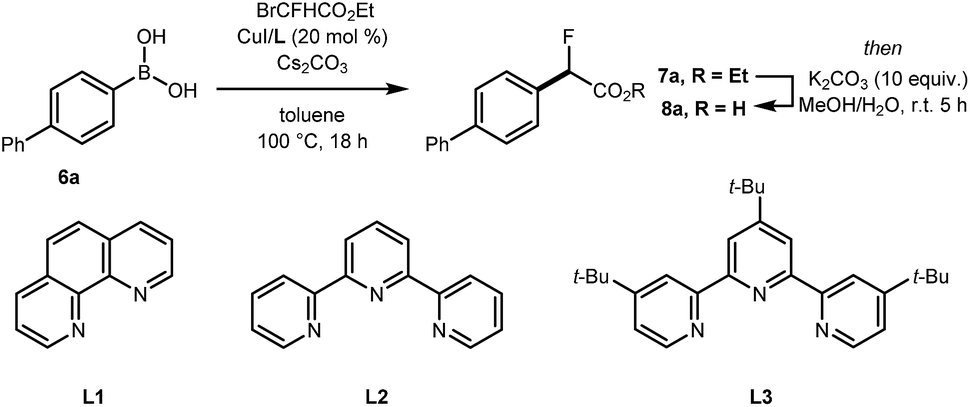
The cross-coupling of arylboronic acids and ethyl bromofluoroacetate has been reported using an excess of boron reagent under Ni or Pd catalysis, but has not been accomplished under Cu catalysis.18–22 Initial studies reacting [1,1′-biphenyl]-4-ylboronic acid 6a (2 equiv.) with ethyl bromofluoroacetate (1 equiv.) in the presence of 1,10-phenanthroline (L1, 20 mol%), CuI (20 mol%) and Cs2CO3 (2 equiv.) in dioxane (0.2 M) under N2 at 100 °C afforded 7a in 7% yield (Table 1, entry 1). When 2,2′:6′,2′′-terpyridine (L2) was used as the ligand, the yield was significantly improved to 58% yield (Table 1, entry 2). When the stoichiometry was altered to 1 equivalent of 6a and 2 equivalents of ethyl bromofluoroacetate in the presence of 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine (L3) in toluene instead of dioxane 7a was obtained in 63% yield (Table 1, entry 3). Further optimisation increasing the concentration led to the optimal protocol consisting of treating 6a (0.1 mmol) with ethyl bromofluoroacetate (0.2 mmol), Cs2CO3 (0.2 mmol), CuI (20 mol%) and L3 (20 mol%) in toluene (0.4 M) at 100 °C. Under these reaction conditions, 7a was isolated in 82% yield (Table 1, entry 4). A one-pot sequence involving cross-coupling followed by hydrolysis with MeOH and aqueous K2CO3 afforded 8a isolated in 75% yield (Table 1, entry 5). In the absence of ligand and/or copper source (Table 1, entries 6, 7), no product formation was observed. Furthermore, no reaction was observed with CuCl2 (Table 1, entry 8), or when the reaction solvent was DMF or DMSO (Table 1, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Cu-source | Ligand | Product | Yieldb |
| a Screening reactions performed on 0.1 mmol scale. b Yield determined by 19F-NMR using α,α,α-trifluorotoluene as internal standard. c 2 equiv. of 6a and 1 equiv. of ethyl bromofluoroacetate. d 1 equiv. of 6a, and 2 equiv. of ethyl bromofluoroacetate. e Yield of isolated product. f One-pot procedure towards 8a. | |||||
| 1c | Dioxane (0.2 M) | CuI | L1 | 7a | 7% |
| 2c | Dioxane (0.2 M) | CuI | L2 | 7a | 58% |
| 3 | Toluene (0.2 M) | CuI | L3 | 7a | 63% |
| 4d | Toluene (0.4 M) | CuI | L3 | 7a | 82%e |
| 5d | Toluene (0.4 M) | CuI | L3 | 8a | 75%e,f |
| 6d | Toluene (0.4 M) | CuI | — | 7a | 0% |
| 7d | Toluene (0.4 M) | — | — | 7a | 0% |
| 8d | Toluene (0.4 M) | CuCl2 | L2 | 7a | 0% |
| 9d | DMF or DMSO (0.2 M) | CuI | L3 | 7a | 0% |
These optimised conditions gave access to a range of 2-fluoro-2-arylacetic acids (Scheme 3). The reaction is broad in scope and tolerates various functional groups, for example alkyl 8c–8e and 8s–8u, alkoxy 8f, 8g, trifluoromethyl 8h, bromo 8p, 8q, iodo 8r, and aldehyde 8i all performed well. Substrates featuring heterocycles such as dibenzofuran 8j, pyridine 8k, triazole 8l, and pyrazoles 8m, 8n are also suitable coupling partners applying our optimised protocol affording the desired products in 40% to 70% yield. Additionally, this cross-coupling chemistry afforded 8o, a derivative of fenofibrate, in 72% yield. Finally, the reaction was amenable to scale-up to 5 mmol (Scheme 3, 8m).
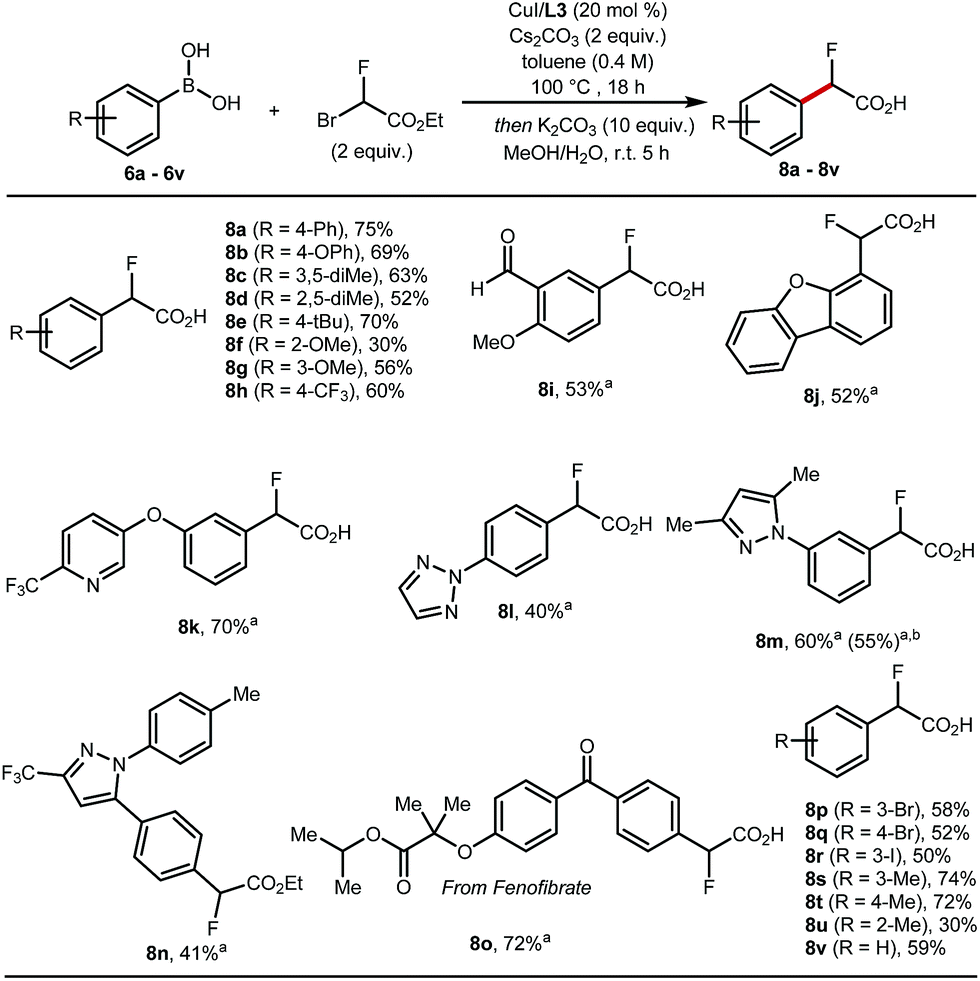 | ||
Scheme 3 Scope of Cu-catalysed cross-coupling. The reactions were performed on a 0.3 mmol scale. Conditions: CuI (20 mol%), L3 (20 mol%), aryl boronic acid (1 equiv.), ethyl bromofluoroacetate (2 equiv.), Cs2CO3 (2 equiv.), toluene (0.4 M) at 100 °C for 18 h then one-pot hydrolysis with K2CO3 (10 equiv.), MeOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 5 h. aHydrolysis performed as a subsequent step with K2CO3 (5 equiv.). bReaction run on 5 mmol scale. All yields are of isolated products. :1), 5 h. aHydrolysis performed as a subsequent step with K2CO3 (5 equiv.). bReaction run on 5 mmol scale. All yields are of isolated products. | ||
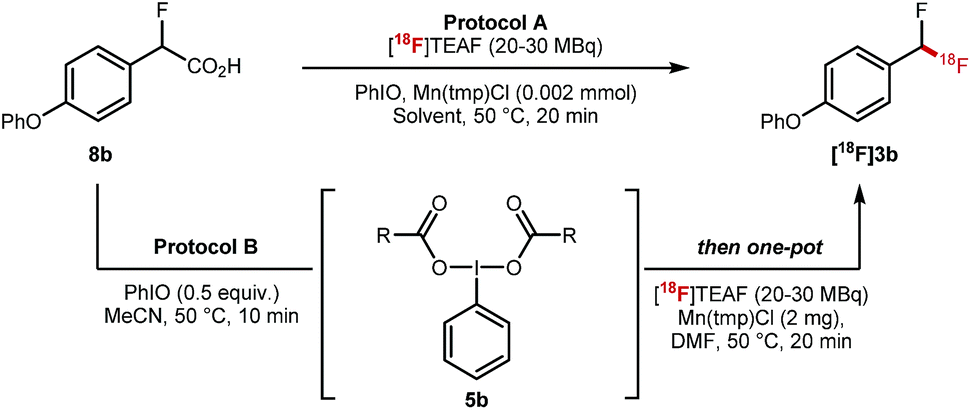
The key 18F-fluorodecarboxylation step was studied next (Table 2). We started our investigation applying protocol A that consists of reacting in one-pot 8b (0.11 mmol) with PhIO (0.33 mmol), Mn(tmp)Cl (2 mg) and [18F]TEAF (20–30 MBq) in MeCN (600 μL) at 50 °C; this protocol led to only traces of [18F]3b (Table 2, entry 1). When the loading of PhIO (0.02 mmol) and MeCN (300 μL) was reduced, [18F]3b was obtained in 6% ± 1% radiochemical conversion (RCC) (Table 2, entry 2). Similar results were obtained in DMF (Table 2, entry 3). Reducing the stoichiometry of 8b led to a significant increase in RCC (22% ± 7%) (Table 2, entry 4). When applying protocol B which consists of mixing 8b with PhIO, a process generating complex 5b, prior to the addition of Mn(tmp)Cl (2 mg) and [18F]TEAF (20–30 MBq) and DMF (300 μL), a drastic improvement was observed, and [18F]3b was obtained in 40% ± 10% RCC (n = 10) (Table 2, entry 5). When the reaction was run at 100 °C, the formation of [18F]3b was not observed (Table 2, entry 6). No 18F-labelled product was obtained when Mn(tmp)OTs was used as catalyst, or in the absence of Mn(tmp)Cl (Table 2, entries 7 and 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Starting material (mmol) | Protocol | Solvent | PhIO (mmol) | RCCa,b (n = 2) |
| a Radiochemical conversion. b n = number of reactions. c 600 μL of MeCN. d 300 μL of MeCN. e MeCN removed at 100 °C after dispensing [18F]TEAF. f (n = 10). g Reaction temperature = 100 °C. h Catalyst is Mn(tmp)OTs. i No Mn Catalyst. | |||||
| 1 | 8b (0.11) | A | MeCNc | 0.33 | 3% ± 1% |
| 2 | 8b (0.11) | A | MeCNd | 0.02 | 6% ± 1% |
| 3 | 8b (0.11) | A | DMFd | 0.02 | 7% ± 2% |
| 4 | 8b (0.055) | A | DMFd,e | 0.02 | 22% ± 7% |
| 5 | 5b (0.014) | B | DMF , | — | 40% ± 10% |
| 6 | 5b (0.014) | B | DMFd,e | — | 0% ± 0%g |
| 7 | 8b (0.014) | A | MeCNd | 0.02 | 0% ± 0%h |
| 8 | 5b (0.014) | B | DMFd,e | — | 0% ± 0%i |
The fluorine substituent is advantageous for 18F-fluorodecarboxylation as demonstrated with a competition experiment subjecting equimolar amount of pre-formed hypervalent iodine(III) complexes 9a and 5a to 18F-fluorination with [18F]TEAF, Mn(tmp)Cl at 50 °C in DMF. Difluoromethylarene [18F]3a was the only product observed in the crude reaction mixture (Scheme 4A). Furthermore, an additional competition experiment showed that the iodine(III) complex 5a is formed preferentially to 9a (Scheme 4B). Fluorine substitution therefore facilitates the two steps of the process leading to fluorodecarboxylation.
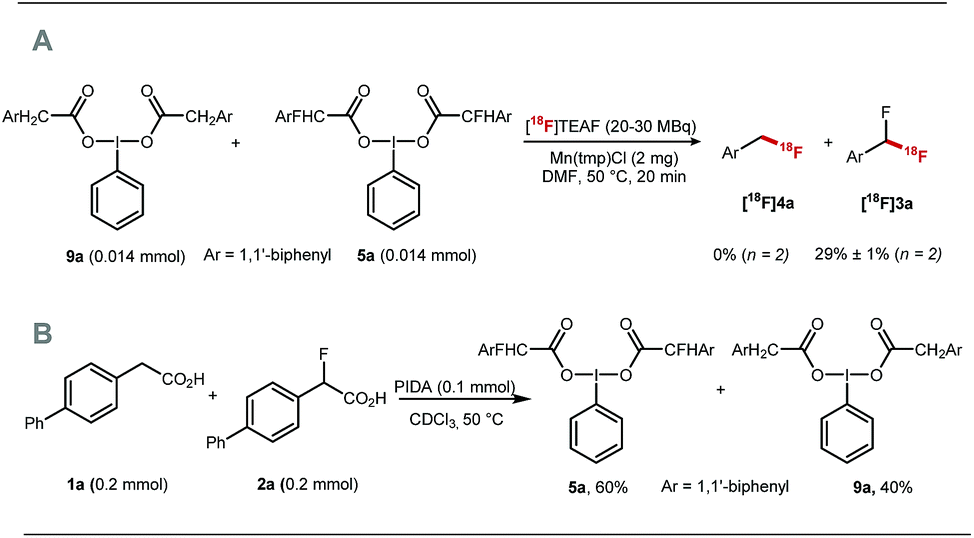 | ||
| Scheme 4 (A) Competition experiment subjecting equimolar amount of 9a and 5a to [18F]fluorodecarboxylation. (B) Competition experiment reacting equimolar amount of 1a and 3a with PIDA. | ||
Protocol B was applied to a selection of arenes using 20–30 MBq of [18F]fluoride (Scheme 5). Ether, alkyl, aldehyde, ketone, pyridine, triazole, pyrazole, dibenzofuran motifs were all tolerated. The highest RCCs were obtained for electron rich arenes. [18F]3o derived from a boronic acid analogue of fenofibrate was successfully labelled in 23% ± 4% (n = 4). The boronic acid derivative of the COX-II inhibitor ZA140 6z was transformed into the labelled difluoromethylated product [18F]3z in 15% ± 2% RCC (n = 3).
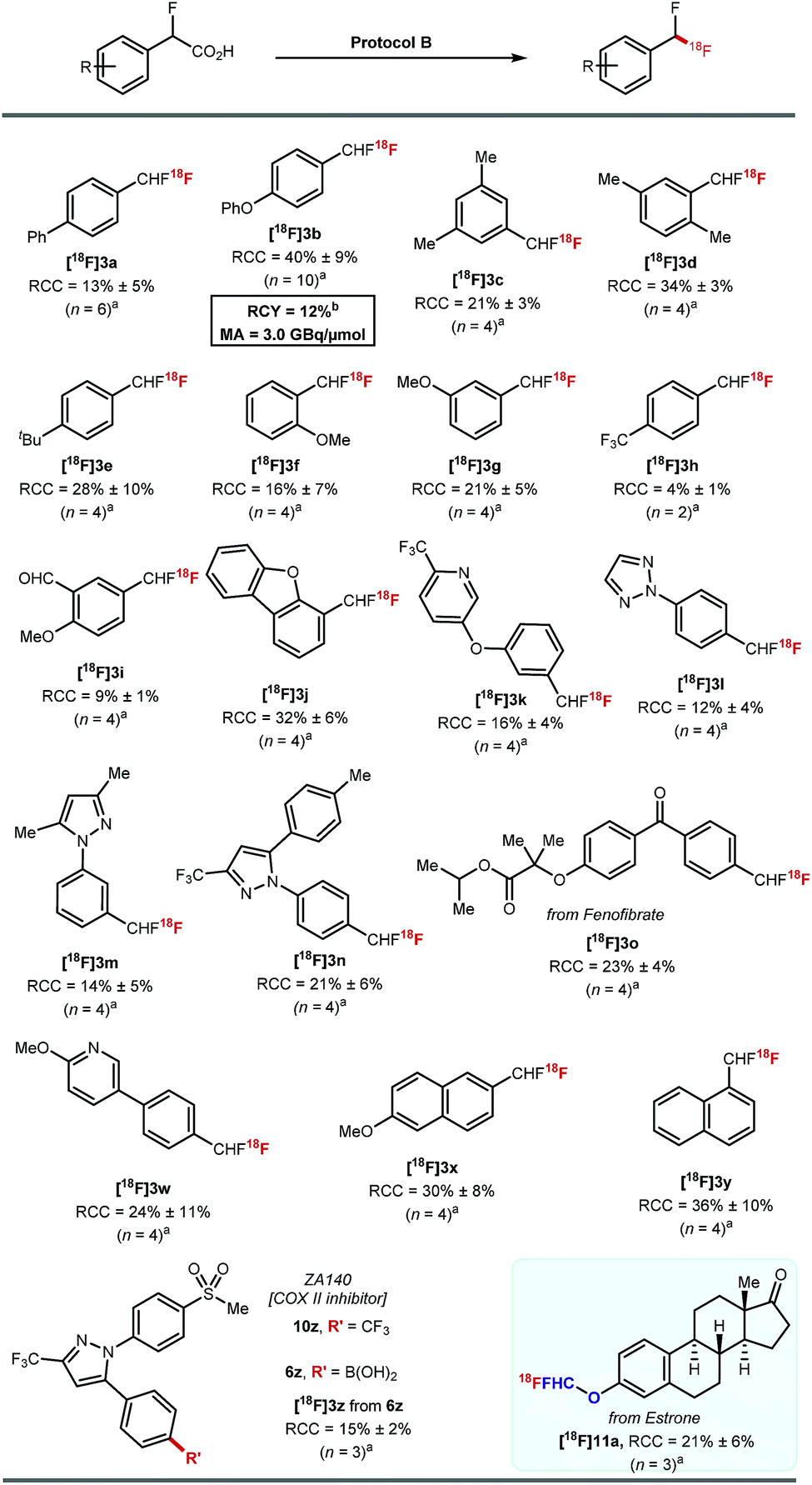 | ||
| Scheme 5 Scope of [18F]fluorodecarboxylation applying protocol B: aArCHFCO2H (0.028 mmol), PhIO (0.5 equiv.), MeCN (1 mL), 50 °C, 10 min then addition of [18F]TEAF (20–30 MBq) Mn(tmp)Cl (2 mg), DMF (300 μL), 50 °C, 20 min. bArCHFCO2H (0.014 mmol), PhIO (0.5 equiv.), MeCN (1 mL), 50 °C, 10 min then addition of [18F]Mn(tmp)F (841 MBq) DCE (300 μL), 60 °C, 20 min. | ||
The 18F-fluorodecarboxylation of 5b performed with 841 MBq of [18F]fluoride required further optimisation. For this experiment, [18F]fluoride was captured on an anion exchange cartridge then eluted using a solution of Mn(tmp)Cl in methanol, resulting in 85% 18F-recovery. Lowering the starting material stoichiometry to 0.007 mmol of 5b and changing the solvent from DMF to DCE afforded the cartridge-purified [18F]3b in a decay corrected RCY of 12% and a molar activity of 3.0 GBq μmol−1 in a total synthesis time of 30 minutes.23
Pleasingly, 18F-fluorodecarboxylation also enabled access to the [18F]ArOCF2H motif. The only known route to label this motif was reported by our group, and required a multi-step synthesis of the ArOCHFCl precursors which were themselves prepared from ArOCHFCO2H.24 The reaction of estrone (1.0 equiv.) with ethyl bromofluoroacetate (1.5 equiv.) and K2CO3 (2.5 equiv.) in DMF (2 mL) at room temperature followed by a subsequent hydrolysis with aqueous NaOH (2.5 equiv.) in 1:1 H2O/Et2O afforded the precursor required for fluorodecarboxylation. 18F-labelling applying protocol B afforded [18F]11a in 21% ± 6% RCC (n = 3).
Conclusions
In summary, a novel method was developed to transform aryl boronic acids to [18F]ArCF2H. Prior to labelling, the cross-coupling with ethyl bromofluoroacetate was accomplished under Cu catalysis followed by in situ hydrolysis. The radioisotope 18F is then introduced in the last step applying a Mn-mediated fluorodecarboxylation with readily available [18F]fluoride. This study has unveiled three key features for this last transformation. Firstly, the fluorine substituent on the carboxylic acid precursor is advantageous for fluorodecarboxylation; secondly, the benefit of preforming the hypervalent iodine complex prior to 18F-fluorination; and thirdly, we have established that Mn-mediated fluorodecarboxylation enables access to [18F]ArOCF2H in addition to [18F]ArCF2H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Pfizer, and the Engineering and Physical Sciences Research Council (EP/N509711/1) (studentship to J. B. I. S.). We also acknowledge the financial support from the Cancer Research UK (C5255/A16466) (T. C. W.), The Agency for Science, Technology and Research (A*STAR, Singapore) (fellowship to C. W. K.), and UCB (N. J. W. S).References
- (a) S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501 CrossRef CAS PubMed; (b) P. M. Matthews, E. A. Rabiner, J. Passchier and R. N. Gunn, Br. J. Clin. Pharmacol., 2012, 73, 175 CrossRef CAS PubMed.
- (a) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS PubMed; (b) Z. Gao, Y. H. Lim, M. Tredwell, L. Li, S. Verhoog, M. Hopkinson, W. Kaluza, T. L. Collier, J. Passchier, M. Huiban and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 6733 CrossRef CAS PubMed; (c) B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 4365 CrossRef CAS PubMed; (d) E. L. Cole, M. N. Stewart, R. Littich, R. Hoareau and P. J. H. Scott, Curr. Top. Med. Chem., 2014, 14, 875 CrossRef CAS; (e) M. Tredwell, S. M. Preshlock, N. J. Taylor, S. Gruber, M. Huiban, J. Passchier, J. Mercier, C. Genicot and V. Gouverneur, Angew. Chem., Int. Ed., 2014, 53, 7751 CrossRef CAS PubMed; (f) A. V. Mossine, A. F. Brooks, K. J. Makaravage, J. M. Miller, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Org. Lett., 2015, 17, 5780 CrossRef CAS PubMed; (g) C. N. Neumann, J. M. Hooker and T. Ritter, Nature, 2016, 534, 369 CrossRef CAS; (h) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS; (i) M. K. Narayanam, G. Ma, P. A. Champagne, K. N. Houk and J. M. Murphy, Angew. Chem., Int. Ed., 2017, 56, 13006 CrossRef CAS PubMed; (j) X. Deng, J. Rong, L. Wang, N. Vasdev, L. Zhang, L. Josephson and S. Liang, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201805501.
- (a) M. Huiban, M. Tredwell, S. Mizuta, Z. Wan, X. Zhang, T. L. Collier, V. Gouverneur and J. Passchier, Nat. Chem., 2013, 5, 941 CrossRef CAS; (b) D. van der Born, C. Sewing, J. D. M. Herscheid, A. D. Windhorst, R. V. Orru and D. J. Vugts, Angew. Chem., Int. Ed., 2014, 53, 11046 CrossRef CAS PubMed; (c) T. Rühl, W. Rafique, V. T. Lien and P. J. Riss, Chem. Commun., 2014, 50, 6056 RSC.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS; (b) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797 CrossRef CAS; (c) C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, J. Am. Chem. Soc., 2017, 139, 9325 CrossRef CAS PubMed; (d) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed; (e) D. Rageot, T. Bohnacker, A. Melone, J. B. Langlois, C. Borsari, P. Hillmann, A. M. Sele, F. Beaufils, M. Zvelebil, P. Hebeisen, W. Löscher, J. Burke, D. Fabbro and M. P. Wymann, J. Med. Chem., 2018, 61, 10084 CrossRef CAS PubMed; (f) G. W. Rewcastle, S. A. Gamage, J. U. Flanagan, R. Frederick, W. A. Denny, B. C. Baguley, P. Kestell, R. Singh, J. D. Kendall, E. S. Marshall, C. L. Lill, W.-J. Lee, S. Kolekar, C. M. Buchanan, S. M. F. Jamieson and P. R. Shepherd, J. Med. Chem., 2011, 54, 7105 CrossRef CAS PubMed; (g) F. Jeppsson, S. Eketjall, J. Janson, S. Karlström, S. Gustavsson, L. L. Olsson, A. C. Radesäter, B. Ploeger, G. Cebers, K. Kolmodin, B. M. Swahn, S. von Berg, T. Bueters and J. Fälting, J. Biol. Chem., 2012, 287, 41245 CrossRef CAS.
- S. Mizuta, I. S. Stenhagen, M. O'Duill, J. Wolstenhulme, A. K. Kirjavainen, S. J. Forsback, M. Tredwell, G. Sandford, P. R. Moore, M. Huiban, S. K. Luthra, J. Passchier, O. Solin and V. Gouverneur, Org. Lett., 2013, 15, 2648 CrossRef CAS.
- S. Verhoog, L. Pfeifer, T. Khotavivattana, S. Calderwood, T. L. Collier, K. Wheelhouse, M. Tredwell and V. Gouverneur, Synlett, 2016, 27, 25 CAS.
- H. Shi, A. Braun, L. Wang, S. H. Liang, N. Vasdev and T. Ritter, Angew. Chem., Int. Ed., 2016, 55, 10786 CrossRef CAS PubMed.
- G. Yuan, F. Wang, N. A. Stephenson, L. Wang, B. H. Rotstein, N. Vasdev, P. Tang and S. H. Liang, Chem. Commun., 2017, 53, 126 RSC.
- (a) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494 CrossRef CAS PubMed; (b) T. T. Tung, S. B. Christensen and J. Nielsen, Chem.–Eur. J., 2017, 23, 18125 CrossRef CAS PubMed; (c) R. Sakamoto, H. Kashiwagi and K. Maruoka, Org. Lett., 2017, 19, 5126 CrossRef CAS PubMed.
- T. C. Wilson, T. Cailly and V. Gouverneur, Chem. Soc. Rev., 2018, 47, 6990 RSC.
- H. Teare, E. G. Robins, A. Kirjavainen, S. Forsback, G. Sandford, O. Solin, S. K. Luthra and V. Gouverneur, Angew. Chem., Int. Ed., 2010, 49, 6821 CrossRef CAS PubMed.
- X. Huang, W. Liu, J. M. Hooker and J. T. Groves, Angew. Chem., Int. Ed., 2015, 54, 5241 CrossRef CAS PubMed.
- X. Huang, W. Liu, H. Ren, R. Neelamegam, J. M. Hooker and J. T. Groves, J. Am. Chem. Soc., 2014, 136, 6842 CrossRef CAS.
- W. R. Dolbier, Chem. Rev., 1996, 96, 1557 CrossRef CAS PubMed.
- Source: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html, accessed on 20/09/18.
- Our attempts to assemble one-pot the aryl boron reagent, ethyl bromofluoroacetate and [18F]fluoride were not fruitful. Details in ESI.†.
- See the ESI.†.
- Y. Wu, H.-R. Zhang, Y.-X. Cao, Q. Lan and X.-S. Wang, Org. Lett., 2016, 18, 5564 CrossRef CAS PubMed.
- C. Guo, X. Yue and F. L. Qing, Synthesis, 2010, 11, 1837 Search PubMed.
- Y. M. Su, G. S. Feng, Z. Y. Wang, Q. Lan and X. S. Wang, Angew. Chem., Int. Ed., 2015, 54, 6003 CrossRef CAS PubMed.
- T. Xia, L. He, Y. A. Liu, J. F. Hartwig and X. Liao, Org. Lett., 2017, 19, 2610 CrossRef CAS PubMed.
- A. Fahandej-Sadi and R. J. Lundgren, Synlett, 2017, 28, 2886 CrossRef CAS.
- All radiochemical yields (RCYs) are decay corrected.
- T. Khotavivattana, S. Verhoog, M. Tredwell, L. Pfeifer, S. Calderwood, K. Wheelhouse, T. L. Collier and V. Gouverneur, Angew. Chem., Int. Ed., 2015, 54, 9991 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05096a |
| This journal is © The Royal Society of Chemistry 2019 |