
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of alkyl-, aryl-, vinyl- and alkynyl-substituted Z-enamides and enol ethers†
Paola
Caramenti‡
 ,
Nina
Declas‡
,
Romain
Tessier
,
Matthew D.
Wodrich
and
Jerome
Waser
*
,
Nina
Declas‡
,
Romain
Tessier
,
Matthew D.
Wodrich
and
Jerome
Waser
*
Laboratory of Catalysis and Organic Synthesis, Institut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Ch-1015, Switzerland. E-mail: jerome.waser@epfl.ch
First published on 4th February 2019
Abstract
Enamides and enol ethers are valuable building blocks in synthetic chemistry, yet their stereoselective synthesis can be challenging. Herein, we report a new stereoselective synthesis of vinyl, aryl, alkynyl, alkyl and thio-substituted Z-enamides and enol ethers based on the use of vinylbenziodoxolone (VBX) reagents. The stable VBX reagents were synthesized by stereoselective addition of N- or O-nucleophiles on the corresponding alkynyl reagents in the presence of a catalytic amount of cesium carbonate. The VBX reagents were used in palladium-catalyzed cross-couplings at room temperature to access Z-enamides and enol ethers.
1. Introduction
The chemistry of carbonyl compounds, which can serve both as nucleophilic or electrophilic synthons, has been described as the backbone of organic synthesis.1 In particular, enolates, enamines and their derivatives have found broad applications as nucleophilic synthons.2 Enamides and enol ethers are especially attractive as nucleophiles due to their enhanced stability (Scheme 1(A)).3 In addition, they are valuable starting materials for the stereoselective synthesis of oxygen- and nitrogen-containing building blocks, especially via hydrogenation,4 as well as important pharmacophores in bioactive compounds, such as the natural product salicylihalamide B (1).5 Due to these numerous applications, the stereoselective synthesis of enamides and enol ethers is an important topic of research (Scheme 1(B)). Methods have been developed for the vinylation of amides, carbamates, alcohols and phenols,6 and the oxidative amidation of conjugated olefins (B1).7 However, these methods present issues of stereoselectivity or are based on the use of stereodefined starting materials, such as halogenated alkenes, which are often difficult to access. The selective synthesis of Z-enamides is particularly challenging.8 Alternatives to C–heteroatom bond formation have been developed,9 but lack convergence, as no new carbon–carbon or carbon–heteroatom bond is formed. Functionalization of enamides and enol ethers via C–C bond formation has recently been achieved when they are used as nucleophiles in C–H functionalization or Heck reactions (B2).10 In contrast, their use as electrophiles in C–C bond forming reactions has been much less exploited due to their low reactivity, limiting the range of available transformations (B3). Only the use of enol carboxylic and phosphonic esters and trans-iodo phthalimides has been reported.11 For these special substrates, the electron-density on oxygen/nitrogen is diminished by either one or two electron-withdrawing groups, making cross-coupling easier.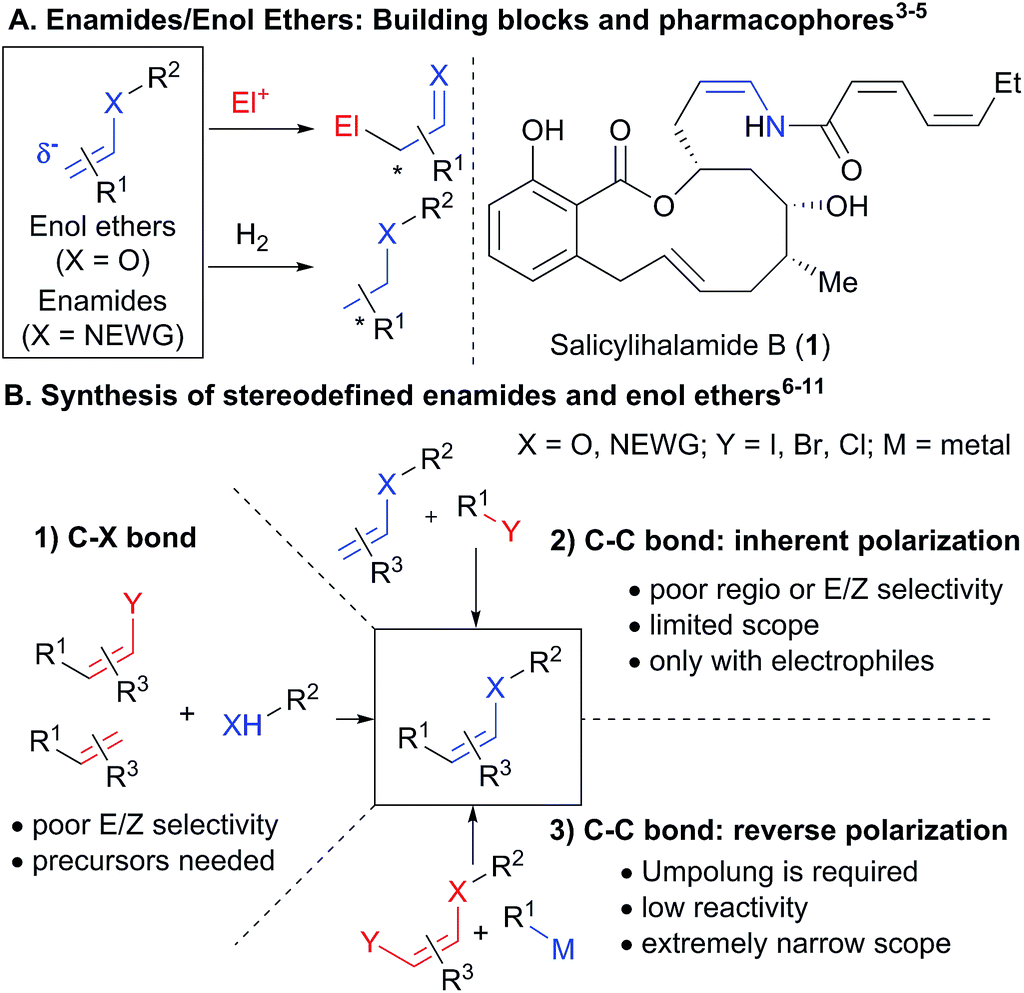 | ||
| Scheme 1 Enamides and enol ethers: importance and stereoselective synthesis. | ||
To succeed in the general use of enamides and enol ethers as electrophiles in C–C bond forming reactions, a more efficient umpolung of their inherent reactivity is therefore required. In this context, hypervalent iodine is well established for its capability to reverse the polarization of nucleophiles.12 Recently, Szpilman and coworkers used iodonium salts for the umpolung of enolates (Scheme 2(A)).13 The enolonium is generated in situ and cannot be isolated. Heteroatom substituted alkenyliodonium salts were reported only in the case of derivatives bearing less electron-rich fluorides and sulphonates substituents.14 Cyclic hypervalent iodine reagents, especially benziodoxoles, display enhanced stability.15 In 2016, Yoshikai and coworkers reported the palladium-catalyzed addition of carboxylates onto EthynylBenziodoXole (EBX) reagents to give the corresponding oxygen-substituted VinylBenziodoXoles (VBX), and used the latter in cross-coupling reactions.16 This was an important breakthrough in the development of stable reagents for the umpolung of enol esters. Nevertheless, the method required the use of a palladium catalyst and more expensive and difficult to access hypervalent iodine reagents bearing two trifluoromethyl groups. Furthermore, no umpolung of enamides was reported. In 2018, Miyake and co-workers demonstrated that phenols can be efficiently added onto aryl-EBX reagents derived from cheaper benzoic acid with high Z selectivity without the need of a transition metal catalyst.17 However, the formed VBX reagents displayed limited stability and only a single example was isolated in low yield. Therefore, they were immediately converted to iodides under light irradiation, preventing the exploitation of the highly reactive hypervalent bond in other transformations. In contrast to these first successes in the umpolung of enol esters and ethers, there is to the best of our knowledge no report for the umpolung of enamides. Only azide-substituted vinyl hypervalent iodine reagents have been reported by Kitamura and co-workers in 1997.18 In fact, it is well-known that the reaction of alkynyliodonium salts19 or EBX reagents20 with amides give directly ynamides as products (Scheme 2(B)). For this reason, hypervalent iodine reagents could not be used for the umpolung of enamides so far.
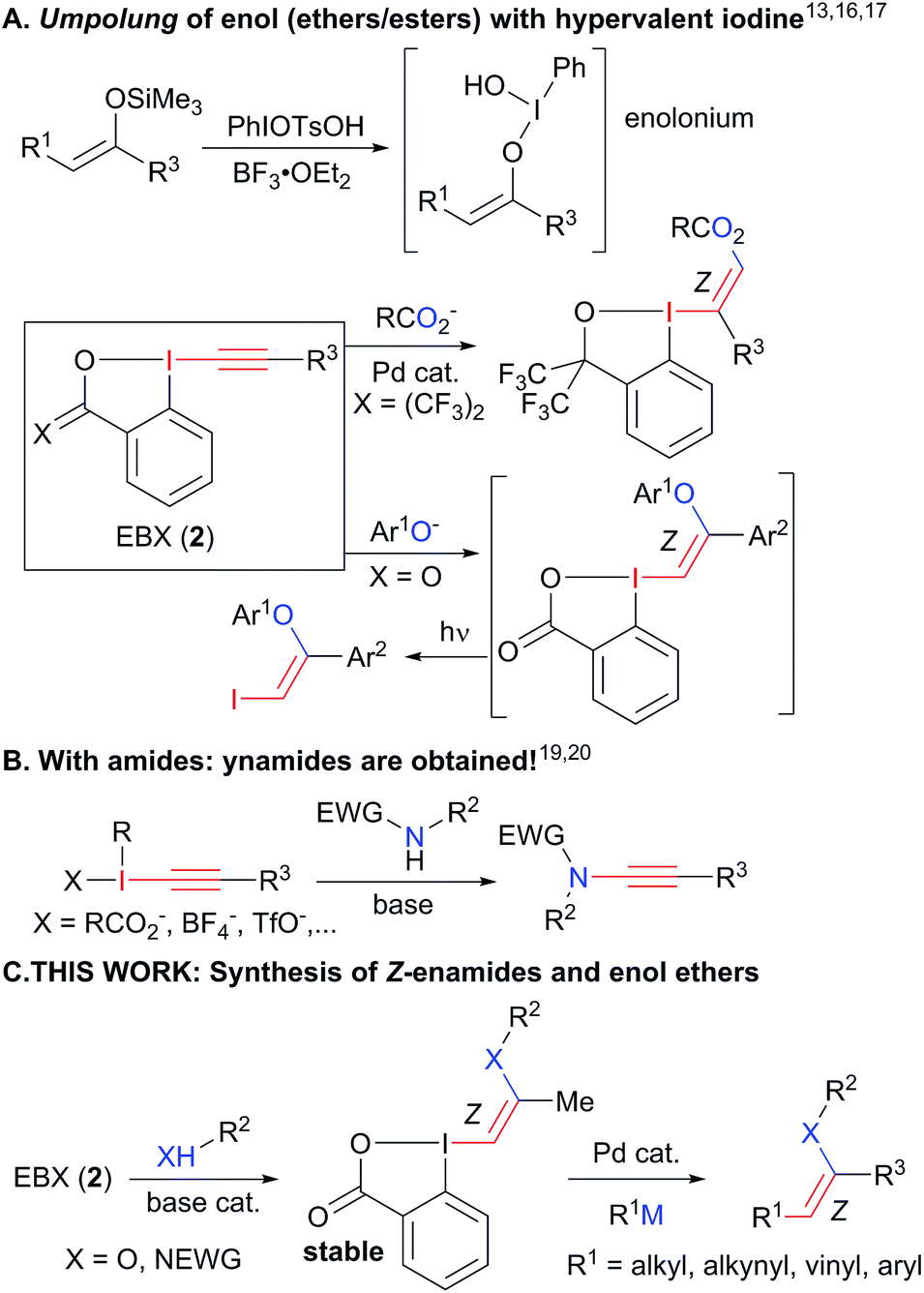 | ||
| Scheme 2 Umpolung with hypervalent iodine reagents: current limitation to enol esters and esters (A). Formation of ynamides with nitrogen nucleophiles (B) and our work on the synthesis of enamides and enol ethers (C). | ||
Herein, we report the first general synthesis of Z-enamides and enol ethers based on an umpolung-cross coupling approach (Scheme 2(C)). Enamide/enol ether-substituted benziodoxolone reagents were obtained by addition of tosyl amides or phenols onto alkyl substituted EBXs using a catalytic amount of base. The mild reaction conditions tolerated numerous functional groups, allowing the modification of drugs and natural products. The reaction proceeded with high Z selectivity. The new reagents are stable and could be engaged in a broad range of cross-coupling reactions, enabling the stereoselective synthesis of alkyl, aryl, vinyl and alkynyl enamides and enol ethers.
2. Results and discussions
Synthesis of new VBX reagents
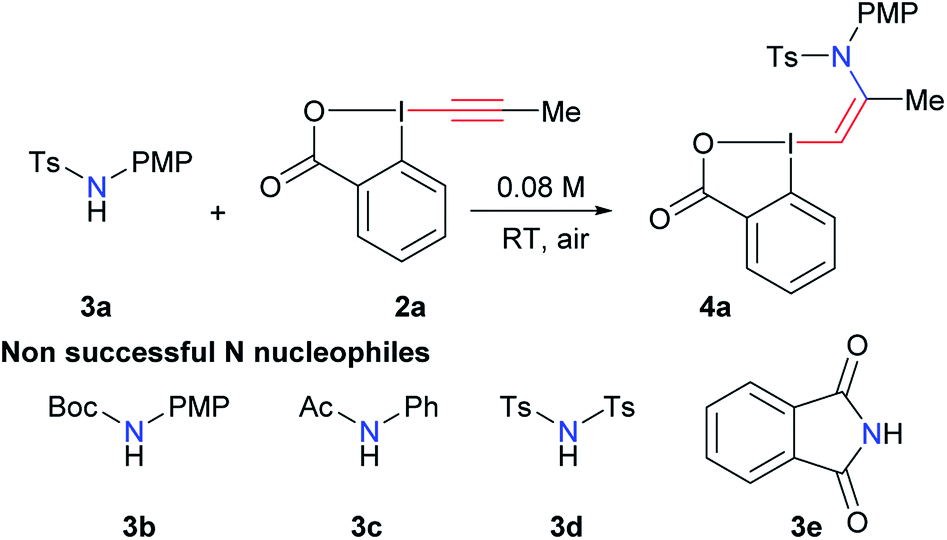
With the aim of accessing nitrogen-substituted VBX reagents, we first investigated the new synthesis recently reported by Olofsson and co-workers involving the reaction of alkenyl boronic acids with in situ generated iodine(III) precursors.21 However, we could never isolate the desired reagents using these reaction conditions. Therefore, we decided to re-investigate the addition of amide nucleophiles onto EBX reagents, despite the negative precedence.19,20 In our previous work on alkynylation of thiols, we observed the formation of sulfur-substituted VBX reagents in trace amounts.22 Interestingly, the amount of this side product could be increased from <5% to 20% using a catalytic amount of base. We therefore decided to use similar conditions (10 mol% tetramethylguanidine (TMG) in THF) in the screening of nitrogen nucleophiles for the addition on EBX reagent 2a (Table 1). Gratifyingly, the desired product 4a could be obtained in 22% yield using para-methoxyphenyl (PMP)-substituted tosyl amide 3a, whereas carbamate 3b, amide 3c, bistosylimide 3d and phthalimide (3e) were not successful (entry 1). In this case, the main issue was decomposition. Besides the desired product, only 2-iodo benzoic acid could be isolated, indicating a reduction at the iodine atom. A slight increase in yield was observed with CH2Cl2 (entry 2) and alcohols, such as MeOH and EtOH (entries 3 and 4) as solvents. Weaker organic bases led to a further increase in yield (entries 5 and 6). Switching to inorganic bases afforded cleaner reactions and higher yields (entries 7–11). The best result was obtained with cesium carbonate (93% NMR yield, entry 11). Compound 4a was stable and could be purified with only minor loss by column chromatography (68% isolated yield). Finally, 10 mol% of cesium carbonate was confirmed as the best base loading (entries 12 and 13). In fact, the use of base in catalytic amount is essential to avoid decomposition or formation of the alkyne product as was observed previously.19,20 The developed optimized conditions are highly convenient, as the reaction can be done in ethanol in an open flask using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of amide 3a and EBX 2a to give 4a with complete Z-stereoselectivity.23
:1 ratio of amide 3a and EBX 2a to give 4a with complete Z-stereoselectivity.23
|
|
|||
|---|---|---|---|
| a Reactions conditions: 0.10 mmol 3a, 0.10 mmol 2a, 10 μmol base, 0.08 M in indicated solvent, RT, air, 14 h. NMR yield using 0.39 mol% of 1,3,5-trimethoxybenzene as internal standard. b Isolated yield after column chromatography on silica gel. | |||
| Entry | Solvent | Base | Yielda |
| 1 | THF | 10 mol% TMG | 22 |
| 2 | CH2Cl2 | 10 mol% TMG | 28 |
| 3 | MeOH | 10 mol% TMG | 23 |
| 4 | EtOH | 10 mol% TMG | 30 |
| 5 | EtOH | 10 mol% NEt3 | 43 |
| 6 | EtOH | 10 mol% pyridine | 38 |
| 7 | EtOH | 10 mol% NaHCO3 | 81 |
| 8 | EtOH | 10 mol% KHCO3 | 79 |
| 9 | EtOH | 10 mol% CsHCO3 | 83 |
| 10 | EtOH | 10 mol% CsOH·H2O | 54 |
| 11 | EtOH | 10 mol% Cs 2 CO 3 | 93(68) |
| 12 | EtOH | 25 mol% Cs2CO3 | 46 |
| 13 | EtOH | 5 mol% Cs2CO3 | 39 |
The scope of the reaction was then investigated (Scheme 3) we focused first on the alkyne substituent on EBX 2 (Scheme 3(A)). In addition to methyl-substituted 4a, the unsubstituted Z-enamide 4b was obtained in 57% yield starting from a silylated EBX reagent. Reagents 4c–e bearing primary alkyl chains and functional groups such as a chlorine and an alkyne were also obtained in good yield. Cyclopropyl, cyclopentyl and cyclohexyl derivatives 4f–h could be isolated in excellent yields (74–94%). A sterically encumbered tertiary substituent was also well tolerated (product 4i). In contrast, an aryl substituent was not tolerated, leading to decomposition. Both a smaller mesyl and a nosyl sulfonamides could be used to give reagents 4k–n in good yields. Nosyl groups are in principle easier to cleave than tosyl groups. The same protocol could be also applied to phenols as nucleophiles (products 5a–f, Scheme 3(B)). The obtained enol ethers bear alkyl substituents in contrast to those reported by Miyake and co-workers and displayed enhanced stability.
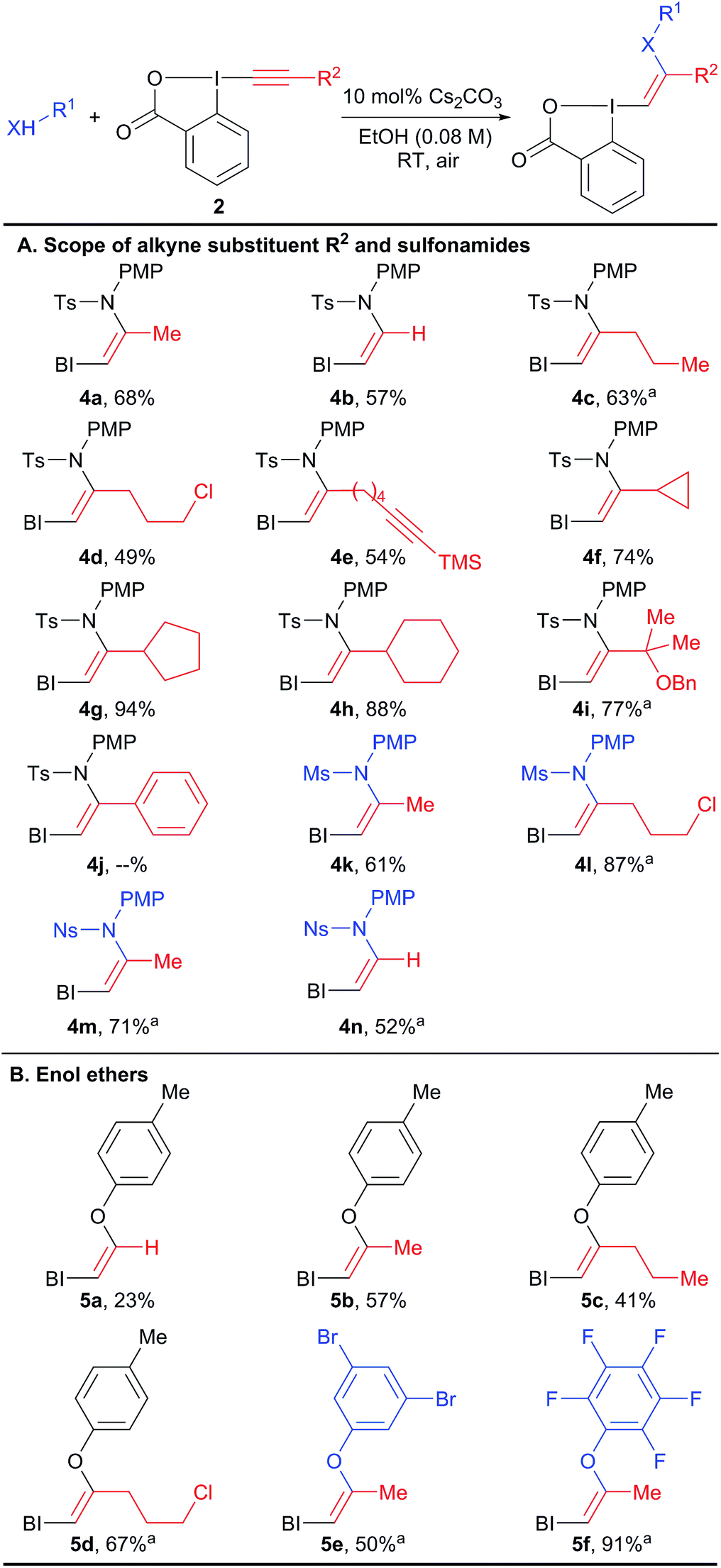 | ||
| Scheme 3 Scope of VBX reagents. Reactions performed on 1.0 mmol scale. BI = benziodoxolone. aReaction performed on 0.10 mmol scale. | ||
In all the previous work involving the synthesis and use of VBX reagents, focus had been restricted to only small organic molecules.16–18,21 When considering the very mild conditions developed in our work, we wondered if the approach could be used for the functionalization of more complex natural products and drugs (Scheme 4). These compounds present multiple heteroatoms and nucleophilic positions, leading to challenges in selectivity. Gratifyingly, our mild protocol allowed the functionalization of the sulfa drug sulfaphenazole in 43% yield to give VBX 6 without reaction of the free aromatic amine. Hypervalent iodine reagents derived from bioactive complex phenols such as tyrosine, α-tocopherol, capsaicin and estradiol derivatives 7–10 were also obtained in 40–79% yield. In this case, both amides and aliphatic alcohols were tolerated. The high selectivity observed is striking and is probably originates from the deprotonation of the most acidic O–H or N–H bond to generate the active nucleophile. The transformation was also successful for other acidic nitrogen functionalities: the tetrazole heterocycle of valsartan reacted as a nucleophile to give benziodoxolone 11. No reaction with the carboxylic acid was observed, in contrast to the work of Yoshikai and co-workers.16 The inertness of the carboxylate is not well-understood at this stage, but it is important to note that Yoshikai's functionalization required a palladium catalyst.
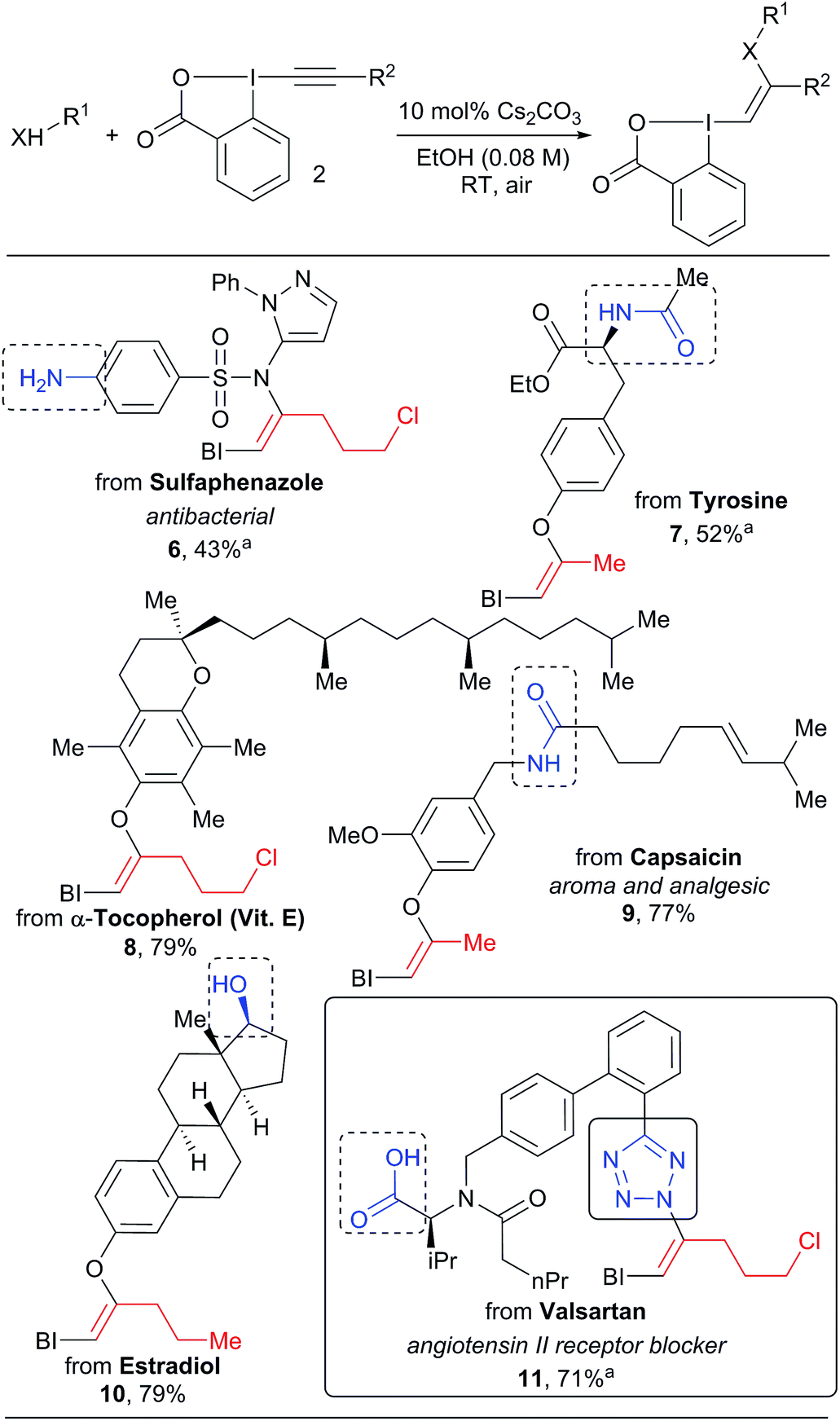 | ||
| Scheme 4 Scope of natural products and drugs. Reactions performed on 1.0 mmol scale. aReaction performed on 0.10 mmol scale. | ||
Speculative reaction mechanism
To better understand the switch in reactivity when using a catalytic amount of base, we turned to computational chemistry. DFT analysis (at the PBE0-dDsC/TZ2P//M06/def2-SVP level) for the addition of amide 3f on Me-EBX (2a) was first performed with TMG as a base and THF as a solvent, the conditions under which the reaction had been discovered. We were able to locate an α and a β-addition transition state aTS1 and bTS1 (Fig. 1), as shown in our previous work on thiol nucleophiles.22,24 In the case of a nitrogen nucleophile, β addition was favored by 10.3 kcal mol−1 leading to intermediate b1. The protonation of b1 is very easy with a barrier of only 3.2 kcal mol−1 to give vinylbenziodoxolone 4k, whereas breaking of the C–I bond requires 17.7 kcal mol−1, leading to formation of carbene intermediate b2. The alkyne product 12 can then be obtained after 1,2-amine shift with a barrier of 18.6 kcal mol−1 in a highly exergonic reaction. Interestingly, the barrier for the deprotonation of 4k back to b1 is only 13.7 kcal mol−1. We then repeated the calculations in ethanol using carbonate as a base (Fig. 2). The energies of both α- and β-additions were slightly higher under these conditions, with β-addition being even more favored (11.2 kcal mol−1). From intermediate b1, protonation was barrierless and the energy for carbon–iodine bond breaking was significantly lower (from 17.7 to 11.9 kcal mol−1). However, the difference between both transition states did not change significantly. Finally, the barrier for 1,2-amine shift was only slightly lower.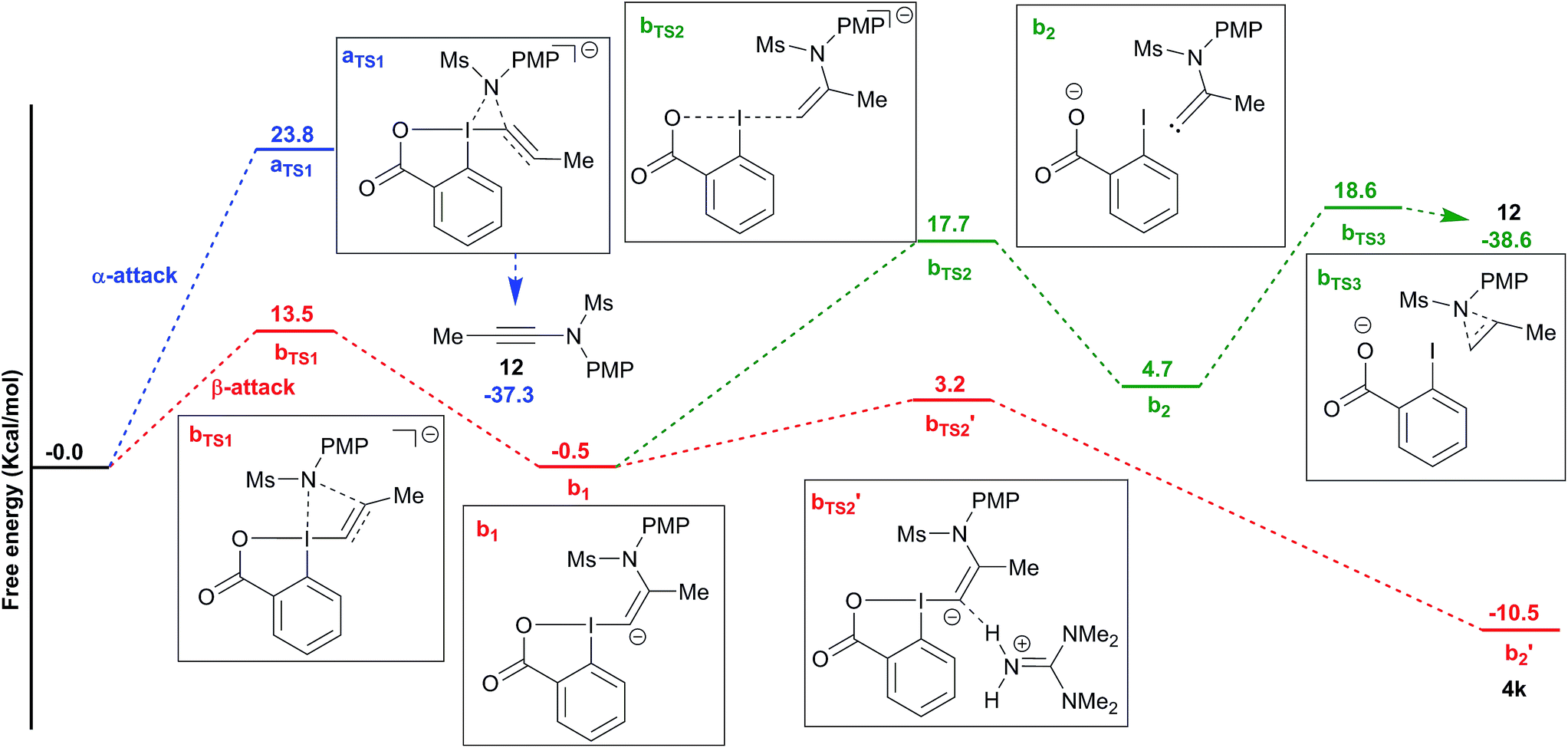 | ||
| Fig. 1 Reaction free energy profile for the addition of amide 3f to EBX 2a with TMG in THF. | ||
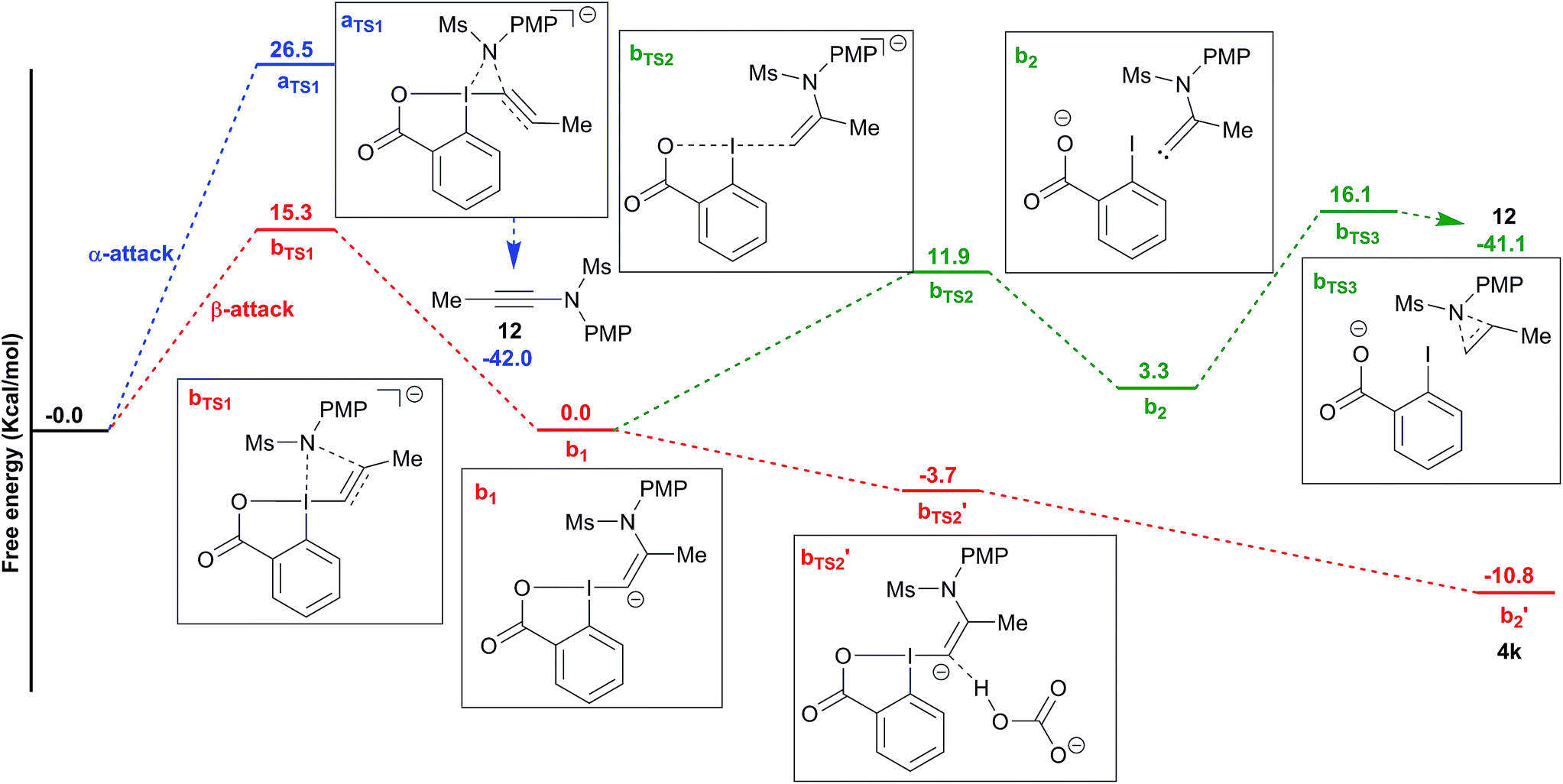 | ||
| Fig. 2 Reaction free energy profile for the addition of amide 3f to EBX 2a with cesium carbonate in ethanol. | ||
Based on these results, a speculative mechanism for the selective formation of VBX 4k can be proposed (Scheme 5). In the presence of a base, a small amount of amide 3f is deprotonated and reacts fast and reversibly with EBX 2a to form anion b1. b1 is itself in equilibrium with VBX 4k by re-protonation. The equilibrium of 2a, b1 and 4k lies strongly in favor of 4k, allowing its isolation once the reaction mixture is neutralized. On the other hand, b1 reacts slowly and irreversibly via carbene b2 to form alkyne 12. Higher base concentration leads to increased amount of b1, resulting finally in full conversion to ynamide 12. According to the computation results, the formation of VBX 4k is favored over ynamides 12 with about the same energy difference under preliminary and optimized conditions. The better yields obtained are probably due to suppression of decomposition pathways.
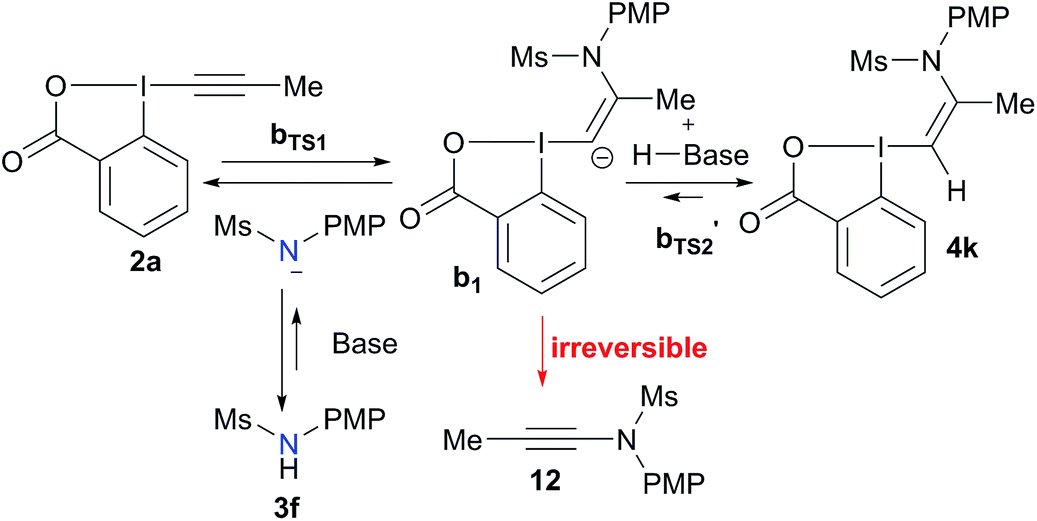 | ||
| Scheme 5 Speculative mechanism for the selective formation of VBX 4k. | ||
Functionalization of the VBX products
With a broad scope of functionalized VBX reagents in hand, we investigated their conversion into the desired enamides (Scheme 6). Palladium-catalyzed Stille cross-coupling was investigated first (Scheme 6(A)). The coupling of vinyl, aryl and alkyl stannyl reagents to give products 13–16 was possible at room temperature. Diene enamides are especially sensitive compounds and only a few synthetic methods have been reported to access them.25 Stille cross-coupling with an enol ether also proceeded smoothly at room temperature (product 17), whereas similar reaction with simple iodides required heating at 80–120 °C.17,26 In all Stille couplings, complete stereospecificity was observed and only the Z products were obtained. Enyne 18 was then obtained in a 6:1 Z:E ratio through a Sonogashira coupling (Scheme 6(B)).27 Finally, the addition of a strong thiol nucleophile was possible without a transition metal catalyst to give thioenamide 19 (Scheme 6(C)).8a–c
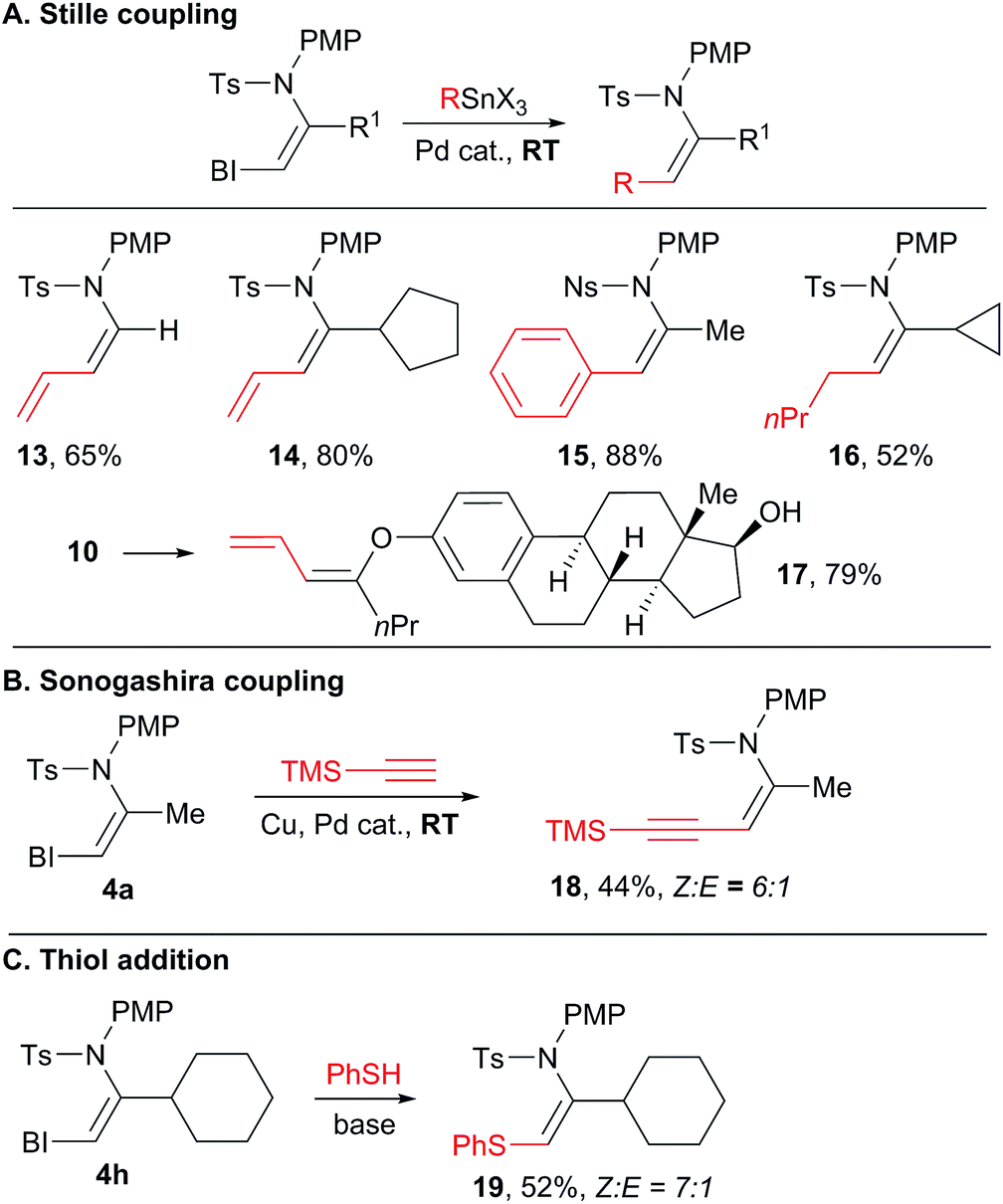 | ||
| Scheme 6 Functionalization of the VBX products. Reaction conditions: (A) 0.10 mmol VBX reagent, 0.20 mmol stannane, 5 mol% Pd(PhCN)2Cl2, 0.1 M in DMF, 10 h, RT. (B) 0.10 mmol VBX reagent 4a, 0.30 mmol trimethylsilylacetylene, 5 mol% Pd(PPh3)2Cl2, 20 mol% CuI, 0.10 mmol NEt3, 0.1 M in DMF, 10 h, RT. (C) 0.10 mmol VBX reagent 4h, 0.10 mmol phenylthiol, 0.12 mmol potassium tert-butoxide, 0.1 M in DME, 16 h, RT. | ||
In general, sulfonyl enamides have been broadly used in synthetic chemistry, for example as partners in cycloaddition reactions,28 in addition to iminiums29 or in oxidative heterofunctionalization reactions.30 Interestingly, most of these studies focused on the use of E-enamides, due to the difficulties in accessing the Z isomers. Easier access to Z-enamides will enhance the utility of these methodologies by enabling the synthesis of other stereoisomers.
Nevertheless, we were missing a direct comparison with the reactivity of simple iodides in the case of enamide substrates. We were unable to design an efficient synthesis of the corresponding iodo enamides using other methods. Although this already demonstrates an important synthetic advantage of our approach, we were still interested in directly comparing the reactivity of standard and hypervalent iodine bonds in the cross-coupling reaction. Fortunately, when the photoredox conditions reported by Miyake and co-workers were applied to VBX 4g, iodide 20 was obtained in 51% yield (Scheme 7(A)). This demonstrated that Miyake's procedure can also be applied to certain alkyl-substituted VBX reagents. Stille cross-coupling was then attempted with iodide 20, but no conversion was observed at room temperature and 50 °C. At 75 °C, less then 10% of the desired product was observed by 1H NMR, together with significant decomposition. This result definitively demonstrated the higher reactivity and synthetic utility of the VBX enamide reagents.
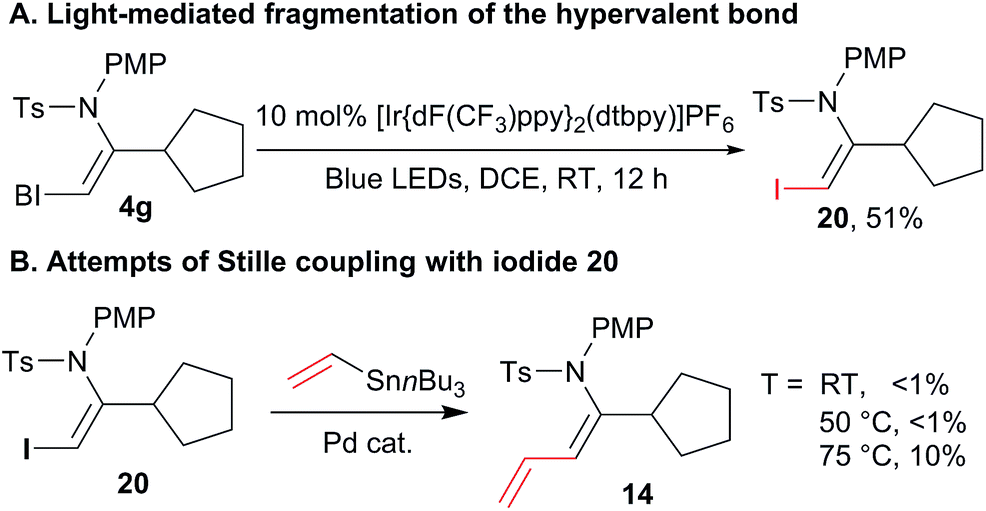 | ||
| Scheme 7 Comparison of the reactivity of enamide iodides and enamide benziodoxolones. Reaction conditions: (A) 0.10 mmol VBX reagent 4g, 10 mol% [Ir{dF(CF3)ppy}2(dtbpy)]PF6, 0.1 M in DCE, blue LEDS irradiation, 12 h, RT. (B) 0.10 mmol VBX reagent, 0.20 mmol stannane, 5 mol% Pd(PhCN)2Cl2, 0.1 M in DMF, 10 h, at the indicated temperature. | ||
3. Conclusions
In summary, we have developed a highly stereoselective synthesis of Z-enamides based on the use of vinylbenziodoxolone reagents. The key for success was the use of a catalytic amount of base, which avoided the formation of the thermodynamically favored ynamides. The obtained nitrogen-substituted VBX reagents were stable even to column chromatography and could be obtained in high yield under very mild reaction conditions, tolerating many functional groups. The reaction could be also extended to phenols as substrates. The high reactivity of the hypervalent iodine bond allowed the formation of aryl, vinyl, alkynyl, alkyl and thio-substituted Z-enamides as well as enol ethers with high stereospecificity at room temperature. This general access to Z-enamides and enol ethers will facilitate their broader use in synthetic chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Swiss National Science Foundation (No. 200021_159920 and 200020_182798) and EPFL. We thank Dr Durga Hari from our group for the synthesis of alkynyl reagents 2g and 2h, and Dr R. Scopelliti and Dr F. F. Tirani from ISIC at EPFL for X-ray analysis. M. D. W. thanks Prof. C. Corminboeuf for financial support and the Laboratory for Computational Molecular Design for providing computational resources.Notes and references
- (a) R. E. Ireland, Organic Synthesis, Prentice-Hall, Englewood Cliffs, 1969 Search PubMed; (b) E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley, New York, 1989 Search PubMed.
- G. Stork, J. Szmuszkovicz, R. Terrell, A. Brizzolara and H. Landesman, J. Am. Chem. Soc., 1963, 85, 207 CrossRef CAS.
- (a) T. Mukaiyama, K. Banno and K. Narasaka, J. Am. Chem. Soc., 1974, 96, 7503 CrossRef CAS; (b) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS PubMed; (c) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed; (d) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455 RSC; (e) T. Courant, G. Dagousset and G. Masson, Synthesis, 2015, 47, 1799 CrossRef CAS.
- (a) M. J. Burk, Y. M. Wang and J. R. Lee, J. Am. Chem. Soc., 1996, 118, 5142 CrossRef CAS; (b) M. J. Burk, C. S. Kalberg and A. Pizzano, J. Am. Chem. Soc., 1998, 120, 4345 CrossRef CAS; (c) A. G. Hu, Y. Fu, J. H. Xie, H. Zhou, L. X. Wang and Q. L. Zhou, Angew. Chem., Int. Ed., 2002, 41, 2348 CrossRef CAS; (d) Q. L. Zhou and J. H. Xie, in Stereoselective Formation of Amines, ed. W. Li and X. Zhang, 2014, vol. 343, pp. 75–101 Search PubMed.
- (a) L. Yet, Chem. Rev., 2003, 103, 4283 CrossRef CAS PubMed; (b) R. B. Andrade, Org. Prep. Proced. Int., 2009, 41, 359 CrossRef CAS; (c) M. J. Martín, L. Coello, R. Fernández, F. Reyes, A. Rodríguez, C. Murcia, M. Garranzo, C. Mateo, F. Sánchez-Sancho, S. Bueno, C. de Eguilior, A. Francesch, S. Munt and C. Cuevas, J. Am. Chem. Soc., 2013, 135, 10164 CrossRef PubMed.
- (a) M. C. Willis, D. Taylor and A. T. Gillmore, Chem. Commun., 2003, 2222 RSC; (b) L. Jiang, G. E. Job, A. Klapars and S. L. Buchwald, Org. Lett., 2003, 5, 3667 CrossRef CAS PubMed; (c) X. H. Pan, Q. Cai and D. W. Ma, Org. Lett., 2004, 6, 1809 CrossRef CAS PubMed; (d) J. R. Dehli, J. Legros and C. Bolm, Chem. Commun., 2005, 973 RSC; (e) Y. Bolshan and R. A. Batey, Angew. Chem., Int. Ed., 2008, 47, 2109 CrossRef CAS PubMed; (f) T. Kuranaga, Y. Sesoko and M. Inoue, Nat. Prod. Rep., 2014, 31, 514 RSC.
- J. M. Lee, D.-S. Ahn, D. Y. Jung, J. Lee, Y. Do, S. K. Kim and S. Chang, J. Am. Chem. Soc., 2006, 128, 12954 CrossRef CAS PubMed.
- Most reports have been so far limited to specific classes of Z-enamides. For Z-thioenamides, see: (a) B. Banerjee, D. N. Litvinov, J. Kang, J. D. Bettale and S. L. Castle, Org. Lett., 2010, 12, 2650 CrossRef CAS PubMed; (b) P. García-Reynaga, A. K. Carrillo and M. S. VanNieuwenhze, Org. Lett., 2012, 14, 1030 CrossRef PubMed; (c) J. A. Lutz, V. Subasinghege Don, R. Kumar and C. M. Taylor, Org. Lett., 2017, 19, 5146 CrossRef CAS PubMed ; For Z-iodoenamides, see: ; (d) H. Li, X. Li, Z. Zhao, T. Ma, C. Sun and B. Yang, Chem. Commun., 2016, 52, 10167 RSC ; For Z-methylenamides, see: ; (e) F. Weber, P. S. Steinlandt, M. Ballmann and G. Hilt, Synthesis, 2017, 49, 440 CAS ; and ref. 9a; For Z-silylenamides, see: ; (f) Y. Kim, R. B. Dateer and S. Chang, Org. Lett., 2017, 19, 190 CrossRef CAS PubMed ; For Z-esterenamides, see ref. 7. For an approach via hydrogenation of ynamides, see: ; (g) X. Zhang, Y. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. Shen and M. R. Tracey, J. Org. Chem., 2006, 71, 4170 CrossRef CAS PubMed.
- (a) S. Lin, Z.-Q. Yang, B. H. B. Kwok, M. Koldobskiy, C. M. Crews and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6347 CrossRef CAS PubMed; (b) K. C. Nicolaou and C. J. N. Mathison, Angew. Chem., Int. Ed., 2005, 44, 5992 CrossRef CAS PubMed; (c) B. M. Trost, J. J. Cregg and N. Quach, J. Am. Chem. Soc., 2017, 139, 5133 CrossRef CAS PubMed.
- Review: (a) N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Chem.–Eur. J., 2014, 20, 7548 CrossRef CAS PubMed ; Selected examples: ; (b) A. L. Hansen and T. Skrydstrup, Org. Lett., 2005, 7, 5585 CrossRef CAS PubMed; (c) K. D. Hesp, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 11430 CrossRef CAS PubMed; (d) Y. Liu, D. Li and C.-M. Park, Angew. Chem., Int. Ed., 2011, 50, 7333 CrossRef CAS PubMed; (e) S. Pankajakshan, Y.-H. Xu, J. K. Cheng, M. T. Low and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 5701 CrossRef CAS PubMed; (f) C. Feng, D. M. Feng and T. P. Loh, Chem. Commun., 2014, 50, 9865 RSC; (g) N. Gigant and I. Gillaizeau, Org. Lett., 2012, 14, 3304 CrossRef CAS PubMed; (h) N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Org. Lett., 2013, 15, 816 CrossRef CAS PubMed; (i) H. Jiang, C. M. Huang, J. J. Guo, C. Q. Zeng, Y. Zhang and S. Y. Yu, Chem.–Eur. J., 2012, 18, 15158 CrossRef CAS PubMed.
- (a) B. C. Chary, S. Kim, D. Shin and P. H. Lee, Chem. Commun., 2011, 47, 7851 RSC; (b) P. Pawluć, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Tetrahedron, 2012, 68, 3545 CrossRef; (c) P. J. González-Liste, F. León, I. Arribas, M. Rubio, S. E. García-Garrido, V. Cadierno and A. Pizzano, ACS Catal., 2016, 6, 3056 CrossRef; (d) F. León, P. J. González-Liste, S. E. García-Garrido, I. Arribas, M. Rubio, V. Cadierno and A. Pizzano, J. Org. Chem., 2017, 82, 5852 CrossRef PubMed.
- (a) V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Applications of Polyvalent Iodine Compounds, John Wiley & Sons, Inc., 2014 Search PubMed; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed.
- (a) S. Arava, J. N. Kumar, S. Maksymenko, M. A. Iron, K. N. Parida, P. Fristrup and A. M. Szpilman, Angew. Chem., Int. Ed., 2017, 56, 2599 CrossRef CAS PubMed; (b) S. Maksymenko, K. N. Parida, G. K. Pathe, A. A. More, Y. B. Lipisa and A. M. Szpilman, Org. Lett., 2017, 19, 6312 CrossRef CAS PubMed; (c) A. A. More, G. K. Pathe, K. N. Parida, S. Maksymenko, Y. B. Lipisa and A. M. Szpilman, J. Org. Chem., 2018, 83, 2442 CrossRef CAS PubMed; (d) K. N. Parida, G. K. Pathe, S. Maksymenko and A. M. Szpilman, Beilstein J. Org. Chem., 2018, 14, 992 CrossRef CAS PubMed.
- Selected examples: (a) M. Ochiai, M. Kunishima, S. Tani and Y. Nagao, J. Am. Chem. Soc., 1991, 113, 3135 CrossRef CAS; (b) M. Yoshida and S. Hara, Org. Lett., 2003, 5, 573 CrossRef CAS PubMed; (c) E. Zawia, D. J. Hamnett and W. J. Moran, J. Org. Chem., 2017, 82, 3960 CrossRef CAS PubMed ; For a review, see: ; (d) D. P. Hari, S. Nicolai and J. Waser, Patai's Chem. Funct. Groups, 2018 DOI:10.1002/9780470682531.pat0951.
- (a) V. V. Zhdankin, Curr. Org. Synth., 2005, 2, 121 CrossRef CAS; (b) J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2015, 54, 8876 CrossRef CAS PubMed; (c) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS PubMed; (d) D. P. Hari, P. Caramenti and J. Waser, Acc. Chem. Res., 2018, 51, 3212 CrossRef CAS PubMed.
- (a) J. Wu, X. Deng, H. Hirao and N. Yoshikai, J. Am. Chem. Soc., 2016, 138, 9105 CrossRef CAS PubMed; (b) J. Wu, K. Xu, H. Hirao and N. Yoshikai, Chem.–Eur. J., 2017, 23, 1521 CrossRef CAS PubMed.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 12829 CrossRef CAS PubMed.
- Y. Fujiwara, T. Fukuoka and T. Kitamura, Synlett, 1997, 7, 659 Search PubMed.
- (a) P. Murch, B. L. Williamson and P. J. Stang, Synthesis, 1994, 1255 CrossRef CAS; (b) B. Witulski and T. Stengel, Angew. Chem., Int. Ed., 1998, 37, 489 CrossRef CAS ; For a review on ynamides synthesis and use, see: ; (c) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 49, 2840 CrossRef CAS PubMed.
- T. Aubineau and J. Cossy, Chem. Commun., 2013, 49, 3303 RSC.
- E. Stridfeldt, A. Seemann, M. J. Bouma, C. Dey, A. Ertan and B. Olofsson, Chem.–Eur. J., 2016, 22, 16066 CrossRef CAS PubMed.
- R. Frei, M. D. Wodrich, D. P. Hari, P. A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563 CrossRef CAS PubMed.
- The structure of 4a was confirmed by X-ray analysis. The data is available at the Cambridge Crystallographic Data Center (CCDC number 1876011†).
- See figures of Section SI6† for a full energy profile with both N and O nucleophiles.
- (a) L. E. Overman, G. F. Taylor, C. B. Petty and P. J. Jessup, J. Org. Chem., 1978, 43, 2164 CrossRef CAS; (b) L. E. Overman, L. A. Clizbe, R. L. Freerks and C. K. Marlowe, J. Am. Chem. Soc., 1981, 103, 2807 CrossRef CAS; (c) D. A. Alonso, E. Alonso, C. Nájera, D. J. Ramón and M. Yus, Tetrahedron, 1997, 53, 4835 CrossRef CAS; (d) K.-Y. Lee, Y.-H. Kim, M.-S. Park, C.-Y. Oh and W.-H. Ham, J. Org. Chem., 1999, 64, 9450 CrossRef CAS; (e) T. J. Greshock and R. L. Funk, J. Am. Chem. Soc., 2006, 128, 4946 CrossRef CAS PubMed; (f) R. Hayashi, R. P. Hsung, J. B. Feltenberger and A. G. Lohse, Org. Lett., 2009, 11, 2125 CrossRef CAS PubMed; (g) M.-G. Zhou, R.-H. Dai and S.-K. Tian, Chem. Commun., 2018, 54, 6036 RSC.
- For selected examples of dienol ether synthesis, see: (a) A. Deagostino, P. Balma Tivola, C. Prandi and P. Venturello, Synlett, 1999, 1999, 1841 CrossRef; (b) M. Pawliczek, T. F. Schneider, C. Maaß, D. Stalke and D. B. Werz, Angew. Chem., Int. Ed., 2015, 54, 4119 CrossRef CAS PubMed . See also ref. 11a.
- For selected examples of enyne amides synthesis, see: (a) R. Martín, M. Rodríguez Rivero and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 7079 CrossRef PubMed; (b) H. Xu, S. Gu, W. Chen, D. Li and J. Dou, J. Org. Chem., 2011, 76, 2448 CrossRef CAS PubMed . See also ref. 10f.
- (a) N. Sarkar, A. Banerjee and S. G. Nelson, J. Am. Chem. Soc., 2008, 130, 9222 CrossRef PubMed; (b) J. Zhu, Y.-J. Cheng, X.-K. Kuang, L. Wang, Z.-B. Zheng and Y. Tang, Angew. Chem., Int. Ed., 2016, 55, 9224 CrossRef CAS PubMed; (c) T. R. Pradhan, H. W. Kim and J. K. Park, Org. Lett., 2018, 20, 5286 CrossRef CAS PubMed.
- L. Andna and L. Miesch, Org. Lett., 2018, 20, 3430 CrossRef CAS PubMed.
- (a) M. Nakanishi, C. Minard, P. Retailleau, K. Cariou and R. H. Dodd, Org. Lett., 2011, 13, 5792 CrossRef CAS PubMed; (b) S. Nocquet-Thibault, P. Retailleau, K. Cariou and R. H. Dodd, Org. Lett., 2013, 15, 1842 CrossRef CAS PubMed; (c) S. Nocquet-Thibault, C. Minard, P. Retailleau, K. Cariou and R. H. Dodd, Tetrahedron, 2014, 70, 6769 CrossRef CAS; (d) S. Nocquet-Thibault, A. Rayar, P. Retailleau, K. Cariou and R. H. Dodd, Chem.–Eur. J., 2015, 21, 14205 CrossRef CAS PubMed; (e) R. Beltran, S. Nocquet-Thibault, F. Blanchard, R. H. Dodd and K. Cariou, Org. Biomol. Chem., 2016, 14, 8448 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational data. CCDC 1876011. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05573d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |