
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced crystallography for structure determination of natural products
Jian-Guo
Song
 abc,
Wen-Cai
Ye
*abc and
Ying
Wang
*abc
abc,
Wen-Cai
Ye
*abc and
Ying
Wang
*abc
aState Key Laboratory of Bioactive Molecules and Druggability Assessment, Guangdong Basic Research Center of Excellence for Natural Bioactive Molecules and Discovery of Innovative Drugs, Jinan University, Guangzhou, 510632, P. R. China. E-mail: wangying_cpu@163.com; chywc@aliyun.com
bGuangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM & New Drugs Research, Guangdong-Hong Kong-Macau Joint Laboratory for Pharmacodynamic Constituents of TCM and New Drugs Research, Jinan University, Guangzhou, 510632, P. R. China
cCenter for Bioactive Natural Molecules and Innovative Drugs Research, College of Pharmacy, Jinan University, Guangzhou, 510632, P. R. China
First published on 3rd February 2025
Abstract
Covering: up to 2025
Crystallographic analysis has become the most reliable method for elucidating the structures of natural products, as it can provide absolute configurations with precise spatial arrangement information at the molecular level. However, obtaining high-quality and suitable-sized single crystals can be challenging for many natural products, making their structure determination difficult through traditional crystallography techniques. Recent advancements in this field have introduced innovative strategies to overcome the obstacle. These cutting-edge strategies include post-orientation of organic molecules within pre-prepared porous crystals (crystalline sponge method), co-crystallization of organic molecules with a crystalline mate through supramolecular interactions (crystalline mate method), encapsulation of organic molecules within inert oil nanodroplets (encapsulated nanodroplet crystallization method), and the use of electron diffraction and microscopy for nanocrystals (microcrystal electron diffraction method). This highlight delves into the fundamental principles, key characteristics, and representative applications of each strategy, as well as their respective advantages and limitations, aiming to guide researchers in choosing the most suitable crystallography approach for analyzing the structures of natural products.
 Jian-Guo Song | Jian-Guo Song obtained his bachelor's degree from Guangzhou University of Chinese Medicine in 2015. Afterwards, he earned his PhD degree from Jinan University and completed postdoctoral training. In 2024, he joined the State Key Laboratory of Bioactive Molecules and Druggability Assessment of Jinan University as an associate professor. His research interest is focused on the targeted isolation and structure determination of bioactive natural products. |
 Wen-Cai Ye | Wen-Cai Ye received his bachelor's degree in Traditional Chinese Medicine from China Pharmaceutical University in 1983. From 1983 to 1988, he was employed as a lecturer at Wuhu Higher Technological School of Traditional Chinese Medicine. Afterwards, he went back to China Pharmaceutical University and began his independent career as an assistant, associate professor, and full professor. He obtained his PhD degree in Chemistry from Hong Kong University of Science & Technology. In 2003, he joined the College of Pharmacy at Jinan University. He works in the field of natural medicinal chemistry, focusing on developing innovative drugs from natural products. |
 Ying Wang | Ying Wang received her bachelor's degree in Traditional Chinese Medicine Preparation from China Pharmaceutical University in 1999. She obtained her master's and PhD degrees in Medicinal Chemistry from the China Pharmaceutical University in 2003 and 2006, respectively. Afterwards, she moved to the College of Pharmacy of Jinan University as a postdoctoral fellow, associate professor, and full professor. Her research interests mainly focus on the discovery of lead compounds from natural products for innovative drugs. |
1 Introduction
Natural products (NPs) play critical roles in drug discovery and serve as molecular probes for exploring therapeutic targets and pharmacological principles. They exhibit a broad range of structural diversity and biological activities. According to a recent survey by Newman and Cragg, 68% of the 1181 FDA-approved small-molecule drugs between 1981 and 2019 were directly or indirectly derived from NPs.1 NPs are specialised metabolites produced by living organisms, often containing lower nitrogen and higher oxygen levels compared to synthetic molecules. Additionally, NPs tend to have more intricate structures with higher molecular weights and more stereogenic centers at sp3-hybridized carbons.2 These distinct features of NPs make them ideal for interacting with biological targets and serving as preferred ligand–protein binding motifs. However, despite these advantages, determining the structure of NPs poses significant challenges.Spectroscopic analysis is a commonly used method for elucidating the structure of NPs. By exhaustively analyzing ultraviolet-visible (UV), infrared (IR), mass spectrometry (MS), and nuclear magnetic resonance (NMR) spectroscopic data, researchers can deduce the structures and even relative configurations of most unknown compounds. However, spectroscopic methods can sometimes lead to seemingly reasonable but incorrect structures due to misleading signals. In contrast, single crystal X-ray diffraction (SCXRD) is considered the most reliable structure elucidation method as it provides detailed information on the spatial arrangement of atoms, bonding types, and absolute configuration of molecules. Nevertheless, obtaining crystals of sufficient size for SCXRD can be challenging, especially with NPs found in vanishingly small quantities. Further, certain compounds, including oily, waxy, and liquid substances, may not crystallize at room temperature, making implementing SCXRD difficult in such cases.
In recent years, advanced methods and techniques have been developed to address challenges associated with traditional organic molecule crystal growth, while still utilizing diffraction-based analytical techniques. For example, crystalline porous materials have been used as crystalline sponges to absorb and align organic molecules inside the cavities, allowing for direct observation through traditional SCXRD analysis.3 Discrete molecular entities have been employed as crystalline mates to co-crystallize with organic molecules, aiding in their assembly into crystalline lattices.4 Encapsulating nanoliter of organic solution in inert oil has also been demonstrated to be beneficial for crystal nucleation and growth.5 Advances in crystallography have enabled fine structure analysis of nanocrystalline samples thanks to the revolutionary 3D acquisition and electron diffraction.6 These advancements represent a set of sophisticated tools that can greatly facilitate crystallization and data collection, opening up new territories for elucidating the structures of NPs.
Given the significance of accurate structure determination, a timely review of these advanced crystallography techniques is essential for researchers in the field of NP research. This article examines four emerging modern crystallography advancements for molecular structure elucidation, including the crystalline sponge method, crystalline mate method, encapsulated nanodroplet crystallization method, and microcrystal electron diffraction method. Through a series of representative examples, the superiorities of these methods were illustrated, particularly for the structure elucidation of natural compounds. In addition, their relative advantages and limitations are summarized to aid researchers in choosing the most suitable method for analyzing the structures of NPs difficult to crystallize on their own.
2 Crystalline sponge method
2.1 The crystalline sponge for molecular structure determination
In 2013, Makoto Fujita and co-workers introduced the crystalline sponge method for crystallographic analysis, bypassing the traditional crystallization process for determining the structures of organic molecules.7 The initially reported crystalline sponge is a porous metal–organic framework (MOF) possessing regular cavities for guest molecule recognition. The MOF can selectively absorb organic molecules, arranging them orderly within the cavities through host–guest interactions. Importantly, the absorption process of the organic molecules does not compromise the crystallinity of the MOF, enabling the molecular structure of the absorbed organic molecules to be visualized alongside the host framework using conventional crystallographic analysis. This method is also described as a “post-crystallization” process, utilizing a pre-prepared MOF as the crystalline matrix for periodically arranging organic molecules within the MOF. With the potential to determine the molecular structure by just using a tiny single crystal, one significant advantage of this crystalline sponge method is its ability to analyze samples on a nanogram to microgram scale.The most well-known crystalline sponge, {[(ZnI2)3(tpt)2]·x(solvent)}n (ZnI2-tpt, tpt = tris(4-pyridyl)-1,3,5-triazine), was synthesized by diffusing a methanol solution of ZnI2 to a nitrobenzene solution of tpt.8 After approximately seven days at room temperature, single crystals of ZnI2-tpt formed at the interface of the two solution layers. SCXRD analysis revealed a porous lattice-like structure with infinite channels of approximately 8 × 5 Å along the crystallographic b-axis (Fig. 1A). The tpt ligand provides electron-deficient planes that facilitate π⋯π and CH⋯π interactions with organic molecules. The channels of ZnI2-tpt are initially occupied by nitrobenzene, which must be replaced by cyclohexane before soaking with organic molecules. This solvent exchange process takes about a week at 50 °C, with only about 5% of crystals remaining suitable for the soaking process. Due to the relatively weak interactions of cyclohexane in the channels of ZnI2-tpt, organic molecules with stronger interactions can diffuse into the ZnI2-tpt even at low concentrations. Over time, organic molecules can enrich and align within the channels, with their electron density observable through X-ray analysis (Fig. 1B and C). The anomalous scattering effect of Zn and I atoms in ZnI2-tpt allows for the determination of the absolute configuration of chiral molecules.
 | ||
| Fig. 1 Fujita's crystalline sponge for molecular structure determination. Reprinted with permission from ref. 7. Copyright 2013 Springer Nature Limited. | ||
Since the first crystalline sponge was reported, this method has undergone a series of operational optimizations aimed at improving the ease of use and reliability of crystallographic analysis.9 Clardy et al. found that using chloroform instead of nitrobenzene as a synthetic solvent can reduce the synthesis time of ZnI2-tpt to three days. In addition, changing the terminal ligand from I to Br/Cl to afford isomorphic ZnBr2-tpt and ZnCl2-tpt can decrease X-ray scattering of the framework and enhance guest visibility.10 During the soaking process, replacing cyclohexane with moderately polar solvents such as ketones and esters can effectively promote the diffusion and ordering of organic molecules by co-crystallizing with absorbed organic molecules to fill the gaps in the channels.11 Furthermore, for diffraction data collection, synchrotron radiation X-ray sources have proven to significantly reduce data collection time.12
In the past decade, ZnI2-tpt and its Br or Cl analogues have shown great potential for the structure elucidation of mass-limited NPs (Fig. 2A). Elatenyne (1), a marine NP isolated in 1986, has a complex pseudo-mirror-symmetric structure and barely any optical rotation, making its absolute configuration difficult to confirm. In 2016, Fujita et al. utilized ZnI2-tpt as a crystalline sponge to investigate the structure of 1, successfully establishing its absolute configuration by observing two distinct alkyl side chains confined within the ZnI2-tpt channel.13 Collimonins A and B (2 and 3) are unstable polyenes isolated from the fungus-feeding bacterium Collimonas fungivorans Ter331. Their absolute configurations were determined through a combination of spectroscopic, chemical, and ZnCl2-tpt crystalline sponge methods.14 Tenebrathin (4), a C-5-substituted γ-pyrone with a nitroaryl side chain, was isolated from Streptoalloteichus tenebrarius NBRC 16177 and had its structure elucidated using a similar approach by Fujita et al.15 Phaseolide A (5), a 12-membered macrolide produced by Aspergillus oryzae, had its absolute stereochemistry established through vibrational circular dichroism spectroscopy and the ZnCl2-tpt crystalline sponge.16Trans-iso-α-acid in fresh beer contributes to its bitterness and undergoes various conversions during aging. Fujita et al. successfully applied the ZnCl2-tpt crystalline sponge method in conjunction with high-performance liquid chromatography (HPLC) to isolate and identify the thirteen products of trans-iso-α-acid transformation (6–18), including eight previously unreported ones, with absolute configuration assignment.17 Vanitaracin A (19), an anti-hepatitis B virus agent isolated from the culture broth of the fungus Talaromyces sp., had its absolute configurations established, including six stereogenic centers, using ZnI2-tpt crystalline sponge method by Kamisuki et al.18 Additionally, Carmalt et al. utilized ZnI2-tpt crystalline sponge for the structural analysis of terpenoids, such as geraniol (20), farnesol (21) and β-damascone (22).19
 | ||
| Fig. 2 Chemical and crystal structures of NPs elucidated by Fujita's crystalline sponge method. | ||
In addition to elucidating the structure of unknown compounds, the crystalline sponge method has also been used for the structure revision of NPs (Fig. 2B). Cycloelatanenes A and B (23 and 24), isomeric marine NPs with a spiro[5.5]undecene skeleton, had their relative structures previously deduced through NMR studies. Fujita et al. utilized the ZnI2-tpt crystalline sponge method to determine the structures of 23 and 24, correcting the chirality assignment of the stereogenic center at the C-4 position.20 Furthermore, using the same crystalline sponge, they revised the structure of fuliginone (25) from a phenyl-substituted phenalenone to a hydroxyl-substituted phenalenone.21
The crystalline sponge method allows for the analysis of samples from nano- to micrograms, on the same scale as the quantity of analytes in analytical chromatography separation, thus making this method compatible with HPLC/GC. In a 2013 report, Fujita et al. demonstrated the tandem use of HPLC and crystalline sponge method. Specifically, 30 mg of orange peel extract was separated by HPLC and each collected chromatographic fraction was directly treated with ZnI2-tpt. As a result, the molecular structures of three polymethoxyflavones were successfully determined through X-ray crystallography analysis.7 In a separate study, the team further developed the tandem use of gas chromatography (GC) and crystalline sponge method for structural elucidation of trace amounts of volatile compounds. In a proof-of-concept experiment, major components of peppermint essential oil were isolated by preparative GC, and eight volatile compounds were characterized, including their stereochemistry.22
The correct identification of NP structures is crucial for understanding the enzymes involved in their biosynthesis. Traditional methods are sometimes insufficient for their structure elucidation due to limited quantities of these compounds. The crystalline sponge method has been instrumental in resolving such issues (Fig. 2C). Fujita et al. used the microgram-scale NMR coupled with ZnI2-tpt crystalline sponge method to determine the absolute stereochemistry of prespatane (26), a sesquiterpene product of LphTPS-A. This makes LphTPS-A the first biochemically characterized bourbonane-producing sesquiterpene synthase from red algae.23 In another study on diosgenin biosynthesis, ZnCI2-tpt helped decipher the structure of biosynthetic intermediate 16S,22S-dihydroxycholesterol (27), assisting in understanding the catalytic sequence of cytochrome P450-mediated spiroketal steroid biosynthesis in plants.24 Dehydrosecodines are considered biosynthetic intermediates for many indole alkaloids, but the high reactivity of dehydrosecodine hinders their isolation and structure elucidation. Fujita et al. achieved the first X-ray structural determination of a dehydrosecodine-type compound 28 by encapsulating it in ZnCI2-tpt.25 Abe et al. used ZnCI2-tpt to characterize the products 29–33 generated by chemical enzymatic methods, facilitating the exploitation of the potential of meroterpenoid cyclases and expansion of the chemical space of fungal meroterpenoids.26 PcCs is a chimeric enzyme of prenyltransferase-diterpene synthase discovered in Penicillium chrysogenum MT-12. Due to the broadened NMR signals of its catalytic product, determining the structure solely by NMR analysis posed challenges. However, the use of ZnCI2-tpt proved successful in revealing the structure of the catalytic product 34, showing a 6/5/5/5 fused ring system.27 In addition, Abe et al. determined the structure of astellifadiene (35), a sesterterpene hydrocarbon with a 6/8/6/5 ring system.28 This compound was obtained from genome mining and heterologous expression of fungal terpene synthase. These results highlight the efficacy of the crystalline sponge method in the structure elucidation of complex hydrocarbon-based NPs.
Understanding the metabolism of new drug candidates is important during drug development, as the metabolites may reflect the efficacy and safety of drugs. In most cases, metabolites are present in trace amounts within complex mixtures, making the full characterization of their structures challenging. Fujita et al. used HPLC coupled with the crystalline sponge method to analyse the structure of microgram metabolites produced by enzymatic reductions in bread yeast. In the case of microbial metabolism, ZnCI2-tpt was used as a crystalline sponge to elucidate the structures of three trace metabolites.29 Using the same crystalline sponge, Badolo et al. identified phase I and phase II metabolites of gemfibrozil generated from in vitro liver microsomes or S9 fractions.30 Oxidation is one of the most common occurrences in drug metabolism, and identifying oxidation sites and newly established stereochemistry during drug metabolism is critical. Fujita et al. treated α-humulene on a microgram scale with various oxidants to simulate the oxidative metabolism of drug molecules and analysed the products using the crystalline sponge method. This led to the successful structure determination of seven oxidation products, including their relative and absolute stereochemical configurations.31
2.2 The advantages and limitations of the crystalline sponge method
As mentioned above, Fujita's crystalline sponge method demonstrated several outstanding advantages, including the ability to analyse samples on a nano- to microgram scale without the need for organic molecule crystallization, and the capability to determine the absolute configuration of chiral molecules. Since the development of ZnCI2-tpt as a crystalline sponge, other porous crystalline materials such as MOFs and supramolecular organic frameworks have also been investigated for structure elucidation.32–38 Nevertheless, the crystalline sponge method also has the following limitations: (i) organic molecules must be small enough to fit within the cavities of the framework, limiting the maximum molecular weight. Up to now, the largest molecular weight resolved using Fujita's crystalline sponge is 428 Da. (ii) The electron-deficient and hydrophobic cavities may not be suitable for analyzing the structures of hydrophilic molecules. (iii) The use of cyclohexane as a soaking solvent might be problematic for analyzing compounds insoluble in cyclohexane. (iv) The sample must have high purity, as impurities can also be absorbed into the cavity of the crystalline sponge, interfering with the analysis. (v) The low occupancy of organic molecules and the co-existence of many solvent molecules may result in poor diffraction data, potentially leading to structural misassignments.3 Crystalline mate method
Inspired by using chaperones to aid protein crystallization through non-covalent binding, a co-crystallization strategy has been developed for the structure elucidation of organic molecules. This alternative method for determining the structure of difficult-to-crystallize compounds involves using discrete molecular entities as crystalline mates to co-crystallize through non-covalent interactions followed by crystallographic analysis. Unlike the crystalline sponge method, which uses a pre-prepared porous crystal to absorb organic molecules, the crystalline mate method involves a crystal growth process of the target molecule and the crystalline mate together. In the obtained co-crystals, the organic molecule and the crystalline mate were packed in a stoichiometric ratio, with 100% occupancy. Therefore, the crystalline mate method often achieves higher structural resolution accuracy and configurational determination capabilities. This is particularly beneficial when dealing with complex chiral NPs that are difficult to crystallize on their own. Triphenylphosphine oxide is a well-known crystalline mate for hydrogen-donor molecules.39 In addition, porphyrins, cyclodextrins, and pillararenes also showed their ability to form crystalline inclusion complexes with simple organic molecules.40,41 It is worth pointing out that the recently reported tetraaryladamantanes (TAAs) and cyclic trinuclear complexes (CTCs) are two promising crystalline mates, showing significant potential in the structure elucidation of NPs.3.1 Tetraaryladamantanes for molecular structure determination
The tetraaryladamantanes (TAAs) are a family of organic molecules with tetrahedral-like symmetry, consisting of four aromatic groups attached to the bridgehead positions of an adamantane core. In an early study, Clemens Richert and co-workers found that 1,3,5,7-tetrakis(2,4-dimethoxyphenyl)adamantane (TDA) readily forms crystalline inclusion complexes with various liquid reagents, such as benzoyl chloride, acetylchloride, and phosphorus trichloride. The process involves heating and dissolving tetraaryladamantane and liquid molecules at high temperatures until the solution is clear and transparent, then slowly cooling to room temperature. After several hours to days, co-crystals containing organic molecules can be obtained. Initially, they utilized the co-crystallization capability of TDA to create a crystalline coating for hazardous and malodorous agents to mask their reactivity and odor.42 In 2020, Richert et al. started to apply TAAs as crystalline mates for the absolute configuration determination of small organic molecules (Fig. 3).43 1,3,5,7-Tetrakis(2,4-diethoxyphenyl)adamantane (TEO), a homologue of TDA, has been demonstrated to have the superior co-crystallization ability. In single-run thermal crystallization experiments with TEO, Richert et al. successfully determined the structures of 21 liquid molecules, including 8 natural compounds (36–43). This success led to the proposal of a spectroscopy-co-crystallization coupled method. In a proof-of-concept demonstration, 3 mg of nicotine (41) was first subjected to 1H NMR data collection. After evaporation of some deuterated solvent, 1 mg of TEO powder was added directly to the NMR tube. The resulting mixture was left standing overnight at 20 °C, producing high-quality co-crystals for X-ray diffraction. Structure refinement resulted in an excellent Flack parameter [0.00 (8)], allowing for the determination of the absolute configuration of nicotine as S. | ||
| Fig. 3 TAA-based crystalline mate method. (A) Chemical structures of TEO, TBrO, TFM, and TMAPA. (B) Packing arrangement in representative co-crystals. (C) Chemical and crystal structures of NPs elucidated by TEO, TBrO, TFM, or TMAPA. | ||
1,3,5,7-Tetrakis(2-bromo-4-phenyl)adamantane (TBrO) was developed to be more compatible with lipophilic compounds, by introducing halogens to strengthen interactions with saturated hydrocarbons. In addition, Br atoms in TBrO generate strong anomalous dispersion signals in X-ray crystallography, facilitating the determination of absolute configurations. Using TBrO as a crystalline mate, Richert et al. successfully determined the structures of five lipophilic organic molecules, including geraniol (44), farnesol (45), α-humulene (46), and muscone (47). In another study, TBrO was used to confirm the relative configurations of the cis–trans isomers of propargyl cyclopropanes.44 So far, the largest organic molecule successfully encapsulated by TBrO is 47 with a molecular weight of 238 g mol−1, however, its structural resolution is insufficient for unambiguously determining the absolute configuration.43
In 2024, Richert et al. developed the second-generation crystalline mate 1,3,5,7-tetrakis(2-fluoro-4-methoxyphenyl)adamantane (TFM) by replacing the Br atoms of TBrO with F atoms. This new crystalline mate allows for the analysis of the structures of larger, linear, and flexible NPs, including myrcene, R-citronellal, geraniol, R-citronellol, decanal, decanoic acid, farnesol, and phytol (48–54, Fig. 3C).45 It should be pointed out that those with hydrogen-bonding donor sites such as hydroxyl and carboxyl groups may not be suitable for co-crystallization with TMF and may require acetylation in advance. The encapsulation of acyclic compounds can be achieved through a temperature jump method, which involves heating to 120–140 °C to form a clear solution, and then transferring the hot solution directly to an 8 or −20 °C refrigerator for co-crystallization. When using TMF for elucidating the structure of 54 (molecular weight: 338 g mol−1), difficulties were encountered due to severe disorder of the molecule, leading to ambiguity in its configuration. Although the R value was still within the acceptable range, the determination of the absolute configuration of the stereogenic center became less reliable. Richert et al. later explored the upper limit of TFM as a crystalline mate by testing its co-crystallization with squalene, a linear triterpene with a molecular weight of 411 g mol−1. The results showed that squalene could be encapsulated within the crystal lattice of TFM, but the lack of ordering hindered further refinement and posed challenges for elucidating its structure.
Richert's method for structure elucidation involves directly heating oily organic molecules and powdered TAA without the need for solvents, where the oily organic molecules act as solvents for dissolving TAA. Therefore, this method may not be suitable when the targeted oily organic molecules have difficulty dissolving TAA. To overcome this limitation, Wang et al. introduced a TAA crystalline mate with anthracene arm, 1,3,5,7-tetrakis(4-(10-methoxyanthracen-9-yl)phenyl)adamantane (TMAPA). TMAPA can co-crystallize with various oily organic molecules (55–62) under solvent-assistance conditions.46 The addition of anthracene moieties leads to the extension of the four arms of TMAPA, allowing for adaptive rotation to capture organic molecules for co-crystallization. Using chlorobenzene or tetrahydrofuran as solvents, 51 co-crystals of organic molecules (chiral or achiral) and TMAPA were determined quickly. The encapsulated molecules had molecular weights ranging from 60.1 to 209.44, and oil–water partition coefficient (log P) values ranging from −0.811 to 4.76. Attempts were made with larger organic molecules, such as α- and β-caryophyllene, δ-catenene, 15-crown-5, 12-crown-4, farnesyl alcohol, methyl L-pyrog-lutamate, muscone, and all-trans-retinol. However, SCXRD analysis of the obtained crystals shows organic molecules were not involved in the co-crystallization, suggesting an upper limit for TMAPA as a crystalline mate.
3.2 The advantages and limitations of tetraaryladamantanes
In co-crystal structures, most organic molecules and TTA do not form strong directional interactions such as hydrogen bonds, which is atypical in host–guest systems. The co-crystallization ability of TAAs can be attributed to the rigidity, symmetry, and effective packing of their core skeleton structures. In addition, the alkoxy group on the aryl group acts as a “molecular clamp” that can capture different organic molecules, while the lipophilic surface of the alkoxy group can provide van der Waals interactions for these molecules. Overall, TAAs possess significant advantages for structure elucidation, particularly for liquid organic molecules. On the one hand, co-crystal growth is a kinetic process, enabling the acquisition of high-resolution structures with 100% occupancy of organic molecules once the crystal nucleus is formed. On the other hand, the co-crystallization process does not involve solvent molecules and does not require solvent screening, making it easy to use. However, some drawbacks were observed for this method: (i) the currently available TAAs are not suitable for complex organic molecules, with the limit for well-resolved structures appearing to be 338 g mol−1. (ii) High-temperature cooling crystallization hinders the analysis of thermally unstable compounds. (iii) Organic molecules act as solvents for powdered TAAs in thermal crystallization, limiting the use of TAAs in the analysis of liquid compounds.3.3 Cyclic trinuclear complexes for molecular structure determination
Cyclic trinuclear complexes (CTCs) are planar coordination compounds composed of three monovalent coinage metal cations and three bridging heterocyclic ligands. They are versatile supramolecular synthons and a tuneable platform that can provide supramolecular interactions with various substrates.47 In 2023, Wang, Lu, Ye, Li, and co-workers explored the application of cyclic trinuclear Ag(I) pyrazolate complex (Ag3Pz3) as an adaptive crystalline mate for a series of challenging organic molecules (Fig. 4).4 Ag3Pz3 was prepared by refluxing a mixture of Ag2O and 3,5-bis(trifluoromethyl)pyrazole in diethyl ether for six hours. Density functional theory calculations on Ag3Pz3 revealed a highly positive electrostatic potential on the central Ag3N6 metallacycle. Researchers hypothesized that the strong π-acidity of Ag3Pz3 could enable it to engage in π-acid⋯base supramolecular interactions with organic molecules bearing electron-rich functionalities. In addition, the trifluoromethyl groups on the exterior of Ag3Pz3 not only rendered the complex soluble in various organic solvents (such as cyclohexane, dichloromethane, acetonitrile, and methanol) but also constrained the flexibility of co-crystallized structures by forming C–H⋯F interactions with neighbouring organic molecules. Notably, the presence of the heavy silver atoms provided strong anomalous scattering effects, which could aid in determining the absolute configuration of chiral molecules during X-ray diffraction analysis. | ||
| Fig. 4 Ag3Pz3-based crystalline mate method. (A) Four types of binding modes observed in the co-crystals of Ag3Pz3 and NPs. (B) Representative crystal structures of four binding modes. (C) Chemical structures of NPs elucidated by Ag3Pz3. | ||
The use of Ag3Pz3 as a crystalline mate resulted in dozens of co-crystals of Ag3Pz3 and organic molecules in their initial crystallization attempts (63–87). Subsequent SCXRD analysis revealed high-resolution structures of these co-crystals, allowing for the unambiguous determination of the absolute configurations of chiral molecules (Fig. 4C). Remarkably, researchers were able to demonstrate the co-crystallization with as little as 5 μg of chrysophanol (68), a yellowish natural anthraquinone. The quality of the obtained co-crystals was sufficient to elucidate the complete structure, suggesting the potential of Ag3Pz3 for characterizing trace amounts of NPs. Ag3Pz3 is highly adaptive and can pack with organic molecules in four different binding modes in accordance with their structural and electronic properties (Fig. 4A). Specifically, planar molecules were found to bind to Ag3Pz3 in a sandwich-like mode, displaying strong π-acid⋯π-base interactions with plane-to-plane distances between the organic molecule and Ag3Pz3 shorter than the sum of their van der Waals radii. In contrast, non-planar molecules with electron-rich functionalities tend to bind to Ag3Pz3 through π-acid··base interactions, forming single-site, dual-site, or multi-site binding modes depending on the number of electron-rich sites on the organic molecules (Fig. 4B).
In the co-crystallization of enantiomers, (±)-, (+)-, and (–)-securinine (76) were successfully co-crystallized with Ag3Pz3 in similar binding modes. This indicates that achiral Ag3Pz3 cannot distinguish enantiomers of chiral compounds during co-crystallization. In addition to volatile liquid compounds such as trans-anethol (64), Ag3Pz3 can also be used for the structure elucidation of complex NPs. Eurycolactone E (85) is a quassinoid featuring a highly oxygenated tetracyclic scaffold and ten contiguous stereocenters. The structure of 85 was previously determined solely through the NMR method, with its absolute configuration remaining unclear. With Ag3Pz3 as a crystalline mate, researchers were able to confirm its structure and determine the absolute configuration for the first time. The conformational flexibility of macrocyclic NPs often makes their structure elucidation and configuration assignment challenging, sometimes leading to errors. For instance, the stereochemistry of the macrocyclic diterpene ester euphornin (86) was initially misassigned in 1981 and later corrected in 1989 by Yamamura et al. using a chemical derivatization method. The co-crystallization of 86 and Ag3Pz3 revealed its correct structure with an unambiguous absolute configuration.
The robustness and adaptability of Ag3Pz3 in the co-crystallization with a wide range of natural compounds in trace quantities prompted researchers to investigate this crystalline mate for the structure elucidation of mixtures directly without separation. They studied mixed samples of molecules that bind in the same mode and mixed samples of molecules with different binding modes. Interestingly, in one-pot co-crystallization, all organic molecules in mixtures were separately co-crystallized with Ag3Pz3 in similar manners as they were in their individual co-crystallization with Ag3Pz3. Crude extracts of medicinal plants are more complicated than artificially mixed samples. By directly adding Ag3Pz3 to a solution containing Cleistocalyx operculatus extract and evaporating it at room temperature, researchers were able to obtain co-crystals with five different polymethylflavonoids (88–92), including two new ones (88 and 92). Compared to traditional NP chemistry workups, this crystalline mate approach eliminates tedious separation and extensive spectroscopic analyses (UV, IR, MS, 1D, and 2D NMR), allowing for direct visualization of the 3D chemical structure of unknown NPs in crude extracts (Fig. 5).
 | ||
| Fig. 5 Ag3Pz3 as crystalline mate for direct structure elucidation of NPs in crude extracts. (A) Workflow for the co-crystallization of Cleistocalyx operculatus crude extract with Ag3Pz3. (B) Crystal structures of Ag3Pz3·88–Ag3Pz3·90 and Ag3Pz3·91·92 (inset: photo of single crystal). | ||
3.4 The advantages and limitations of cyclic trinuclear complexes
Notably, Ag3Pz3 has been shown to form co-crystals with substances other than NPs, such as triethylammonium nitrate, fullerene, and organometallics, demonstrating its broad applicability as a crystalline mate.48 Other CTCs also have the potential to serve as crystalline mates.49 As the first metal–organic complexes for co-crystallization with organic compounds, the use of CTCs for molecular structure elucidation offers several outstanding merits. Firstly, the strong anomalous scattering of CTCs and their high adaptability to diverse organic molecules enable high-resolution structure determination and stereochemical assignment of chiral molecules. Secondly, CTCs as crystalline mates are not restricted by the size and shape of organic molecules, allowing for the structure elucidation of large-sized and oddly-shaped molecules. Additionally, CTCs can be used directly for the structure elucidation of organic molecules in mixtures and even in crude extracts, which has never been reported before. Nevertheless, CTCs have the following limitations: (i) organic molecules with strong oxidizing or reducing potential (e.g., ascorbic acid and 3-chloroperoxybenzoic acid) may compromise the structure of CTCs. (ii) Ag3Pz3 primarily works for flat or oxygenated organic molecules, with limited exploration of molecules lacking electron-rich functionalities. Further modification of CTCs, such as adjusting the acidity/basicity of CTCs or introducing chiral substituents on the ligands, may expand their applicability for the structure elucidation and enantiomeric recognition of different types of compounds.4 Encapsulated nanodroplet crystallization method
4.1 Encapsulated nanodroplet crystallization for molecular structure determination
In 2019, Bernhard Spingler and co-workers discovered a method for inducing crystal growth of water-soluble organic salts by placing microliters of these salts in a porous plate and covering them with inert oil, allowing for continuous evaporation of water from the solution.50 They demonstrated the effectiveness of this method for screening active pharmaceutical ingredients in salt forms. Traditional microbatch under-oil technique necessitates the use of aqueous solutions and paraffin oils to maintain phase separation and droplet mobility, posing challenges for organic molecules with poor solubility in water. To address these limitations, Michael R. Probert and co-workers developed a high-throughput encapsulated nanodroplet crystallization (ENaCt) technique for crystal growth.5 This innovative method involves encapsulating nanoliter-scale droplets of organic solvent in inert viscous oils with low miscibility in common organic solvents. Oil encapsulation causes a slow evaporation of solvent in the nanoliter-scale droplets. As solvent slowly evaporates from the droplet, the concentrations of organic molecules reach their saturation points, leading to the formation of single crystals (Fig. 6A). It is worth pointing out that the ENaCt experiments can be set up and performed using a liquid-handling robot, enabling semi-automated operation, in which hundreds of individual crystallization experiments can be initiated within a few minutes. This high-throughput parallel screening approach allows for rapid exploration of crystallization space and provides reliable access to crystals suitable for diffraction-based characterization.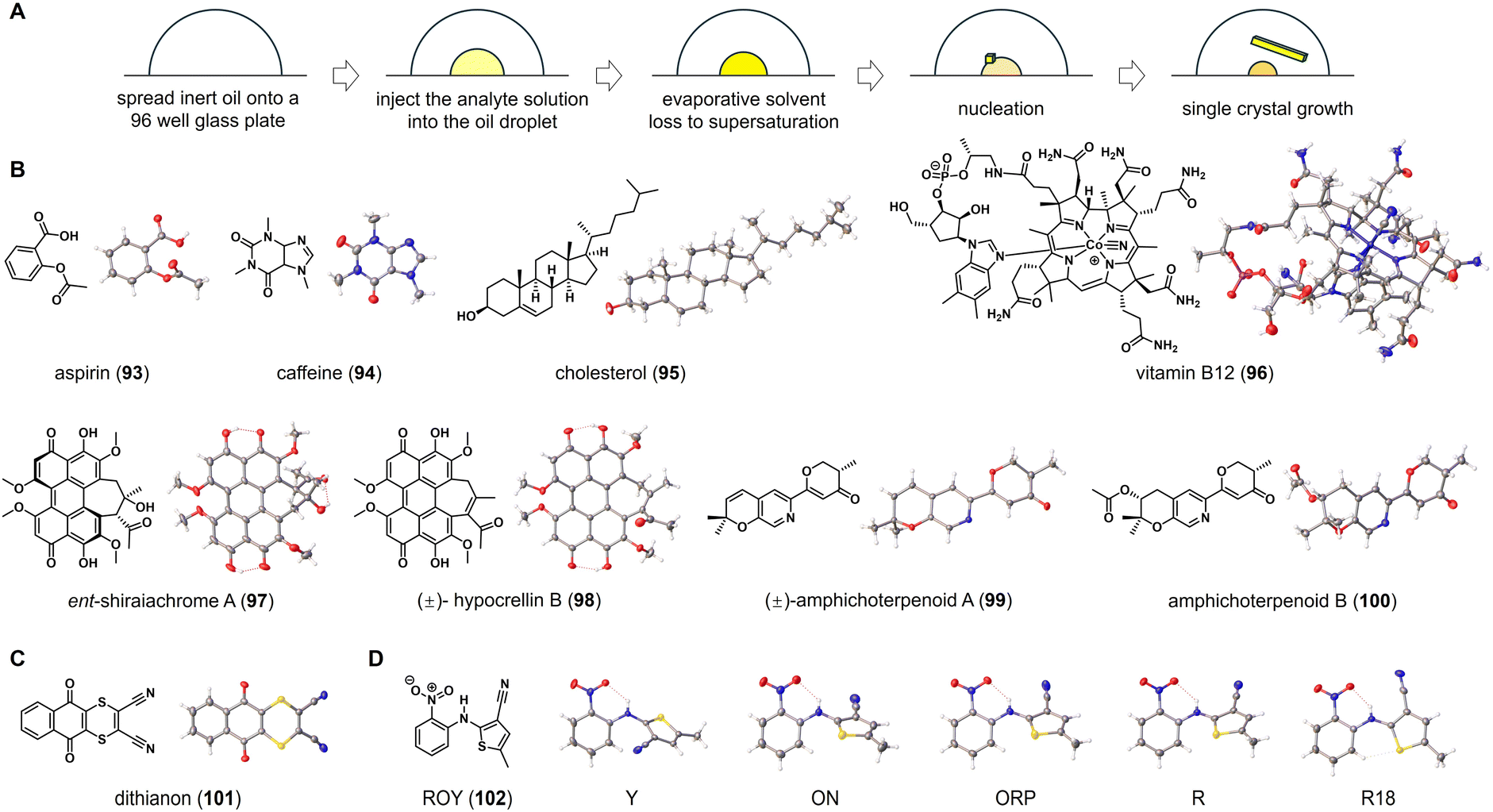 | ||
| Fig. 6 ENaCt for structure elucidation of organic molecules. (A) The workflow and principle of ENaCt method. (B–D) Chemical and crystal structures of organic molecules elucidated by ENaCt. | ||
As a general procedure of using a liquid-handling robot, 250 nL of inert oil droplets are placed into a 96-well glass plate. Then, solutions of 50 nL of organic molecules in suitable organic solvents are added to the droplets. The resulting 96-well glass plate is sealed and stored at room temperature. After a few days, single crystals suitable for X-ray diffraction can be observed under cross-polarizing light microscopy. In the crystallization process, it has been found that fluorous (FC-40) and non-fluorous (PDMSO) are ideal oils for encapsulating nanodroplets, while using organic solvents such as acetone, ethyl acetate, ethanol, 1,2-dichloroethane, N,N-dimethylformamide, and dimethyl sulfoxide. Single crystals of various organic molecules, including aspirin (93), caffeine (94), cholesterol (95), and vitamin B12 (96), have been obtained from their respective 96-well plates. The subsequent SCXRD analysis allows for the determination of their molecular structure, including absolute configuration (Fig. 6B).
Ent-shiraiachrome A (97) and hypocrellin B (98) are two fungal perylenequinones isolated from Shiraia-like sp. (strain MSX60519). Due to the limited sample quantities, Shabnam et al. conducted a study on the crystallization of 97 and 98 using the ENaCt method.51 Out of 288 individual crystallization experiments for 97, 96 wells in the glass plate (33%) appeared microcrystals, and 1 well (0.3%) contained single crystals suitable for SCXRD. In the case of 98, 10 wells (3%) of the 288 individual ENaCt experiments contained single crystals suitable for SCXRD. Crystallographic analysis revealed that 97 crystallizes with two solvent molecules, dimethylformamide and water, while 98 is a pair of P/M racemic mixtures that crystallize with dimethyl sulfoxide molecules. In a separate study of fungal metabolites by Chen et al., the modified ENaCt method was applied to the crystallization of amphichoterpenoids, picoline-derived meroterpenoids with a pyrano[3,2-c]pyridinyl-γ-pyranone scaffold. The crystal growth was successful and SCXRD was used to establish the molecular structures of amphichoterpenoids A and B (99 and 100).52 In Probert's original report, ENaCt has also been applied to the challenging crystallization of dithianon (101), previously considered “uncrystallizable”. In 384 individual crystallization experiments, block-like crystals of 101 were obtained, and the structure of 101 was successfully elucidated through SCXRD (Fig. 6C). This, along with the others discussed above, indicated that the ENaCt can effectively solve the challenging crystallization problem of organic molecules.
Polyforms are particularly important in the pharmaceutical industry, as different polymorphs of a drug molecule might exhibit significantly different physical properties (such as solubility and stability). ENaCt has been proven to be useful for screening polymorphic forms of organic molecules. By using 5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile (ROY, 102) as a test molecule, nanodroplets of ROY solution were encapsulated in a series of inert oils for crystal growth. After extensive screening of crystallization conditions, all four known ROY polymorphs (Y, R, ON, and ORP) were obtained. In addition, a new polymorph, R18, was also crystallized and successfully analyzed by SCXRD, making it the thirteenth discovered polymorph (Fig. 6D).
4.2 The advantages and limitations of encapsulated nanodroplet crystallization method
In short, the ENaCt method is an efficient crystallization screening technique that allows for simultaneously running hundreds of parallel experiments to investigate crystal formation. This enables quick discovery of crystallization conditions. The main advantages of this method include semi-automated operation with a liquid-handling robot and high-throughput crystallization screening that only requires microgram samples. In addition, unlike crystalline sponge and crystalline mate, this method enables the organic molecules to crystallize on their own, thus providing insight into molecular packing and interactions. However, there are some drawbacks associated with this method: (i) extensive exploration of crystallization conditions are required for growing high-quality single crystals, (ii) preparing encapsulated nanodroplets and maintaining crystallization equipment can be technically challenging, (iii) the ENaCt method is not suitable for organic molecules that are oily or liquid at room temperature.5 Microcrystal electron diffraction method
5.1 Microcrystal electron diffraction for molecular structure determination
Electron cryo-microscopy (CryoEM) is an advanced crystallographic technique that utilizes electron diffraction to analyse the structures of micro- or nanocrystals. Compared to X-rays, electron beams, with their shorter wavelengths and stronger interactions with matter, are able to generate high-resolution diffraction patterns from smaller crystals, while depositing less energy into them. Microcrystal electron diffraction (MicroED) was initially applied in the field of structural biology. Since the successful elucidation of the protein structure of lysozyme in 2013,53 MicroED has been widely used for structural analysis of proteins, protein–ligand complexes, organometallics, NPs, and drug molecules.54–56 The samples for MicroED analysis are typically thin, with a thickness of less than 100 nm and a volume of approximately 1/6000 of normal single crystals, making them difficult to identify using traditional optical microscopes. Preliminary evaluation of the crystallinity of samples can be done using powder X-ray diffraction (PXRD), with negative-stain EM further confirming the presence of nanocrystals. After loading the crystalline sample into CryoEM, a grid screening was first performed under a low magnification setting to identify promising areas, with the identified nanocrystals positioned at the centre of the electron beam for diffraction analysis. Cryogenic techniques are employed to minimize sample damage from electron beams during grid preparation and data collection. A finely focused electron beam passing through a rotating nanocrystal generates a diffraction pattern, which was captured by a highly sensitive electron detector. Advanced computational techniques are then used to index diffraction spots, integrate their intensity, and solve phase problems, providing information on the 3D arrangement of atoms within the crystal (Fig. 7A). | ||
| Fig. 7 MicroED for structure elucidation of NPs. (A) The workflow of MicroED method. (B) Chemical and crystal structures of NPs resolved by MicroED. Reprinted with permission from ref. 57. Copyright 2018 American Chemical Society. Reprinted with permission from ref. 58. Copyright 2021 The Author(s), under exclusive licence to Springer Nature America, Inc. Reprinted with permission from ref. 59. Copyright 2021 American Chemical Society. Reprinted with permission from ref. 60. Copyright 2024 Published by Elsevier B.V. on behalf of Chinese Chemical Society and Institute of Materia Medica, Chinese Academy of Medical Sciences. | ||
Tamir Gonen and co-workers demonstrated that seemingly amorphous materials, such as solid powders isolated by silica gel chromatography and rotary evaporation, actually contain microcrystals that can be used for MicroED analysis. This enables rapid and high-quality elucidation of their molecular structures at an atomic resolution (Fig. 7B).57 Initially, MicroED was tested on the natural steroid progesterone (103), obtained in powdered form from a chemical supplier. The diffraction data collection took less than 3 minutes, resulting in a high-resolution (1 Å) crystal structure of 103. Encouraged by the above results, researchers expanded their exploration of MicroED to include a variety of NPs and drugs. As a result, MicroED analysis successfully elucidated the structures of compounds such as cinchonine (104), limaspermidine (105), brucine (106), and thiostrepton (107) from their powdered forms. In addition, Gonen et al. conducted direct MicroED analysis on four seemingly amorphous solids collected from the column chromatography and determined the atomic-resolution structures of two of the compounds. In cases where samples are heterogeneous mixtures, MicroED can directly identify microcrystals of different compounds within the mixtures and perform structure elucidation on these compounds separately.57
Nelson et al. reported a combination of MicroED and genome mining to accelerate the identification of NPs (Fig. 7B).58 By integrating multiple genes into the heterologous biosynthetic host, they successfully isolated a new metabolite named Py-469 (108). Without the tedious spectroscopic characterization, MicroED was used to elucidate the crystal structure of 108 with a resolution of 0.85 Å and its relative configuration within a few hours. Subsequently, researchers investigated the structure of fischerin (109), a metabolite isolated from Neosartorya fischeri 25 years ago. Previous attempts to determine its relative stereochemistry were inconclusive, with only speculations based on quantum chemical calculations. Electron micrographs of pale-yellow particles precipitated from a CH3CN–H2O solution of 109 revealed the presence of microcrystals. After hundreds of crystallization condition optimizations, high-quality microcrystals were obtained for MicroED analysis and data collection, providing a 1.05 Å ab initio resolution. The refinement of MicroED data unambiguously determined the structure of 109 and corrected the previously misassigned relative stereochemistry. In addition, they were able to elucidate the structure of austinol (110), an impurity associated with 109, with a resolution exceeding 1.00 Å. These results showcase the collaborative potential of synthetic biology and MicroED in the field of NP discovery.
Lomaiviticins are a group of dimeric genotoxic metabolites with unusual diazocyclopentadiene functional groups and 2–4 deoxyglycoside residues. Due to the fact that only 6 out of 19 carbon atoms in the monomeric aglycon unit are proton-attached, their structure determination by NMR spectroscopic analysis is difficult. In 2021, Herzon et al. used MicroED to successfully determine the structure of lomaviticin C (111) (Fig. 7B), leading to a revision of the original core of lomaiviticins A–C.59 In a recent phytochemical investigation of Plumeria rubra, Wang et al. discovered three [2 + 2] cyclobutane-containing iridoid glycoside dimers with molecular weight of more than 1200. Through MicroED analysis, the structure of plumerubradin B (112) was completely resolved, showcasing the potential of this technique in elucidating complex structures, including the ones with multiple stereogenic centers and highly substituted sugar moieties (Fig. 7B).60
5.2 The advantages and limitations of microcrystal electron diffraction method
Compared to traditional X-ray analysis, the MicroED method offers multiple advantages, such as the ability to provide detailed structural information of complex molecules from small crystals that cannot be analysed by in-house single-crystal diffraction instruments. Additionally, MicroED accelerates the efficiency of structure elucidation by collecting diffraction data within minutes and accurately observing hydrogen atoms due to the strong interaction between electrons and light elements. Despite the exciting achievement with MicroED, it is important to acknowledge its limitations: (i) a micro- and nanocrystal with sufficient diffraction is a prerequisite for MicroED, limiting the analysis of non-crystalline compounds. (ii) The intensity of the electron beam may lead to sample degradation. (iii) Samples must maintain the crystalline form under vacuum conditions, as electrons can only propagate in a vacuum, restricting the study of hydrates and solvates. (iv) Stereochemical elucidation is often limited to dynamical refinement or a known chiral reference, which is not yet routine. (v) High equipment cost of MicroED limits its accessibility.6 Conclusions and prospects
This article focuses on four advanced crystallography techniques that have emerged in recent years, highlighting their applications in determining the structures of difficult-to-crystallize organic molecules and accelerating the discovery of NPs. The crystalline sponge and crystalline mate methods fall under the category of molecule/material-assisted crystallization techniques, where exogenous substances are used to facilitate the ordered arrangements of organic molecules. In principle, the crystalline sponge method entails the post-orientation of organic molecules within porous crystals, often referred to as a “post-crystallization” process. A tiny single crystal is sufficient for this method, with sample mass ranging from micrograms to nanograms. On the other hand, the crystalline mate method involves in situ crystal growth of organic molecules and crystalline mate. As a notable example of crystalline mate, Ag3Pz3 is viewed as a new hope for crystallization,61 showcasing its versatility and adaptability in accommodating organic compounds of different sizes and shapes. ENaCt is a self-crystallization method of organic molecules that produces crystals without the use of exogenous molecules or materials. It is a robot-assisted high-throughput technique for exploring the crystallization space and polymorphic forms of organic-soluble small molecules. MicroED is a crystallographic technique that allows for the structure elucidation of nanocrystals, reducing the need for lengthy crystallization experiments typically associated with SCXRD and shortening the time for diffraction data collection. From the perspective of material availability, crystalline sponges (ZnI2-tpt) and crystalline mates (TAAs and Ag3Pz3) can be synthesized from simple materials through one or several reaction steps, while inert oils used for ENaCt are commercially available. Therefore, these methods can be smoothly implemented by researchers in their laboratories. However, the high cost of equipment required for MicroED might limit its accessibility. In summary, each method has its own advantages and limitations (Table 1), and researchers are encouraged to choose the most suitable method based on the specific challenges in their structural analysis.| Method | Molecular properties of analyte | Crystallographic information of analyte | |||||
|---|---|---|---|---|---|---|---|
| Liquid molecules | MW > 500 Da | Trace analysis | Mixture analysis | Hydrophilic molecules | Absolute configuration | Supramolecular interactions | |
| Crystalline sponge | √ | × | √ | × | × | √ | × |
| TAA-crystalline mate | √ | × | × | × | × | √ | × |
| CTC-crystalline mate | √ | √ | √ | √ | × | √ | × |
| ENaCt | × | √ | √ | × | √ | √ | √ |
| MicroED | × | √ | √ | √ | √ | Not routine | √ |
It is anticipated that new types of auxiliary crystallization molecules/materials with properties similar to crystalline sponge or crystalline mate will continue to be developed. The potential is unlimited for molecular structure elucidation through the precise regulation and optimization of these crystallization methods, enabling customized host systems for the crystallization of specific compounds. Some initial exploratory efforts have already been undertaken. For example, Scheer et al. developed BTB-MOF-24 for elucidating fatty-acid-based compounds.62 Fujita et al. used a saccharide-based crystalline sponge for hydrophilic compounds.63 The use of crystalline mates for in situ structure elucidation from natural extracts has greatly accelerated the discovery of NPs and is expected to continue to pique the interest of scientists. MicroED could also be utilized for analyzing crystals obtained from the crystalline sponge, crystalline mate, and ENaCt methods. Additionally, the integration of artificial intelligence and machine learning in the discovery of host systems, crystal growth, and structure prediction is poised to usher into a more automated and intelligent era of crystallographic structure analysis.
7 Data availability
No new data or code were created for this article. All data used can be obtained from the original publications. Crytallographic data can be downloaded from the Cambridge Crystallographic Data Center (https://www.ccdc.cam.ac.uk/structures/?) using the published CCDC numbers.8 Author contributions
The manuscript was written by all authors, and all authors have given approval to the final version of the manuscript.9 Conflicts of interest
There are no conflicts to declare.10 Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82293681 (82293680), 82204234, 82321004, and 82430108), the Guangdong Basic and Applied Basic Research Foundation (No. 2022B1515120015), the Guangdong Major Project of Basic and Applied Basic Research (No. 2023B0303000026), and the Science and Technology Projects in Guangzhou (No. 202102070001).11 References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- N. Zigon, V. Duplan, N. Wada and M. Fujita, Angew. Chem., Int. Ed., 2021, 60, 25204–25222 CrossRef CAS PubMed.
- J. G. Song, J. Zheng, R. J. Wei, Y. L. Huang, J. Jiang, G. H. Ning, Y. Wang, W. Lu, W. C. Ye and D. Li, Chem, 2024, 10, 924–937 CAS.
- A. R. Tyler, R. Ragbirsingh, C. J. McMonagle, P. G. Waddell, S. E. Heaps, J. W. Steed, P. Thaw, M. J. Hall and M. R. Probert, Chem, 2020, 6, 1755–1765 CAS.
- E. Danelius, S. Halaby, W. A. van der Donk and T. Gonen, Nat. Prod. Rep., 2021, 38, 423–431 RSC.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246–252 CrossRef CAS PubMed.
- M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139–151 CrossRef CAS PubMed.
- T. R. Ramadhar, S. L. Zheng, Y. S. Chen and J. Clardy, Chem. Commun., 2015, 51, 11252–11255 RSC.
- N. Wada, K. Kageyama, Y. Jung, T. Mitsuhashi and M. Fujita, Org. Lett., 2021, 23, 9288–9291 CrossRef CAS PubMed.
- T. R. Ramadhar, S. L. Zheng, Y. S. Chen and J. Clardy, Acta Crystallogr., Sect. A, 2015, 71, 46–58 CrossRef CAS PubMed.
- S. Urban, R. Brkljaca, M. Hoshino, S. Lee and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 2678–2682 CrossRef CAS PubMed.
- K. Kai, M. Sogame, F. Sakurai, N. Nasu and M. Fujita, Org. Lett., 2018, 20, 3536–3540 CrossRef CAS PubMed.
- S. Hoshino, T. Mitsuhashi, T. Kikuchi, C. P. Wong, H. Morita, T. Awakawa, M. Fujita and I. Abe, Org. Lett., 2019, 21, 6519–6522 CrossRef CAS PubMed.
- Y. Morishita, T. Sonohara, T. Taniguchi, K. Adachi, M. Fujita and T. Asai, Org. Biomol. Chem., 2020, 18, 2813–2816 RSC.
- Y. Taniguchi, T. Kikuchi, S. Sato and M. Fujita, Chem.–Eur. J., 2022, 28, e202103339 CrossRef CAS PubMed.
- S. Kamisuki, H. Shibasaki, K. Ashikawa, K. Kanno, K. Watashi, F. Sugawara and K. Kuramochi, J. Antibiot., 2022, 75, 92–97 CrossRef CAS PubMed.
- F. Habib, D. A. Tocher, N. J. Press and C. J. Carmalt, Microporous Mesoporous Mater., 2020, 308, 110548 CrossRef CAS.
- S. Lee, M. Hoshino, M. Fujita and S. Urban, Chem. Sci., 2017, 8, 1547–1550 RSC.
- S. Lee, M. Hoshino, M. Fujita and S. Urban, Molecules, 2017, 22, 211/1–211/8 CrossRef PubMed.
- N. Zigon, T. Kikuchi, J. Ariyoshi, Y. Inokuma and M. Fujita, Chem.–Asian J., 2017, 12, 1057 CrossRef CAS PubMed.
- R. D. Kersten, S. Lee, D. Fujita, T. Pluskal, S. Kram, J. E. Smith, T. Iwai, J. P. Noel, M. Fujita and J. K. Weng, J. Am. Chem. Soc., 2017, 139, 16838–16844 CrossRef CAS PubMed.
- B. Christ, C. Xu, M. Xu, F. S. Li, N. Wada, A. J. Mitchell, X. L. Han, M. L. Wen, M. Fujita and J. K. Weng, Nat. Commun., 2019, 10, 1–11 CrossRef CAS PubMed.
- G. Tay, T. Wayama, H. Takezawa, S. Yoshida, S. Sato, M. Fujita and H. Oguri, Angew. Chem., Int. Ed., 2023, 62, e202305122 CrossRef CAS PubMed.
- T. Mitsuhashi, L. Barra, Z. Powers, V. Kojasoy, A. Cheng, F. Yang, Y. Taniguchi, T. Kikuchi, M. Fujita, D. J. Tantillo, J. A. Porco and I. Abe, Angew. Chem., Int. Ed., 2020, 59, 23772–23781 CrossRef CAS PubMed.
- T. Mitsuhashi, T. Kikuchi, S. Hoshino, M. Ozeki, T. Awakawa, S. P. Shi, M. Fujita and I. Abe, Org. Lett., 2018, 20, 5606–5609 CrossRef CAS PubMed.
- Y. Matsuda, T. Mitsuhashi, S. Lee, M. Hoshino, T. Mori, M. Okada, H. Zhang, F. Hayashi, M. Fujita and I. Abe, Angew. Chem., Int. Ed., 2016, 55, 5785–5788 CrossRef CAS PubMed.
- Y. Inokuma, T. Ukegawa, M. Hoshino and M. Fujita, Chem. Sci., 2016, 7, 3910–3913 RSC.
- L. Rosenberger, C. von Essen, A. Khutia, C. Kuehn, K. Urbahns, K. Georgi, R. W. Hartmann and L. Badolo, Drug Metab. Dispos., 2020, 48, 587–593 CrossRef CAS PubMed.
- N. Zigon, M. Hoshino, S. Yoshioka, Y. Inokuma and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 9033–9037 CrossRef CAS PubMed.
- A. D. Cardenal and T. R. Ramadhar, ACS Cent. Sci., 2021, 7, 406–414 CrossRef CAS PubMed.
- S. Lee, E. A. Kapustin and O. M. Yaghi, Science, 2016, 353, 808–811 CrossRef CAS PubMed.
- X. Pei, H. B. Buergi, E. A. Kapustin, Y. Liu and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 18862–18869 CrossRef CAS PubMed.
- Y. Wada, P. M. Usov, B. Chan, M. Mukaida, K. Ohmori, Y. Ando, H. Fuwa, H. Ohtsu and M. Kawano, Nat. Commun., 2024, 15, 81 CrossRef CAS PubMed.
- C. Chen, Z. Di, H. Li, J. Liu, M. Wu and M. Hong, CCS Chem., 2021, 3, 1352–1362 Search PubMed.
- Y. Li, S. Tang, A. Yusov, J. Rose, A. N. Borrfors, C. T. Hu and M. D. Ward, Nat. Commun., 2019, 10, 4477 CrossRef PubMed.
- J. C. Liu, W. P. Huang, Y. X. Tian, W. Xu, W. C. Ye and R. W. Jiang, J. Mater. Chem. A, 2024, 12, 12609–12618 RSC.
- M. C. Etter and P. W. Baures, J. Am. Chem. Soc., 1988, 110, 639–640 CrossRef CAS.
- M. P. Byrn, C. J. Curtis, Y. Hsiou, S. I. Khan, P. A. Sawin, S. K. Tendick, A. Terzis and C. E. Strouse, J. Am. Chem. Soc., 1993, 115, 9480–9497 CrossRef CAS.
- M. Ceborska, Chem. Phys. Lett., 2016, 651, 192–197 CrossRef CAS.
- A. Schwenger, W. Frey and C. Richert, Angew. Chem., Int. Ed., 2016, 55, 13706–13709 CrossRef CAS PubMed.
- F. Krupp, W. Frey and C. Richert, Angew. Chem., Int. Ed., 2020, 59, 15875–15879 CrossRef CAS PubMed.
- F. Krupp, M. I. Picher, W. Frey, B. Plietker and C. Richert, Synlett, 2021, 32, 350–353 CrossRef CAS.
- T. Berking, J. Hartenfels, C. Lenczyk, S. Q. Gustavo, W. Frey and C. Richert, Angew. Chem., Int. Ed., 2024, 63, e202402976 CrossRef CAS PubMed.
- H. Li, J. Jiao, W. Xie, Y. Zhao, C. Lin, J. Jiang and L. Wang, ACS Mater. Lett., 2023, 5, 2673–2682 CrossRef CAS.
- J. Zheng, Z. Lu, K. Wu, G. H. Ning and D. Li, Chem. Rev., 2020, 120, 9675–9742 CrossRef CAS PubMed.
- A. A. Titov, O. A. Filippov, L. M. Epstein, N. V. Belkova and E. S. Shubina, Inorg. Chim. Acta, 2018, 470, 22–35 CrossRef CAS.
- M. A. Omary, A. A. Mohamed, M. A. Rawashdeh-Omary and J. P. Fackler, Coord. Chem. Rev., 2005, 249, 1372–1381 CrossRef CAS.
- M. Babor, P. P. Nievergelt, J. Cejka, V. Zvonicek and B. Spingler, IUCrJ, 2019, 6, 145–151 CrossRef CAS PubMed.
- Z. Y. Al Subeh, A. L. Waldbusser, H. A. Raja, C. J. Pearce, K. L. Ho, M. J. Hall, M. R. Probert, N. H. Oberlies and S. Hematian, J. Org. Chem., 2022, 87, 2697–2710 CrossRef CAS PubMed.
- M. Jiang, Z. Wu, Q. Wu, H. Yin, H. Guo, S. Yuan, Z. Liu, S. Chen and L. Liu, Chin. Chem. Lett., 2021, 32, 1893–1896 CrossRef CAS.
- D. Shi, B. L. Nannenga, M. G. Iadanza and T. Gonen, eLife, 2013, 2, e01345 CrossRef PubMed.
- M. D. Purdy, D. Shi, J. Chrustowicz, J. Hattne, T. Gonen and M. Yeager, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 13258–13263 CrossRef CAS PubMed.
- C. G. Jones, M. Asay, L. J. Kim, J. F. Kleinsasser, A. Saha, T. J. Fulton, K. R. Berkley, D. Cascio, A. G. Malyutin, M. P. Conley, B. M. Stoltz, V. Lavallo, J. A. Rodriguez and H. M. Nelson, ACS Cent. Sci., 2019, 5, 1507–1513 CrossRef CAS PubMed.
- P. Brazda, L. Palatinus and M. Babor, Science, 2019, 364, 667–669 CrossRef CAS PubMed.
- C. G. Jones, M. W. Martynowycz, J. Hattne, T. J. Fulton, B. M. Stoltz, J. A. Rodriguez, H. M. Nelson and T. Gonen, ACS Cent. Sci., 2018, 4, 1587–1592 CrossRef CAS PubMed.
- L. J. Kim, M. Ohashi, Z. Zhang, D. Tan, M. Asay, D. Cascio, J. A. Rodriguez, Y. Tang and H. M. Nelson, Nat. Chem. Biol., 2021, 17, 872–877 CrossRef CAS PubMed.
- L. J. Kim, M. Xue, X. Li, Z. Xu, E. Paulson, B. Mercado, H. M. Nelson and S. B. Herzon, J. Am. Chem. Soc., 2021, 143, 6578–6585 CrossRef CAS PubMed.
- X. Yu, L. J. Hu, J. G. Song, D. Zhang, Y. S. Peng, X. J. Huang, J. Hong, B. Zhu, W. C. Ye and Y. Wang, Chin. Chem. Lett., 2024 DOI:10.1016/j.cclet.2024.110149.
- J. T. F. Dobson, H. Kurz and J. R. Nitschke, Chem, 2024, 10, 1038–1040 CAS.
- T. N. Tu and M. Scheer, Chem, 2023, 9, 227–241 CAS.
- G. H. Ning, K. Matsumura, Y. Inokuma and M. Fujita, Chem. Commun., 2016, 52, 7013–7015 RSC.
| This journal is © The Royal Society of Chemistry 2025 |