Amorphous molybdenum sulfide films as catalysts for electrochemical hydrogen production in water†
Daniel
Merki
,
Stéphane
Fierro
,
Heron
Vrubel
and
Xile
Hu
*
Laboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), EPFL-ISIC-LSCI, BCH 3305, Lausanne, CH 1015, Switzerland. E-mail: xile.hu@epfl.ch; Fax: +41 216939305; Tel: +41 216939781
First published on 14th April 2011
Abstract
Amorphous molybdenum sulfide films are efficient hydrogen evolution catalysts in water. The films are prepared via simple electro-polymerization procedures and are characterized by XPS, electron microscopy and electronic absorption spectroscopy. Whereas the precatalysts could be MoS3 or MoS2, the active form of the catalysts is identified as amorphous MoS2. Significant geometric current densities are achieved at low overpotentials (e.g., 15 mA cm−2 at η = 200 mV) using these catalysts. The catalysis is compatible with a wide range of pHs (e.g., 0 to 13). The current efficiency for hydrogen production is quantitative. A 40 mV Tafel slope is observed, suggesting a rate-determining ion+atom step. The turnover frequency per active site is calculated. The amorphous molybdenum sulfide films are among the most active non-precious hydrogen evolution catalysts.
Introduction
Hydrogen is proposed as a major energy carrier for the future world.1,2 The most desirable source of hydrogen is water, as it is abundant and contains no carbon.1 The production of hydrogen and oxygen from water, or “water splitting”, consists of two half cell reactions (eqn (1)–(3)).| H2O → H2 + 0.5 O2 | (1) |
| 2H+ + 2e → H2 | (2) |
| OH− → 0.5 O2 + H+ + 2e | (3) |
The hydrogen evolution reaction (eqn (2)) requires catalysts. Whereas Ni based catalysts are often employed in commercial alkaline electrolyzers,3Pt and its composites are the most active catalysts for hydrogen evolution in an acidic medium.4Pt-type catalysts are more compatible with the conditions envisioned for photochemical water splitting, especially in direct photo-electrochemical approaches.5 However, the large scale application of Pt catalysts is limited by their high cost and low abundance. Extensive efforts have been devoted to the search of alternative catalysts containing only non-precious elements under heterogeneous conditions.6–10 Yet functional and robust catalysts operating with reasonable current densities (J) at low overpotentials (η) in water are scarce.7–10
Recently, MoS2nanoparticles have been identified as promising hydrogen evolution catalysts. Bulk MoS2 is a poor catalyst;11 nano-crystals of MoS2 and related metal sulfides, however, are more active.8,12,13 The best catalysts are crystalline, single-layered MoS2 polygons deposited on Au(111), with J ≈ 0.2 mA cm−2 at η = 150 mV and pH = 0.8 Notwithstanding the impressive advances, the practical implementation of these systems is hindered by their sophisticated and/or energy intensive preparation procedures, such as ultra-high vacuum conditions, reduction by H2S streams and annealing at elevated temperatures. Here we report that amorphous molybdenum sulfide films are active hydrogen evolution catalysts. The catalysts are prepared at room temperature and atmospheric pressure, and in a simple, rapid, and scalable manner. Furthermore, compared to MoS2nanoparticles, the amorphous molybdenum sulfide films exhibit higher activity.
Results and discussion
Deposition of MoS3 film by cyclic voltammetry and characterization of this film
When studying the electrochemistry of [MoS4]2−, we noticed that thin films were deposited onto the working electrodes by potential cycling experiments. For example, when the potential was cycled continuously from 0.3 V to −0.8 V vs.SHE (SHE = standard hydrogen electrode) at a rate of 50 mV s−1 in a 2.0 mM aqueous solution of (NH4)2[MoS4], one oxidation and one reduction peak grew in at −0.1 V and −0.6 V, respectively, concomitant with film formation (Fig. 1). After 5 scans, the film was visible; after 25 scans, the heights of the two redox peaks approached saturation. The deposition worked on different conducting substrates such as fluorine-doped tin oxide (FTO), indium tin oxide (ITO), and glassy carbon.![Deposition of MoS3-CV film on a FTO coated glass by repeated cyclic voltammetry (25 cycles) with a solution of [MoS4]2− in water. The arrows point to the growth of peaks during the deposition.](/image/article/2011/SC/c1sc00117e/c1sc00117e-f1.gif) | ||
| Fig. 1 Deposition of MoS3-CV film on a FTO coated glass by repeated cyclic voltammetry (25 cycles) with a solution of [MoS4]2− in water. The arrows point to the growth of peaks during the deposition. | ||
The X-ray photoelectron spectroscopy (XPS) survey spectrum of this film is dominated by the characteristic Mo and S peaks in addition to some smaller peaks of C and O from adventitious impurities (Fig. S2, ESI†). The binding energy of Mo 3d5/2 in the films is 228.6 eV, indicating a +4 oxidation state for the Mo ion (Fig. 2A).14,15 The S 2p spectrum (Fig. 2B) is best fit with two doublets, with S 2p3/2 energies of 162.4 and 163.9 eV, respectively. The spectrum indicates the presence of both S2− and S22−ligands.14,15 The spectrum is distinct from that of commercial MoS2 particles (Fig S2, ESI†). Quantification by XPS gave a Mo/S ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.9. Both the Mo and S XPS spectra are similar to those of amorphous MoS3,14 for which no hydrogen evolution activity was studied. Thus, we tentatively assign the material as MoS3 (MoS3-CV)16 with a formula of [Mo(IV)(S2)2−S2−]. There might be some amount of amorphous MoS2 whose XPS spectra is buried under those of MoS3.
:2.9. Both the Mo and S XPS spectra are similar to those of amorphous MoS3,14 for which no hydrogen evolution activity was studied. Thus, we tentatively assign the material as MoS3 (MoS3-CV)16 with a formula of [Mo(IV)(S2)2−S2−]. There might be some amount of amorphous MoS2 whose XPS spectra is buried under those of MoS3.
 | ||
| Fig. 2 XPS spectra of MoS3-CV film on a FTO coated glass. (A) Mo 3d and S 1s region. (B) S 2p region. | ||
The formation of MoS3-CV film from [MoS4]2− must result from an oxidation process. Bélanger et al. previously reported the electrodeposition of amorphous molybdenum sulfide from (NH4)2[MoS4].17 It was shown that anodic electrolysis of an aqueous solution of (NH4)2[MoS4] at ca. 0.55 V vs.SHE gave amorphous MoS3 films which were identified by SEM,18 chemical analysis, XAS and XPS.19 The XPS data of those MoS3 films resemble those in Fig. 2.
Fig. 3 shows typical transmission and scanning electron microscopy images of a film deposited on ITO. The film has a thickness of less than 100 nm, and is amorphous. No electron diffraction pattern was observed. The lack of crystallinity of the MoS3-CV film is further confirmed by powder X-ray diffraction which shows no peak besides those from the tin oxide substrate (not shown).
 | ||
| Fig. 3 Electron micrographs of the MoS3-CV film on ITO. | ||
It is possible to control the MoS3-CV film thickness by the number of scan cycles and/or varying the concentration of the solutes (Fig. 4). Most films have a thickness between 40 and 150 nm. Increasing the number of scan cycles significantly increases the thickness, up until about 35 scan cycles. Then the thickness of the film approaches an upper limit (Fig. 4A). If deposited with the same number of scans (e.g., 25), the film is thicker when the concentration of [MoS4]2− in the starting solution is higher.
 | ||
| Fig. 4 Thickness of MoS3-CV films on FTO as a function of scanning cycles and concentration of precursors. The measurements have been repeated multiple times to give the averaged values and error bars. (A) Thickness of MoS3-CV films as a function of scanning cycles; the concentration of MoS42− is 2.0 mM. (B) Thickness of MoS3-CV films as a function of MoS42− concentrations; 25 scanning cycles were applied for each deposition. | ||
Preparation and characterization of various MoSx films
The MoS3-CV film reported in this study is prepared by cyclic voltammetry at both anodic and cathodic potentials. While MoS3 could be formed anodically (vide supra), amorphous MoS2 film might be formed cathodically. Indeed, Lévy-Clément et al. reported that amorphous MoS2 film was formed when an aqueous solution of (NH4)2[MoS4] was electrolyzed at −0.75 to 1.15 V vs.SHE.20 The MoS2 film was X-ray amorphous, and electron probe microanalysis gave a S to Mo ratio of 1.9 to 2.1. Annealing of the MoS2 at 550 °C in Ar then gave the crystalline MoS2 particles.20 Judging from the potential window of our cyclic voltammetric experiments in Fig. 1, we suspected that a small amount of amorphous MoS2 was also produced together with MoS3 in the MoS3-CV film. To evaluate the catalytic property of different amorphous MoSx films, we decided to prepare MoS3 and MoS2 films according to the methods of Bélanger and Lévy-Clément. Furthermore, we also prepared a molybdenum sulfide film by the same continuous cyclic voltammetry as in Fig. 1, with the exception that the scans started from −0.8 to 0.3 V, and the final scan finished at −0.8 V. This way, the formation of film was terminated at the cathodic potential.XPS was used to identify the chemical compositions of these new films. The XPS spectra of MoS3 film prepared by the method of Bélanger (MoS3-AE, Fig. S3, ESI†)16 are very similar to those in Fig. 2. The S to Mo ratio is 3.2. The XPS spectra of MoS2 film (MoS2-CE, Fig. S3, ESI†)16 prepared by the method of Lévy-Clément are similar to those of commercial MoS2 particles, and do not contain the S peaks from S22−. The S to Mo ratio is 1.9. Interestingly, the molybdenum sulfide film prepared by cyclic voltammetry finishing at the cathodic potential gives XPS spectra similar to the MoS2 film and MoS2 particles (Fig. S3, ESI†). The S to Mo ratio is 2. We assign this film to MoS2-CV,16 although it probably contains a small amount of MoS3 that cannot be detected by XPS in the presence of a large amount of MoS2.
In addition to XPS, UV-Vis absorption spectroscopy was used to characterize the four different types of MoSx films. The films exhibit similar spectra with absorption peaks at ca. 310, 375, 480, and 630 nm (Fig. S4, ESI†), probably because they all contain Mo(IV) ions and S2−ligands. No absorption peaks associated with the S22−ligands in MoS3-CV and MoS3-AE films could be located.
Hydrogen evolution activity of four MoSx films
The catalytic activity of the four MoSx films was studied by electrochemistry. Fig. 5 shows the polarization curves of MoS3-CV on a rotating glassy carbon disk electrode at pH = 0 to 5; Fig. S5, ESI,† shows its polarization curves at pH = 7, 9, 11, 13, and the curve for MoS3-CV deposited on FTO at pH = 2. The MoS3-CV films display high catalytic activity for hydrogen evolution at a wide range of pH values. As expected, the apparent current densities decrease with an increase of pHs (Fig S5, ESI†). At low overpotentials (η < 250 mV), the current is independent of rotating rates and therefore kinetic-controlled. The amorphous MoS3-CV is more active than the MoS2 single crystals deposited on Au(111).8 The apparent current density (J) for the MoS3-CV film at η = 150 mV is ca. 0.4 mA cm−2 at pH = 0, higher than that for the MoS2 crystals at the same overpotential (J ≈ 0.2 mA cm−2).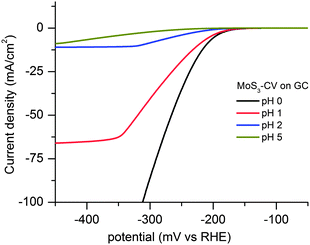 | ||
| Fig. 5 Polarization curves of MoS3-CV film on a rotating glassy carbon disk electrode recorded at pH = 0, 1, 2, and 5. Scan rate: 2 mV s−1; rotating rate: 4500 rpm. | ||
The catalytic activity of MoS3-AE film is nearly identical to that of MoS3-CV film (Fig. 6), except that the activity decreases gradually during consecutive scans (Fig. S6, ESI†). In contrast, the activity of MoS3-CV film remains constant during consecutive scans (Fig. S6, ESI†). The higher stability of MoS3-CV film compared to MoS3-AE film suggests an advantage for the potential-cycling process.
 | ||
| Fig. 6 Polarization curves of MoSx-films on FTO recorded at pH = 0. Scan rate: 2 mV s−1. The curves are from the second polarization scans for the freshly prepared samples, some of which need a pre-activation process in the first polarization scan (see main text for details). | ||
Interestingly, the MoS2-CV film, made also through the potential-cycling process, is as active and stable as the MoS3-CV film (Fig. 6 and Fig. S6, ESI†). This observation raised the question: what is the active catalyst for all the amorphous MoSx films? It prompted us to study the MoSx films after catalysis (vide infra). The MoS2-CE film, prepared by potentiostatic cathodic electrolysis, is the least active catalyst (Fig. 6). Its activity also decreases during consecutive scans (Fig. S6, ESI†).
Active catalyst
All four MoSx films exhibited hydrogen evolution activity. These films are better considered as pre-catalysts. To verify if these pre-catalysts were activated to form the same catalyst under the conditions for hydrogen evolution, we carefully examined the first polarization curves for the freshly prepared films. Indeed a pre-activation process was found for MoS3-CV, MoS3-AE, MoS2-CV, but not for MoS2-CE.For MoS3-CV and MoS3-AE, a reduction peak was observed prior to hydrogen evolution (Fig. 7). The same reduction peak was observed for MoS2-CV, but it was significantly smaller. No reduction was observed for MoS2-CE. For all MoSx films, no reduction peak before hydrogen evolution was observed in the second and following polarization scans (Fig. 6).
 | ||
| Fig. 7 First polarization curves of freshly prepared MoSx films on FTO at pH = 0. The arrows point to the reduction prior to hydrogen evolution. | ||
We hypothesized that the reduction peak in the first scan originated from the reduction of MoS3 to MoS2, whereby the S22− accepted two electrons to form S2−. The MoS2 species is then responsible for the hydrogen evolution catalysis. To verify this hypothesis, we carried out a XPS study on the MoS3-CV film after several polarization measurements. The XPS spectra of the film have changed, and are similar to those of MoS2 (Fig. S7, ESI†). The peaks of S22− disappeared. We recognize that XPS is a surface technique, and plan to carry out a similar study using a technique that can probe the bulk composition of the thin films, e.g., XAS.
The UV-Visspectra of the four MoSx films after multiple polarization scans were measured and are similar. They are also similar to the spectra of freshly prepared samples (Fig. S8, ESI†). While the UV-Vis spectra of these films are not sensitive to the presence or absence of S22−ligand, they suggest that the bulk compositions of the films are MoSx, and most likely MoS2.
It thus appears that amorphous MoS2 is the real catalyst for all four MoSx films. The MoS3-CV and MoS2-CV films reported here are the best precatalysts in terms of activity and stability and they are indistinguishable. The MoS3-AE film is as active, but some MoS3 in this film might be re-dissolved at cathodic potentials to form the soluble species [MoS4]2−. For this reason it loses activity during consecutive scans. This problem is alleviated in the potential cycling procedures used to make the MoS3-CV and MoS2-CV films. The MoS2-CE film is the least active and least stable catalyst. We suspect that when deposited at a very negative potentials, the film is more rapidly formed and is structurally inferior.
Only MoS3-CV film is used for the further studies in this paper.
Tafel analysis and bulk electrolysis
Tafel behavior was observed for the polarization curve of MoS3-CV on glassy carbon at η = 120 to 200 mV and pH = 0 (Fig. 8). Analysis of the data at η = 170 to 200 mV gave a Tafel slope of 40 mV per decade and an apparent Jo of ca. 1.3 × 10−7 A cm−2 for a film made from 25 scanning cycles (Fig. S9, ESI†). | ||
| Fig. 8 Tafel plot of the polarization curve of MoS3-CV film on glassy carbon at pH = 0. The film was made with 25 scanning cycles. Scan rate for polarization: 1 mV s−1. | ||
Bulk electrolysis was carried out to determine the current efficiency of hydrogen evolution. In a 1 M solution of H2SO4, the MoS3-CV film deposited on a glassy carbon disk (25 cycles) gave current densities of 160 and 14 mA cm−2 at η = 300 and 200 mV, respectively. These current densities are among the highest reported for non-noble catalysts in acidic or neutral conditions.10,21 For comparison, a recently reported and very active Ni-bisphosphine/carbon nanotube based H2 production catalyst gave a current density of 20 mA cm−2 at η = 300 mV.9 The Faraday yields for H2 production were found to be quantitative within experimental errors. By measuring pressure change during water splitting, it was possible to monitor H2 production in situ. Fig. 9 and Fig. S10, ESI,†show that the current efficiency remains quantitative for hours. Thus, while it was reported that vacuum-dried MoS3 could absorb H2 to form H2S,22 under the electrolysis conditions, the MoS3-CV film reported here remains stable and active during H2 production.
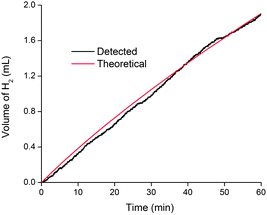 | ||
| Fig. 9 Current efficiency for H2 production catalyzed by a MoS3-CV film on glassy carbon at pH = 0 and 200 mV overpotential. The theoretical line was calculated according to the cumulative charge, assuming a 100% Faraday's yield for H2 production. The current density was ca. 14 mA cm−2. | ||
The catalytic activity of the MoS3-CV films depends on the thickness. Fig. 10A shows the polarization curves of films deposited on a rotating glassy carbon electrode with different scanning cycles. Films deposited by higher numbers of cycles are more active. The intrinsic catalytic activity is however measured by the turnover frequency (TOF) for each active site. We attempted to quantify the active sites by electrochemistry. Fig. 10B shows the cyclic voltammograms in the region of −0.2 V to 0.6 V vs. RHE for the MoS3-CV films at pH = 7. While it is difficult to assign the observed peaks to a given redox couple, the integrated charge over the whole potential range should be proportional to the total number of active sites. Assuming a one-electron process for both reduction and oxidation, the upper limit of active MoS2 sites could be calculated for each film. Fig. 10C shows the polarization curves at pH = 0 normalized by the active sites, and expressed in terms of TOF (see ESI†). Up to η = 200 mV, a fairly uniform activity was observed for films made from different scan cycles, suggesting that even though there is a significant uncertainty in the estimation of the active sites, self-consistent information on the site-averaged catalytic activity can be extracted from this analysis. At η >200 mV, some deviations were observed, probably due to the influence of substrate diffusion. Fig. S11, ESI,†shows the calculated TOFs at pH = 7.
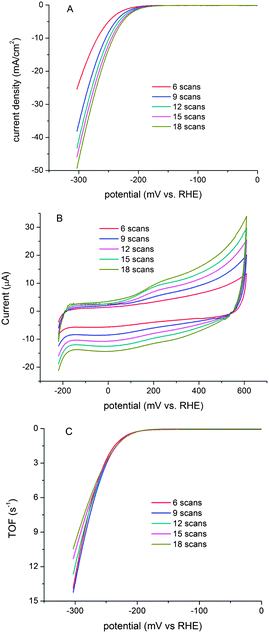 | ||
| Fig. 10 (A) Polarization curves of MoS3-CV films made from different numbers of scanning cycles recorded at pH = 0. The films were deposited on a rotating glassy carbon electrode. Scan rate: 1 mV s−1; rotating rate: 2000 rpm. (B) Cyclic voltammograms of the same MoS3-CV films recorded at pH = 7 and between −0.2 V and 0.6 V vs. RHE; scan rate: 50 mV s−1. (C) Calculated turnover frequencies for the MoS3-CV films at pH = 0. | ||
The rough estimation of TOFs makes it possible to compare the activity of MoS3-CV film with other catalysts. The most active catalyst, Pt, has a TOF of 0.8 s−1 at η = 0 and pH = 0;8 to reach the same TOF, MoS3-CV films need an overpotential of ca. 220 mV. An interesting Mo-oxo system is reported to work on a mercury pool electrode on which it might be adsorbed.23 It has a TOF of 2 s−1 at η ≈ 1000 mV at pH = 7. The TOF of MoS3-CV film reaches the same value at η ≈ 240 mV and pH = 0. The Mo-oxo system uses water as the substrate, while the MoS3-CV system uses protons as the substrate. To make a fair comparison, we also measured the TOFs of a MoS3-CV film at pH = 7 (Fig. S11, ESI†). The Mo-oxo system has a TOF of 0.3 s−1 at η ≈ 600 mV,23 while the MoS3-CV film reaches the same TOF at η ≈ 340 mV. Thus, amorphous MoS3-CV compares favorably with known non-precious catalysts in terms of bulk catalytic properties.
Mechanism of hydrogen evolution
Given that single crystals and nanoparticles of MoS2 but not bulk MoS2 are active H2 evolution catalysts, the discovery that the four amorphous MoSx films are active catalysts was unexpected. The overall catalytic activity per geometric area of MoS3-CV and MoS2-CV film is higher than that of MoS2 single crystals (vide infra) and nanoparticles (J ≈ 160 mA cm−2 at η ≈ 300 mV for MoS3-CV film and J < 2 mA cm−2 at η ≈ 300 mV for MoS2nanoparticles).13H2 evolution by amorphous MoS2 (which is the active species in all four MoSx films) seems to proceed via a different mechanism from that by MoS2 single crystals and nanoparticles. MoS2 films have a Tafel slope of 40 mV per decade. This Tafel slope is different from those of MoS2 crystals8 (55 to 60 mV per decade) or MoS2 nanoparticulate13 (120 mV per decade). According to the classic theory on the mechanism of hydrogen evolution,24 a Tafel slope of 40 mV indicates that the surface coverage of adsorbed hydrogen is less than 10%, and hydrogen production occurs via a fast discharge reaction (eqn (4)) and then a rate-determining ion+atom reaction (eqn (5)).
| discharge reaction H3O+ + e + cat ⇌ cat-H + H2O | (4) |
| ion + atom reaction H3O+ + e + cat-H ⇌ cat + H2 + H2O | (5) |
| combination reaction cat-H + cat-H ⇌ 2cat + H2 | (6) |
A Tafel slope of 60 mV per decade indicates a larger surface coverage of adsorbed hydrogen and a rate-determining recombination (eqn (6)) or ion+atom reaction.25 A Tafel slope of 120 mV could arise from various reaction pathways depending on the surface coverage.24 The different Tafel slopes point to a unique catalytic property for amorphous MoSx films when compared to crystalline forms of MoS2. For MoS2 single crystals and nanoparticles, the active sites are proposed to be on the edges which have coordinatively unsaturated Mo and/or S atoms.8 The MoS2 films described here may have more such unsaturated sites thanks to their amorphous nature. We suspect that this is the reason why the films are more active than the single crystals and nanoparticles. More work is required to shed light on the mechanism of H2 evolution at the molecular level. Given that both Mo and S are capable of accepting electrons and protons, a ‘bifunctional’, metal–ligand cooperative mode of catalysis is probable.26
Conclusions
In summary, we show that amorphous MoSx films are highly active hydrogen evolution catalysts. While the precatalysts can exist in various forms of molybdenum sulfide, the real catalyst is amorphous MoS2. The catalyst is made of relatively cheap and abundant elements and offers significant advantages over noble metal catalysts. The catalyst can be easily prepared in a procedure that is amenable to large scale manufacture. The catalyst works in water and is compatible with a wide range of pHs. Significant current densities could be obtained at low overpotentials and the current efficiency for H2 production is quantitative. Our results thus provide new opportunities for the development of renewable and economic hydrogen production technologies. Further characterization and application of the catalyst, such as further stability studies and impedance analysis are currently underway.Acknowledgements
This work is supported by the EPFL, the Swiss National Science Foundation (project number 200021_119663), and a starting grant from the European Research Council under the European Community's Seventh Framework Programme (FP7 2007–2013)/ERC Grant agreement n° 257096. X.L. Hu thank Prof. Nathan Lewis (Caltech) and Profs. Michael Grätzel, Hubert Girault and Christos Comninellis (EPFL) for discussion and suggestions. Nicolas Xanthopoulos is acknowledged for help with XPS measurements.Notes and references
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- J. Ivy Summary of electrolytic hydrogen production. Milestone completion report NREL/MP-560–36734. National Renewable Energy Laboratory: Golden, Colorado; 2004; G. Schiller, R. Henne, P. Mohr and V. Peinecke, Int. J. Hydrogen Energy, 1998, 23, 761–765 Search PubMed.
- D. E. Bartak, B. Kazee, K. Shimazu and T. Kuwana, Anal. Chem., 1986, 58, 2756–2761 CrossRef CAS; P. Millet, F. Andolfatto and R. Durand, Int. J. Hydrogen Energy, 1996, 21, 87–93 CrossRef CAS.
- H. B. Gray, Nat. Chem., 2009, 1, 7–7 CrossRef CAS; W. J. Youngblood, S. H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS.
- F. A. Armstrong, N. A. Belsey, J. A. Cracknell, G. Goldet, A. Parkin, E. Reisner, K. A. Vincent and A. F. Wait, Chem. Soc. Rev., 2009, 38, 36–51 RSC; M. M. Jaksic, Electrochim. Acta, 2000, 45, 4085–4099 CrossRef CAS.
- L. A. Berben and J. C. Peters, Chem. Commun., 2010, 46, 398–400 RSC; B. Keita and L. Nadjo, J. Electroanal. Chem., 1985, 191, 441–448 CAS; B. Keita and L. Nadjo, Mater. Chem. Phys., 1989, 22, 77–103 CrossRef CAS; B. Winther-Jensen, K. Fraser, C. Ong, M. Forsyth and D. R. MacFarlane, Adv. Mater., 2010, 22, 1727–1730 CrossRef CAS.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS; P. D. Tran, A. Le Goff, H.J., B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS.
- E. Navarro-Flores, Z. W. Chong and S. Omanovic, J. Mol. Catal. A: Chem., 2005, 226, 179–197 CrossRef CAS.
- W. Jaegermann and H. Tributsch, Prog. Surf. Sci., 1988, 29, 1–167 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Norskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS; X. Zong, H. J. Yan, G. P. Wu, G. J. Ma, F. Y. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS.
- J. Bonde, P. G. Moses, T. F. Jaramillo, J. K. Norskov and I. Chorkendorff, Faraday Discuss., 2009, 140, 219–231 RSC.
- T. Weber, J. C. Muijsers and J. W. Niemantsverdriet, J. Phys. Chem., 1995, 99, 9194–9200 CrossRef CAS.
- J. C. Muijsers, T. Weber, R. M. vanHardeveld, H. W. Zandbergen and J. W. Niemantsverdriet, J. Catal., 1995, 157, 698–705 CrossRef.
- In this paper, we abbreviate the films by their preparative methods. CV designates the films made by cyclic voltammetry, AE designates the films made by anodic electrolysis and CE designates the films made by cathodic electrolysis.
- G. Laperriere, B. Marsan and D. Belanger, Synth. Met., 1989, 29, 201–F206 CrossRef.
- D. Belanger, G. Laperriere and B. Marsan, J. Electroanal. Chem., 1993, 347, 165–183 CrossRef CAS.
- D. Belanger, G. Laperriere, F. Girard, D. Guay and G. Tourillon, Chem. Mater., 1993, 5, 861–868 CrossRef CAS.
- E. A. Ponomarez, M. Neumann-Spallart, G. Hodes and C. Levy-Clement, Thin Solid Films, 1996, 280, 86–89 CrossRef CAS.
- NiMo alloys were reported to have similar activity, but largely due to the high surface roughness. The normalized geometric current density is ca. 0.015 mA cm−2 at η = 150 mV, smaller than 0.4 mA cm−2 found for the MoS3-CV film. See ref. 10.
- P. Afanasiev, H. Jobic, C. Lorentz, P. Leverd, N. Mastubayashi, L. Piccolo and M. Vrinat, J. Phys. Chem. C, 2009, 113, 4139–4146 CrossRef CAS.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS.
- J. O. M. Bockris and E. C. Potter, J. Electrochem. Soc., 1952, 99, 169–186 CrossRef CAS.
- J. G. Thomas, Trans. Faraday Soc., 1961, 57, 1603–1611 RSC.
- M. R. DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC; A. M. Appel, D. L. DuBois and M. R. DuBois, J. Am. Chem. Soc., 2005, 127, 12717–12726 CrossRef CAS; T. B. Rauchfuss, Inorg. Chem., 2004, 43, 14–26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: 10.1039/c1sc00117e |
| This journal is © The Royal Society of Chemistry 2011 |