Expanding the pleuromutilin class of antibiotics by de novo chemical synthesis†
Stephen D.
Lotesta
a,
Junjia
Liu
a,
Emma V.
Yates
a,
Inna
Krieger
b,
James C.
Sacchettini
b,
Joel S.
Freundlich
b and
Erik J.
Sorensen
*a
aDepartment of Chemistry, Princeton University, Frick Chemical Laboratory, Princeton, New Jersey 08544, USA. E-mail: ejs@princeton.edu; Fax: +1-608-258-1980; Tel: +1-609-258-8135
bDepartment of Biochemistry and Biophysics, Texas A&M University, College Station, Texas, 77843-2128, USA
First published on 14th April 2011
Abstract
New pleuromutilin-like compounds were synthesized in approximately 11 steps from 3-allylcyclopent-2-enone by a strategy featuring sequential carbonyl addition reactions. Several analogs possessing the C14 tiamulin ester side chain displayed activity in a Mycobacterium tuberculosis mc27000 assay. The results described herein provide a basis for further efforts to expand the structural and stereochemical diversity of the pleuromutilin class of bacterial protein synthesis inhibitors through advances in chemical synthesis.
Introduction
The rate of new antibiotic approvals is declining in an era when drug resistant and extremely drug resistant strains of bacteria are posing greater threats to human health.1–3 This already serious situation is worsened by the dearth of new antibiotic drug candidates in the pipelines of pharmaceutical companies.4 Historically, a handful of families of natural products, such as the penicillins, cephalosporins, erythromycins, and tetracyclines have given rise to a large fraction of the approved antibiotics, and the process of semi-synthesis continues to be the predominant avenue to new ones.4 In a noteworthy departure from the tried-and-true strategy of producing new antibiotics through systematic synthetic tailoring of the structures of known antibacterial natural products, the Myers laboratory demonstrated the power of de novo chemical synthesis in their expansion of the tetracycline class of antibiotics;5 their efforts led to the discovery of new tetracyclines that are unavailable from the naturally occurring tetracyclines.Our laboratory was drawn to the problem of achieving an analogous expansion of the pleuromutilin class of antibiotics through the development of direct syntheses of functionalized, pleuromutilin-like molecular scaffolds. Pleuromutilin (1), the flagship member of the antibiotic class bearing its name, possesses eight stereogenic centers and an intriguing “propellane-like” fusion of 5-, 6-, and 8-membered rings wherein the three rings share a common carbon–carbon bond (Fig. 1).6 This unique and rather rigid molecular architecture permits the C11 hydroxyl group and the C3 and C21 carbonyl oxygens to make bonding interactions within the peptidyl transferase center of the 50S subunit of bacterial ribosomes.7 Through this molecular association, which is reinforced by hydrophobic bonding interactions, pleuromutilin (1) and its relatives retapamulin (2) and tiamulin (3) inhibit bacterial protein synthesis and display activity against bacterial strains that have become resistant to the macrolide class. Retapamulin (2) and tiamulin (3) were invented by scientists at GlaxoSmithKline and Sandoz Pharma, respectively, in the course of a substantial effort to develop new antibiotics from the pleuromutilin class by the strategy of semi-synthesis.8,9 Nabriva Therapeutics is also pursuing the development of new, semi-synthetic derivatives of pleuromutilin.10 The aim of the research described herein was the development of a complementary strategy for expanding the depth and diversity of the pleuromutilins that would be founded on direct laboratory syntheses of the structurally complex molecular scaffold that defines this underdeveloped class of antibiotics.11–13 This report addresses our continued advances in this area,13 with a specific focus on application to tuberculosis—a disease presenting significant challenges to the discovery of novel therapeutics.14
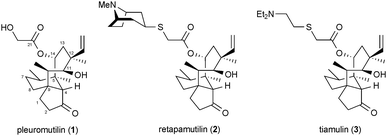 | ||
| Fig. 1 The molecular structures of pleuromutilin (1), retapamulin (2), and tiamulin (3). | ||
Design for synthesis
In the planning phase, we conceived tricyclic scaffolds lacking the methyl groups found in compounds 1–3 since their contribution to the antibacterial properties of the pleuromutilins was unclear and their inclusion would lengthen our synthesis effort. However, the designed scaffolds would retain the pleuromutilin-like hydroxyl group at C11 as well as the C3 and C21 carbonyl functions since these groups make bonding interactions with the bacterial ribosome.7b This reasoning led to the conception of the pleuromutilin-like compound 4 (Scheme 1) as a target for synthesis, and we were especially interested in the prospects for a direct annulation of the 8-membered ring system through chemoselective additions of a hypothetical dianion to the aldehyde functions in a compound of type 5. While there were concerns about the stereochemical outcomes of these carbonyl addition events, we embraced the possibility that this strategy for synthesis might provide direct access to an expanded set of screening candidates and that compounds with unnatural stereochemical configurations may still show useful margins of antibacterial activity. Ultimately, we were able to approximate the “linchpin” tactic outlined in Scheme 1 through the sequential application of the Nozaki–Hiyama–Kishi (NHK) bond construction method.15 The established utility of Cr(II) or Cr(II)/Ni(II)-mediated bond formations in syntheses of challenging medium-sized rings was the principal justification for our decision to rely on this process in this context. | ||
| Scheme 1 Derivation of a simplified, pleuromutilin-like scaffold 4 and the concept of a linchpin cyclization strategy. | ||
Results and discussion
Our path to the desired tricyclic scaffolds utilized bicyclic cyano alkene 9, a substance that was readily available from 3-allylcyclopent-2-enone (6) and Grignard reagent 7 by the reaction sequence shown in Scheme 2.13 The introduction of the nitrile function via a conjugate addition to enone 8 was both efficient and highly diastereoselective. After a high-yielding ozonolysis of the alkene in 9 (Scheme 3), the C11–C12 bond was constructed by a union of aldehyde 10 with vinyl bromide 11 under the conditions of a NHK reaction. This procedure afforded compound 12 as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of C11 epimers. A tert-butyldimethylsilylation of the epimeric mixture of alcohols produced 1316 and a subsequent, selective desilylation under mildly acidic conditions gave rise to compounds 14a and 14b as a separable mixture of diastereoisomers.
:1 mixture of C11 epimers. A tert-butyldimethylsilylation of the epimeric mixture of alcohols produced 1316 and a subsequent, selective desilylation under mildly acidic conditions gave rise to compounds 14a and 14b as a separable mixture of diastereoisomers.
 | ||
| Scheme 2 Synthesis of precursor 9. DMAP = 4-dimethylaminopyridine; p-TsOH = p-toluene sulfonic acid. | ||
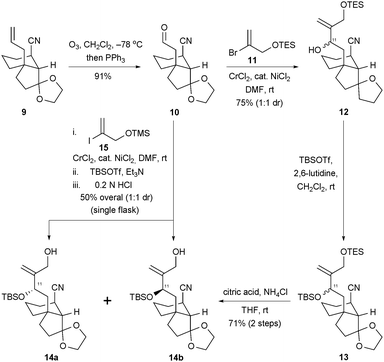 | ||
| Scheme 3 The elaboration of compounds 14a and 14b from 9 featuring an intermolecular NHK bond construction. TBS = tert-butyldimethylsilyl; TES = triethylsilyl; TMS = trimethylsilyl; DMF = N,N-dimethylformamide; THF = tetrahydrofuran. | ||
Interestingly, it was also possible to achieve a one-flask synthesis of 14a and 14b from aldehyde 10 and vinyliodide 15 by an in situ tert-butyldimethylsilylation of the intermediate chromium alkoxides and a simple quench with aqueous HCl to cleave the primary trimethylsilyl ether. This direct strategy for simultaneously achieving a key bond formation and a solution to the hydroxyl group differentiation problem was enabled by Fürstner and Shi's method17 for performing in situ σ-bond metatheses of chromium alkoxides and trialkylsilyl chlorides; it also served our effort by providing access to both epimers at C11.
Epimer
14a was transformed to the pleuromutilin-type tricyclic architecture by the following three-step sequence of reactions (Scheme 4): (1) conversion of the alcohol to the corresponding bromide by the action of carbon tetrabromide and triphenylphosphine; (2) reduction of the nitrile function with diisobutylaluminum hydride (DIBAL-H) and subsequent hydrolysis to afford the desired aldehyde 16a; and (3) the pivotal chromium-mediated cyclization of 16a, which afforded a 3:2 ratio of diastereoisomers 17a and 17b in 73% yield.
 | ||
| Scheme 4 Construction of functionalized pleuromutilin-like scaffolds featuring reductive cyclizations. DIBAL-H = diisobutylaluminum hydride; TBS = tert-butyldimethylsilyl; nOe = nuclear Overhauser effect. | ||
Epimer
14b was advanced in an analogous fashion, and it was observed that the reductive cyclization of 16b to a 2:1 mixture of compounds 17c and 17d was significantly slower than the cyclization of 16a; at room temperature, the reductive cyclization of 16a required only 5 min, while compound 16b required 24 h. After chromatographic separations of the two sets of diastereoisomers (i.e.17a/17b and 17c/17d), NOESY spectroscopic analyses were performed to establish the relative stereochemistry of each stereoisomer. The nOe correlations that supported our assignments are indicated by the two-headed arrows.
Since prior SAR studies established the importance of the C14 ester side chain to antibacterial efficacy within the pleuromutilin class,7a, 9a,b, 10 we conducted the two-stage transformations shown in Scheme 5. The couplings of compounds 17a and 17c with either the pleuromutilin-like glycolic acid derivative 18 or the tiamulin-like carboxylic acid 19 were mediated by dicyclohexylcarbodiimide (DCC) and followed by straightforward acid-induced deprotections to yield the novel screening candidates 4, 20a, and 20c.
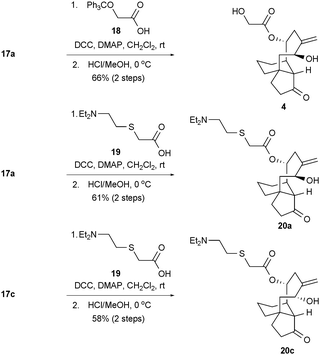 | ||
| Scheme 5 Syntheses of pleuromutilin-like compound 4 and tiamulin-like compounds 20a and 20c. DCC = N,N′-dicyclohexylcarbodiimide; DMAP = 4-dimethylaminopyridine. | ||
Our previously developed synthesis of the molecular framework of pleuromutilin featuring the method of ring-closing olefin metathesis13 invited an opportunity to produce additional antitubercular screening candidates from tricyclic alkenes 21a and 21b (Scheme 6). A straightforward, two-fold deprotection of compound 21a afforded tricyclic alcohol 24, which was subsequently joined with carboxylic acid 19 in the presence of DCC to give the novel, tiamulin-like ester 25. It was also possible to epoxidize the trisubstituted alkenes in compounds 21a and 21b with high margins of diastereoface selectivity; however, our subsequent effort to effect a base-induced epoxide ring opening18 of 22a to compound 23a was unsuccessful and an attempt to achieve an analogous conversion of 22b to 23b was only partially successful.19 In our experience, the chromium-mediated, reductive cyclization strategy described herein is especially effective at producing compounds with the type of constitution embodied in compound 4 and its relatives.
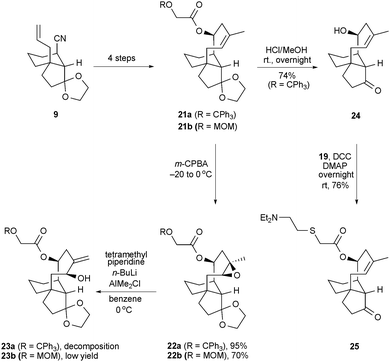 | ||
| Scheme 6 A synthesis of the simplified, tiamulin-like ester 25. m-CPBA = 3-chloroperoxybenzoic acid; MOM = methoxymethyl. | ||
To evaluate the antitubercular efficacies of the tricyclic esters 4, 20a, 20c, and 25, we measured their respective effects on the growth of M. tuberculosis mc27000 (M. tuberculosis H37Rv ΔRD1 ΔpanCD),20 utilizing the Microplate Alamar Blue assay (Table 1).21 Our results with pleuromutilin and tiamulin showed low micromolar MIC values, in accord with a previous report with Mtb strains.22 Expectedly, the compounds with the tiamulin-like ester side chain displayed higher inhibitory activity against Mtb mc27000 (1vs.3; 4vs.20a). In relation to tiamulin (3), the tiamulin-like tricycle 20a displayed an attenuated activity. However, we were intrigued by the promising efficacy of analog 20c, a compound with the tiamulin-type C14 ester side chain and unnatural relative stereochemistry at C11; this scaffold also lacks the peripheral methyl groups as well as the C12 quaternary stereocenter and yet it compares favorably to tiamulin (3) in its inhibitory action against Mtb mc27000. Another unanticipated finding was the moderate, yet significant, level of activity of compound 25, a substance that lacks the polar hydroxyl function at C11 and possesses unnatural relative stereochemistry at C14.
| Compound | mc27000 7H9 MIC (ug mL−1)a |
|---|---|
| a Each MIC value was reported as a range of experimentally assayed compound concentrations that bounds the mean for three independent determinations. Compounds 4, 20a, 20c, and 25 were tested as racemates. | |
| Pleuromutilin (1) | 25–50 |
| Tiamulin (3) | 12–25 |
| 4 | >200 |
| 20a | 50–100 |
| 20c | 12–25 |
| 25 | 50 |
The behaviors of compounds 20c and 25 in this whole-cell assay suggest that it is difficult to extrapolate the antibacterial potentials of simplified, pleuromutilin-like compounds from a knowledge of the structural details of the pleuromutilin/50S ribosome complex.23 Further studies are needed to determine if the new members of the pleuromutilin family described herein are targeting the peptidyl transferase center of the 50S subunit of bacterial ribosomes and to evaluate their activities against a broader range of bacterial strains.
Through the development of a flexible design for synthesis, we accomplished concise syntheses of new compounds that approximate the unique structure of the well known natural product pleuromutilin and show promise as leads for the development of new, pleuromutilin-like antibiotics. The tricyclic molecular scaffold that defines the pleuromutilin family is complex and yet our preliminary studies show that effective syntheses of biologically active compounds having this scaffold are possible. As we move forward in our effort to expand the structural and stereochemical diversity of the pleuromutilin class of antibiotics, there is a high likelihood that we will discover additional, new compounds with promising activity against M. tuberculosis and possibly resistant strains of TB as well as other bacteria.
Acknowledgements
We gratefully acknowledge financial support from the Grand Challenges Initiative at Princeton University, the National Institute of General Medical Sciences (GM065483), a NRSA fellowship (F32 GM083456) from the National Institutes of Health, USA (S.D.L), and the Global Alliance for TB Drug Development for helpful discussions. We also thank Dr István Pelczer for assistance with NMR spectroscopic analyses, Dr John F. Eng for assistance with mass spectrometric analyses, and Lotus Separations (preparative SFC).Notes and references
- C. Nathan, Nature, 2004, 431, 899 CrossRef CAS.
- G. Taubes, Science, 2008, 321, 356 CrossRef CAS.
- D. N. Gilbert, R. J. Guidos, H. W. Boucher, G. H. Talbot, B. Spellberg, J. E. Edwards, Jr., W. M. Scheld, J. S. Bradley and J. G. Bartlett, Clin. Infect. Dis., 2010, 50, 1081 CrossRef.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089 CrossRef CAS and references cited therein.
- M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395 CrossRef CAS.
- (a) F. Kavanagh, A. Hervey and W. Robbins, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 570 CAS; (b) D. Arigoni, Gazz. Chim. Ital., 1962, 92, 884 CAS; (c) D. Arigoni, Pure Appl. Chem., 1968, 17, 331 CrossRef CAS; (d) A. J. Birch, D. W. Cameron, C. W. Holzapfel and R. W. Rickards, Chem. Ind. (London), 1963, 374 CAS; (e) A. J. Birch, C. W. Holzapfel and R. W. Rickards, Tetrahedron, 1966, 22(Suppl. 8 Part II), 359 CrossRef; (f) M. Dobler and B. G. Durr, Cryst. Struct. Commun., 1975, 4, 259 CAS.
- (a) H. Egger and H. Reinshagen, J. Antibiot., 1976, 29, 923 CAS; (b) C. Davidovich, A. Bashan, T. Auerbach-Nevo, R. D. Yaggie, R. R. Gontarek and A. Yonath, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4291 CrossRef CAS.
- For more information, see: GSK developed the first semi-synthetic pleuromutilin antibiotic (Altabax) and continues to explore pleuromutilin targets for anti-tubercular activity in conjunction with Global Alliance for TB Drug Development. http://www.tballiance.org.
- (a) E. Hunt, Drugs Fut., 2000, 25, 1163 CAS; (b) O. A. Phillips and L. H. Sharaf, Expert Opin. Ther. Pat., 2007, 17, 429 Search PubMed; (c) C. H. Hu and Y. Zou, Mini-Rev. Med. Chem., 2009, 9, 1397 CrossRef CAS; (d) Y. Andemichael, J. Chen, J. S. Clawson, W. Dai, A. Diederich, S. V. Downing, A. J. Freyer, P. Liu, L. M. Oh, D. B. Patience, S. Sharpe, J. Sisko, J. Tsui, F. G. Vogt, J. Wang, L. Wernersbach, E. C. Webb, J. Wertman and L. Zhou, Org. Process Res. Dev., 2009, 13, 729 Search PubMed.
- R. Novak and D. M. Shlaes, Curr. Opin. Invest. Drugs, 2010, 11, 182 Search PubMed.
- For total syntheses of pleuromutilin, see: (a) E. G. Gibbons, J. Am. Chem. Soc., 1982, 104, 1767 CrossRef CAS; (b) R. K. Boeckman Jr, D. M. Springer and T. R. Alessi, J. Am. Chem. Soc., 1989, 111, 8284 CrossRef.
- For studies toward syntheses of pleuromutilin, see: (a) L. A. Paquette, P. E. Wiedeman and P. C. Bulman-Page, J. Org. Chem., 1988, 53, 1441 CrossRef CAS; (b) L. A. Paquette, P. C. Bulman-Page, P. D. Pansegrau and P. E., J. Org. Chem., 1988, 53, 1450 CrossRef CAS; (c) L. A. Paquette, P. D. Pansegrau, P. E. Wiedeman and J. P. Springer, J. Org. Chem., 1988, 53, 1461 CrossRef CAS; (d) A. H. Patten and B. M. Trost, Diss. Abstr. Int. B, 1987, 47, 3361 Search PubMed (Chem. Abstr., 106, 156709); (e) M. Kahn, Tetrahedron Lett., 1980, 21, 4547 CrossRef CAS; (f) E. Bacque, F. Pautrat and S. Zard, Org. Lett., 2003, 5, 325 CrossRef CAS; (g) M. D. Helm, M. Da Silva, D. Sucunza, T. J. K. Findley and D. J. Procter, Angew. Chem., Int. Ed., 2009, 48, 9315 CrossRef CAS; (h) T. J. K Findley, D. Sucunza, L. C. Miller, M. D. Helm, M. Helliwell, D. T. Davies and D. J. Procter, Org. Biomol. Chem., 2011, 9, 2433 RSC.
- J. Liu, S. D. Lotesta and E. J. Sorensen, Chem. Commun., 2011, 47, 1500 RSC.
- J. C. Sacchettini, E. J. Rubin and J. S. Freundlich, Nat. Rev. Microbiol., 2008, 6, 41 Search PubMed.
- For excellent reviews of carbon–carbon bond formations via the chemistry of organochromium(III) reagents, including the NHK reaction, see: (a) A. Fürstner, Chem. Rev., 1999, 99, 991 CrossRef; (b) L. A. Wessjohann and G. Scheid, Synthesis, 1999, 1, 1 CrossRef.
- The use of a relatively bulky protecting group (e.g. tert-butyldimethylsilyl or triphenylsilyl) for the oxygen at C11 enabled an easier separation of the epimers that formed in the intermolecular NHK reaction; in contrast, methoxymethyl-protected epimers at C11 could not be resolved by SiO2 chromatography.
- (a) A. Fürstner and N. Shi, J. Am. Chem. Soc., 1996, 118, 2533 CrossRef; (b) A. Fürstner and N. Shi, J. Am. Chem. Soc., 1996, 118, 12349 CrossRef; (c) M. Kurosu, M. Lin and Y. Kishi, J. Am. Chem. Soc., 2004, 126, 12248 CrossRef CAS.
- A. Yasuda, H. Yamamoto and H. Nozaki, Bull. Chem. Soc. Jpn., 1979, 52, 1705 CAS.
- Attempted conversions of epoxide 22 to the corresponding allylic alcohols were problematic. Substrate 22a underwent decomposition under strongly basic conditions. Substrate 22b could be converted to 23b, although the yields were very low and 23b was isolated in trace quantities.
- V. K. Sambandamurthy, S. C. Derrick, T. Hsu, B. Chen, M. H. Larsen, K. V. Jalapathy, M. Chen, J. Kim, S. A. Porcelli, J. Chan, S. L. Morris and W. R. JacobsJr., Vaccine, 2006, 24, 6309 CrossRef CAS.
- L. A. Collins and S. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004 CAS.
- G. Ascher, F. Stauffer, H. Berner and R. Mang, International Publication No. WO 03/082260 A2, Sandoz GMBH, October 2003, 9 Search PubMed.
- D. M. Springer, A. Bunker, B. Luh, M. E. Sorensen, J. T. Goodrich, J. J. Bronson, K. DenBleyker, T. J. Dougherty and J. Fung-Tomc, Eur. J. Med. Chem., 2007, 42, 109 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/c1sc00116g |
| This journal is © The Royal Society of Chemistry 2011 |