
Construction of a metal–organic tetrahedral cage for boosting selective electrocatalytic reduction of nitrite to ammonium†
Chang
Liu
a,
Yang
Yang
 *a,
Xinyu
Li
a,
Jinfeng
Wang
a,
Xu
Jing
*a and
Chunying
Duan
ab
*a,
Xinyu
Li
a,
Jinfeng
Wang
a,
Xu
Jing
*a and
Chunying
Duan
ab
aSchool of Chemistry, Dalian University of Technology, 116024, China. E-mail: yangyang@dlut.edu.cn; xjing@dlut.edu.cn
bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing, 210093, China
First published on 2nd June 2025
Abstract
Electrocatalytic reduction of nitrite to ammonium (NO2RR) presents a promising approach for removing harmful NO2− and efficiently synthesizing ammonium but still faces difficulties, due to the complex six-electron transfer reaction, the formation of various by-products and competition from the hydrogen evolution reaction (HER). Herein, we constructed a metal–organic tetrahedral cage H1 (FeII4L6) with a large inner cavity and Fe metal centers surrounded by hydrazide groups that act as hydrogen bond acceptor sites. This enables the cage to efficiently recognise nitrite and thermodynamically activate it under electrocatalytic conditions for its efficient reduction to ammonium. The kinetic experiments demonstrated that the catalytic process followed the Michaelis–Menten mechanism, which further verified the key role of host–guest interactions in the mimetic activation of nitrite and the enzyme-like catalytic behavior.
Introduction
Metal–organic cages (MOCs) are a class of molecular structures built from organic ligands and metal centres through self-assembly reactions.1 The structural features of MOCs with enzyme-like space-confined domain microenvironments enable them to serve as catalyst carriers and provide efficient catalytically active sites, offering the advantage of mimicking enzyme catalysts for catalytic reactions.2–4 The cavity microenvironments of metal–organic cages selectively bind to specific substrate molecules through weak interactions, such as hydrogen bonding and π–π interactions, encapsulating them within their cavities to create a unique confined environment that enhances molecular recognition and activation.5,6 Moreover, the cavity environment modulates the chemical and physical properties of the substrate, thereby influencing the redox behaviour of the electroactive species.7 The restricted domain microenvironment of metal–organic cages has garnered significant attention due to its promising applications in molecular recognition,8 trapping, asymmetric catalytic synthesis,9 and substance separation, among others.10,11Nitrogen, as a core element of Earth's living system, plays an irreplaceable role in maintaining ecological balance and the progress of human civilisation.12 Ammonium salts are high-value chemical raw materials widely used in the production of nitrogen-rich fertilizers and play a crucial role in various chemical reactions and industrial applications.13 Nitrite (NO2−) emissions from industrial production are a major source of water pollution and pose a significant threat to the environment.14 Electrocatalytic reduction of nitrite to ammonium (NO2RR) presents a promising approach for removing harmful NO2− and efficiently synthesizing ammonium.15,16 However, the NO2RR is a complex six-electron transfer process that involves the generation of multiple intermediates, often accompanied by by-products such as NO, N2O and N2H4.17–19 Additionally, the hydrogen evolution reaction (HER) competes with the NO2RR, which reduces the efficiency of the target reaction. This competition not only diminishes the catalyst's selectivity for the nitrite reduction reaction but also may lead to hydrogen gas as a by-product, further lowering overall catalytic efficiency.20–22 According to previous studies, MOCs, due to host–guest interaction properties, can efficiently regulate electron and proton transfer processes, recognize substrates through inclusion, and specifically activate certain substrates.23–25 Therefore, we speculate that metal–organic cages may exhibit unique catalytic advantages in the nitrite reduction reaction, enabling a highly efficient nitrite reduction process.
Herein, we have designed an Fe-based tetrahedral cage H1 (FeII4L6), formed through the self-assembly of iron(II) ions and organic ligands L containing hydrazide groups. The tetrahedral cage has a large inner space and amide groups evenly distributed around the metal site can act as hydrogen bond acceptors with the substrate. We hypothesize that the hydrogen-bond acceptor amide groups in the internal cavity of H1 can interact with nitrite to facilitate its activation. By introducing metal–organic domain-limited systems into the electrocatalytic reduction of nitrite to ammonium (NO2RR), host–guest interactions facilitate the rapid transfer of electrons and protons from the active sites to the substrates (Scheme 1). This strategy may provide new ideas for electrocatalytic reduction of nitrite to ammonium.
 | ||
| Scheme 1 The tetrahedral cage can selectively recognize nitrite and thermodynamically activate it, enabling efficient electrocatalytic reduction of nitrite to ammonium. | ||
Experimental
Materials and methods
All chemicals and solvents used in the study were of reagent grade, sourced commercially, and utilized without additional purification. 1H NMR spectra were recorded on Bruker AVANCE NEO 400 M and NEO 500 M spectrometers (in DMSO-d6, with TMS as the internal standard) with chemical shifts reported in ppm. DOSY spectra were recorded using a Bruker AVANCE NEO 400 M spectrometer. UV-vis spectra of the solution were obtained using a SHIMADZU UV 2600 UV-vis spectrophotometer. The ESI mass spectra were acquired using an Agilent G6224A HPLC-ESI-TOF/MS spectrometer in an acetonitrile/methanol solution. All electrochemical measurements were performed using a CHI 760E electrochemical workstation with a three-electrode setup, where an Ag/AgCl electrode served as the reference, a platinum wire (0.5 mm in diameter) was used as the counter electrode, and a glassy carbon electrode acted as the working electrode. Prior to all measurements, the solutions were degassed with argon to remove any oxygen interference. The FT-IR reflectance spectra were recorded on a Thermo Fisher iS50 Fourier transform infrared spectrometer. Gas chromatography was performed with a GC 7890T. Gas chromatography mass spectra were acquired using an Agilent 7000B Triple Quadrupole GC/MS.Synthesis of H1
Ligand L (78.9 mg, 0.15 mmol) and Fe(OTF)2 (35.4 mg, 0.1 mmol) were dissolved in 20.0 mL of DMF. The mixture was stirred for 12 h under inert gas protection. After the reaction mixture was cooled to room temperature, the supernatant was filtered twice with a filter. Purple crystals of H1 were obtained by slowly diffusing diethyl ether into the abovementioned DMF solution of H1. The crystals were obtained by static growth at room temperature for 2 weeks. Yield: 43%.Synthesis of M1
2,2′-Dipyridyl (46.8 mg, 0.3 mmol) and Fe(OTF)2 (35.0 mg, 0.1 mmol) were dissolved in 20.0 mL of CH3CN. The mixture was stirred and refluxed over 12 h under inert gas protection. The reaction solution was then recrystallized, yielding dark purple block-shaped crystals. Yield: 63%.The method of electrocatalytic NO2− reduction by H1
Controlled potential electrolysis experiments were conducted in a custom-designed single-chamber electrolytic cell using a CHI 760E electrochemical workstation. The working electrode was a hydrophobic carbon paper electrode (j = 10.0 mA cm−2, electrode area A = 0.25 cm2), the reference electrode was an Ag/AgCl electrode with a 3.0 M KCl solution, and the counter electrode was a platinum sheet with a diameter of 5.0 mm. For the tests, the single-chamber electrolytic cell was filled with 12.0 mL of DMSO/MOPS (v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0.1 M LiCl, 0.1 M NaNO2, and 0.1 mM H1. Before adding the 0.1 mM H1 catalyst, the cell was purged with argon gas for 20 minutes. After 6 hours of reaction, the yield of the target NH4+ was determined by UV-visible spectroscopy.
:1), 0.1 M LiCl, 0.1 M NaNO2, and 0.1 mM H1. Before adding the 0.1 mM H1 catalyst, the cell was purged with argon gas for 20 minutes. After 6 hours of reaction, the yield of the target NH4+ was determined by UV-visible spectroscopy.
Single-crystal analysis
Single crystal X-ray diffraction data for compound H1 were collected using a Bruker D8 VENTURE Metaljet PHOTON II diffractometer with Ga-Ka (λ = 1.34138 Å) radiation at 170 K. Crystal data: C215.5H249.5F24Fe4N56.5O72S8, Mr = 5719.08, cubic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . CCDC 2429704.† The data were collected and reduced using APEX4 programs. The structure was solved using ShelXT and refined by full-matrix least-squares in ShelXL. Disordered anions and solvent molecules could not be modeled and were handled using the SQUEEZE routine in PLATON.26–28 The SQUEEZE process accounted for 8 OTF− anions, one diethyl ether molecule, approximately 8.5 DMF molecules, and 26.5 water molecules. The validity of the squeezed content was further supported by TGA. Although the data resolution and completeness are suboptimal, the connectivity and geometry of the core structure remain unambiguous.
. CCDC 2429704.† The data were collected and reduced using APEX4 programs. The structure was solved using ShelXT and refined by full-matrix least-squares in ShelXL. Disordered anions and solvent molecules could not be modeled and were handled using the SQUEEZE routine in PLATON.26–28 The SQUEEZE process accounted for 8 OTF− anions, one diethyl ether molecule, approximately 8.5 DMF molecules, and 26.5 water molecules. The validity of the squeezed content was further supported by TGA. Although the data resolution and completeness are suboptimal, the connectivity and geometry of the core structure remain unambiguous.
Results and discussion
Construction of metal–organic tetrahedral cage H1
The ligand L was synthesized by reacting dimethyl terephthalate with 2,2-bipyridine-5-carbaldehyde in methanol. The mixture formed the metal–organic cage FeII4L6 (H1) with a yield of 43%.The single crystal structural analysis revealed the formation of an FeII4L6 tetrahedral cage and H1 crystallized in the P space group. The six ligands acted as the edges of the tetrahedral cage and each iron ion was coordinated to three different 2,2-bipyridine units in a mer position with three delocalized NN chelators, showing the robust geometry and high stability of H1. The adjacent Fe ions were separated by an Fe⋯Fe distance of about 22.4 Å. The cage was equipped with an internal cavity with a volume of about 1298.3 Å3, and a triangular window with a height of about 20.3 Å (Fig. 1b), indicating the ability of H1 for substrate encapsulation. The hydrazide groups embellished on the tetrahedral edges were supposed to offer the hydrogen bonding sites around the tetrahedral cage, which would benefit the interactions in the host–guest combination, enabling fast electron and proton transfer from the active sites to substrates.29
 | ||
| Fig. 1 (a) The self-assembly process of H1 and the structure of the tetrahedral metal–organic cage and the inner space of H1 (pink ball). (b) The triangle-shaped window of H1. (c) The space filling pattern of H1. | ||
The 1H NMR spectrum showed a single set of ligand signals, evidencing the high symmetry of the H1 molecule. The complex exclusively contains low-spin iron(II) centres, as confirmed by sharp diamagnetic 1H NMR signals and its characteristic dark purple colour.30 The diffusion-ordered NMR spectrum (DOSY) showed that all resonances corresponded to a single species, with a diffusion coefficient of 5.01 × 10−11 m2 s−1 (Fig. 2a).
 | ||
| Fig. 2 (a) 1H NMR spectrum of L (black line) and 1H NMR and DOSY of H1 (red line) (400 MHz, DMSO-d6,298K). (b) The ESI-MS spectrum of H1 (1.0 mM) in CH3CN solution. (c) The ESI-MS spectrum of H1 (1.0 mM) with the addition of NO2− (3.0 mM) in CH3CN solution. | ||
The ESI-MS spectrum of H1 (1.0 mM) in CH3CN solution exhibited an intense peak at m/z = 422.8595, corresponding to the [FeII4L6]8+ species, which proved the stability of H1 in solution and the presence of divalent iron ions in H1 (Fig. 2b). These results collectively verified the successful formation of the target cage structure with high purity.
The cyclic voltammogram (CV) of H1 in DMSO solution displayed quasi-reversible FeI/Fe0, FeII/FeI and FeIII/FeII couples at −1.46 V, −0.44 V and 0.27 V (vs. Ag/AgCl), respectively.31 Upon the addition of Et3N·HCl to the H1 solution, cyclic voltammograms showed new catalytic waves at approximately −1.16 V (vs. Ag/AgCl). The intensity of these waves increased linearly with increasing concentration of Et3N·HCl, indicating that the H1 hydrogen evolution reaction (HER) occurs at approximately −1.16 V (vs. Ag/AgCl) (Fig. S18 and S19, ESI†).
Encapsulation and activation of NO2− by H1
Upon the addition of NO2− (3.0 mM) to an acetonitrile solution of H1 (1.0 mM), a new peak emerged at m/z = 486.8825. Comparison with simulations based on natural isotopic abundance confirmed the identity of the species as [FeII4L6·6NO2−·6Na+]8+ (Fig. 2c), showing a strong capability for binding NO2−. Meanwhile, upon the addition of NO2− (0.05 mM), the 1H NMR spectrum of H1 (0.01 mM) solution showed downfield shifts for the N–H protons in H1 (from 12.1 ppm to 12.5 ppm), suggesting a hydrogen bonding binding interaction between H1 and NO2− around the NH sites on the cage edges (Fig. 3a). In DMSO solution, H1 exhibited ligand-to-ligand and metal-to-ligand charge transfer transitions at 351 nm and 558 nm. The addition of NO2− (0.05 mM) to H1 (0.1 mM) solution resulted in noticeable changes in the absorption bands, with two isosbestic points observed at 341 and 453 nm, suggesting the formation of interactions between cage H1 and NO2− (Fig. 3b). The Fourier transform infrared (FT-IR) reflectance spectrum revealed that upon the addition of NO2− (5.0 mM) to H1 (1.0 mM) solution, the N–H stretching bond shifted from 3400 cm−1 to 3450 cm−1 (Fig. 3c). These results indicated that the amide group of H1 could interact with NO2− through hydrogen bonding interactions, which would probably enable fast electron and proton transfer from the active sites to substrates in the subsequent reduction process.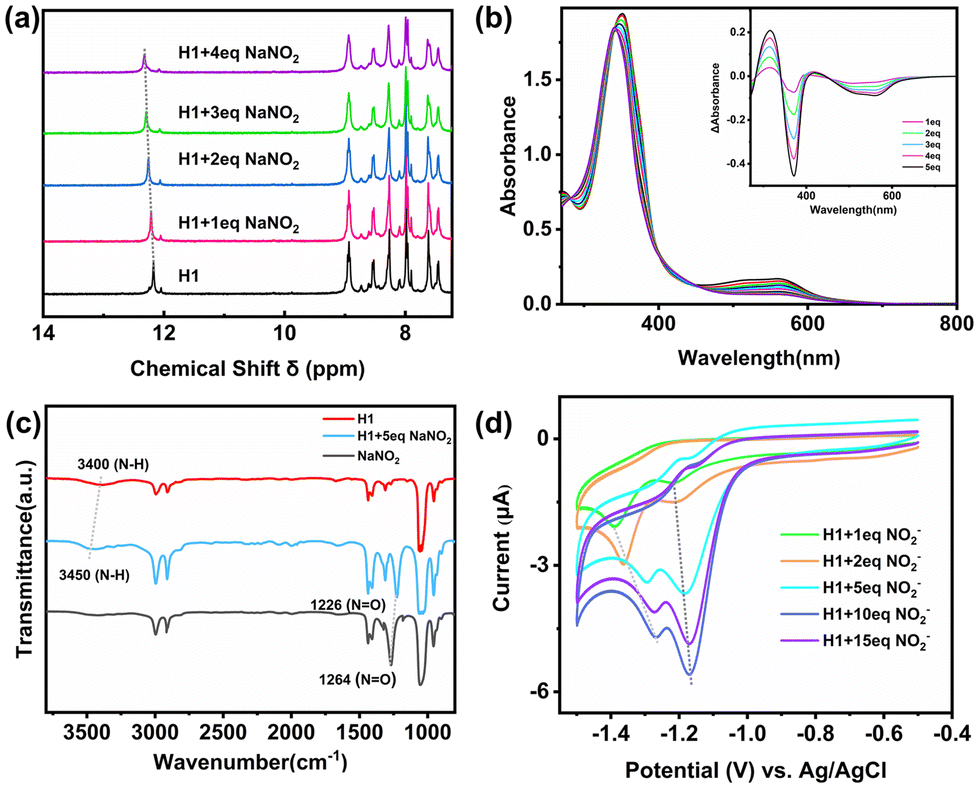 | ||
| Fig. 3 (a) Nuclear magnetic titration experiments of H1 and NO2−. 1H NMR spectrum of H1 (0.01 mM) in DMSO-d6 solution upon addition of NO2−. (b) UV/vis spectra of H1 (0.1 mM) in DMSO solution upon addition of NO2−. Inset: UV-vis absorption difference spectra of H1 with sequential addition of NO2− solution. (c) FT-IR reflectance spectra of H1 (1.0 mM), NO2− (5.0 mM) and H1 (1.0 mM) with the addition of NO2− (5.0 mM) in DMSO solution. (d) Cyclic voltammograms of H1 (0.01 mM) in DMSO solution with the sequential addition of NO2−. | ||
The cyclic voltammograms (CVs) showed the reduction potential of NO2− at near −1.4 V (vs. Ag/AgCl) (Fig. S16, ESI†). However, the addition of NO2− into the H1 solution at increasing concentrations caused new catalytic waves at approximately −1.3 V (vs. Ag/AgCl) besides the reduction potential of the cage itself. Notably, the catalytic wave exhibited a shift from −1.3 V (vs. Ag/AgCl) to near −1.1 V (vs. Ag/AgCl) as the NO2− concentration increased (Fig. 3d). This anode shifted reduction potential of the encapsulated NO2− compared with the free NO2− suggested that H1 could activate NO2− in a thermodynamic way and expand the interval between NO2− reduction and proton reduction, which may play a key role in the further selective electrocatalytic reduction of NO2−.23,32,33
Selective electrocatalytic reduction of NO2− by H1
Due to the solubility limitations of nitrite, its reduction process usually requires an aqueous solution.34 To minimize the pH fluctuations during the catalytic process, a buffer solution was used, with 4-morpholinepropanesulfonic acid (MOPS) selected as the buffering agent.35 UV-vis absorption spectroscopy confirmed that H1 remained stable in a DMSO/MOPS (v/v = 2:1) solution (Fig. S24 and S25, ESI†). Our catalytic performance was initially evaluated in a single-chamber electrochemical cell, where hydrophobic carbon paper served as the working electrode, an Ag/AgCl solid-state electrode was used as the reference electrode, and a graphite electrode with a diameter of 5.0 mm functioned as the counter electrode. Controlled potential electrolysis experiments were conducted using the H1 (0.1 mM) catalyst in a DMSO/MOPS (v/v = 2:1) solution containing 0.1 M LiCl as the electrolyte and 0.1 M NaNO2 as the guest molecules and carried out over 6 hours. The ammonium ion concentration was determined using a standard colorimetric method (Fig. S26 and S27, ESI†).36 Linear sweep voltammograms demonstrated a linear relationship between the current intensity of the reaction mixture and the NO2− concentration (Fig. 4b). The UV-vis absorption spectroscopy test on the electrolyte solution after the reaction and the XPS analysis of the hydrophobic carbon paper after the CPE indicated that the catalyst demonstrated excellent stability throughout the reaction process (Fig. S29 and S42, ESI†).
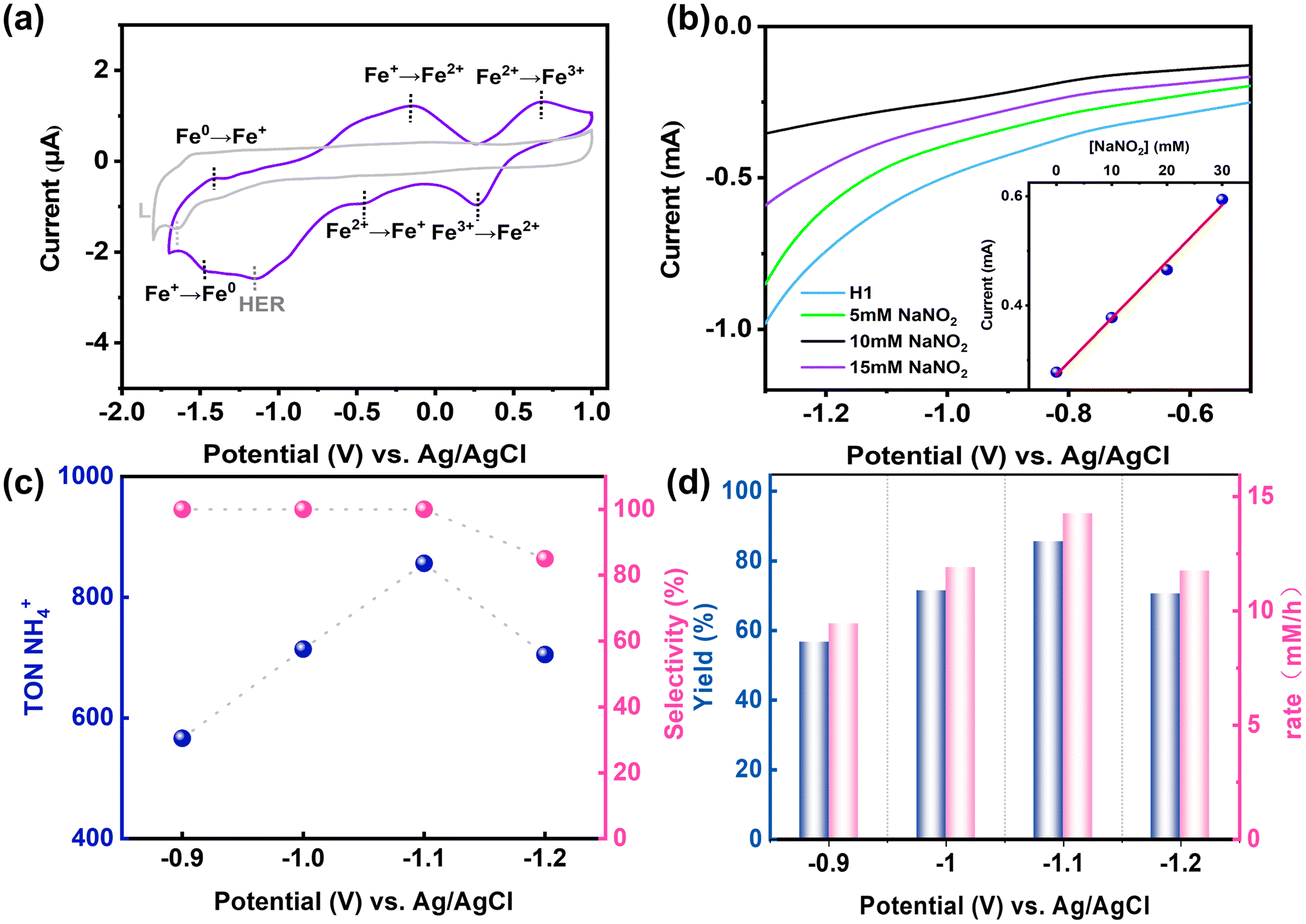 | ||
| Fig. 4 (a) Cyclic voltammograms of H1 and ligand L in DMSO solution. (b) Linear sweep voltammograms of H1 (0.1 mM) with 0.1 M LiCl in DMSO/MOPS (v/v = 2:1) solution were recorded with the addition of NO2−. Inset: current intensity (−1.1 V) versus the concentration of NO2−. (c) The TON and selectivity of NH4+ under standard conditions. (d) Yield and rate of NH4+ produced by electrocatalytic NO2− reduction over the H1 catalyst. | ||
In order to fully analyse the products and by-products of the reaction process, we used a variety of analytical techniques. Gas chromatography (GC) and gas chromatography–mass spectrometry (GC–MS) were used to detect and analyse the gases produced during the reaction. The results showed that at an applied potential of −1.2 V, a small amount of hydrogen gas was generated (Fig. S33 and S34, ESI†). Immediately after electrolysis, all gases in the reaction chamber were purged into a sealed absorption system containing a diluted NaOH aqueous solution. The resulting solution was then analysed using ion chromatography (IC). The results showed no detectable formation of nitrogen oxide by-products (Fig. S31, ESI†). Furthermore, to probe for the potential formation of hydrazine (N2H4) as a side product, UV-vis spectroscopy was employed using the Watt and Christo method. The results indicated that no hydrazine was formed during the reaction (Fig. S32, ESI†). These findings confirm the high selectivity of the electrocatalytic reduction of nitrite by H1.
Under optimal conditions, the nitrite reduction reaction catalysed by H1 achieved a maximum faradaic efficiency of 35.1% and a TON of 856, and only NH4+ was detected as the reduction product, indicating its high selectivity. Moreover, the NH4+ yield reached 85.6%, with a production rate of 14.27 mM h−1 at an applied potential of −1.1 V (vs. Ag/AgCl). When the applied potential was increased from −0.9 V to −1.1 V (vs. Ag/AgCl), the NH4+ yield significantly increased from 56.3% to 85.6%, and the production rate increased from 9.43 mM h−1 to 14.27 mM h−1. Additionally, the TON value increased from 563 to 856, the faradaic efficiency slightly improved, and no hydrogen generation was detected. However, at −1.2 V (vs. Ag/AgCl), the NH4+ yield dropped to 70.5%, the conversion rate decreased to 11.75 mM h−1, and the TON decreased to 705, with only a trace amount of H2 observed (Fig. 4c and d). In addition, in the CPE experiment, the current remained stable, demonstrating that H1 remained stable in solution and maintained its catalytic performance over extended reaction periods without a decline in activity that could affect reaction efficiency.
To determine the source of nitrogen, isotope labeling experiments were conducted using Na15NO2 in the CPE experiment. In the 1H NMR spectrum of the treated reaction solution, two hydrogen signals were observed with a coupling constant of 75 Hz. In the 15N NMR spectrum, a clear signal was detected at 19.6 ppm. These isotope labeling experiments further confirmed that the nitrogen in the ammonium originates from nitrite (Fig. S35 and S36, ESI†).
Control experiments were performed under identical conditions. The mononuclear complex M1 was synthesized by reacting 2,2′-bipyridine with Fe(OTF)2. M1 closely resembles a vertex of tetrahedral cage H1, but M1 lacks the cavity microenvironment. The ESI-MS spectrum of M1 (1.0 mM) in CH3CN solution exhibited a prominent peak at m/z = 262.0872. Comparison with simulation based on natural isotopic abundance indicated that the peak corresponds to the [FeIIL′3]2+ species (Fig. S8, ESI†), demonstrating the physical phase purity and stability of M1. The coordination environments of the metal ions in M1 and H1 are identical. However, the FT-IR reflectance spectrum revealed no interaction between M1 and nitrite (Fig. S14, ESI†). The UV-vis spectroscopy titration experiments showed that no iso-absorption point was created between M1 and nitrite (Fig. S12, ESI†). The cyclic voltammograms showed that the addition of NO2− affected only the change in current, with no new catalytic waves produced (Fig. S22, ESI†). When M1 was used as a catalyst, the ammonium yield was found to be low, at only 14%. We deduce that the mononuclear compound M1, lacking a cavity-limited domain centre, cannot specifically recognize and activate nitrite, despite possessing a similar iron-reducing centre.
In contrast, no catalytic products were observed when Fe(OTF)2 and L were used as catalysts. It is likely that host–guest interactions within the inner cavity of the metal–organic tetrahedral cage H1 (primarily hydrogen bond acceptor sites and electrostatic interactions arising from the high cationic charge of H1) played a crucial role in the electrochemical catalytic conversion, rather than the simple FeII centres and amide group of L. Further comparative experiments also demonstrated that MOPS buffer and passage of argon gas were necessary for product detection (Table 1).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
| Standard conditions: H1 (0.1 mM) and NaNO2 (0.1 M) in DMSO/MOPS (v/v = 2:1) containing LiCl (0.1 M) for 6 h. Yields were determined using the standard colorimetric method. N.D. = not detected. |
||
| 1 | None | 85.6% |
| 2 | Under air | N.D. |
| 3 | No MOPS | N.D. |
| 4 | No stirring | 42% |
| 5 | Fe(OTF)2 | N.D. |
| 6 | L | N.D. |
| 7 | M1 instead of H1 | 14% |
When the concentration of NO2− was fixed and the concentration of H1 varied between 0.02 and 0.1 mM, the initial reaction rate increased with H1 concentration from 0.158 mM h−1 to 0.283 mM h−1, showing a first-order linear relationship (Fig. 5b).37 Kinetic studies of the reduction of NO2− to ammonium by H1 revealed that the catalytic process follows a primary reaction pathway. To further validate the catalytic process of NO2− reduction by the H1 electrocatalytic system as a mimicked enzyme catalysis, the reaction kinetics were studied under different applied potential conditions by varying the substrate concentration. The double reciprocal fitting of the initial reaction rate and the corresponding substrate concentration showed a linear relationship (Fig. 5c). Lineweaver–Burk analysis shows that under different voltages, the initial reaction rate exhibits saturation behaviour with respect to NO2− concentration.38 The fitting results align with the Michaelis–Menten mechanism in enzyme-catalysed reactions. The nearly identical Km (ca. 4.1 mM) across different applied potentials, along with the linear relationship between Kcat and applied potentials, supported the Michaelis–Menten mechanism.39 Therefore, we suggested that host–guest activation interactions, primarily hydrogen bonding and electrostatic interactions between the hydrogen-bonded acceptors and nitrite in the internal cavity of the tetrahedral cage H1, played a crucial role in thermodynamically activating and facilitating its efficient electrocatalytic reduction to ammonium (Fig. 5a).
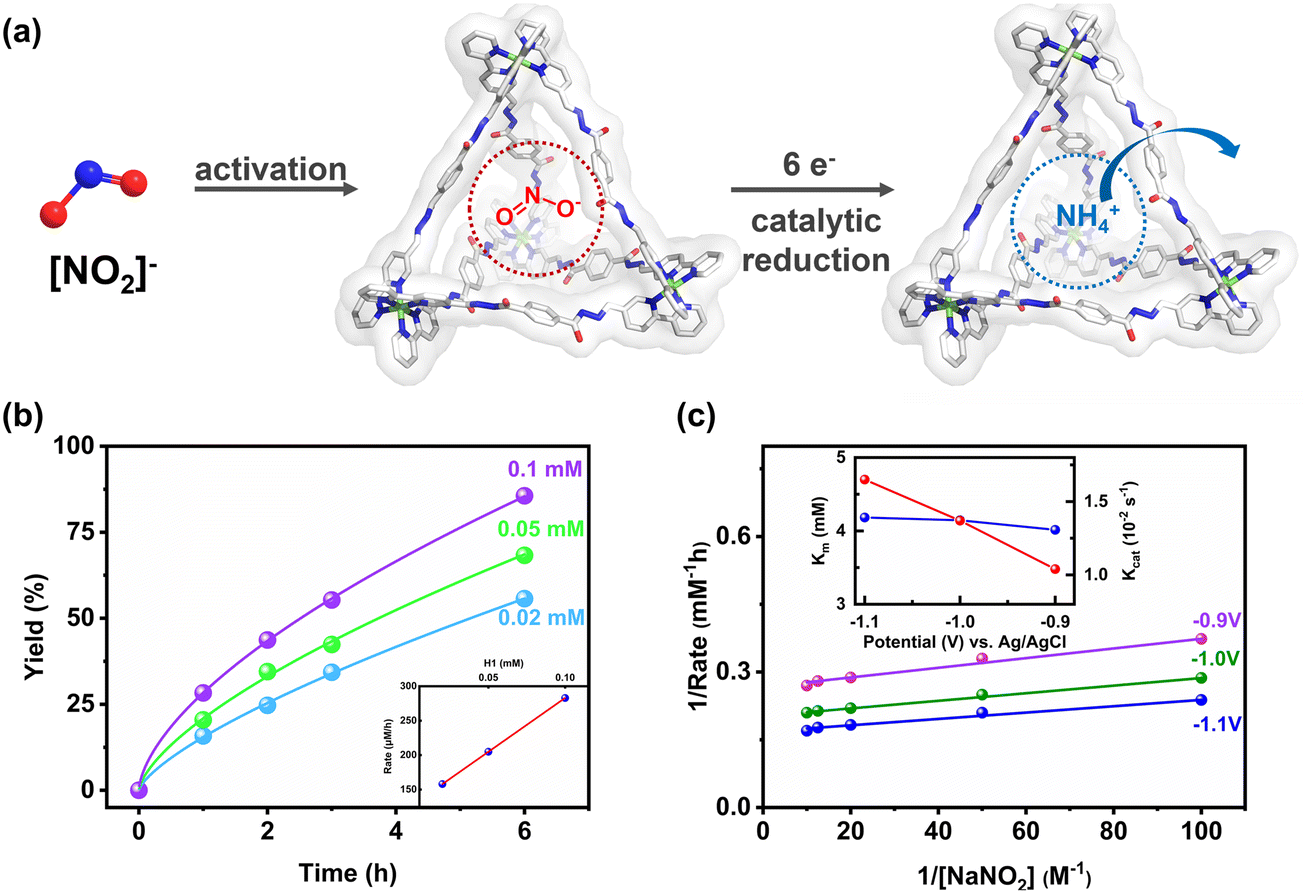 | ||
| Fig. 5 Enzymatic kinetics of the activation of NO2− by H1. (a) Proposed enzymatic mechanism of H1 for the electroreduction of NO2−. (b) Kinetics with different concentrations of H1. Inset: dependence of the initial rate on the concentration of H1. (c) Reciprocal plot of the initial rate at different potentials with the concentration of NO2−. Inset: Km and Kcat at different potentials. | ||
Conclusions
In summary, we reported the synthesis of an Fe-based tetrahedral metal–organic cage with a large internal cavity and an iron-reducing centre, surrounded by hydrazide groups that serve as hydrogen bond acceptor sites. This enables the cage to efficiently recognize nitrite and thermodynamically activate it for effective reduction to ammonium under electrocatalytic conditions. Under optimal conditions, an NH4+ yield of 85.6% and a conversion rate of 14.27 mM h−1 were achieved, with H1 maintaining good catalytic activity throughout the process. Kinetic studies confirmed that the catalytic process followed the Michaelis–Menten mechanism, further validating the crucial role of host–guest interactions in nitrite activation and demonstrating that the process falls under the category of mimetic enzyme catalysis. This offers new insights for the development of mimetic enzymes to catalyse the reduction of nitrite and other substrate molecules.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for H1 have been deposited at the CCDC under 2429704† and can be obtained from https://www.ccde.cam.ac.uk/structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFA1503302), the National Natural Science Foundation of China (22401033), and the China Postdoctoral Science Foundation (2023M740485).References
- Y. Fang, J. Li, T. Togo, F. Jin, Z. Xiao, L. Liu, H. Drake, X. Lian and H.-C. Zhou, Chem, 2018, 4, 555–563 CAS.
- Y.-H. Huang, Y.-L. Lu, X.-D. Zhang, C.-H. Liu, J. Ruan, Y.-H. Qin, Z.-M. Cao, J. Jiang, H.-S. Xu and C.-Y. Su, Angew. Chem., Int. Ed., 2024, 63, e202315053 CrossRef CAS PubMed.
- J. Cai, L. Zhao, C. He, Y. Li and C. Duan, Nat. Commun., 2021, 12, 5092 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, C. Mu, L. Ma, H. Yuan, S. Ling, H. Wang, X. Li and M. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202207289 CrossRef CAS PubMed.
- J. Guan, J. Du, Q. Sun, W. He, J. Ma, S. U. I. Hassan, J. Wu, H. Zhang, S. Zhang and J. Liu, Sci. Adv., 2025, 11, eads0583 CrossRef CAS PubMed.
- G. Li, Z. Du, C. Wu, Y. Liu, Y. Xu, R. Lavendomme, S. Liang, E.-Q. Gao and D. Zhang, Nat. Commun., 2025, 16, 546 CrossRef CAS PubMed.
- D. Liu, H. Ma, C. Zhu, F. Qiu, W. Yu, L.-L. Ma, X.-W. Wei, Y.-F. Han and G. Yuan, J. Am. Chem. Soc., 2024, 146, 2275–2285 CrossRef CAS PubMed.
- S.-C. Li, L.-X. Cai, L.-P. Zhou, F. Guo and Q.-F. Sun, Sci. China:Chem., 2019, 62, 713–718 CrossRef CAS.
- M.-S. Yue, N. Luo, X.-D. Wang, Y.-F. Ao, D.-X. Wang and Q.-Q. Wang, J. Am. Chem. Soc., 2025, 147, 2303–2308 CrossRef CAS PubMed.
- Y.-H. Huang, Y.-L. Lu, Z.-M. Cao, X.-D. Zhang, C.-H. Liu, H.-S. Xu and C.-Y. Su, J. Am. Chem. Soc., 2024, 146, 21677–21688 CrossRef PubMed.
- L.-J. Wang, Z.-E. Zhang, Y.-Z. Zhang and Y.-F. Han, Angew. Chem., Int. Ed., 2024, 63, e202407278 CrossRef CAS PubMed.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed.
- X. Zhao, G. Hu, G.-F. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 Search PubMed.
- F. Wang, S. Shang, Z. Sun, X. Yang and K. Chu, ACS Nano, 2024, 18, 13141–13149 CrossRef CAS PubMed.
- G. Wen, J. Liang, Q. Liu, T. Li, X. An, F. Zhang, A. A. Alshehri, K. A. Alzahrani, Y. Luo, Q. Kong and X. Sun, Nano Res., 2022, 15, 972–977 Search PubMed.
- S.-L. Meng, C. Zhang, C. Ye, J.-H. Li, S. Zhou, L. Zhu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Energy Environ. Sci., 2023, 16, 1590–1596 RSC.
- S. Partovi, Y. Losovyj, N. Yamamoto and J. M. Smith, Inorg. Chem., 2025, 64, 6293–6300 CrossRef CAS PubMed.
- P. H. van Langevelde, S. Engbers, F. Buda and D. G. H. Hetterscheid, ACS Catal., 2023, 13, 10094–10103 CrossRef CAS PubMed.
- S. E. Braley, H.-Y. Kwon, S. Xu, E. Z. Dalton, E. Jakubikova and J. M. Smith, Inorg. Chem., 2022, 61, 12998–13006 CrossRef CAS PubMed.
- H. Wang, F. Zhang, M. Jin, D. Zhao, X. Fan, Z. Li, Y. Luo, D. Zheng, T. Li, Y. Wang, B. Ying, S. Sun, Q. Liu, X. Liu and X. Sun, Sci. China:Chem., 2023, 30, 100944 CAS.
- X. He, Z. Li, J. Yao, K. Dong, X. Li, L. Hu, S. Sun, Z. Cai, D. Zheng, Y. Luo, B. Ying, M. S. Hamdy, L. Xie, Q. Liu and X. Sun, iScience, 2023, 26, 107100 CrossRef CAS PubMed.
- J. Xiang, C. Qiang, S. Shang, K. Chen, C. Kang and K. Chu, Adv. Funct. Mater., 2024, 34, 2401941 CrossRef CAS.
- Y. Yang, X. Jing, Y. Shi, Y. Wu and C. Duan, J. Am. Chem. Soc., 2023, 145, 10136–10148 Search PubMed.
- G. Zhang, N. Xu, M. Yang, W. Wang, K. Su and D. Yuan, Angew. Chem., Int. Ed., 2025, e202423226 Search PubMed.
- Y. Hou, C. Mu, Y. Shi, Z. Zhang, H. Liu, Z. Zhou, S. Ling, B. Shi, X. Duan, C. Yang and M. Zhang, Aggregate, 2024, 5, e628 Search PubMed.
- SMART Data collection software (version 5.629), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- SAINT Data reduction software (version 6.45), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- G. M. Sheldrick, SHELXTL97 Program for crystal structure solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- Y. Yang, H. Li, X. Jing, Y. Wu, Y. Shi and C. Duan, Chem. Commun., 2022, 58, 807–810 RSC.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed.
- M. Cheng, Y. Yu, X. Zhou, Y. Luo and M. Wang, ACS Catal., 2019, 9, 768–774 CrossRef CAS.
- J. Wei, L. Zhao, Y. Zhang, G. Han, C. He, C. Wang and C. Duan, J. Am. Chem. Soc., 2023, 145, 6719–6729 CrossRef CAS PubMed.
- F. J. Rizzuto, D. M. Wood, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 11008–11011 CrossRef CAS PubMed.
- J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, Q. Liu, M. S. Hamdy, B. Tang and X. Sun, Chem, 2023, 9, 1768–1827 CAS.
- J. Ferguson, J. Brown and D. Richeson, ChemCatChem, 2024, 16, e202301168 CrossRef CAS.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- E. Bakalis, A. Soldà, M. Kosmas, S. Rapino and F. Zerbetto, Anal. Chem., 2016, 88, 5790–5796 CrossRef CAS PubMed.
- D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235–1238 CrossRef CAS PubMed.
- Y. Yang, X. Jing, J. Zhang, F. Yang and C. Duan, Nat. Commun., 2022, 13, 1940 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2429704. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00766f |
| This journal is © The Royal Society of Chemistry 2025 |