
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer–drug conjugates as nano-sized multi-targeting systems for the treatment of Alzheimer's disease
Nuruddin
Mahadik
 ,
Gemma A.
Barron
,
Paul
Kong Thoo Lin
and
Colin J.
Thompson
*
,
Gemma A.
Barron
,
Paul
Kong Thoo Lin
and
Colin J.
Thompson
*
School of Pharmacy and Life Sciences, Robert Gordon University, Aberdeen, UK. E-mail: c.thompson@rgu.ac.uk
First published on 29th February 2024
Abstract
Alzheimer's disease (AD) is a progressive, neurodegenerative condition. There are clear markers for the presence and progression of the disease, including β-amyloid (Aβ) plaques and Tau tangles, with many potential causes debated in the scientific community. Most existing treatments only provide symptomatic solutions. Due to poor aqueous solubility and possibly limited uptake across the blood–brain barrier (BBB), medications targeting the hallmarks of AD are still under study despite enormous efforts. Recently, nanoparticle-based drug delivery systems have demonstrated remarkable promise as precision medicines that may effectively increase bioavailability, permeate the BBB, and improve the targeting ability of a variety of pharmaceuticals. Polymer therapeutics have made tremendous progress in recent years, particularly in cancer treatment. Polymer–drug conjugates (PDCs) typically have a longer half-life, higher stability, and enhanced water solubility. Polymers serve as carriers for the administration of drugs, proteins, targeting moieties, and imaging agents in polymeric and macromolecular prodrugs. Numerous commercially viable PDCs for the treatment of various diseases have already proved their potential. This paper focuses mainly on the rationale for the design, synthesis, and potential use of PDCs as a multi-target treatment for neurodegenerative diseases.
1. Introduction
Neurological disorders such as Alzheimer's disease and diseases resulting in dementia, and epilepsy are the most significant global health and social care concerns.1 Alzheimer's disease (AD), the most prevalent type of dementia, is a progressive, neurodegenerative disease.2 Over 27 million people worldwide, or 70% of all dementia cases, are affected by AD, and most of these cases are sporadic with age being the primary risk factor.3,4 As the World Health Organisation (WHO) reported, AD and various forms of dementia ranked seventh among the leading causes of mortality in 2019.5 Several drug therapies based on small molecules, monoclonal antibodies, and CRISPR/CAS9 are being developed or assessed in clinical trials based on two well-established AD mechanisms, namely the Amyloid β (Aβ) plaques and Neurofibrillary Tangles (NFTs), which are regarded as key pathological hallmarks in disease progression.2,6–8 As with contemporary AD therapy, however, symptomatic treatment is the primary countermeasure due to the disease's complex aetiology, relating to many endogenous (genetic) factors, and the environment.2 The Food and Drug Administration (FDA) has approved just seven drugs for the treatment of AD, four of which are acetylcholinesterase inhibitors (AChEIs): tacrine (withdrawn due to hepatotoxicity), donepezil, galantamine, and rivastigmine, and approved an N-methyl-D-aspartate (NMDA) receptor antagonist known as memantine.9,10 Even though the marketed AChE inhibitors have been shown to improve cognitive performance, none of these treatments can delay or reverse AD progression, underscoring the complexity of the disease's genesis.11 Recently the FDA approved monoclonal antibodies-based therapy, aducanumab (approved on June 7, 2021) and lecanemab (approved on January 6, 2023). They were authorised under the accelerated approval pathway and are only disease-modifying treatments for patients with mild AD.12,13 It is noteworthy to mention that approval of aducanumab is still contentious due to its low efficacy in phase III trials.12 Lecanemab is undergoing regulatory review in the European Union, Japan, and China, and clinical development is ongoing in several countries throughout the world.13 However, the benefits of cholinesterase inhibitors surpass those of the amyloid antibody-based therapy. Many drugs are also under development to treat various AD-related causes, including neuroinflammation, neurotransmitter imbalance, oxidative stress, mitochondrial dysfunction, and neuron loss and degeneration.9However, there are various limitations connected with existing therapy. AChEIs are associated with gastrointestinal adverse effects like stomach pain, nausea, weight loss, and vomiting, which most often result in the termination of treatment.14,15 In addition, the therapeutic efficacy of the medicine may be hindered by biological barriers that prevent drugs from reaching the brain. These include the drug's poor physicochemical properties, resulting in low bioavailability, poor pharmacokinetics, and pharmacodynamics and due to adverse effects.16
Advanced drug-delivery systems are being developed to guide drugs more precisely towards the site of action and away from sites of toxicity, and/or to maintain drugs at a therapeutic concentration for extended periods of time.17 Controlled drug delivery systems based on nanotechnology, such as polymeric nanoparticles, offer an intriguing strategy to bypass these reported and recognised disadvantages. Polymeric nanoparticles can preserve drugs from early degradation, enhance their water solubility, and ease-controlled drug release, consequently enhancing plasma circulation time, oral bioavailability, and efficacy. Synthetic polymer materials are employed in a growing number of applications and have several advantages over natural polymers. Synthetic polymers are chosen over natural polymers for the development of nanoparticles due to their superior repeatability and purity. Knowledge of the physicochemical properties of nanoparticles is crucial because it may be used to predict their physical and chemical stability, interactions between drug and polymer, in vivo behaviour, and trajectory of nanoparticles.18
Recently, polymer therapeutics have made tremendous progress in the treatment of cancer.17 Ruth Duncan19 invented the phrase “polymer therapeutics” to describe a novel class of drugs/advanced drug delivery systems that include “polymeric drugs, polymer–drug conjugates, polymer–protein conjugates, polymeric micelles to which drugs are covalently bound, and multicomponent polyplexes being developed as non-viral vectors” (see Fig. 1).
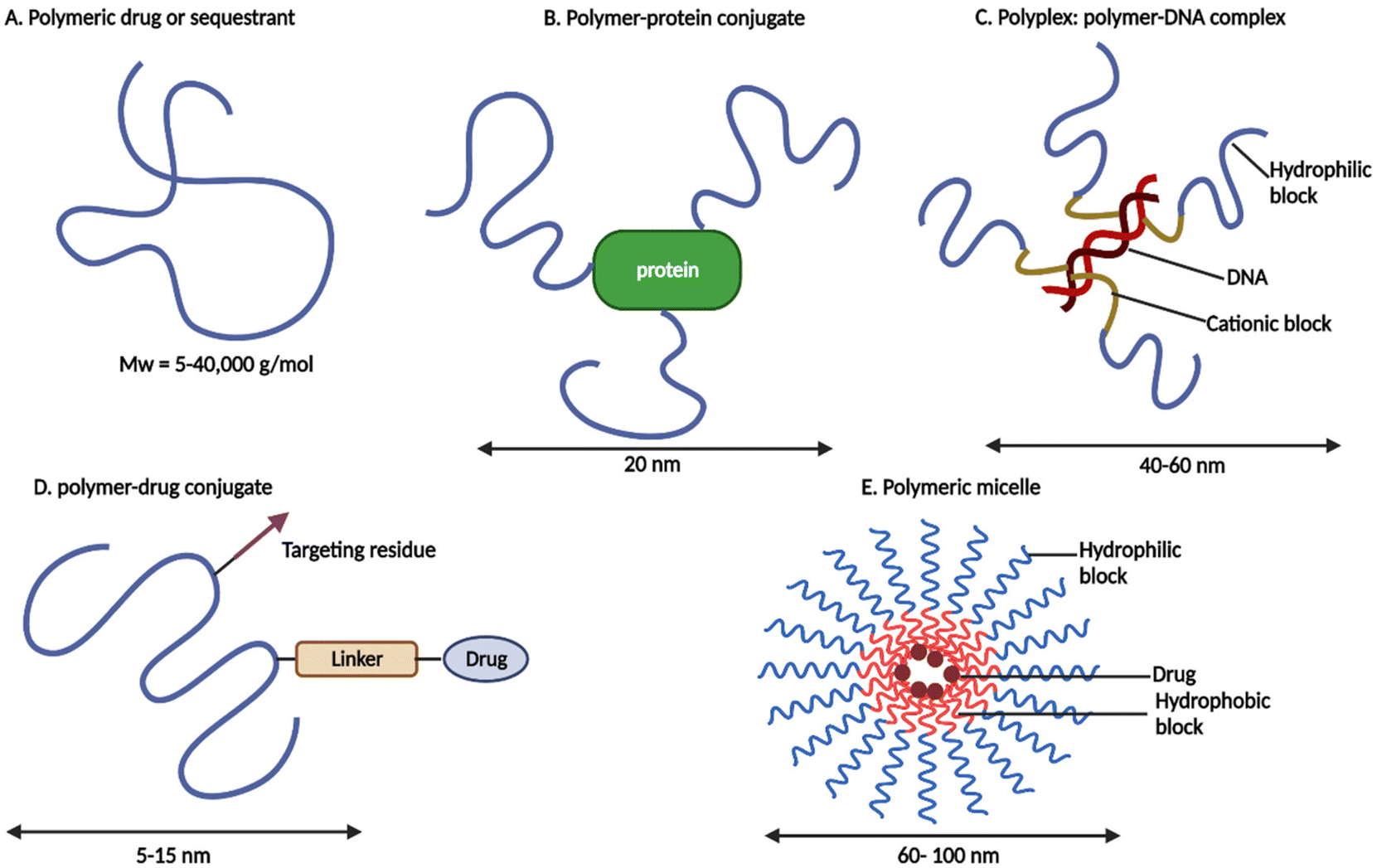 | ||
| Fig. 1 Schematic representation of polymer therapeutics (redrawn using BioRender from ref. 19). | ||
For enhanced drug, protein, or gene delivery, all subclasses employ specialised hydrophilic polymers and sometimes amphiphilic polymers, either as the bioactive itself or as an inert functional component of a multifunctional construct.19,20 From an industrial aspect, these nanosized medications are more akin to novel chemical entities than traditional “drug delivery systems or formulations” that merely entrap, solubilize, or control drug release without the need for chemical conjugation.21 Conjugation of the therapeutics to the polymer enhances the pharmacokinetic and pharmacodynamic properties of pharmaceuticals in many ways. This includes improved plasma half-life, protection of the therapeutic from proteolytic enzymes, reduction in immunogenicity, improved protein stability, enhanced solubility of low molecular weight (Mw) drugs, and the potential for targeted delivery. Most polymer therapeutics are intended for anticancer treatments, although other ailments, such as rheumatoid arthritis, diabetes, and hepatitis B and C, have also been targeted22 (see Table 1).
| Trade name and company | Polymeric carrier | Drug | Application | Year of approval | Ref. |
|---|---|---|---|---|---|
| EMPAVELI-Apellis Pharmaceuticals, Inc. | Polyethylene glycol (PEG) | Cyclic peptide complements C3 & C3b inhibitor | Paroxysmal nocturnal hemoglobinuria | 2021 | 23 |
| SKYTROFA-Ascendis Pharma | mPEG | Human growth hormone | Growth hormone deficiency | 2021 | 24 |
| ESPEROCT™-Novo Nordisk | PEG (40 kDa branched PEG) | Factor VIII | Hemophilia A | 2019 | 25 |
| ELOCTATE™-Biogen Idec, Inc. | PEG | Factor VIII | Hemophilia A | 2019 | 26 |
| JIVI™-Bayer HealthCare LLC | PEG | Factor VIII | Hemophilia A | 2018 | 27 |
| ASPARLAS™-Servier Pharmaceuticals LLC | mPEG | L-Asparaginase | Acute lymphoblastic leukemia | 2018 | 28 |
| REVCOVI™-Leadiant Biosciences Inc. | mPEG | Recombinant adenosine deaminase | Adenosine deaminase severe combined immunodeficiency | 2018 | 29 |
| PALYNZIQ-BioMarin Pharmaceutical Inc. | PEG | Pegvaliase (phenylalanine ammonia-lyase) | Phenyl ketonuria | 2018 | 30 |
| ADYNOVATE-Baxalta US Inc. | PEG | Factor VIII | Hemophilia A | 2015 | 31 |
| MOVANTIK™-AstraZeneca Pharmaceuticals | PEG | Naloxone | Opioid-induced constipation | 2014 | 32 |
| PLEGRIDY™-Biogen Idec Inc. | PEG | Interferon β1a | Relapsing multiple sclerosis | 2014 | 33 |
| KRYSTEXXA®-Savient Pharmaceuticals, Inc. | PEG | Uricase | Chronic gout | 2010 | 34 |
| OPAXIO™-Cell Therapeutics, Inc. | Poliglumex | Paclitaxel | Non-small cell lung cancer | 2008 (withdrawn). In 2013 FDA approved trials on pancreatic cancer as an orphan drug | 35 |
| CIMZIA®-UCB, Inc. | PEG (40 kDa branched PEG) | Anti-TNF Fab’ | Rheumatoid arthritis, psoriatic arthritis, Crohn's disease, & ankylosing spondylitis | 2008 | 36 |
| MIRCERA®-Vifor, Roche | PEG (30 kDa branched PEG) | β-Epoetin | Anemia associated with chronic kidney disease | 2007 | 37 |
| Macugen (Bausch & Lomb) | PEG | Anti-VEGF aptamer | Neovascular age-related macular degeneration | 2004 | 38 |
| SOMAVERT-Pfizer (Pharmacia and Upjohn Company LLC) | PEG | Human growth hormone receptor antagonist; pegvisomant | Acromegaly | 2003 | 39 |
| NEULASTA®-Amgen Inc. | PEG (20 kDa branched PEG) | Granulocyte colony-stimulating factor | Chemotherapy-induced neutropenia | 2002 | 40 |
| PEGASYS®-Hoffmann-La Roche Inc. | PEG (40 kDa branched PEG) | Interferon α2a | Hepatitis B and hepatitis C | 2002 | 41 |
| PEGINTRON®-Merck & Co., Inc. | PEG | Interferon α2b | Hepatitis C | 2001 | 42 |
| ONCASPAR®-Sigma-Tau Pharmaceuticals, Inc. | L-Asparaginase-PEG | L-Asparaginase | Acute lymphoblastic leukemia | 1994 | 43 |
| SMANCS-Astellas Pharma | Poly(styrene-co-maleic acid) | Neocarzinostatin | Advanced and recurrent hepatocellular carcinoma | 1993 | 44 |
| ADAGEN®-ENZON Pharmaceuticals, Inc. | PEG (5 kDa MW) | L-Asparaginase | Adenosine deaminase severe combined immunodeficiency | 1990 | 45 |
However, polymer therapeutics have not been very well studied in the treatment of neurodegenerative diseases. Therefore, looking at the various advantages of polymer therapeutics, the current work here, provides a review on how polymer–drug conjugates can be used as an advanced drug delivery system to improve the treatment of AD and other neurodegenerative diseases.
2. Polymer–drug conjugates
With the clinical and pre-clinical development of polymer-based nanomedicines and other biomedical applications, polymers, and polymer-based drug delivery systems have seen immense growth in the past few years. The main characteristic of polymer–drug conjugates (PDCs) is that the drug is covalently conjugated to the polymeric carrier instead of being non-covalently encapsulated within the polymeric structure. In this context, difficulties related to “burst drug release” can be resolved to a considerable extent. The drug can be covalently bonded to the polymer via linkers designed to release drugs within specified structures, or at a predetermined rate in vivo.46PDCs were first proposed approximately sixty-five years ago. In 1955, Jatzkewitz47 attached mescaline to water-soluble poly(vinyl pyrrolidone) via a dipeptide spacer to create the first PDC. In the mid-1970s, Ringsdorf was the first to provide a clear analysis of a comprehensive polymer-conjugated drug delivery system.48 An ideal PDC is characterised by a water-soluble polymer backbone as a vehicle and bioactive agent(s) typically attached to the polymeric scaffold via a biological response linker, as per the proposed method49 (see Fig. 1(D)). The choice of linkers is crucial as it enables the likelihood of influencing the percentage loading of the drug to the carrier, drug stability, controlling the site, and the rate of release of the drug from the conjugates either by hydrolysis (such as a change in pH), enzymatic degradation, or sensitivity to overexpressed diseased organ/tissue groups, % loading of the drug to the carrier, drug stability.50,51 Several linkers (see Fig. 2), such as hydrazine, azo, peptides, disulphides, and Schiff bases have been utilised for the conjugation of drugs to polymers.51,52
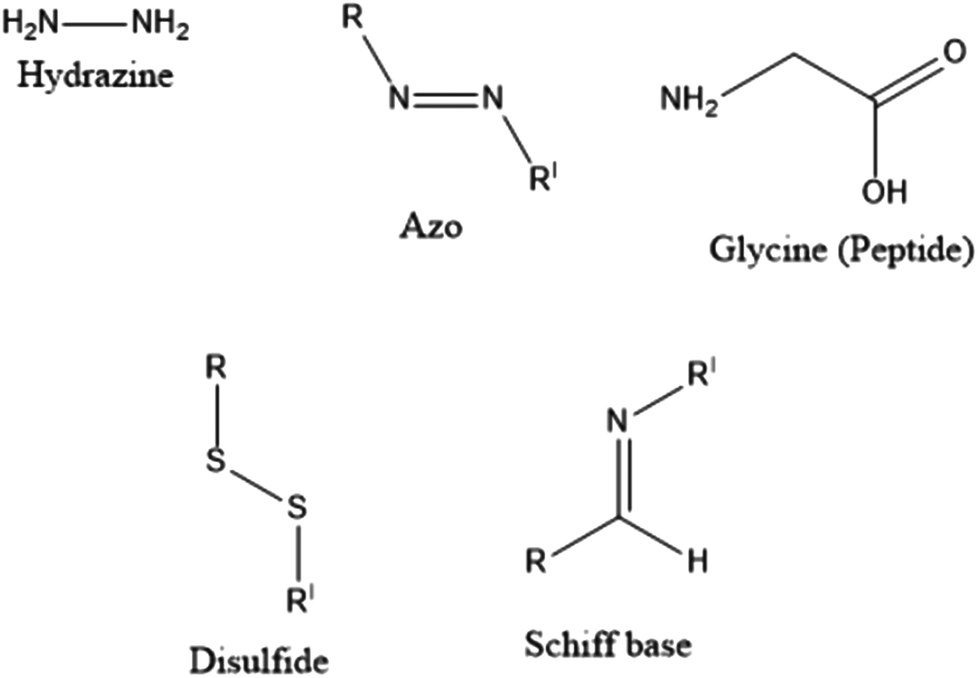 | ||
| Fig. 2 Some examples of linkers utilised for the conjugation of drugs to polymers. | ||
Occasionally, a targeting moiety, solubilizer, or imaging agent is also added to the conjugate to improve its therapeutic efficacy49 (see Fig. 3). Targeting moiety plays a role in delivering the conjugate to desired active site, where the targeting moiety binds to the biological target and releases the drug. Solubilizer increases the water-solubility of the conjugates.51,53 The addition of imaging agents increases the retention time of the conjugates in the biological target with increased concentrations, allowing for precise disease detection and characterisation.54 The drug's mechanism of action is the primary consideration in the design of administration routes for PDCs.49
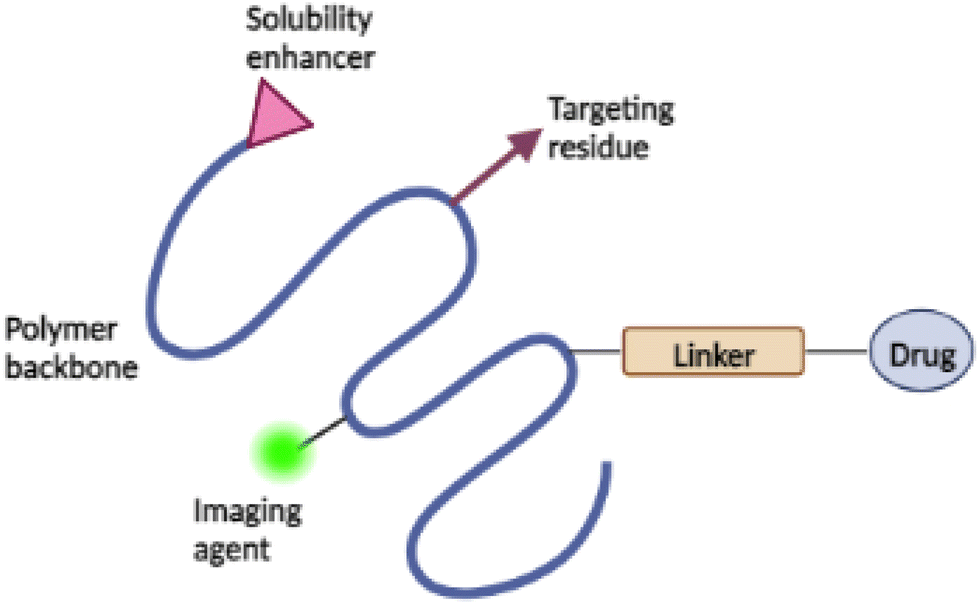 | ||
| Fig. 3 Schematic representation of PDC with a targeting moiety, solubilizer, and imaging agent. | ||
PDCs are commonly synthesised using one of the following three methods (see Fig. 4): (1) conjugating the drug to a synthetic polymer, (2) conjugation of the drug to a monomer, which leads to polymerisation followed by reversible addition–fragmentation transfer (RAFT), ring-opening metathesis polymerisation (ROMP), or ring-opening polymerisation (ROP), and (3) using a drug with two or more functional groups as a monomer for poly-drug polymerisation (permitting polycondensation reaction).46,55 The final two methods for synthesizing PDCs were recently designed to overcome the problems of uncontrolled conjugation to the polymer backbone leading to increased drug loading, and steric hindrance during conjugation.51,55
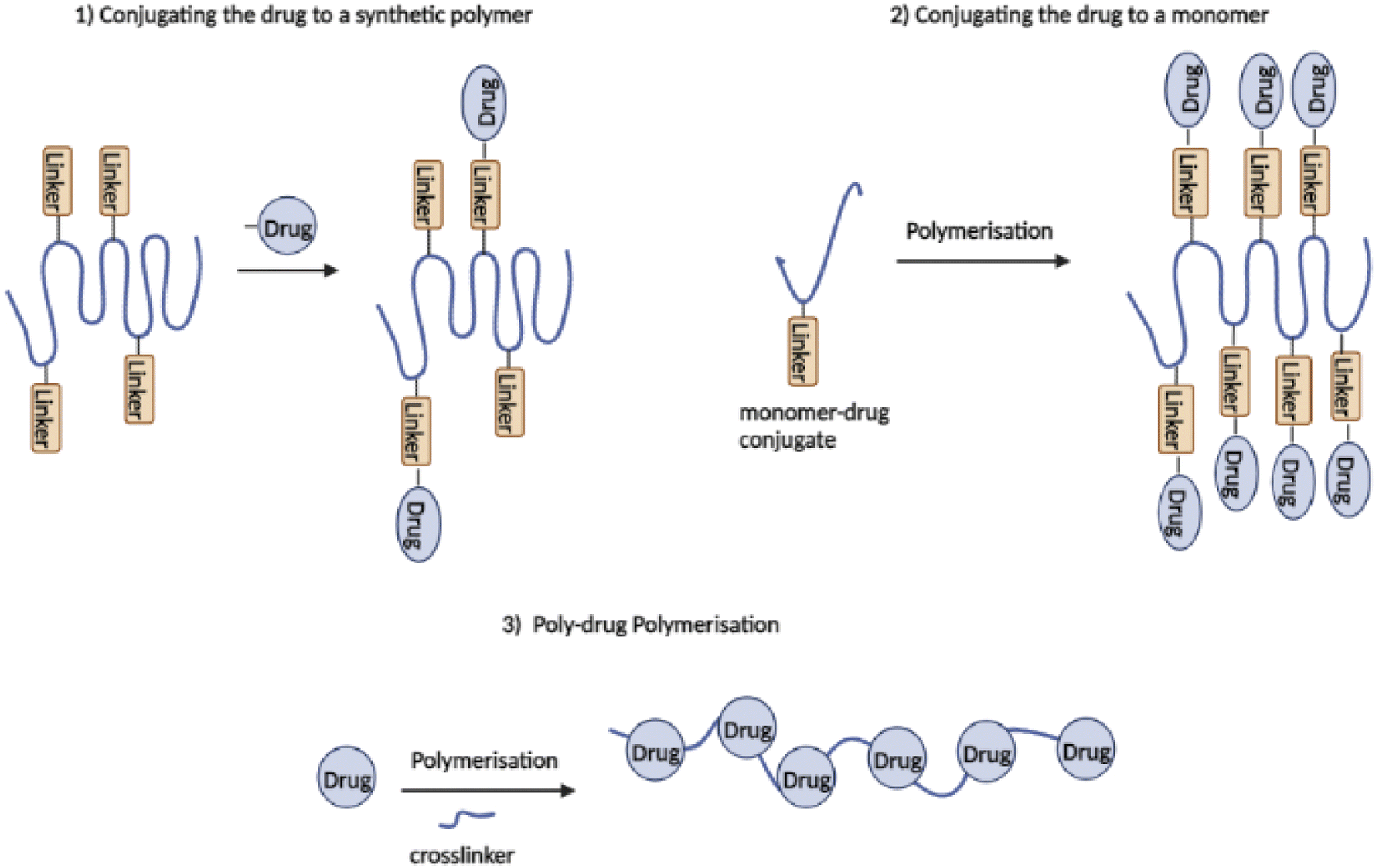 | ||
| Fig. 4 Three synthetic strategies of polymer–drug conjugates (redrawn using BioRender from ref. 55). | ||
Several characteristics are needed for the successful design of PDCs. The polymer should be non-toxic and non-immunogenic, and its molecular weight (Mw) must be high enough to ensure long circulation (for non-biodegradable polymeric carriers, this must be <40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 or below to ensure renal elimination of the carrier) but low enough to ensure endocytic internalisation (typically <100000 g mol−1). It must carry a payload equivalent to the drug's potency, and the linker must be stable throughout transport while releasing the drug at an optimal pace upon arrival at the target site.56 It also improves in vivo drug stability and controlled drug release rate and site.46 This objective is closely tied to the choice of the proper linker or spacer, which must be stable during circulation and capable of releasing the drug under specific conditions. Despite these benefits, the complete ability of PDCs as drug delivery platforms has yet to be realised, as most of the existing “nanomedicine” drug delivery systems continue to employ the less expensive drug encapsulation technique.46
000 g mol−1 or below to ensure renal elimination of the carrier) but low enough to ensure endocytic internalisation (typically <100000 g mol−1). It must carry a payload equivalent to the drug's potency, and the linker must be stable throughout transport while releasing the drug at an optimal pace upon arrival at the target site.56 It also improves in vivo drug stability and controlled drug release rate and site.46 This objective is closely tied to the choice of the proper linker or spacer, which must be stable during circulation and capable of releasing the drug under specific conditions. Despite these benefits, the complete ability of PDCs as drug delivery platforms has yet to be realised, as most of the existing “nanomedicine” drug delivery systems continue to employ the less expensive drug encapsulation technique.46
2.1. PDC-based combination therapy
Due to the complex aetiology of diseases such as cancer, HIV/AIDS, AD, and other neurodegenerative diseases, PDCs may also be used in the development of combination/multi-target therapies. “Combination therapy” typically refers to the concurrent administration of two or more active drugs or the combination of multiple methods of therapy for the treatment of a disease (e.g., chemotherapy and radiotherapy). Combination therapy, as opposed to single-agent therapy, can affect many signalling pathways in diseased cells, improve the therapeutic index by either improved efficacy or equivalent efficacy with decreased toxicity and potentially prevent the development of drug resistance.57 To date, four types of PDC-based combination therapy have been proposed (see Fig. 5). These include the administration of (i) PDC + free drug(s) (Type I). (ii) PDC + PDC (Type II), (iii) multiple drugs conjugated to a single polymeric carrier (Type III), and (iv) polymer-directed enzyme prodrug therapy (PDEPT) and polymer enzyme liposome therapy (PELT) (Type IV). In PDEPT, a combination of PDC along with a polymer carrying the enzyme handles the release of the drug at the target site, while PELT is based on the combination of a liposome-encapsulated drug delivery system and a polymeric carrier carrying the enzyme responsible for liposome degradation and drug release.57,58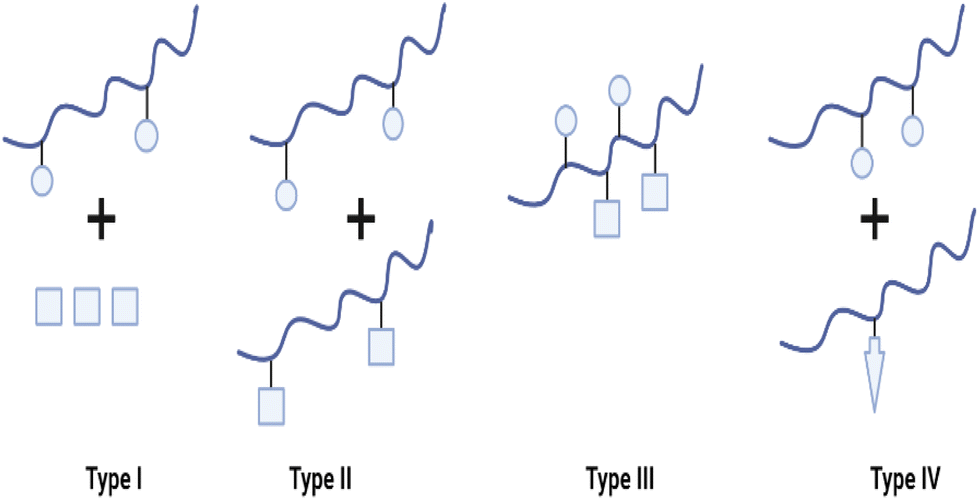 | ||
| Fig. 5 Diverse types of polymer-based combination therapy: (I) a PDC carrying a single drug administered with a small Mw drug or another type of therapy (e.g. radiotherapy), (II) a combination of two different PDC each containing a single therapeutic agent, (III) two or more drugs conjugated to the same polymer, and (IV) PDEPT is based on the combination of a PDC with a PELT is an analogous technique in which a polymer–enzyme conjugate is administered in conjunction with the liposome to stimulate its breakdown, thereby allowing the encapsulated medication to be released (redrawn using BioRender from ref. 57). | ||
The administration of low Mw drug combinations is well-established clinically, especially in the treatment of cancer. However, there is no assurance that the co-administered medication reaches the active target at the same time and in the same proportion as the initially administered intravenous doses. Possibly they may not reach the active target due to differences in the whole body and cellular pharmacokinetics. In the case of PDC-based combinations, if the PDC reaches the active site, the drug release rate can be tuned to maximise the synergistic effect.20 The rationale for developing PDC-based combination therapy is straightforward. Vicent et al.59 designed the first PDC combination HPMA copolymer–aminoglutethimide–doxorubicin conjugate (see Fig. 6). The aim of that research was to develop a polymer conjugate that would combine endocrine therapy and chemotherapy for the first time with the hope of evoking enhanced antitumor activity in breast cancer. The conjugate HPMA copolymer–aminoglutethimide–doxorubicin demonstrated significantly greater in vitro cytotoxicity activity against MCF-7 breast cancer cells than the parent drugs, combinations of parent drugs, or parent drug conjugated HPMA individually and mixed. For example, in vitro cytotoxicity (IC50) of HPMA copolymer–aminoglutethimide–doxorubicin conjugate was 40.9 μM which was greater than HPMA copolymer–aminoglutethimide (IC50 ≥ 1377 μM) and doxorubicin (IC50 = 5238 μM), respectively. Its enhanced activity may be attributable to several variables, including the kinetics of drug release rate, conjugate structure in solution, and the activation of specific biochemical pathways (e.g. induction of apoptosis downregulation of Bcl-2 protein).57
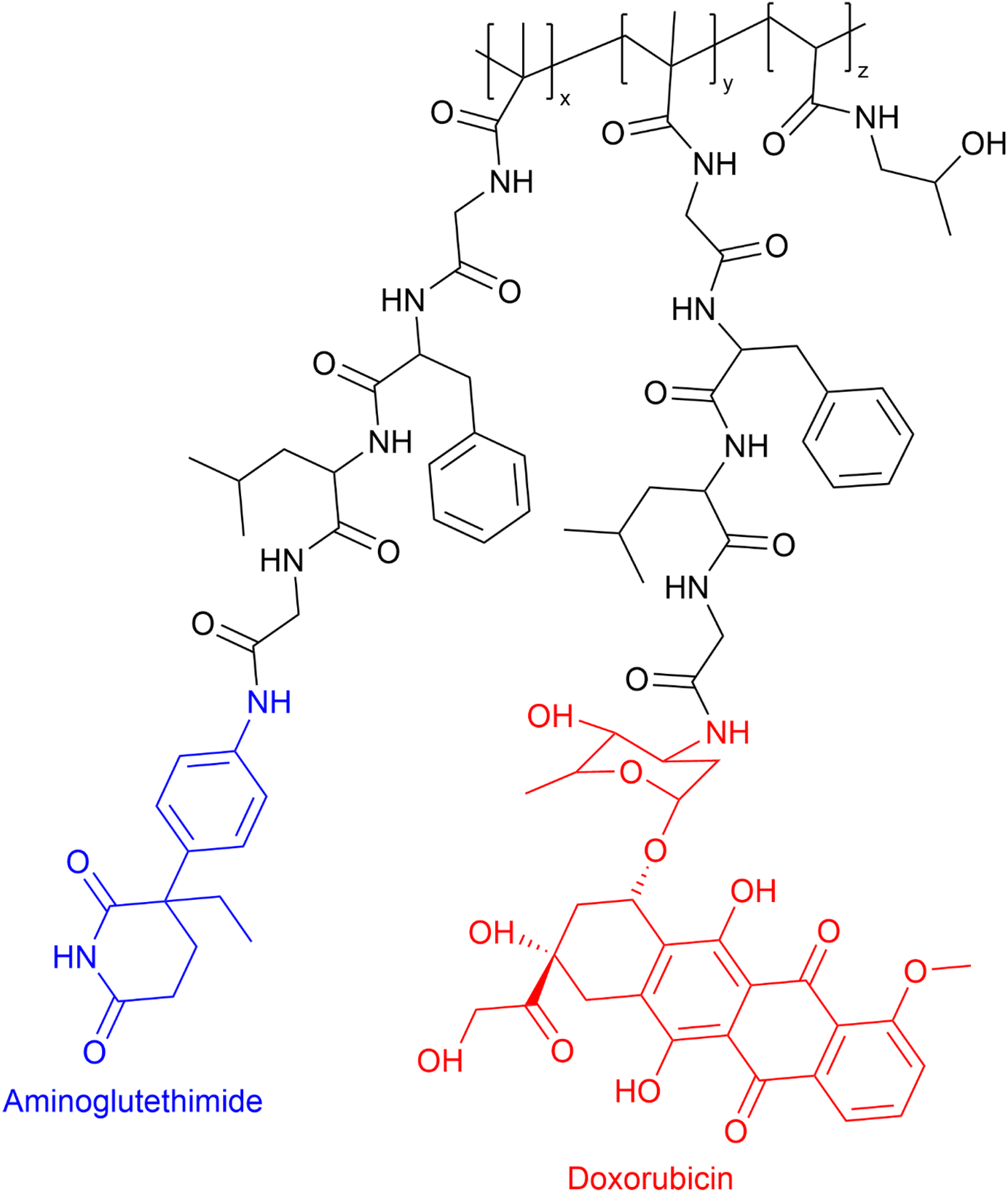 | ||
| Fig. 6 First PDC combination: HPMA copolymer–aminoglutethimide–doxorubicin conjugate. | ||
Another strategy for developing a Type II PDC combination is to develop a polyelectrolyte complex (PEC) between two individual polymers with opposing charges (cationic polymers, such as polyallylamine hydrochloride, polyethylenimine, chitosan, and anionic polymer, such as polyacrylic acid, sodium alginate), in which the individual drug is conjugated to two different polymers. The opposing charges on each conjugate will allow the formation of nano-polyplexes between them and facilitate both active moieties to be delivered in a single formulation60 (see Fig. 7).
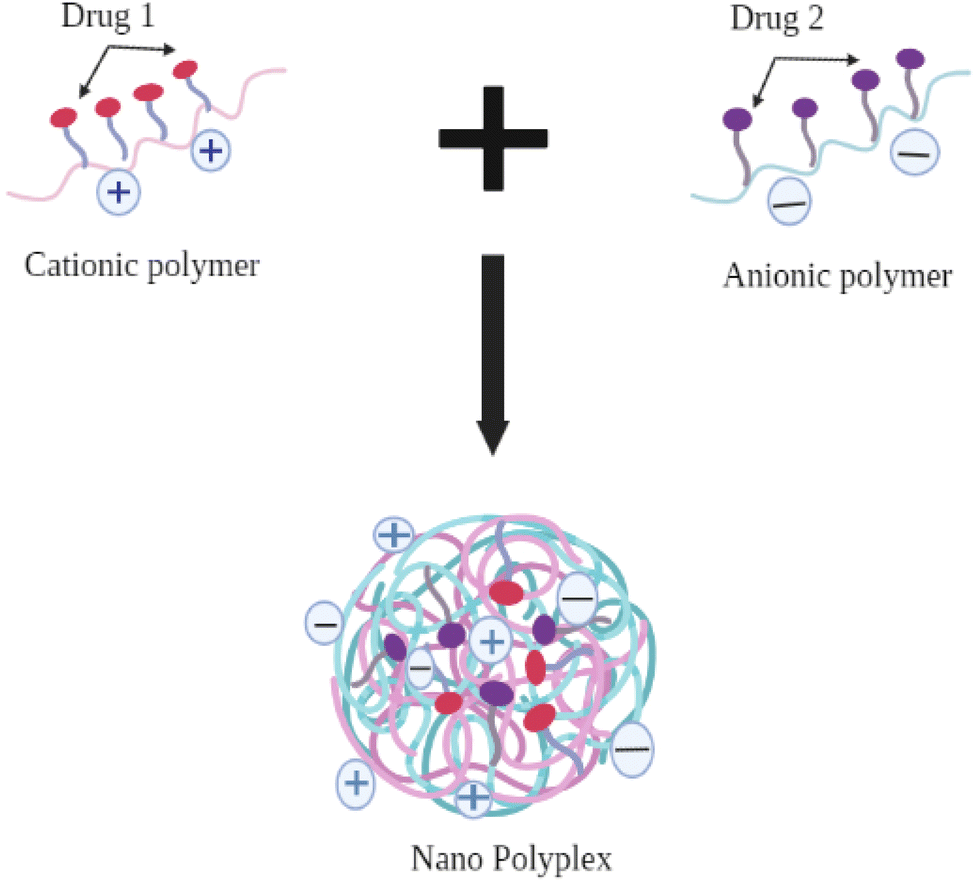 | ||
| Fig. 7 Formation of nano polyplex between cationic and anionic polymers in solution. | ||
The PECs are formed because of strong electrostatic (Coulomb's) interactions between charged counterparts of at least two oppositely charged polyelectrolytes.61 Quantitatively, PECs are categorized as either stoichiometric (S-PECs; PECs prepared by polymers in equimolar ratio), or non-stoichiometric (N-PECs; one polymer in excess compared to another, so that the resulting complex becomes more hydrophilic and soluble). A range of factors such as the mode of mixing polyelectrolyte solutions, ionic strength, pH, Mw, should be taken into consideration while developing a PEC complex.61 PECs have been discovered to have potential applications in the formulation of oral protein delivery systems such as polymer–insulin polyelectrolyte complex as well as in the delivery of polymer–sRNA polyplex.60,62
Curcumin (see Fig. 8), a hydrophobic polyphenol, has triggered a lot of interest among scientists. Curcumin's anti-inflammatory and anti-oxidant properties have shown potential in the prevention and treatment of a variety of diseases, including cancer, cardiovascular disease, neurodegenerative disorders, and inflammatory disorders.63 Regardless of its remarkable advancements in pharmaceutical applications, the therapeutic value of curcumin remains uncertain due to its poor aqueous solubility, low absorption rate, rapid metabolism, and poor bioavailability.64
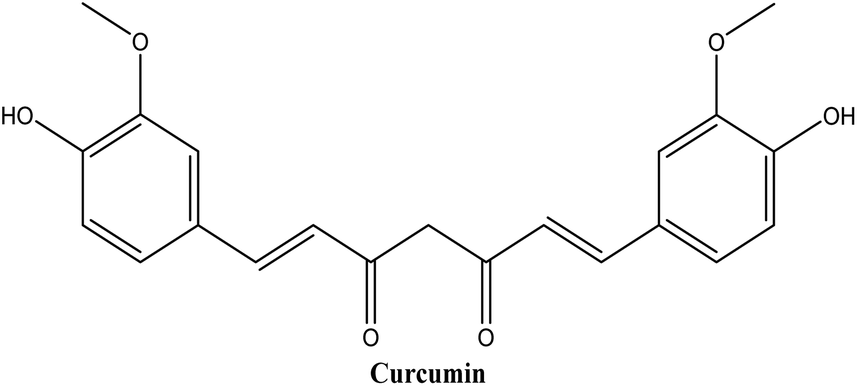 | ||
| Fig. 8 Chemical structure of curcumin. | ||
Szczepanowicz et al.65 developed nanodroplets of curcumin encapsulated in biocompatible polyelectrolyte shells: cationic polymer poly-L-lysine (PLL) and anionic polymer poly-L-glutamic acid (PGA). The average particle size of the polyelectrolyte nano-capsules was approximately 100 nm, and their zeta potential was −40 mV. The MTT assay revealed that PLL-coated nano-capsules were significantly toxic to the SH-SY5Y human neuroblastoma cell line (20–60% cell viability), whereas PLL/PGA-coated nano-capsules had no toxic effects on SH-SY5Y cells (cell viability above 80%). Curcumin encapsulated in PLL/PGA demonstrated comparable neuroprotective activity against hydrogen peroxide (H2O2)-induced cell injury as 5 μM curcumin pre-dissolved in DMSO (approximately 16% protection). Determination of curcumin concentration in cell lysate confirmed that curcumin in nano-capsules has a cell-protective effect at concentrations (at least 20-fold) lower than when given alone. Intracellular mechanisms of encapsulated curcumin-mediated protection involved the prevention of the H2O2-induced decrease in mitochondrial membrane potential (MMP) but did not inhibit the formation of reactive oxygen species (ROS). These findings imply that polyelectrolyte shell nano-capsules could be an alternative method for the delivery of hydrophobic drugs like curcumin for Alzheimer's and other neurogenerative disorders.
Lately, polyethyleneimines (PEIs) (see Fig. 9A), a hydrophilic, weak polyelectrolyte (polycationic), and highly basic aliphatic polymer with a 1:2:1 ratio of primary, secondary, and tertiary amino groups, has gained considerable interest.66 As a non-viral gene delivery technology, PEI has been regarded as the gold standard for polymer-based gene carriers due to the exceptional transfection efficiencies of its polyplexes (complex of nucleic acid and polymer) in both in vitro and in vivo models.66 However, PEI is known to have varying degree of toxic effects on cells based on its chemical structure (i.e., amount of primary and secondary amine).67,68 Various methods have been utilised for reducing the toxicity of cationic polymers, such as PEGylation,69 glycosylation,70 and quaternisation71 of the amino group. Hwang et al.72 developed encapsulated anionic miRNA into thiolated disulphide-crosslinked branched PEI (SSPEI) along with rabies virus glycoprotein (RVG) peptide labelled to SSPEI polymer mediated by an NHS-PEG-MAL linker. This polyplexes structure resulted in the formation of nanosized particles (290.5 nm) by electrostatic interaction. The Cell Counting Kit 8 (CCK-8) assay revealed that the branched PEI/miRNA complex caused severe dose-dependent cellular toxicity, whereas the RVG-SSPEI/miRNA-treated group was less toxic in acetylcholine receptor-positive Neuro2a cells. This study provides evidence that the disulphide linkage-modified SSPEI exhibited less cellular toxicity when reacting with the synthetic miRNA compared to the toxicity of the branched PEI polymer. In conclusion, polymer polyplex systems can be developed as potential drug delivery systems for the treatment of neurological diseases.
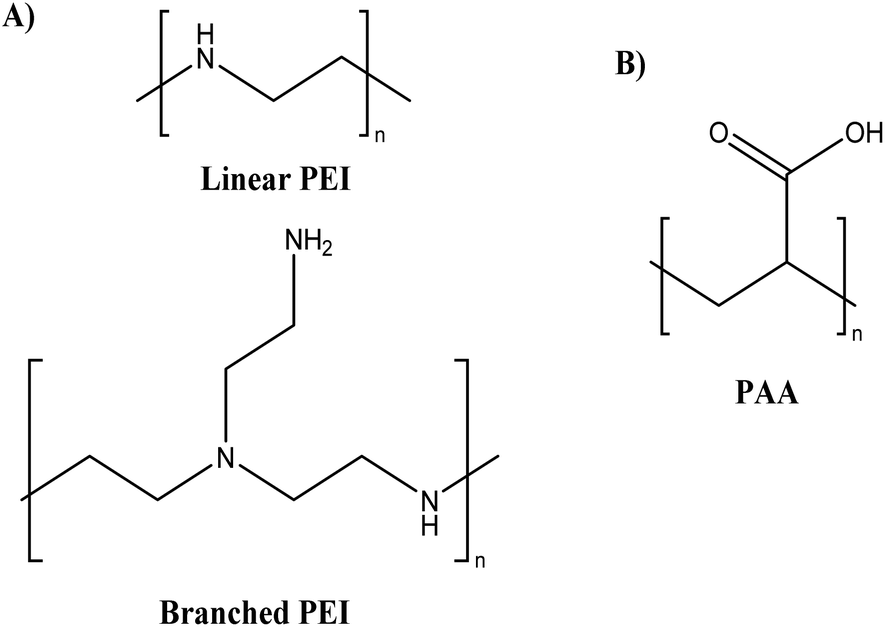 | ||
| Fig. 9 Chemical structure of (A) PEI and (B) PAA. | ||
Another strategy to reduce toxicity is the introduction of anionic polymers, such as poly(acrylic acid) (PAA) (see Fig. 9B), which can reduce the positive charge density by electrostatic interaction with the cationic polymers (development of PEC).73 PAA is an anionic synthetic polymer having a wide range of Mw composed of acrylic acid monomers. PAA is a synthetic weak polyelectrolyte (polyanionic), non-toxic, biocompatible, and biodegradable polymer that has attracted significant attention in recent years. Due to their stability and biocompatibility, nanoparticles derived from PAA derivatives can be employed to deliver drugs.74 Tripathi et al.75 developed nanocomposite/pDNA complexes by electrostatically and covalently linked nanocomposites of polyethylenimine (PEI) and PAA and evaluated them for their cytotoxicity on HEK293 and CHO cell lines. The MTT results revealed that both complexes of nanostructures showed no cytotoxicity (cell viability ≥ 80%) at their best working w/w ratios compared to the complexes of branched PEI/pDNA (cell viability = 53%) and Lipofectamine (cell viability = 47%), respectively, in both cell lines. These results showed that a reduction in the density of primary amines through electrostatic and covalent interaction with PAA significantly reduced the toxicity of the complexes.
Attaching poorly soluble drugs to these water-soluble polymers will increase their aqueous solubility.76 Regarding uptake, this can be improved by increasing the circulation time of the active compounds and by easing the crossing of the BBB. For the former, it is well established that increasing the molar mass of active compounds will increase their circulation time as this class of drugs accumulates at the targeting site.56,76 Thereby, reducing the amount of drug which is cleared/metabolised by, e.g., reducing glomerular filtration.77 To promote transport across the Blood–Brain Barrier (BBB), five fundamental transport pathways that allow solute molecules to cross the BBB can be utilised as per the conjugates mechanism which is discussed in Section 3.1 in detail.78 Additionally, the efficacy of some drugs is their propensity for degradation in vivo.79 This means that, although they produce promising data in vitro, those are not translated in vivo. By conjugating the drug to a polymer and its formulation as a polyplex, it should be shielded from degradation, whether by changes in pH, or enzymatic.60
PDC-based-PEC combination can provide various advantages such as controlled drug delivery, reduced toxicity, increased bioavailability, and prevent degradation of drugs. However, this strategy is still yet to be explored as a multitarget treatment for neurodegenerative and other diseases.
3. PDCs for the treatment of neurodegenerative diseases
Nanotechnology has the potential to treat neurodegenerative diseases by providing multiple solutions to overcome BBB and to deliver drugs to a specific target in the brain and release them in a controlled manner.80 PDCs are potential options for the treatment of neurodegenerative diseases because they possess important qualities such as (i) enhanced bioavailability and stability, (ii) reduced toxicity (iii) enhanced circulation time (iv) addition of targeting moiety and imaging agent for specific active targeting, and (v) controlled release using cleavable linkers under specific environments. These characteristics allow PDCs to circumvent the primary limitations of low Mw drugs. PDCs are additionally more chemically stable in plasma, less immunogenic, and contain particle sizes below 50 nm for enhanced penetration in comparison to other nanocarriers such as micelles and liposomes.81N-Acetylcysteine (NAC) (see Fig. 10A), a glutathione precursor with strong antioxidant, pro-neurogenesis, and anti-inflammatory properties, has acquired considerable potential in clinical applications for stroke, neuroinflammation, and neurodegenerative diseases.82,83 However, NAC must be administered in higher and repetitive doses. Due to the presence of unbound sulfhydryl groups in NAC, which are capable of spontaneous oxidation and forming disulphide bonds with plasma proteins, NAC has poor bioavailability and blood stability.82 Early pharmacokinetic studies have demonstrated limited oral bioavailability of NAC (in the range of 6% and 10%), which was attributed to low blood concentrations of NAC.84,85 The administration of large doses can result in cytotoxicity and adverse effects, such as elevated blood pressure.83 Navath et al.86 developed conjugates of NAC with thiol-terminated multi-arm (6 and 8) poly(ethylene-glycol) (PEG) with disulfide linkages involving sulfhydryls of NAC for the treatment of neuroinflammation in maternal-fetal applications (see Fig. 10B for the structure of 6-arm-PEG-S-S-NAC).
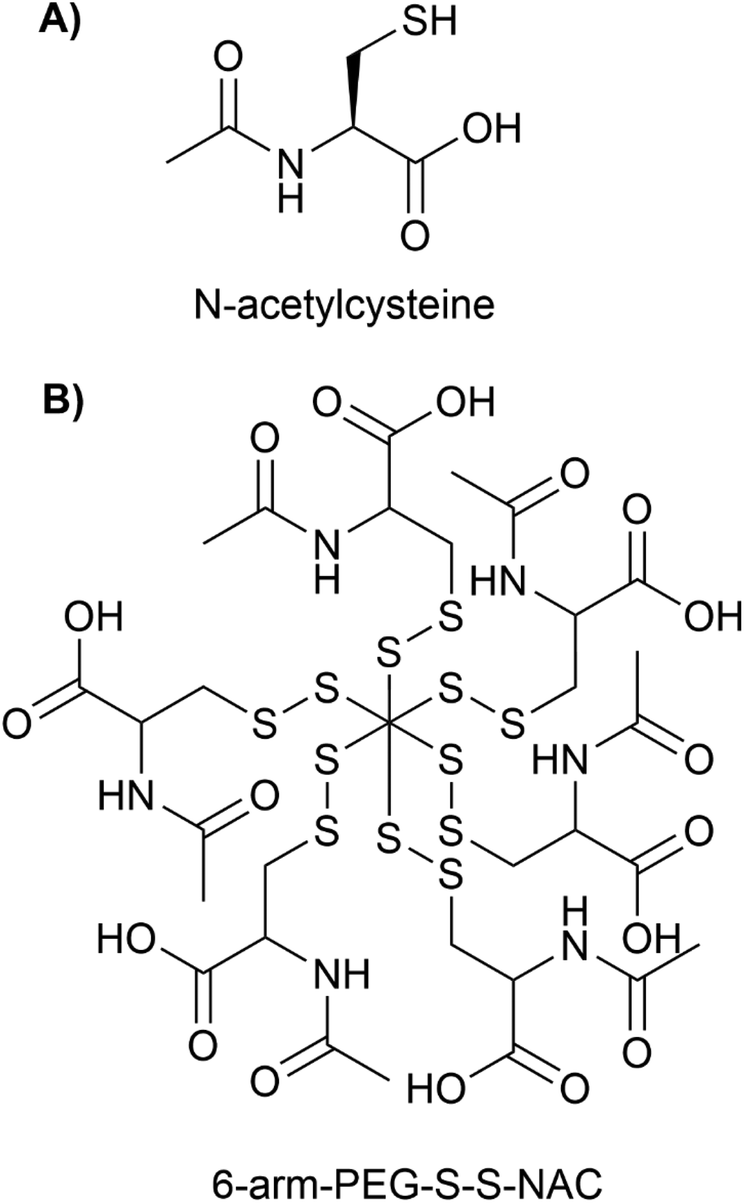 | ||
| Fig. 10 Chemical structure of (A) N-acetylcysteine and (B) 6-arm-PEG-S-S-NAC. | ||
The conjugates 6-arm-PEG-S-S-NAC and 8-arm-PEG-S-S-NAC had particle size and zeta-potential of 21–28 nm, −12.68 mV and 34–43 nm, −10.75 mV respectively. Both the conjugates released 74% of NAC within 2 hours via thiol exchange reactions in the intracellular redox milieu provided by glutathione (GSH) (2–10 mM). Both conjugates were stable at physiological extracellular glutathione concentrations (2 μM) and did not release NAC. The MTT results revealed that both conjugates at concentrations varying from 0.05 to 5 mM exhibited no cytotoxicity (cell viability > 80%) in BV-2 microglial cell lines. The conjugates inhibited lipopolysaccharide (LPS)-induced ROS, free radical nitrile (NO), GSH, and tumor necrosis factor-alpha (TNF-α) production in BV-2 cells by 2-fold as effectively as the free NAC. In conclusion, conjugates surmount the limitations of free NAC and demonstrated greater anti-inflammatory efficacy at lower doses than free NAC. To demonstrate its promise in treating neuroinflammation, however, extensive in vivo testing and evidence of transport across the BBB, are still required to ensure the therapeutic usefulness of this novel PDC.
Donepezil (see Fig. 11A), is a piperidine-derived cholinergic medication that is reversible and non-competitively inhibits acetylcholinesterase. Furthermore, donepezil functions at the molecular and cellular level in the pathogenesis of AD, inhibiting different aspects of glutamate-induced excitotoxicity, reducing early production of inflammatory cytokines, inducing a neuroprotective isoform of AChE, and decreasing oxidative stress-induced effects. It was authorised for the treatment of mild to moderate cases of AD in 1996.87 Singh et al.88 synthesised dendrimer–donepezil conjugates to treat AD with enhanced brain delivery and enhanced in vivo pharmacokinetics. It has been documented in the scientific literature that positively charged NPs in conjunction with mucus have improved cellular uptake.89 This suggests that positively charged dendrimers may aid in the transfer of drugs to the brain through parenteral administration. This strategy is based on the premise that polyamidoamine (PAMAM) dendrimers may be suitable for improved brain delivery of donepezil via conjugation. Amine-terminated 4.0 G PAMAM dendrimers with an ethylenediamine core were produced. After conjugating the synthesised PAMAM dendrimer with the donepezil-ester, the donepezil (ester)–PAMAM conjugate (PDZ) was produced (see Fig. 11B). Approximately 26% of donepezil molecules were coupled with PAMAM molecules, and 16 donepezil molecules were attached to each PAMAM molecule. The reported PDZ conjugate size and zeta-potential were 122 ± 1.88 nm and 0.434 ± 0.322 mV, respectively. Release tests (in vitro) indicated that donepezil was released in a sustained manner for 120 hours at physiological pH. The in vitro AChE inhibitory activity of PDZ formulation at 1 μM dosage was substantially greater than that of donepezil alone (p < 0.05). In in vivo investigations, the brain absorption of PDZ following intravenous treatment in Sprague-Dawley rats was significantly greater than that of the parent drug. Improved pharmacokinetic characteristics were the result of plasma drug concentration investigations. Half-life, volume of distribution, and clearance were determined to be 5.75 ± 0.41 h−1, 0.135 ± 0.02 L, and 0.016 ± 0.0021 L h−1 for the PDZ formulation and 1.09 ± 0.10 h−1, 0.172 ± 0.016 L, and 0.108 ± 0.014 L h−1 for the donepezil solution, respectively. In the case of the dendrimer-conjugated formulation, a longer half-life and a fourfold increase in brain absorption were reported, indicating that the synthesised conjugates give much more donepezil brain delivery. The developed PDZ-conjugated formulation enhanced both in vitro AChE inhibition and in vivo brain delivery.
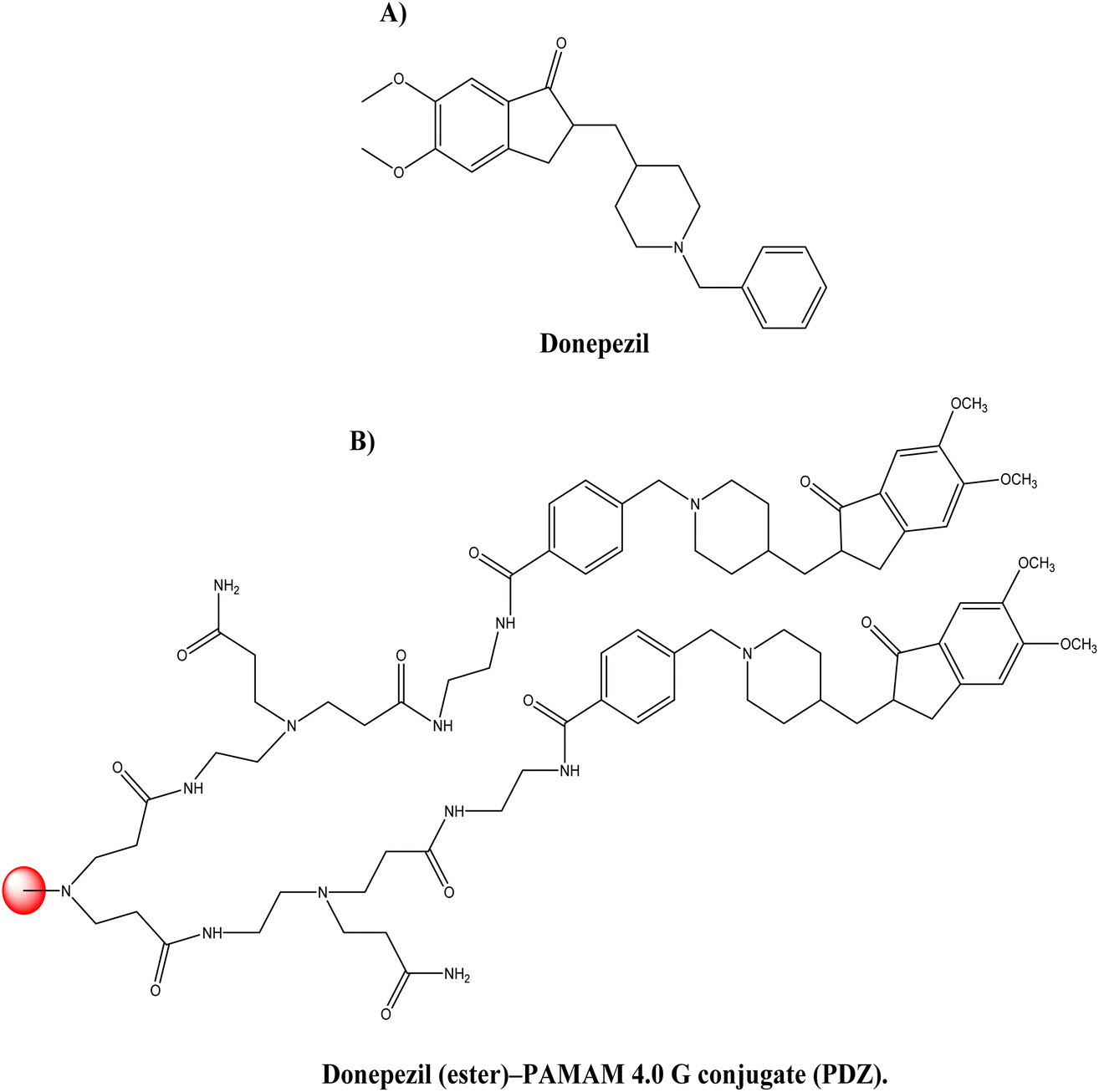 | ||
| Fig. 11 Chemical structure of (A) donepezil and (B) donepezil (ester)–PAMAM 4.0 G conjugate (PDZ). | ||
Various combination therapies for AD are under clinical trials that target different pathways, such as cromolyn and ibuprofen (ALZT-OP1, AZTherapies Inc., phase 3) targeting amyloid and inflammation, dextromethorphan and quinidine (AVP-923/Nuedexta, Avanir Therapeutics, phase 4) targeting neurotransmitters.90–92 Following this strategy, Castano et al.92 modified St-Cl-polyglutamic acids (PGAs) by postpolymerization through rationally designed linkers (amide, disulphide, and ester linkers) to bear a combination of neuroprotective propargylamine residues (Pr) (see Fig. 12A) and bisdemethoxycurcumin (BDMC) (see Fig. 12B), a polyphenolic curcuminoid that targets different AD pathological pathways.
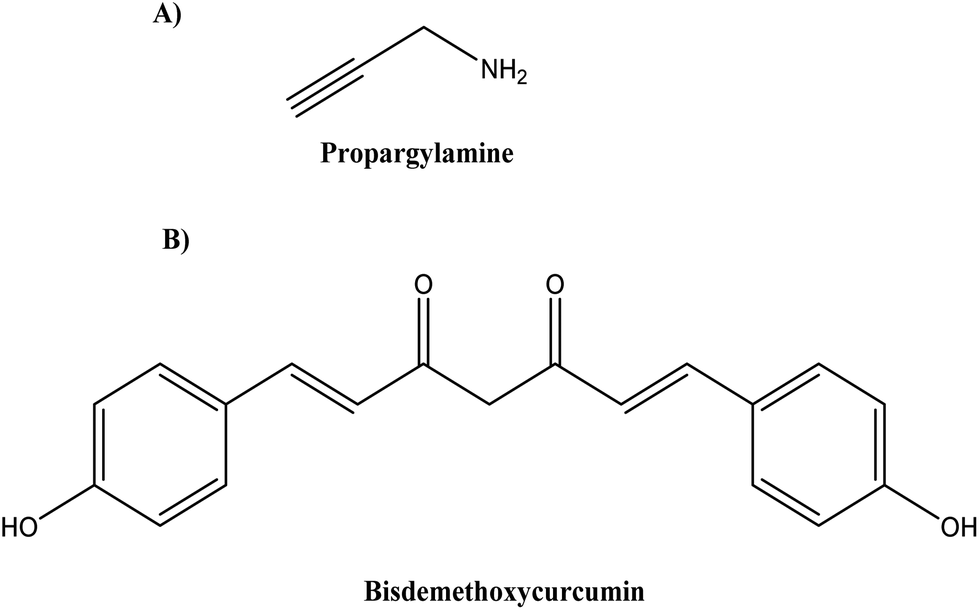 | ||
| Fig. 12 Chemical structure of (A) propargylamine and (B) bisdemethoxycurcumin. | ||
To improve the ability of the combination polypeptide-based nanoconjugates to cross the BBB, the conjugates were modified to carry Angiopep-2 (ANG), a specific peptide ligand for the low-density lipoprotein receptor-related protein 1 (LRP1) that promotes transcytosis from blood to brain and thus serves as a target for substance delivery into the CNS (Central Nervous System). In AD, LRP1 expression in the brain endothelium mediates the rapid elimination of Aβ from the brain via transport across the BBB, making LRP1 a potential therapeutic target. The particle size and zeta-potential of the St-Cl-Pr–BDMC–ANG conjugate were 22 ± 4 nm and −30 ± 2 mV, respectively. On the SH-SY5Y neuroblastoma cell line and primary neuronal cells, the toxicity of the St-Cl-Pr–BDMC–ANG conjugate was insignificant. In organotypic hippocampal cultures, an increase in the dendritic density of pyramidal neurons demonstrated the neuroprotective and neurotrophic effects of nanoconjugate (P > 0.001). The targeting moiety of Angiopep-2 improved nanoconjugate passage through the BBB and modulated brain distribution, with nanoconjugate accumulation in neurogenic regions, such as the olfactory bulb. Nanoconjugate treatment effectively decreased neurotoxic amyloid aggregate levels (P ≤ 0.05) and reversed impairments in olfactory memory and object recognition (P ≤ 0.0001) in APP/PS1 transgenic AD model mice. Overall, this study provides a description of a targeted combination polyglutamate-based nanoconjugate with neuroprotective and neurotrophic potential for AD treatment. Zhang et al.93 designed thermodynamically ultra-stable insulin-containing dextran–nateglinide conjugated prodrug micelles (NA/INS) (see Fig. 13A and B) and dextran–vitamin E succinate conjugated prodrug micelles (VES/INS) (see Fig. 13C and D) to synergistically treat dual symptoms of oxidative stress and glycometabolic abnormality by activating the PI3K/AKT and AMPK signal pathways. In comparison to macromolecular micelles, dextran-conjugated prodrug micelles with ultra-low CMC exhibit superior anti-electrolyte and anti-surfactant stability. The particle size of the conjugates VES/INS and NA/INS was 114.1 ± 1.2 nm and 105.7 ± 2.5 nm, respectively. According to the cell viability results, neither conjugate at a concentration of 1 mg mL−1 was cytotoxic to SH-SY5Y cell lines. NA/INS and VES/INS conjugates were efficiently cleaved by ROS, resulting in the release of over 80% of the encapsulated and conjugated drugs. NA/INS conjugates increased glucose uptake and insulin sensitivity via activation of the AKT and AMPK1 pathways, whereas VES/INS conjugates decreased neuron oxidative stress via reduction of ROS. Additionally, the conjugates improved metabolic dysfunctions in SAMP8 mice by repairing damaged mitochondria, lowering insulin resistance, and reducing oxidative stress. This study demonstrated a study for AD combination therapies with high specificity and efficacy.
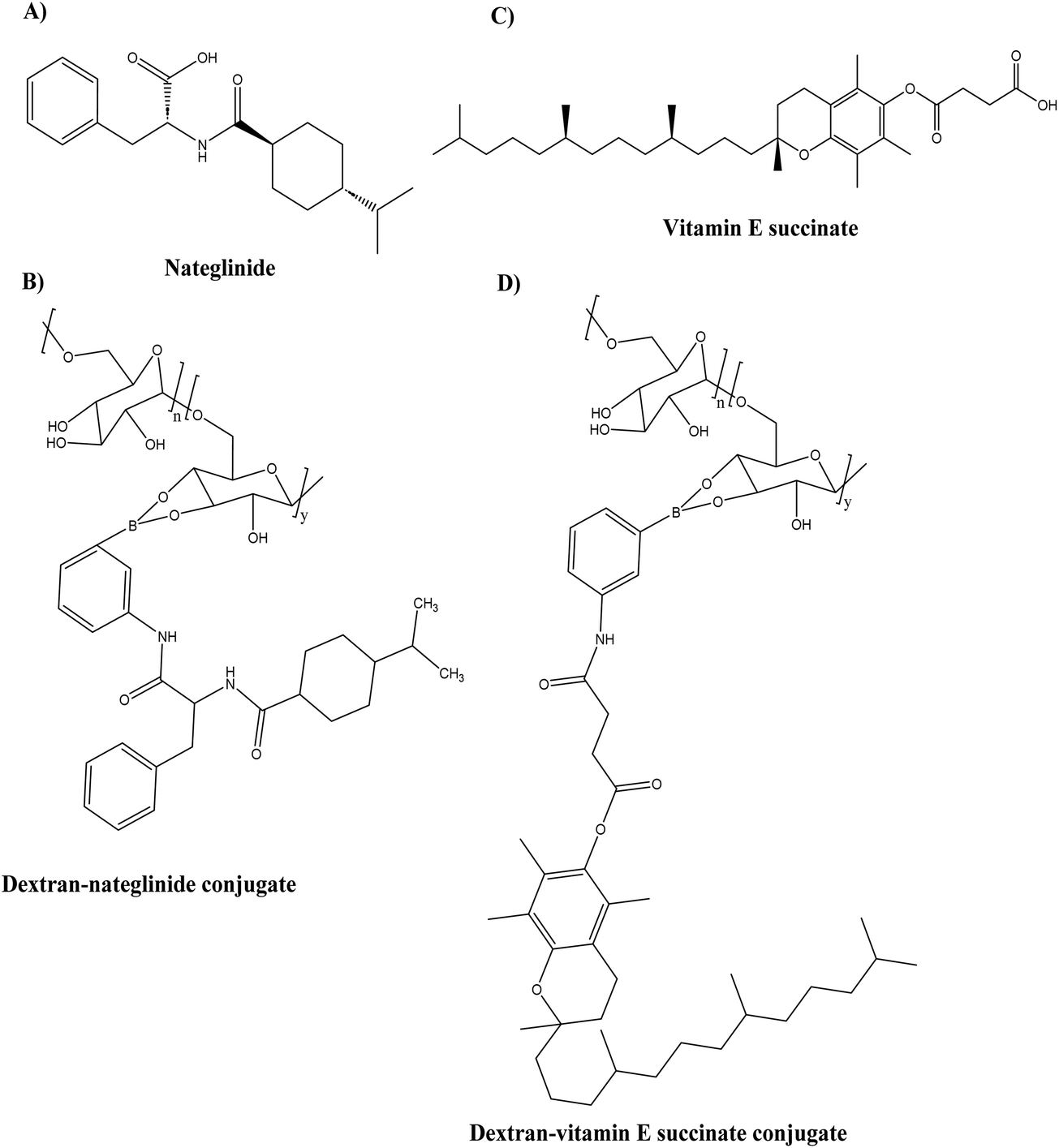 | ||
| Fig. 13 Chemical structure of (A) nateglinide, (B) dextran–nateglinide conjugate (C) vitamin E succinate, and (D) dextran–vitamin E succinate conjugate. | ||
Naki et al.94 synthesised a class of polyamidoamine-based PDCs containing tacrine, memantine and (E)-N-(3-aminopropyl)cinnamide alone as well in a combination of memantine and tacrine, tacrine and (E)-N-(3-aminopropyl)cinnamide, and memantine and (E)-N-(3-aminopropyl)cinnamide (see Fig. 14). All the conjugates exhibited particle size below 400 nm except for memantine conjugate (3075.0 ± 831 nm), tacrine, and N-(3-aminopropyl)cinnamide combined conjugate (499.4 ± 74 nm) while the tacrine conjugate particle size could not be determined. The drug release from the conjugates was sustained for 10 h in the range of 20–36%. All conjugates were more effective at inhibiting AChE (IC50 = 13–44.4 μM) than tacrine (IC50 = 1698.8 μM). Docking analyses confirmed that conjugates enhanced anti-acetylcholinesterase activity. However, extensive in vivo testing is still necessary to determine the therapeutic utility of this novel PDC in the treatment of AD.
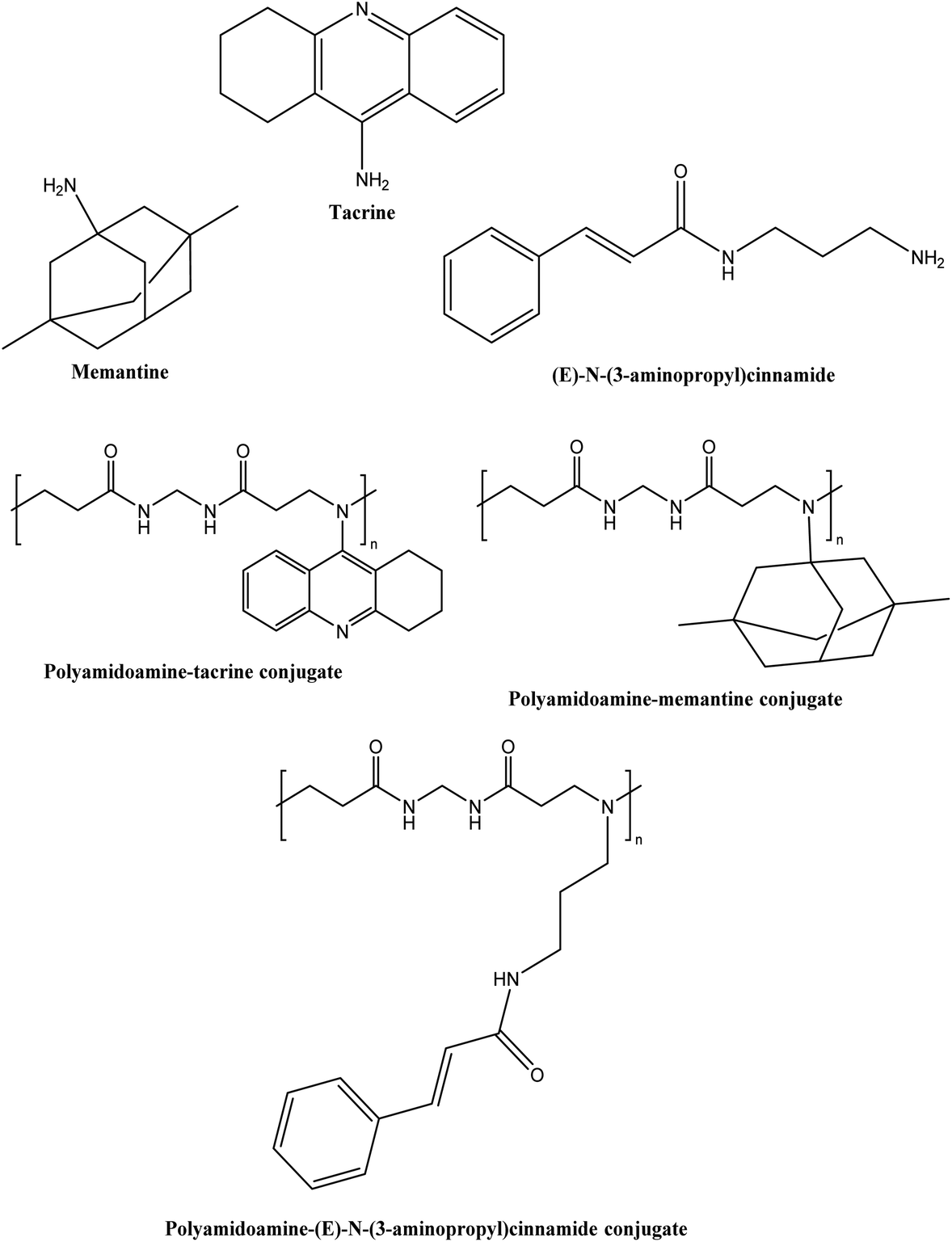 | ||
| Fig. 14 Chemical structure of tacrine, memantine, (E)-N-(3-aminopropyl)cinnamide and their conjugates. | ||
It is important to note that the PDC studies described above are still in the initial stages of development, but the results are encouraging for the future prospects of achieving successful drug delivery for neurodegenerative disease.
3.1. Transport mechanism of PDCs in the blood–brain barrier
There are promising treatments against Aβ toxicity;95,96 however, nanocarriers are needed to explore their maximum effect on CNS cells. The availability of medications in the CNS is the greatest challenge in AD therapy. The fundamental cause is the presence of a fully functional, semipermeable BBB, which impedes the migration of neurotherapeutic molecules (such as drugs, peptides, and vectors) into the CNS.97 The BBB is primarily composed of the tight brain endothelium, which is surrounded by the basal lamina and regulated by the pericytes, glial cells, and neurons of the neurovascular unit. Neurovascular unit-affiliated cells influence the permeability of the brain endothelium, which is affected by the majority of CNS pathologies.98 Numerous CNS pathologies are characterised by dysfunctional BBB endothelial cells. This dysfunction is frequently associated with neuroinflammation, which alters the BBB's permeability. Therefore, the tight junctions between endothelial cells are disrupted, allowing additional substances to pass through the BBB or macrophages to penetrate the CNS. This permeability alteration can be seen in neuroinflammation caused by bacterial lipopolysaccharides (LPS). In the context of AD, the permeability of BBB is modified as a result of reduced expression of P-glycoprotein. This reduction in expression leads to a decrease in the effectiveness of efflux pumps in facilitating the transport of neurotoxic substances from the brain back into the bloodstream.99 BBB dysfunction is typically only significant in the later stages of the disease and at the most severely affected sites.98 Effective delivery of therapeutic agents is impeded by the BBB and its selective transport of molecules into the brain. In addition, the BBB has a negative impact on therapeutic efficacy and tolerance, as substantial dosages of medicines are needed to reach concentrations above the brain's minimal effective concentration. Nanotechnology, including nanoparticulate systems, can be employed as ‘Trojan horses’ to carry active compounds across the BBB, thereby decreasing toxicity and enhancing therapeutic efficacy.97The BBB blocks many drugs from entering the CNS. Not only are large molecule drugs unable to cross the BBB, but 98% of all small compounds are also unable to easily traverse the BBB.78,100 The BBB holds a variety of highly selective mechanisms for nutrition delivery into the brain. There are five fundamental transport pathways that allow molecules to cross the BBB (see Fig. 15).
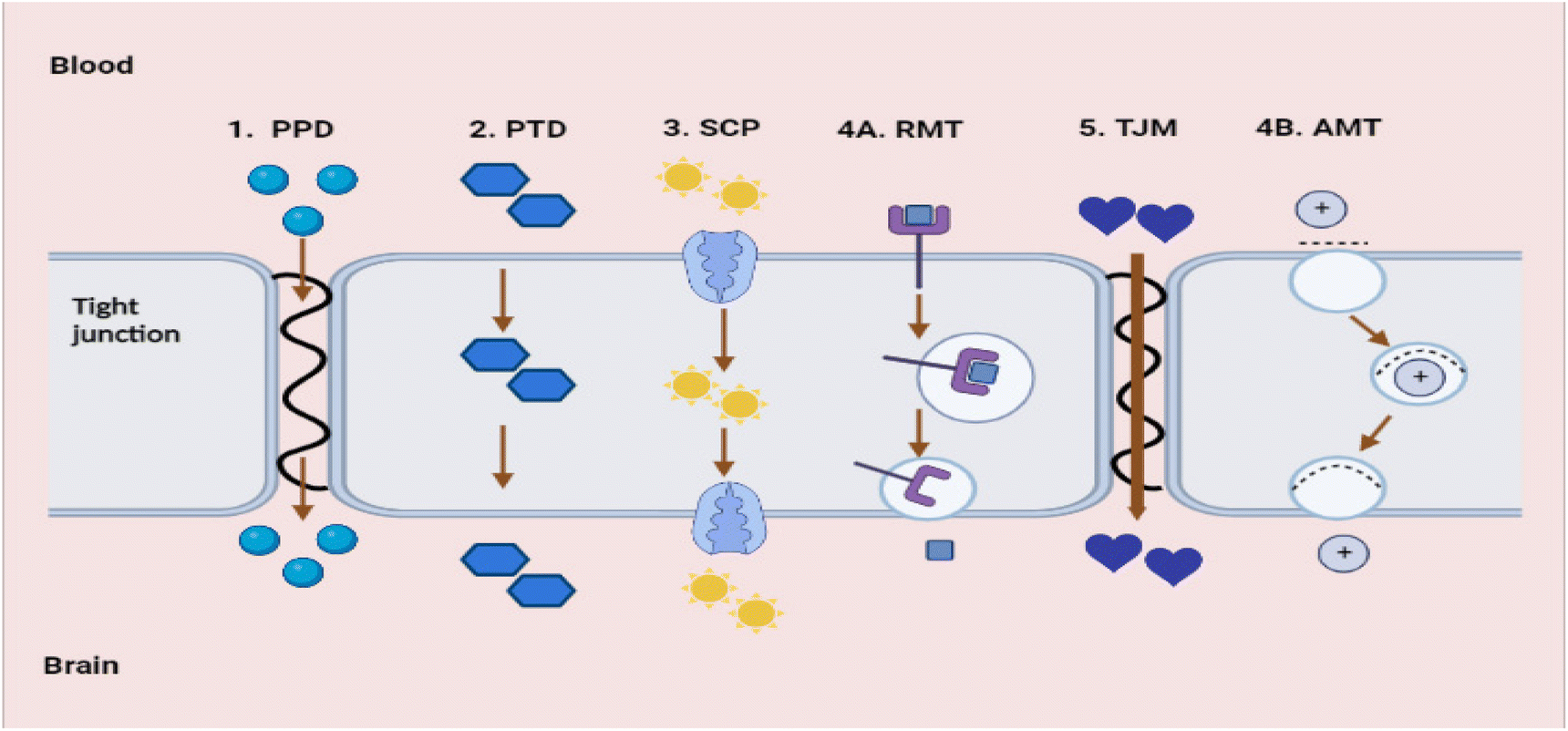 | ||
| Fig. 15 Transport pathways allow molecules to cross the BBB (redrawn using BioRender from ref. 78). (1) PPD – passive paracellular diffusion. (2) PTS – passive transcellular diffusion. (3) SCP – solute carrier protein influx. (4A) RMT – receptor-mediated transcytosis. (4B) AMT – adsorptive-mediated transcytosis. (5) TJM – tight junction modulation. | ||
The first is passive paracellular diffusion (PPD), which is the movement of molecules through epithelial cells via intercellular gaps. Due to the existence of tight junctions, this pathway is non-existent in a healthy BBB. The second process is passive transcellular diffusion (PTD), in which molecules diffuse past the bilayer cell membrane into the intracellular space. The third is solute carrier proteins (SCP) are a type of carrier-mediated endocytosis whereby solute molecules bind to certain membrane protein carriers, also from high to low concentration.100 Fourth is transcytosis, a process by which big molecules traverse the BBB and enter the CNS. This process is dynamic, saturable, and pH- and temperature-dependent. There are two mechanisms to carry out transcytosis: receptor-mediated transcytosis (RMT) and adsorptive-mediated transcytosis (AMT). Electrostatically charged and polycationic AMT molecules or nanoparticles, such as protein and peptides, bind to the anionic molecules of the luminal surface of the endothelial cells, which are then internalised in vesicles and transported through the membrane. On the luminal surface of the endothelial cells. During RMT, specialised receptors (transferrin receptors, insulin receptors) exist for specific substances. These receptors, unlike other receptors, bind to a set of extremely specific ligands; upon binding, the receptor molecules are internalised, and the ligand is delivered into the brain. This is the major method through which macromolecules including insulin, low-density lipoproteins, transferrin, and growth factors penetrate the BBB. Regarding RMT, there have been many breakthroughs in the field of medication administration; many receptors have been exploited to boost brain delivery. In AD, transferrin receptors carrying antibodies,101 conjugated dopamine,102 and insulin receptors targeting enhanced insulin delivery have been explored.103 These are just a few instances of how RMT mechanisms can be used to distribute therapeutic nanoparticles throughout the CNS.104
Tight junction modulation (TJM) is the final mechanism, which does not readily occur in a healthy BBB and whose mechanism of action is not entirely understood.78 Furthermore, small molecule compounds can be tailored to penetrate the BBB via endogenous carrier-mediated transport mechanisms expressed in the brain capillary endothelium. Utilizing molecular Trojan horse technology that targets the endogenous RMT systems produced within the brain capillary endothelium, large molecule drugs, like recombinant proteins, peptides, and antisense radiopharmaceuticals can be transported over the BBB.78
3.2. PDC delivery route targeting CNS
The intravenous (IV) route is the most commonly used in clinical settings, and the majority of polymer therapeutics developed to date are intended for this route. As discussed earlier, for IV treatment to be effective, PDCs must be designed with sufficient blood stability and compatibility (low interaction with plasma proteins and cells) and extended circulation times. In the case of brain disorders, targeting relevance remains essential to enhance brain accumulation after intravenous administration and prevent the majority of the administered dose from reaching the clearance organs.105Regarding CNS drug delivery, two additional obstacles must be considered: crossing the BBB and diffusing throughout the brain parenchyma for the drug to reach its target once inside the brain.105 Zheng et al.106 developed an intravenous hybrid siRNA delivery system for AD based on polyethylene glycol-modified poly(2-(N,N-dimethylamino)ethyl methacrylate), an ideal carrier with relatively low toxicity and high transfection efficiency, conjugated with CGN peptide for brain targeting, and Aβ-targeting peptide and tested whether the complexes could overcome the obstacles described above. In transgenic AD mice, the hybrid complex entered the brain in an intact manner and localised around the plaques after an IV injection. The precise delivery of amyloid plaques led to an increase in therapeutic activities, as evidenced by the strong mRNA (36.4%) knockdown of BACE1 (a therapeutic target of AD), reduction of soluble amyloid precursor protein (sAPPβ) (42.6%) and resulted in better neurons protection than the single component complexes.
An additional alternative route to directly target therapeutics to the CNS while completely circumventing the BBB is an Intranasal (IN) administration.105 Drug delivery after IN administration is based on the idea that drugs can exit the nose submucous space and cross the arachnoid membrane, entering olfactory CSF. This could potentially enter the brain from the CSF flow tracts. Two factors to consider are any drug that enters into olfactory CSF will exit the CSF flow tracts and enter the peripheral bloodstream. Another one is the high resistance of the arachnoid membrane, which separates olfactory CSF from the nasal spaces, and the volume of drug administered into the nose. Lipid-soluble small molecules can only cross the membrane without disruption, while physically or chemically disrupted membranes may allow drug entry. The human nasal cavity can only receive about 100 μL per nostril without local injury.100 Alternately, drugs may utilise axonal transport via the trigeminal nerves, which innervate the respiratory region, the largest region of the nasal cavity, thereby facilitating access to the caudal and rostral regions of the brain via the pons. IN permits increased brain targeting (≈10-fold) compared to IV administration, necessitating lower drug doses for effective clinical outcomes and minimising off-target effects. However, not all drugs/biologicals can cross the nasal barriers, and only 1% of the total dose of those can reach the brain, demonstrating the need for novel drug delivery systems to increase this proportion.105 In the case of PDCs, this route is still yet to be explored for neurodegenerative diseases.
3.3. Targeted delivery using PDC
For PDC design, it is essential to figure out the location of the target. Whether the target is intracellular in the brain capillary endothelial cells, extracellular in the brain's extracellular space and/or the neuron/glial cell surface, or intracellular in brain cells (neurons, astrocyte, dendritic cells, and pericytes) (see Fig. 16(A)).107 Based on the pathological hallmarks of AD i.e. Aβ plaques (extracellular insoluble protein deposits) and NFTs (intracellular aggregation of tau), the target can be extracellular in the brain extracellular space or intracellular in brain cells108,109 (see Fig. 16(B)).110 Therefore, the PDCs should be able to reach the targeted location, and this can be achieved by attaching an imaging agent to the PDC also known as theranostic agents such as curcumin,111 and Congo red112 (see Fig. 17).113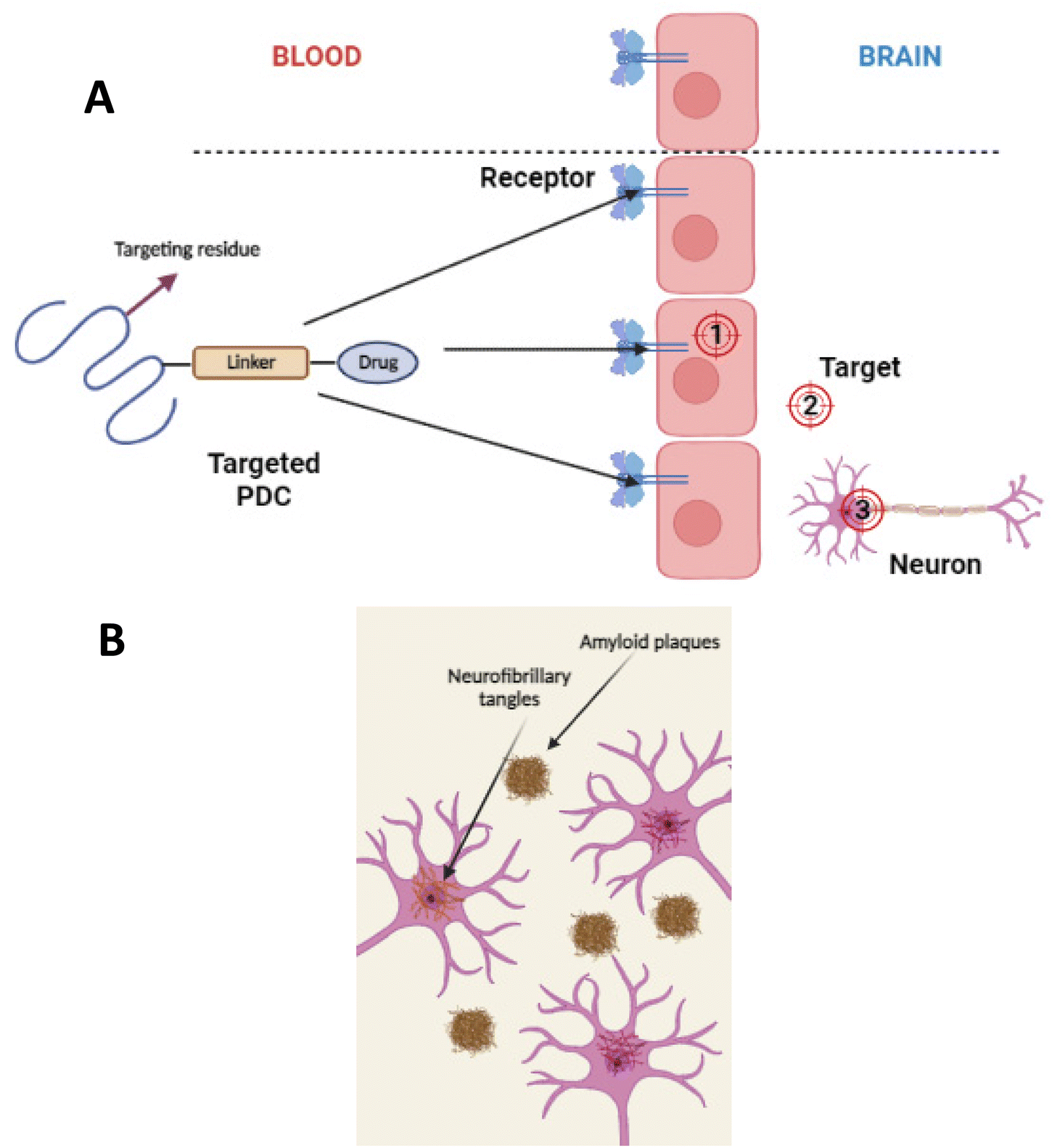 | ||
| Fig. 16 (A) Divergent targeting of macromolecular drugs to the BBB/brain. (1) Intracellular targets in the capillary endothelial cells of the brain. (2) Extracellular targets in the extracellular space or neuronal/glial cell surface of the brain. (3) Intracellular targets in brain cells, including neurons, astrocytes, dendritic cells, and pericytes (redrawn using BioRender from ref. 107). (B) Formation of Aβ plaques in the extracellular space of the brain and intracellular formation of NFTs in the neurons (redrawn using BioRender from ref. 110). | ||
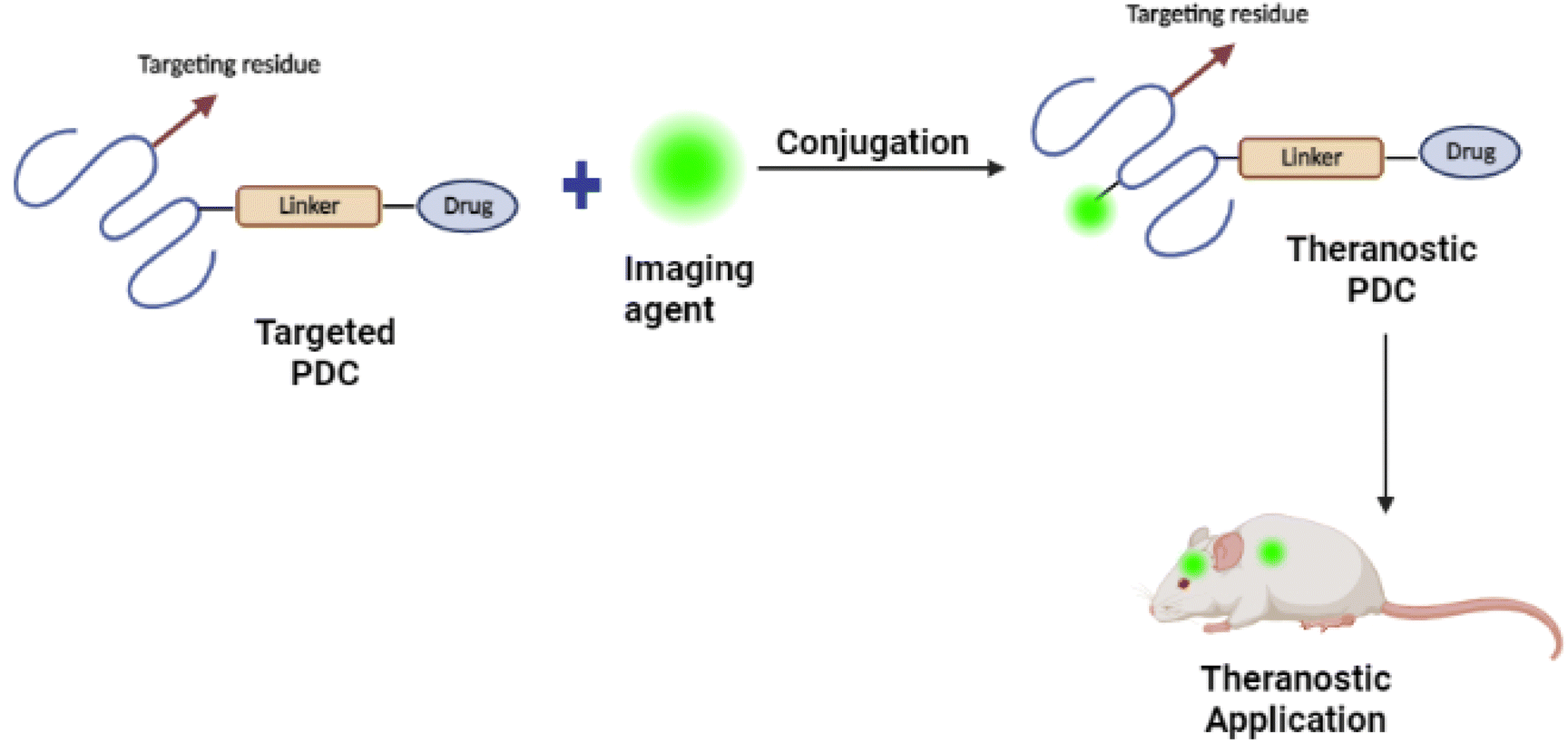 | ||
| Fig. 17 Schematic representation of the development of PDC-based theranostic nanoparticles (redrawn using BioRender from ref. 113). | ||
The optical anisotropy of amyloids upon binding with Congo red and the resulting signature of apple-green birefringence makes this technique one of the most reliable for detecting amyloids in tissues and clinical samples.114 Hu et al.112 successfully developed Congo red/Rutin-magnetic nanoparticles as nano theranostics that could detect Aβ plaques by magnetic resonance imaging (see Fig. 17). The nanoparticles achieved targeted delivery of AD therapeutic agents, controlled drug release by H2O2 responses, and prevented oxidative stress in both in vitro and in vivo studies. In general, conjugating Congo red with PDCs as nano theranostics could provide a new avenue for applications in AD theranostics.
3.4. In vitro BBB models
The BBB continues to pose the greatest obstacles to drug development for neurological diseases. Despite the use of animal models, it is difficult to predict drug responses in humans due to species differences. In addition, the complex physiology of animal models makes it challenging to conduct mechanistic studies and direct quantitative analysis of nanoparticles with the barrier at the molecular and cellular levels in real-time. To overcome these obstacles, it is of utmost importance in developing an in vitro model that mimics the human BBB and reproduces the key relationships between healthy and dysfunctional barrier functions under controlled conditions.115In vitro models are viable options for gaining a comprehensive understanding in the progression of neurological diseases and for facilitating drug discovery.116 Petri dish models and transwell models are traditional in vitro BBB models.116–118 The Petri dish model is highly effective in assessing the cytotoxicity of potential drug candidates, as well as elucidating the mechanism and function of transporter proteins. However, they are unsuitable for evaluating drug transport.117,118 Alternatively, transwell models are commonly used in investigations on drug transportation across the BBB by culturing endothelial cells on semi-permeable microporous inserts.118Table 2 provides information on traditional in vitro BBB models used in AD research.| In vitro BBB model | Application | Advantages | Disadvantages |
|---|---|---|---|
| Petri dish models | Study of neuronal death through Aβ-induced damage | • Inexpensive | • No shear stress |
| • Easy to standardize | • Limited to monolayers | ||
| • Large quantities | • Limited in understanding the mechanisms of AD pathology including AD-related BBB dysfunction and inflammation as well as neural damage | ||
| • Simple fabrication | • Unrealistic in terms of how they mimic the complex features of AD pathology | ||
| • Control over the microenvironment optically | • Lack features such as controlled physical stimuli such as direct cell–cell interactions, controlled flow dynamics, circulating blood cells, and a brain-specific microenvironment and extracellular matrix (ECM) | ||
| Transwell models | AD-related BBB dysfunction, AD-inflammation linked study, and Aβ-associated microglial activation | • Uses brain endothelial cells | • No shear stress |
| • Inexpensive as low fabrication cost | • Limited cell differentiation | ||
| • Allows co-culture | • Unrealistic in terms of how they mimic the complex features of AD pathology | ||
| • Simple fabrication | • Lack features such as controlled physical stimuli such as direct cell–cell interactions, controlled flow dynamics, circulating blood cells, and a brain-specific microenvironment and ECM | ||
| • Moderate scalability | |||
| • Highly convenient for high-throughput screens |
Recently, microfluidic models of the BBB have garnered significant interest due to their ability to incorporate various properties, including, flow-induced shear stress, selective permeability, fluid dynamics, cellular tension, measurement of transendothelial electrical resistance (TEER – which helps to detect the barrier integrity of the brain), and cultivation of multiple cell types. The effective cultivation of cells that can replicate in vivo physiological conditions involves the utilisation of various membrane materials, a particular type of culture media, and hydrogels. These factors significantly impact the in vitro BBB model.119
Choi et al.120 developed a bio-mimetic microfluidic system that generates spatial gradients of diffusible oligomeric assemblies to study the neurotoxicity of diffusible moieties by modulating microenvironments and evaluating their effects on cultured neurons. Integration of an osmotic pump into the system was used to induce osmotic pressure and generate a fluid flow comparable to brain interstitial flow in the study. It was discovered that amyloid beta exposure induced an atrophying effect and observed neurite extension during neural progenitor cell differentiation increased when cells were cultured with continuous flow. The outcomes demonstrated the potential neurotoxicity of oligomeric assemblies and established a promising microfluidic platform for investigating the neurotoxicity of amyloid beta.
In addition, studies were performed to investigate molecular transport pathways through the BBB using a particular type of transmembrane proteins. Ahn et al.115 developed a micro-physiological platform that replicates the fundamental structure and function of the human BBB and enables 3D mapping of high-density lipoprotein (HDL)-mimetic nanoparticles with apolipoprotein A1 (eHNP-A1) which replicate the physiologically pertinent size and composition of discoidal HDL nanoparticle distributions in the vascular and perivascular regions. They tested the hypothesis that eHNP-A1 primarily uses scavenger receptor class B type 1 (SR-B1) on brain endothelial cells to cross the BBB via transcytosis. Blocking SR-B1 activity reduces eHNP-A1 uptake by immortalized Human Brain Microvascular Endothelial Cells (HBMECs), whereas it may induce a compensatory upregulation of other receptors or determinants of endothelial transcytosis such as SNAREs. The results revealed that treatment with block lipid transporter-1 (BLT-1) reduced the quantity of eHNP-A1 entering the tissue (EC uptake and perivascular channel) by a factor of three. SR-B1 mediates the cellular absorption of eHNP-A1 in BBB models, with perivascular cells decreasing by 38.5% with BLT-1 treatment and 53.8% without. Whereas there was no change in the proportion of HBMECs with eHNP-A1. In conclusion, the model accurately reproduced 3D nanoparticle distributions at the cellular level and showed distinct cellular uptakes and BBB penetrations via RMT.
Microfluidic models of the BBB could be used as an alternative to animal models for pre-screening lead compounds and could serve as a valuable resource for translational medicine research, particularly in the treatment of neurodegenerative diseases.
4. Future insight and conclusion
The design of new drug delivery systems utilising the many properties of polymers has been one of the most prominent areas of study for several decades. Polymers are commonly employed nowadays to increase the bioavailability of therapeutic molecules in the blood and to provide site-specific drug delivery. As opposed to others, PDCs are regarded as a novel field in the drug delivery system. The amount of PDCs on the market has increased significantly in recent years. In addition, given the increasing promise of PDCs in the treatment of cancer, it is a viable strategy to investigate the use of PDCs in the treatment of neurodegenerative diseases. Due to the complex aetiology of neurological disorders, it requires a multitargeted approach in its treatment strategy. However, this can be accomplished through PDC-based combination/multi-target therapies as opposed to low Mw drug combinations where there is no guarantee that the drug will reach the active target at the same time and in the same proportion. Similarly, the advantages provided by more advanced approaches, such as the incorporation of molecular targeting and an imaging agent on the same carrier, can aid in circumventing the BBB and releasing the drug at the targeted site. In addition, the use of in vitro BBB models that can simulate the human BBB can provide additional benefits for understanding the mechanism of PDCs despite the use of animal models in which it is challenging to predict drug responses in humans due to species differences. There is a tremendous deal of studies that can be conducted on future insight. Several intriguing avenues are likely to be explored: (1) design and development of multi-targeting systems in which PDCs are altered with targeting ligands that can recognise particular cell receptors; (2) PDCs for the treatment of neurodegenerative diseases and their transport mechanism across the BBB. The use of PDCs can provide new insights into the delivery of drugs that can circumvent the BBB and treat neurodegenerative diseases.Conflicts of interest
There are no conflicts of interest disclosed by the authors. The author is solely accountable for the article's content and writing.Acknowledgements
The authors would like to acknowledge the School of Pharmacy and Life Sciences at Robert Gordon University for providing their support for the completion of this work.References
- C. Ding, Y. Wu, X. Chen, Y. Chen, Z. Wu, Z. Lin, D. Kang, W. Fang and F. Chen, Global, regional, and national burden and attributable risk factors of neurological disorders: The Global Burden of Disease study 1990–2019, Front. Public Health, 2022, 10(952161) DOI:10.3389/fpubh.2022.952161.
- H. Zhang, Y. Wang, Y. Wang, X. Li, S. Wang and Z. Wang, Recent advance on carbamate-based cholinesterase inhibitors as potential multifunctional agents against Alzheimer's disease, Eur. J. Med. Chem., 2022, 240, 114606, DOI:10.1016/j.ejmech.2022.114606.
- R. Guerreiro and J. Bras, The age factor in Alzheimer's disease, Genome Med., 2015, 7, 106, DOI:10.1186/s13073-015-0232-5.
- D. Ferreira, L. Perestelo-Pérez, E. Westman, L.-O. Wahlund, A. Sarría and P. Serrano-Aguilar, Meta-Review of CSF Core Biomarkers in Alzheimer’s Disease: The State-of-the-Art after the New Revised Diagnostic Criteria, Front. Aging Neurosci., 2014, 6(47) DOI:10.3389/fnagi.2014.00047.
- World Health Organization, The Top 10 Causes of Death [Internet], World Health Organization, 2020, accessed on 2023 Oct 28, Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- E. Siemers, J. Hitchcock, K. Sundell, R. Dean, J. Jerecic, E. Cline, K. Iverson, J. Moore, C. Edgar, R. Manber, N. Fuin, T. Poppe and R. Barton, ACU193, a Monoclonal Antibody that Selectively Binds Soluble Aβ Oligomers: Development Rationale, Phase 1 Trial Design, and Clinical Development Plan, J. Prev. Alzheimer’s Dis., 2022, 10(1), 19–24, DOI:10.14283/jpad.2022.93.
- M. Ortiz-Virumbrales, C. L. Moreno, I. Kruglikov, P. Marazuela, A. Sproul, S. Jacob, M. Zimmer, D. Paull, B. Zhang, E. E. Schadt, M. E. Ehrlich, R. E. Tanzi, O. Arancio, S. Noggle and S. Gandy, CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer's PSEN2 N141I neurons, Acta Neuropathol. Commun., 2017, 5, 77, DOI:10.1186/s40478-017-0475-z.
- S. T. DeKosky, Pathology and Pathways of Alzheimer’s Disease with an Update on New Developments in Treatment, J. Am. Geriatr. Soc., 2003, 51(5 Suppl Dementia), S314–S320, DOI:10.1046/j.1532-5415.5157.x.
- C. Zhu, S. Fu, Y. Chen, L. Li, R. Mao, J. Wang, R. Liu, Y. Liu and X. Wang, Advances in Drug Therapy for Alzheimer's Disease, Curr. Med. Sci., 2020, 40, 999–1008, DOI:10.1007/s11596-020-2281-2.
- J. Folch, O. Busquets, M. Ettcheto, E. Sánchez-López, R. D. Castro-Torres, E. Verdaguer, M. L. Garcia, J. Olloquequi, G. Casadesús, C. Beas-Zarate, C. Pelegri, J. Vilaplana, C. Auladell and A. Camins, Memantine for the Treatment of Dementia: A Review on its Current and Future Applications, J. Alzheimer's Dis., 2018, 62, 1223–1240, DOI:10.3233/JAD-170672.
- L. Blaikie, G. Kay and P. Kong Thoo Lin, Current and emerging therapeutic targets of alzheimer's disease for the design of multi-target directed ligands, MedChemComm, 2019, 10, 2052–2072, 10.1039/C9MD00337A.
- T. Behl, I. Kaur, A. Sehgal, S. Singh, N. Sharma, H. A. Makeen, M. Albratty, H. A. Alhazmi, S. G. Felemban, A. M. Alsubayiel, S. Bhatia and S. Bungau, “Aducanumab” making a comeback in Alzheimer's disease: An old wine in a new bottle, Biomed. Pharmacother., 2022, 148, 112746, DOI:10.1016/j.biopha.2022.112746.
- S. M. Hoy, Lecanemab: First Approval, Drugs, 2023, 83, 359–365, DOI:10.1007/s40265-023-01851-2.
- B. Fonseca-Santos, M. Chorilli and M. Palmira Daflon Gremião, Nanotechnology-based drug delivery systems for the treatment of Alzheimer's disease, Int. J. Nanomed., 2015, 4981, DOI:10.2147/IJN.S87148.
- T. Pardo-Moreno, A. González-Acedo, A. Rivas-Domínguez, V. García-Morales, F. J. García-Cozar, J. J. Ramos-Rodríguez and L. Melguizo-Rodríguez, Therapeutic Approach to Alzheimer's Disease: Current Treatments and New Perspectives, Pharmaceutics, 2022, 14, 1117, DOI:10.3390/pharmaceutics14061117.
- D. Nunes, J. A. Loureiro and M. C. Pereira, Drug Delivery Systems as a Strategy to Improve the Efficacy of FDA-Approved Alzheimer's Drugs, Pharmaceutics, 2022, 14, 2296, DOI:10.3390/pharmaceutics14112296.
- R. Duncan, Polymer conjugates as anticancer nanomedicines, Nat. Rev. Cancer, 2006, 6, 688–701, DOI:10.1038/nrc1958.
- L. F. Dalmolin, N. M. Khalil and R. M. Mainardes, Delivery of vanillin by poly(lactic-acid) nanoparticles: Development, characterization and in vitro evaluation of antioxidant activity, Mater. Sci. Eng., C, 2016, 62, 1–8, DOI:10.1016/j.msec.2016.01.031.
- R. Duncan, The dawning era of polymer therapeutics, Nat. Rev. Drug Discovery, 2003, 2, 347–360, DOI:10.1038/nrd1088.
- R. Duncan and M. J. Vicent, Polymer therapeutics-prospects for 21st century: The end of the beginning, Adv. Drug Delivery Rev., 2013, 65, 60–70, DOI:10.1016/j.addr.2012.08.012.
- R. Duncan, H. Ringsdorf and R. Satchi-Fainaro, Polymer therapeutics—polymers as drugs, drug and protein conjugates and gene delivery systems: Past, present and future opportunities, J. Drug Targeting, 2006, 14, 337–341, DOI:10.1080/10611860600833856.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Polymers for Drug Delivery Systems, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173, DOI:10.1146/annurev-chembioeng-073009-100847.
- D. El Mehdi, F. V. Grossi, P. Deschatelets, H. Pullon, L. Tan, C. A. Vega and C. G. Francois, APL-2, a complement C3 inhibitor, may potentially reduce both intravascular and extravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria, Mol. Immunol., 2017, 89, 115, DOI:10.1016/j.molimm.2017.06.039.
- P. Chatelain, O. Malievskiy, K. Radziuk, G. Senatorova, M. O. Abdou, E. Vlachopapadopoulou, Y. Skorodok, V. Peterkova, J. A. Leff and M. Beckert, A Randomized Phase 2 Study of Long-Acting TransCon GH vs Daily GH in Childhood GH Deficiency, J. Clin. Endocrinol. Metab., 2017, 102, 1673–1682, DOI:10.1210/jc.2016-3776.
- A. M. Ebied, K. H. Patel and R. M. Cooper-DeHoff, New Drugs Approved in 2019, Am. J. Med., 2020, 133, 675–678, DOI:10.1016/j.amjmed.2020.01.030.
- P. Chowdary, E. Fosbury, A. Riddell and M. Mathias, Therapeutic and routine prophylactic properties of rFactor VIII Fc (efraloctocog alfa, Eloctate®) in hemophilia A, J. Blood Med., 2016, 7, 187–198, DOI:10.2147/JBM.S80814.
- E. Santagostino, S. Lalezari, M. T. Reding, J. Ducore, H. J. Ng, L. H. Poulsen, L. A. Michaels and C. C. G. Linardi, Safety and efficacy of BAY 94-9027, an extended-half-life factor VIII, during surgery in patients with severe hemophilia A: Results of the PROTECT VIII clinical trial, Thromb. Res., 2019, 183, 13–19, DOI:10.1016/j.thromres.2019.08.023.
- R.-J. Li, R. Jin, C. Liu, X. Cao, M. L. Manning, X. M. Di, D. Przepiorka, F. Namuswe, A. Deisseroth, K. B. Goldberg, G. M. Blumenthal and R. Pazdur, FDA Approval Summary: Calaspargase Pegol-mknl For Treatment of Acute Lymphoblastic Leukemia in Children and Young Adults, Clin. Cancer Res., 2020, 26, 328–331, DOI:10.1158/1078-0432.CCR-19-1255.
- D. H. Kim, H. S. Lee, T.-W. Kwon, Y.-M. Han, N.-W. Kang, M. Y. Lee, D.-D. Kim, M. G. Kim and J.-Y. Lee, Single enzyme nanoparticle, an effective tool for enzyme replacement therapy, Arch. Pharmacal Res., 2020, 43, 1–21, DOI:10.1007/s12272-020-01216-3.
- J. Thomas, H. Levy, S. Amato, J. Vockley, R. Zori, D. Dimmock, C. O. Harding, D. A. Bilder, H. H. Weng, J. Olbertz, M. Merilainen, J. Jiang, K. Larimore, S. Gupta, Z. Gu and H. Northrup, Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM), Mol. Genet. Metab., 2018, 124, 27–38, DOI:10.1016/j.ymgme.2018.03.006.
- B. A. Konkle, O. Stasyshyn, P. Chowdary, D. H. Bevan, T. Mant, M. Shima, W. Engl, J. Dyck-Jones, M. Fuerlinger, L. Patrone, B. Ewenstein and B. Abbuehl, Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A, Blood, 2015, 126, 1078–1085, DOI:10.1182/blood-2015-03-630897.
- K. P. Garnock-Jones, Naloxegol: A Review of Its Use in Patients with Opioid-Induced Constipation, Drugs, 2015, 75, 419–425, DOI:10.1007/s40265-015-0357-2.
- B. C. Kieseier, D. L. Arnold, L. J. Balcer, A. A. Boyko, J. Pelletier, S. Liu, Y. Zhu, A. Seddighzadeh, S. Hung, A. Deykin, S. I. Sheikh and P. A. Calabresi, Peginterferon beta-1a in multiple sclerosis: 2-year results from ADVANCE, Mult. Scler. J., 2015, 21, 1025–1035, DOI:10.1177/1352458514557986.
- H. S. Baraf, M. A. Becker, S. R. Gutierrez-Urena, E. L. Treadwell, J. Vazquez-Mellado, C. D. Rehrig, F. D. Ottery, J. S. Sundy and R. A. Yood, Tophus burden reduction with pegloticase: results from phase 3 randomized trials and open-label extension in patients with chronic gout refractory to conventional therapy, Arthritis Res. Ther., 2013, 15, R137, DOI:10.1186/ar4318.
- S. Svenson, Clinical translation of nanomedicines, Curr. Opin. Solid State Mater. Sci., 2012, 16, 287–294, DOI:10.1016/j.cossms.2012.10.001.
- P. J. Mease, R. Fleischmann, A. A. Deodhar, J. Wollenhaupt, M. Khraishi, D. Kielar, F. Woltering, C. Stach, B. Hoepken, T. Arledge and D. van der Heijde, Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a Phase 3 double-blind randomised placebo-controlled study (RAPID-PsA), Ann. Rheum. Dis., 2014, 73, 48–55, DOI:10.1136/annrheumdis-2013-203696.
- S. Lamon, S. Giraud, L. Egli, J. Smolander, M. Jarsch, K.-G. Stubenrauch, A. Hellwig, M. Saugy and N. Robinson, A high-throughput test to detect C.E.R.A. doping in blood, J. Pharm. Biomed. Anal., 2009, 50, 954–958, DOI:10.1016/j.jpba.2009.06.038.
- R. Autrata, I. Krejčířová, K. Šenková, M. Holoušová, Z. Doležel and I. Borek, Intravitreal pegaptanib combined with diode laser therapy for stage 3+ retinopathy of prematurity in zone I and posterior zone II, Eur. J. Ophthalmol., 2012, 22, 687–694, DOI:10.5301/ejo.5000166.
- P. U. Freda, M. B. Gordon, N. Kelepouris, P. Jonsson, M. Koltowska-Haggstrom and A. J. van der Lely, Long-Term Treatment with Pegvisomant as Monotherapy in Patients with Acromegaly: Experience from Acrostudy, Endocr. Pract., 2015, 21, 264–274, DOI:10.4158/EP14330.OR.
- Y. Kosaka, Y. Rai, N. Masuda, T. Takano, T. Saeki, S. Nakamura, R. Shimazaki, Y. Ito, Y. Tokuda and K. Tamura, Phase III placebo-controlled, double-blind, randomized trial of pegfilgrastim to reduce the risk of febrile neutropenia in breast cancer patients receiving docetaxel/cyclophosphamide chemotherapy, Support. Care Cancer, 2015, 23, 1137–1143, DOI:10.1007/s00520-014-2597-1.
- X. Chen, X. Chen, W. Chen, X. Ma, J. Huang and R. Chen, Extended peginterferon alfa-2a (Pegasys) therapy in Chinese patients with HBeAg-negative chronic hepatitis B, J. Med. Virol., 2014, 86, 1705–1713, DOI:10.1002/jmv.24013.
- G. Scotto, E. Palumbo, V. Fazio, D. C. Cibelli, A. Saracino and G. Angarano, Peginterferon alfa-2b Treatment for Patients Affected by Acute Hepatitis C: Presentation of Six Case Reports, Infection, 2005, 33, 30–32, DOI:10.1007/s15010-005-4032-5.
- M. Graham, Pegaspargase: a review of clinical studies, Adv. Drug Delivery Rev., 2003, 55, 1293–1302, DOI:10.1016/S0169-409X(03)00110-8.
- H. Maeda, SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy, Adv. Drug Delivery Rev., 2001, 46, 169–185, DOI:10.1016/S0169-409X(00)00134-4.
- M. S. Hershfield, R. H. Buckley, M. L. Greenberg, A. L. Melton, R. Schiff, C. Hatem, J. Kurtzberg, M. L. Markert, R. H. Kobayashi, A. L. Kobayashi and A. Abuchowski, Treatment of Adenosine Deaminase Deficiency with Polyethylene Glycol-Modified Adenosine Deaminase, N. Engl. J. Med., 1987, 316, 589–596, DOI:10.1056/NEJM198703053161005.
- N. Marasini, S. Haque and L. M. Kaminskas, Polymer-drug conjugates as inhalable drug delivery systems: A review, Curr. Opin. Colloid Interface Sci., 2017, 31, 18–29, DOI:10.1016/j.cocis.2017.06.003.
- H. Jatzkewitz, Peptamin (glycyl-L-leucyl-mescaline) bound to blood plasma expander (polyvinylpyrrolidone) as a new depot form of a biologically active primary amine (mescaline), Z. Naturforsch., 1955, 10, 27–31 CrossRef.
- H. Ringsdorf, Structure and properties of pharmacologically active polymers, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153, DOI:10.1002/polc.5070510111.
- X. Pang, X. Yang and G. Zhai, Polymer-drug conjugates: recent progress on administration routes, Expert Opin. Drug Delivery, 2014, 11, 1075–1086, DOI:10.1517/17425247.2014.912779.
- J. Khandare and T. Minko, Polymer–drug conjugates: Progress in polymeric prodrugs, Prog. Polym. Sci., 2006, 31, 359–397, DOI:10.1016/j.progpolymsci.2005.09.004.
- S. Alven, X. Nqoro, B. Buyana and B. A. Aderibigbe, Polymer-Drug Conjugate, a Potential Therapeutic to Combat Breast and Lung Cancer, Pharmaceutics, 2020, 12, 406, DOI:10.3390/pharmaceutics12050406.
- X. Feng, D. Li, J. Han, X. Zhuang and J. Ding, Schiff base bond-linked polysaccharide–doxorubicin conjugate for upregulated cancer therapy, Mater. Sci. Eng., C, 2017, 76, 1121–1128, DOI:10.1016/j.msec.2017.03.201.
- N. Larson and H. Ghandehari, Polymeric Conjugates for Drug Delivery, Chem. Mater., 2012, 24, 840–853, DOI:10.1021/cm2031569.
- Z.-R. Lu, F. Ye and A. Vaidya, Polymer platforms for drug delivery and biomedical imaging, J. Controlled Release, 2007, 122, 269–277, DOI:10.1016/j.jconrel.2007.06.016.
- Q. Feng and R. Tong, Anticancer nanoparticulate polymer–drug conjugate, Bioeng. Transl. Med., 2016, 1, 277–296, DOI:10.1002/btm2.10033.
- R. Duncan, Designing polymer conjugates as lysosomotropic nanomedicines, Biochem. Soc. Trans., 2007, 35, 56–60, DOI:10.1042/BST0350056.
- F. Greco and M. J. Vicent, Combination therapy: Opportunities and challenges for polymer–drug conjugates as anticancer nanomedicines, Adv. Drug Delivery Rev., 2009, 61, 1203–1213, DOI:10.1016/j.addr.2009.05.006.
- M. J. Vicent and R. Duncan, Polymer conjugates: nanosized medicines for treating cancer, Trends Biotechnol., 2006, 24, 39–47, DOI:10.1016/j.tibtech.2005.11.006.
- M. J. Vicent, F. Greco, R. I. Nicholson, A. Paul, P. C. Griffiths and R. Duncan, Polymer Therapeutics Designed for a Combination Therapy of Hormone–Dependent Cancer, Angew. Chem., 2005, 117, 4129–4134, DOI:10.1002/ange.200462960.
- C. Ibie, R. Knott and C. J. Thompson, In-vitro evaluation of the effect of polymer structure on uptake of novel polymer-insulin polyelectrolyte complexes by human epithelial cells, Int. J. Pharm., 2015, 479, 103–117, DOI:10.1016/j.ijpharm.2014.12.058.
- A. D. Kulkarni, Y. H. Vanjari, K. H. Sancheti, H. M. Patel, V. S. Belgamwar, S. J. Surana and C. V. Pardeshi, Polyelectrolyte complexes: mechanisms, critical experimental aspects, and applications, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1615–1625, DOI:10.3109/21691401.2015.1129624.
- P. Pereira, A. F. Jorge, R. Martins, A. A. C. C. Pais, F. Sousa and A. Figueiras, Characterization of polyplexes involving small RNA, J. Colloid Interface Sci., 2012, 387, 84–94, DOI:10.1016/j.jcis.2012.07.088.
- S. S. Patel, A. Acharya, R. S. Ray, R. Agrawal, R. Raghuwanshi and P. Jain, Cellular and molecular mechanisms of curcumin in prevention and treatment of disease, Crit. Rev. Food Sci. Nutr., 2020, 60, 887–939, DOI:10.1080/10408398.2018.1552244.
- K. Gayathri, M. Bhaskaran, C. Selvam and R. Thilagavathi, Nano formulation approaches for curcumin delivery – a review, J. Drug Delivery Sci. Technol., 2023, 82, 104326, DOI:10.1016/j.jddst.2023.104326.
- K. Szczepanowicz, D. Jantas, M. Piotrowski, J. Staroń, M. Leśkiewicz, M. Regulska, W. Lasoń and P. Warszyński, Encapsulation of curcumin in polyelectrolyte nanocapsules and their neuroprotective activity, Nanotechnology, 2016, 27, 355101, DOI:10.1088/0957-4484/27/35/355101.
- P. Vicennati, A. Giuliano, G. Ortaggi and A. Masotti, Polyethylenimine In Medicinal Chemistry, Curr. Med. Chem., 2008, 15, 2826–2839, DOI:10.2174/092986708786242778.
- A. Calarco, M. Bosetti, S. Margarucci, L. Fusaro, E. Nicolì, O. Petillo, M. Cannas, U. Galderisi and G. Peluso, The genotoxicity of PEI-based nanoparticles is reduced by acetylation of polyethylenimine amines in human primary cells, Toxicol. Lett., 2013, 218, 10–17, DOI:10.1016/j.toxlet.2012.12.019.
- R. K. Oskuee, A. Dehshahri, W. T. Shier and M. Ramezani, Alkylcarboxylate grafting to polyethylenimine: a simple approach to producing a DNA nanocarrier with low toxicity, J. Gene Med., 2009, 11, 921–932, DOI:10.1002/jgm.1374.
- A. K. Lam, E. L. Moen, J. Pusavat, C. L. Wouters, H. Panlilio, M. J. Ferrell, M. B. Houck, D. T. Glatzhofer and C. V. Rice, PEGylation of Polyethylenimine Lowers Acute Toxicity while Retaining Anti-Biofilm and β-Lactam Potentiation Properties against Antibiotic-Resistant Pathogens, ACS Omega, 2020, 5, 26262–26270, DOI:10.1021/acsomega.0c04111.
- O. Boussif, T. Delair, C. Brua, L. Veron, A. Pavirani and H. V. J. Kolbe, Synthesis of Polyallylamine Derivatives and Their Use as Gene Transfer Vectors in Vitro, Bioconjugate Chem., 1999, 10, 877–883, DOI:10.1021/bc9900439.
- C. O. Ibie, C. J. Thompson and R. Knott, Synthesis, characterisation and in vitro evaluation of novel thiolated derivatives of polyallylamine and quaternised polyallylamine, Colloid Polym. Sci., 2015, 293, 1737–1748, DOI:10.1007/s00396-015-3561-4.
- D. W. Hwang, S. Son, J. Jang, H. Youn, S. Lee, D. Lee, Y.-S. Lee, J. M. Jeong, W. J. Kim and D. S. Lee, A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA, Biomaterials, 2011, 32, 4968–4975, DOI:10.1016/j.biomaterials.2011.03.047.
- F. Richter, K. Leer, L. Martin, P. Mapfumo, J. I. Solomun, M. T. Kuchenbrod, S. Hoeppener, J. C. Brendel and A. Traeger, The impact of anionic polymers on gene delivery: how composition and assembly help evading the toxicity-efficiency dilemma, J. Nanobiotechnol., 2021, 19, 292, DOI:10.1186/s12951-021-00994-2.
- H. Arkaban, M. Barani, M. R. Akbarizadeh, N. Pal Singh Chauhan, S. Jadoun, M. Dehghani Soltani and P. Zarrintaj, Polyacrylic Acid Nanoplatforms: Antimicrobial, Tissue Engineering, and Cancer Theranostic Applications, Polymers, 2022, 14, 1259, DOI:10.3390/polym14061259.
- S. K. Tripathi, Z. Ahmadi, K. C. Gupta and P. Kumar, Polyethylenimine-polyacrylic acid nanocomposites: Type of bonding does influence the gene transfer efficacy and cytotoxicity, Colloids Surf., B, 2016, 140, 117–120, DOI:10.1016/j.colsurfb.2015.12.007.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Polymer–drug conjugate therapeutics: advances, insights and prospects, Nat. Rev. Drug Discovery, 2019, 18, 273–294, DOI:10.1038/s41573-018-0005-0.
- X. Pang, H.-L. Du, H.-Q. Zhang, Y.-J. Zhai and G.-X. Zhai, Polymer–drug conjugates: present state of play and future perspectives, Drug Discovery Today, 2013, 18, 1316–1322, DOI:10.1016/j.drudis.2013.09.007.
- T. T. Nguyen, T. T. D. Nguyen, T. K. O. Nguyen, T. K. Vo and V. G. Vo, Advances in developing therapeutic strategies for Alzheimer's disease, Biomed. Pharmacother., 2021, 139, 111623, DOI:10.1016/j.biopha.2021.111623.
- J. E. de Souza, L. M. Casanova and S. S. Costa, Biodisponibilidade de compostos fenólicos: um importante desafio para o desenvolvimento de fármacos?, Rev. Fitos, 2015, 9(1) DOI:10.5935/2446-4775.20150006.
- M. Pinto, V. Silva, S. Barreiro, R. Silva, F. Remião, F. Borges and C. Fernandes, Brain drug delivery and neurodegenerative diseases: Polymeric PLGA-based nanoparticles as a forefront platform, Ageing Res. Rev., 2022, 79, 101658, DOI:10.1016/j.arr.2022.101658.
- B. Kateb, K. Chiu, K. L. Black, V. Yamamoto, B. Khalsa, J. Y. Ljubimova, H. Ding, R. Patil, J. A. Portilla-Arias, M. Modo, D. F. Moore, K. Farahani, M. S. Okun, N. Prakash, J. Neman, D. Ahdoot, W. Grundfest, S. Nikzad and J. D. Heiss, Nanoplatforms for constructing new approaches to cancer treatment, imaging, and drug delivery: What should be the policy?, NeuroImage, 2011, 54, S106–S124, DOI:10.1016/j.neuroimage.2010.01.105.
- R. S. Navath, Y. E. Kurtoglu, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Dendrimer–Drug Conjugates for Tailored Intracellular Drug Release Based on Glutathione Levels, Bioconjugate Chem., 2008, 19, 2446–2455, DOI:10.1021/bc800342d.
- D. R. Skvarc, O. M. Dean, L. K. Byrne, L. Gray, S. Lane, M. Lewis, B. S. Fernandes, M. Berk and A. Marriott, The effect of N-acetylcysteine (NAC) on human cognition – A systematic review, Neurosci. Biobehav. Rev., 2017, 78, 44–56, DOI:10.1016/j.neubiorev.2017.04.013.
- L. Borgström, B. Kågedal and O. Paulsen, Pharmacokinetics of N-acetylcysteine in man, Eur. J. Clin. Pharmacol., 1986, 31, 217–222, DOI:10.1007/BF00606662.
- K. Teder, L. Maddison, H. Soeorg, A. Meos and J. Karjagin, The Pharmacokinetic Profile and Bioavailability of Enteral N-Acetylcysteine in Intensive Care Unit, Medicina, 2021, 57, 1218, DOI:10.3390/medicina57111218.
- R. S. Navath, B. Wang, S. Kannan, R. Romero and R. M. Kannan, Stimuli-responsive star poly(ethylene glycol) drug conjugates for improved intracellular delivery of the drug in neuroinflammation, J. Controlled Release, 2010, 142, 447–456, DOI:10.1016/j.jconrel.2009.10.035.
- T. Pardo-Moreno, A. González-Acedo, A. Rivas-Domínguez, V. García-Morales, F. J. García-Cozar, J. J. Ramos-Rodríguez and L. Melguizo-Rodríguez, Therapeutic Approach to Alzheimer's Disease: Current Treatments and New Perspectives, Pharmaceutics, 2022, 14, 1117, DOI:10.3390/pharmaceutics14061117.
- A. K. Singh, A. Gothwal, S. Rani, M. Rana, A. K. Sharma, A. K. Yadav and U. Gupta, Dendrimer Donepezil Conjugates for Improved Brain Delivery and Better in Vivo Pharmacokinetics, ACS Omega, 2019, 4, 4519–4529, DOI:10.1021/acsomega.8b03445.
- A. Mistry, S. Stolnik and L. Illum, Nanoparticles for direct nose-to-brain delivery of drugs, Int. J. Pharm., 2009, 379, 146–157, DOI:10.1016/j.ijpharm.2009.06.019.
- ALZFORUM, ALZT-OP1 [Internet], ALZFORUM, 2023, accessed on 2023 Oct 28, Available from: https://www.alzforum.org/therapeutics/search?fda_statuses=&target_types=&therapy_types%5B%5D=36536&conditions%5B%5D=145&keywords-entry=&keywords=.
- ALZFORUM, AVP-923 [Internet], ALZFORUM, 2023, accessed on 2023 Oct 28, Available from: https://www.alzforum.org/therapeutics/search?fda_statuses=&target_types=&therapy_types%5B%5D=36536&conditions%5B%5D=145&keywords-entry=&keywords=.
- A. Duro-Castano, C. Borrás, V. Herranz-Pérez, M. C. Blanco-Gandía, I. Conejos-Sánchez, A. Armiñán, C. Mas-Bargues, M. Inglés, J. Miñarro, M. Rodríguez-Arias, J. M. García-Verdugo, J. Viña and M. J. Vicent, Targeting Alzheimer’s disease with multimodal polypeptide-based nanoconjugates, Sci. Adv., 2021, 7(13), eabf9180, DOI:10.1126/sciadv.abf9180.
- B. Zhang, Y. Gao, X. Zhang, J. Jiang, J. Ren, S. Wang, H. Hu, Y. Zhao, L. Chen, K. Zhao and F. Dai, Ultra-stable dextran conjugated prodrug micelles for oxidative stress and glycometabolic abnormality combination treatment of Alzheimer's disease, Int. J. Biol. Macromol., 2022, 203, 430–444, DOI:10.1016/j.ijbiomac.2022.01.154.
- T. Naki, W. M. R. Matshe, M. O. Balogun, S. Sinha Ray, S. A. Egieyeh and B. A. Aderibigbe, Polymer drug conjugates containing memantine, tacrine and cinnamic acid: promising nanotherapeutics for the treatment of Alzheimer's disease, J. Microencapsulation, 2023, 40, 15–28, DOI:10.1080/02652048.2023.2167011.
- L. Blaikie, G. Kay and P. Kong Thoo Lin, Synthesis and in vitro evaluation of vanillin derivatives as multi-target therapeutics for the treatment of Alzheimer's disease, Bioorg. Med. Chem. Lett., 2020, 30, 127505, DOI:10.1016/j.bmcl.2020.127505.
- M. Scipioni, G. Kay, I. Megson and P. Kong Thoo Lin, Novel vanillin derivatives: Synthesis, anti-oxidant, DNA and cellular protection properties, Eur. J. Med. Chem., 2018, 143, 745–754, DOI:10.1016/j.ejmech.2017.11.072.
- S. Tiwari, V. Atluri, A. Kaushik, A. Yndart and M. Nair, Alzheimer's disease: pathogenesis, diagnostics, and therapeutics, Int. J. Nanomed., 2019, 14, 5541–5554, DOI:10.2147/IJN.S200490.
- B. Oller-Salvia, M. Sánchez-Navarro, E. Giralt and M. Teixidó, Blood–brain barrier shuttle peptides: an emerging paradigm for brain delivery, Chem. Soc. Rev., 2016, 45, 4690–4707, 10.1039/C6CS00076B.
- F. Zhang, Y.-A. Lin, S. Kannan and R. M. Kannan, Targeting specific cells in the brain with nanomedicines for CNS therapies, J. Controlled Release, 2016, 240, 212–226, DOI:10.1016/j.jconrel.2015.12.013.
- W. M. Pardridge, The blood-brain barrier: Bottleneck in brain drug development, NeuroRx, 2005, 2, 3–14, DOI:10.1602/neurorx.2.1.3.
- Y. J. Yu, Y. Zhang, M. Kenrick, K. Hoyte, W. Luk, Y. Lu, J. Atwal, J. M. Elliott, S. Prabhu, R. J. Watts and M. S. Dennis, Boosting Brain Uptake of a Therapeutic Antibody by Reducing Its Affinity for a Transcytosis Target, Sci. Transl. Med., 2011, 3(84), 84ra44, DOI:10.1126/scitranslmed.3002230.
- Y. Kang, H. Jung, J. Oh and D. Song, Use of PEGylated Immunoliposomes to Deliver Dopamine Across the Blood–Brain Barrier in a Rat Model of Parkinson's Disease, CNS Neurosci. Ther., 2016, 22, 817–823, DOI:10.1111/cns.12580.
- L.-H. Dieu, D. Wu, C. G. Palivan, V. Balasubramanian and J. Huwyler, Polymersomes conjugated to 83-14 monoclonal antibodies: In vitro targeting of brain capillary endothelial cells, Eur. J. Pharm. Biopharm., 2014, 88, 316–324, DOI:10.1016/j.ejpb.2014.05.021.
- B. Sharma, K. Luhach and G. T. Kulkarni, In vitro and in vivo models of BBB to evaluate brain targeting drug delivery, in Brain targeted drug delivery systems: a focus on nanotechnology and nanoparticulates, ed. H. Gao and X. Gao, Elsevier, 2019, pp. 53–101. DOI:10.1016/B978-0-12-814001-7.00004-4.
- F. Rodriguez-Otormin, A. Duro-Castano, I. Conejos-Sánchez and M. J. Vicent, Envisioning the future of polymer therapeutics for brain disorders, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11(1), e1532, DOI:10.1002/wnan.1532.
- X. Zheng, X. Pang, P. Yang, X. Wan, Y. Wei, Q. Guo, Q. Zhang and X. Jiang, A hybrid siRNA delivery complex for enhanced brain penetration and precise amyloid plaque targeting in Alzheimer's disease mice, Acta Biomater., 2017, 49, 388–401, DOI:10.1016/j.actbio.2016.11.029.
- J. Rip, G. Schenk and A. de Boer, Differential receptor-mediated drug targeting to the diseased brain, Expert Opin. Drug Delivery, 2009, 6, 227–237, DOI:10.1517/17425240902806383.
- G. K. Gouras, T. T. Olsson and O. Hansson, β-Amyloid Peptides and Amyloid Plaques in Alzheimer's Disease, Neurotherapeutics, 2015, 12, 3–11, DOI:10.1007/s13311-014-0313-y.
- B. Eftekharzadeh, J. G. Daigle, L. E. Kapinos, A. Coyne, J. Schiantarelli, Y. Carlomagno, C. Cook, S. J. Miller, S. Dujardin, A. S. Amaral, J. C. Grima, R. E. Bennett, K. Tepper, M. DeTure, C. R. Vanderburg, B. T. Corjuc, S. L. DeVos, J. A. Gonzalez, J. Chew, S. Vidensky, F. H. Gage, J. Mertens, J. Troncoso, E. Mandelkow, X. Salvatella, R. Y. H. Lim, L. Petrucelli, S. Wegmann, J. D. Rothstein and B. T. Hyman, Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer's Disease, Neuron, 2018, 99, 925–940, DOI:10.1016/j.neuron.2018.07.039 .e7.
- A. De Loof and L. Schoofs, Alzheimer's Disease: Is a Dysfunctional Mevalonate Biosynthetic Pathway the Master-Inducer of Deleterious Changes in Cell Physiology?, OBM Neurobiol., 2019, 3, 1, DOI:10.21926/obm.neurobiol.1904046.
- Á. Sarabia-Vallejo, P. López-Alvarado and J. C. Menéndez, Small-molecule theranostics in Alzheimer's disease, Eur. J. Med. Chem., 2023, 255, 115382, DOI:10.1016/j.ejmech.2023.115382.
- B. Hu, F. Dai, Z. Fan, G. Ma, Q. Tang and X. Zhang, Nanotheranostics: Congo Red/Rutin–MNPs with Enhanced Magnetic Resonance Imaging and H2O2-Responsive Therapy of Alzheimer's Disease in APPswe/PS1dE9 Transgenic Mice, Adv. Mater., 2015, 27, 5499–5505, DOI:10.1002/adma.201502227.
- S. Manandhar, E. Sjöholm, J. Bobacka, J. M. Rosenholm and K. K. Bansal, Polymer-Drug Conjugates as Nanotheranostic Agents, J. Nanotheranostics, 2021, 2, 63–81, DOI:10.3390/jnt2010005.
- N. Sinha, T. Zahra, A. Y. Gahane, B. Rout, A. Bhattacharya, S. Basu, A. Chakrabarti and A. K. Thakur, Protein reservoirs of seeds are amyloid composites employed differentially for germination and seedling emergence, Plant J., 2023, 116, 329–346, DOI:10.1111/tpj.16429.
- S. I. Ahn, Y. J. Sei, H.-J. Park, J. Kim, Y. Ryu, J. J. Choi, H.-J. Sung, T. J. MacDonald, A. I. Levey and Y. Kim, Microengineered human blood–brain barrier platform for understanding nanoparticle transport mechanisms, Nat. Commun., 2020, 11, 175, DOI:10.1038/s41467-019-13896-7.
- T. Osaki, Y. Shin, V. Sivathanu, M. Campisi and R. D. Kamm, In Vitro Microfluidic Models for Neurodegenerative Disorders, Adv. Healthc. Mater., 2018, 7(2) DOI:10.1002/adhm.201700489.
- E. Jagtiani, M. Yeolekar, S. Naik and V. Patravale, In vitro blood brain barrier models: An overview, J. Controlled Release, 2022, 343, 13–30, DOI:10.1016/j.jconrel.2022.01.011.
- Y. Liang and J.-Y. Yoon, In situ sensors for blood-brain barrier (BBB) on a chip, Sens. Actuators Rep., 2021, 3, 100031, DOI:10.1016/j.snr.2021.100031.
- S. Reshma, K. B. Megha, S. Amir, S. Rukhiya and P. V. Mohanan, Blood brain barrier-on-a-chip to model neurological diseases, J. Drug Delivery Sci. Technol., 2023, 80, 104174, DOI:10.1016/j.jddst.2023.104174.
- Y. J. Choi, S. Chae, J. H. Kim, K. F. Barald, J. Y. Park and S.-H. Lee, Neurotoxic amyloid beta oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow, Sci. Rep., 2013, 3, 1921, DOI:10.1038/srep0192.
| This journal is © The Royal Society of Chemistry 2024 |