Nanoengineered parallelogram-NiFe2O4/rGO nanocomposite-based biosensing interface for highly efficient electrochemical detection of neurodegenerative disorders via dopamine monitoring
Received
30th August 2023
, Accepted 11th October 2023
First published on 8th November 2023
Abstract
A low-temperature hydrothermal method was used to synthesize a unique parallelogram (Pg) morphology based on Pg-NiFe2O4 and Pg-NiFe2O4/reduced graphene oxide (rGO) nanocomposite. This unique Pg morphology provided high surface area, loading of a biomolecule, and high charge transfer between the substrate and analyte. This nanocomposite enabled creation of an efficient selective electrochemical biosensor for 3,4-dihydroxy phenylalanine (dopamine (DA)). DA is a key monoamine neurotransmitter that causes numerous disorders, including Parkinson's disease, Alzheimer's disease, dementia, and hyperactivity disorder due to an imbalanced concentration of DA in biological fluids. Thus, the in situ-prepared Pg-NiFe2O4/rGO nanocomposite was utilized for immobilization of the enzyme tyrosinase (Tyr) and for electrochemical estimation of DA. In addition, the structural, morphological, and electrochemical characteristics of the synthesized Pg nanocomposite were investigated by X-ray diffraction analysis, Fourier transform infrared spectroscopy, Raman spectroscopy, UV-visible spectroscopy, X-ray photoelectron spectroscopy, transmission electron microscopy, scanning electron microscopy, atomic force microscopy, and electrochemical method. The Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode had a excellent linear detection range of 1–300 μM, high sensitivity (9.456 × 10−4 mA μM−1 cm−2), low response time of 10 s, long stability of 40 days, good repeatability, and low limit of detection (0.0456 μM). Hence, the in situ-prepared nanocomposite, with its unique parallelogram morphology, offers an efficient means for monitoring DA, that can be essential for managing neurodegenerative disorders, by providing excellent sensitivity, stability, and detection capabilities.
1 Introduction
Ferrite is a ferromagnetic compound having iron oxide as a principal element, and mixes readily with other metals. Ferrites are commonly represented as AB2O4, especially in electrochemistry. Iron and nickel are regularly considered as two eco-friendly and inexpensive elements in electrochemical applications.1,2 Spinel ferrites can be represented as MFe2O4. M corresponds to a divalent fricative metal ion (e.g., Ni, Zn, Co) and Cu nanoparticles have an eight face-centered cubic (FCC) arrangement formed by oxygen atoms, in which only 1/8 of the 64 tetrahedral sites are occupied by Ni ions and 1/2 of octahedral sites by Fe ions. In an inverse spinel structure (parallelogram (Pg)-NiFe2O4, n-type semiconductor), one trivalent ferric ion (Fe3+) in tetrahedral lattice sites and a divalent metal ion (Ni2+) with a trivalent ferric ion (Fe3+) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio are present on octahedral lattice sites.3 The magnetic ferrite-based metal oxides Fe3O4, ZnFe2O4, CoFe2O4, and NiFe2O4 are used in various applications, such as catalysts,4 energy-storage devices, sensing and medical applications,5 electrochemical sensing,6 controlled drug release,7 semiconductors, and hydrogen-evolution reaction.8 Among these nano-ferrites, Pg-NiFe2O4 is one of the most important spinel soft ferrites. It has a significant role in biosensing applications due to good frequency and electrical conductivity, little loss of eddy currents, good permeability, good mechanical and electrochemical stabilities, and because the target molecule can bind readily on its surface.9 The conductivity of a material has a crucial role in amperometry-based biosensors by improving the response current. However, Pg-NiFe2O4 cannot be used directly in biosensing due to poor conductivity10 as compared with Pg-NiFe2O4/reduced graphene oxide (rGO) nanocomposites.
:1 ratio are present on octahedral lattice sites.3 The magnetic ferrite-based metal oxides Fe3O4, ZnFe2O4, CoFe2O4, and NiFe2O4 are used in various applications, such as catalysts,4 energy-storage devices, sensing and medical applications,5 electrochemical sensing,6 controlled drug release,7 semiconductors, and hydrogen-evolution reaction.8 Among these nano-ferrites, Pg-NiFe2O4 is one of the most important spinel soft ferrites. It has a significant role in biosensing applications due to good frequency and electrical conductivity, little loss of eddy currents, good permeability, good mechanical and electrochemical stabilities, and because the target molecule can bind readily on its surface.9 The conductivity of a material has a crucial role in amperometry-based biosensors by improving the response current. However, Pg-NiFe2O4 cannot be used directly in biosensing due to poor conductivity10 as compared with Pg-NiFe2O4/reduced graphene oxide (rGO) nanocomposites.
Graphene is a two-dimensional (2D) carbon material and the “mother” of all carbon nanostructures having sp2 hybrid single-layer carbon atoms. Graphene exhibits four important properties: (1) finite dimensions; (2) morphological (e.g., edge and dislocation) flaws; (3) 5-6-8 member rings in honeycomb and hexagon shapes; (4) holes in the plane (these sheets can aggregate through weak van der Waal contact and pi–pi interactions).11 Reduced graphene oxide (rGO) is a supreme derivative of graphene. It has a layered structure with oxygen-containing functional groups on its edges and basal plane.12 The surface of rGO contains several functional groups, such as carboxyl, epoxide, and hydroxyl, which are responsible for hydrophilicity and dispersibility in aqueous solution.13 It also exhibits good thermal, optical, electrical, mechanical, and conducting properties that make it ideal material in several domains, such as electrochemical applications,14 sensors,15 photocatalysts,16 solar cells, and photochemistry.17 However, the oxygen functional group present on the rGO surface decreases the electrical conductivity of the material and controls its direct application in electro-functional materials and devices.18 Therefore, to improve the electrical conductivity of the rGO surface, some modifications (e.g., chemical modification,19 doping,20 and composite formation with other conductive materials21) have been carried out to improve its conductive efficiency.
Dopamine (DA) is an essential monoamine neurotransmitter in the human body. DA is directly related to pathways that control mobility, motivation, metabolic activity, and the central nervous system in mammals.22 Its concentration in whole human blood and serum for healthy persons ranges from 10−6 to 10−9 mol L−1. A low concentration of DA in the physiological system creates serious health issues, such as hypertension, the death of heart muscles, and neurological disorders such as Parkinson's disease (which results in the loss of dopaminergic cells), and schizophrenia.23,24 Thus, the dysfunctional metabolism of DA in the brain must be treated by L-dopamine. To resolve these above-mentioned health-related problems, a rapid and effective method is required for DA measurement.
In recent years, several methods have been reported to measure the DA concentration, such as electrochemical methods, photoelectrochemical analysis,25 chemiluminescence,26 localized surface plasmon resonance (LSPR),27 chromatography, and mass spectrometry.28 Among all of these methods, the electrochemical approach is exquisite due to quick detection, high sensitivity, high specificity, low limit of detection, handleability, cost-effectiveness, eco-friendliness, on-site and real-time monitoring, and portability.29–32 The electrochemical approach allows recording the real-time response by changing the DA concentration. However, the selectivity of DA can be poor due to the presence of ascorbic acid and uric acid, which have a close redox potential to that of DA. Thus, carbon-based materials are used to improve the selectivity response of DA. In carbon-based materials, GO improves the selectivity significantly and can detect a low concentration of DA.33 A literature survey indicated that the mixing of NiFe2O4 into corban materials improved the electrocatalytic activity and performance of electrochemical biosensors for many analytes, such as acetaminophen, codeine,34 DA,35 and epinephrine.36
Herein we report, for the first time, an amperometric-based biosensor for the detection of DA using a tyrosinase (Tyr)/Pg-NiFe2O4/rGO/ITO bioelectrode. We synthesized a nanocomposite via a hydrothermal method for the detection of DA. Furthermore, the prepared nanocomposite material was applied to the ITO surface through electrophoretic deposition (EPD). Then, Tyr was immobilized on the surface of the modified Pg-NiFe2O4/rGO/ITO electrode by drop-casting. The prepared Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode showed outstanding amperometric results, such as excellent linearity, high sensitivity, low limit of detection, high stability, and good response time. These features make this bioelectrode an excellent candidate for electro-oxidation and detection of DA. The reason for effective enhancement of the electrochemical response in the Pg-NiFe2O4/rGO nanocomposite was due to the decoration of rGO with Pg-NiFe2O4. The intercalation between rGO nanosheets and oxygen-containing functional groups in rGO interacted electrostatically with the unique Pg-metal ferrite to enhance the amperometric response, surface area, and stability of the composite material for use in biosensing applications. Hence, the newly developed bioelectrode presents a pioneering amperometric biosensor for DA detection, with exceptional linearity, sensitivity, low detection limit, stability, and rapid response time. These attributes are crucial for precise DA monitoring in the management of neurodegenerative disorders. Further, its prospects include broader application in disease diagnosis and treatment optimization. These sensors are needed to provide accurate and timely data for tailoring interventions and therapies, which ultimately improves patient health in neurodegenerative disorders.
2 Experimental
2.1 Chemicals
Ni(NO3)2·6H2O, Fe(NO3)3·9H2O, H2O2 (30% solution), and NH3 (25% solution) were purchased from Merck (Whitehouse Station, NJ, USA). Graphite flakes were obtained from Millipore Sigma (Burlington, MA, USA). Cetyl-trimethyl-ammonium-bromide (CTAB, 99% purity) was from SRL Sisco Research Laboratories (Maharashtra, India). H2SO4 (98%) was sourced from Loba-Chemie (Mumbai, India). Sodium dihydrogen phosphate-dihydrate ([NaH2PO4]·2H2O), disodium hydrogen phosphate-dihydrate ([Na2HPO4]·2H2O, 99%), potassium hexacyano-ferrate(III) (K3[Fe(CN)6], 98.5%), and potassium hexacyanoferrate(II)trihydrate (K4[Fe(CN)6]·3H2O) were purchased from Merck. Dopamine hydrochloride (C8H12ClO2N) and enzyme dihydroxy-phenylalanine (Tyr extract from mushroom, 8503 unit per mg lyophilized solid powder) were purchased from Millipore Sigma. Sodium chloride (NaCl, 99%; Merck) and ITO sheets with surface resistivity (20 Ω sq−1) were procured from Millipore Sigma and used as a working electrode.
2.2 Synthesis of Pg-NiF2O4/rGO nanocomposite
GO was synthesized following the method reported by Hummer and colleagues37 with some modifications. Graphite flakes (2 g) were mixed with H2SO4 (46 ml) solution into a round-bottom flask by slow addition of KMnO4 (6 g) at 0–5 °C and continuously stirred for 2 h. The reaction temperature was maintained at 35 °C (overnight). Then, 92 ml of distilled water and 30% H2O2 solution (25 ml) were added with continued stirring to give a yellowish solution. Washing and drying were done to obtain rGO sheets. The hydrothermal synthesis of the NiFe2O4/rGO nanocomposite has been reported.38 We modified the reported synthetic process to prepare the Pg-NiF2O4/rGO nanocomposite. First, 0.4 M Ni(NO3)2·6H2O and 0.8 M Fe(NO3)3·9H2O were mixed in 50 ml of distilled water and stirred continuously to prepare a uniform solution. To this solution was added 25% NH3 to maintain a constant pH of 10 and a clear-brown solution appeared. After that, 30 mg of rGO was added to 25 ml of distilled water, and this solution was sonicated for 45 min at room temperature. A 50 ml mixture of 0.4 M Ni(NO3)2·6H2O and 0.8 M Fe(NO3)3·9H2O was transferred to 25 ml of rGO solution. Next, 0.1 M CTAB was added as a surfactant to the solution stated above, and the whole solution was stirred continuously at 60 °C for 2 h. The same solution was transferred to a hydrothermal autoclave (180 °C, 18 h) and the autoclave cooled down naturally at room temperature. A mixture of distilled water and ethanol was used for washing the precipitate, which removed unreacted materials and maintained a neutral pH. This precipitate was dried at 90 °C for 6 h. The material was obtained in powder form, and was calcinated at 500 °C for 5 h. The resulting material was the Pg-NiF2O4/rGO nanocomposite. To confirm this material, we undertook X-ray diffraction (XRD) analysis, Fourier transform infrared (FTIR) spectroscopy, and transmission electron microscopy (TEM). A similar method was used for the synthesis of Pg-NiFe2O4 without using rGO and surfactant. Eqn (1)–(3) show how the Pg-NiF2O4/rGO nanocomposite was synthesized.| | | 2Fe(NO3)3 + Ni(NO3)2 + 8NH4OH → 2Fe(OH)3 + Ni(OH)2 + 8NH4NO3 | (1) |
| | | 2Fe(OH)3 + Ni(OH)2 → NiO + Fe2O3 + 4H2O | (2) |
| | | NiO + Fe2O3 → Pg-NiFe2O4 | (3) |
2.3 Fabrication of Pg-NiFe2O4/rGO/ITO and immobilization of Tyr on the prepared electrode
Fabrication of the Pg-NiFe2O4/rGO/ITO electrode was done using an EPD technique. A 30 mg uniform suspension of Pg-NiFe2O4/rGO was mixed in double-distilled water (12 ml) and ethanol solution (3 ml) following ultrasonication for 60 min before EPD. A copper electrode was used as an anode material and functionalized ITO glass plate was employed as the cathode. The two electrodes were dipped in a suspension of Pg-NiFe2O4/rGO and hung vertically with a 1 cm gap between the two electrodes. A uniform thin film of the Pg-NiFe2O4/rGO nanocomposite was prepared on the ITO surface (0.25 cm2) by applying a 30 mV potential for 25 s. The prepared Pg-NiFe2O4/rGO/ITO electrode was removed from the EPD solution, washed with distilled water to remove unbound particles, and stored in a cool place for future utilization. A 20 μL solution of Tyr was immobilized on the Pg-NiFe2O4/rGO/ITO electrode surface using the drop-casting method and dried in a humid chamber at room temperature for 12 h. Tyr was immobilized on the surface of the Pg-NiFe2O4/rGO/ITO electrode and stored at 4 °C. Atomic force microscopy (AFM) was done to confirm the immobilization of Tyr on the surface of the Pg-NiFe2O4/rGO/ITO electrode.
3 Characterization
The characterization of hydrothermally synthesized Pg-NiFe2O4, and Pg-NiFe2O4/rGO nanocomposite was done through various methods. A powder X-ray diffractometer (λ = 1.5418; Cu Kα; D8 Advance; Bruker, Billerica, MA, USA) with a scan rate of 2° min−1 in the range of 0° to 90° was used for analyses of the crystallite nature and phase. A FT-IR spectrometer (FTIR-4700; Jasco, Tokyo, Japan) in the range of 400–4000 cm−1 at 25 °C was used for structural confirmation. Optical properties were checked by a UV-visible absorbance spectroscopy using a spectrometer (UV-1700; Shimadzu, Tokyo, Japan). A Raman spectrophotometer (LabRam HR Evolution; Horiba, Tokyo, Japan) was employed to detect in the range of 200–3500 cm−1 using a high-performance charged-coupled-device detector. A laser emitting a He–Ne beam at 633 nm was used to illuminate the sample. The surface morphology of the powder Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite was determined by scanning electron microscopy (SEM) using an EVO18 system (Zeiss, Oberkochen, Germany). AFM was done using a Solver Nano (NT-MDT Spectrum Instruments, Tempe, AZ, USA) system to confirm the surface morphology of Pg-NiFe2O4/ITO, Pg-NiFe2O4/rGO/ITO, and Tyr/Pg-NiFe2O4/rGO/ITO. X-ray photoelectron spectroscopy (XPS) was undertaken with a PHI 5000 VersaProbe-III Scanning XPS Microprobe (ULVAC-PHI, Tokyo, Japan) to investigate elemental composition and the level of reduction in rGO and Pg-NiFe2O4/rGO. A multi-purpose surface-area and pore-size analyzer (As-i Q-C; Quantachrome Instruments, Boynton Beach, FL, USA) was used to calculate pore size and surface area by the Brunauer–Emmett–Teller (BET) method. The internal morphology of the proposed materials was confirmed by TEM using a JEM 1400 setup (Jeol, Tokyo, Japan). Elemental composition was determined by energy-dispersive spectroscopy (EDS) using the EVO18 setup (Zeiss). NaCl solution (0.9%) was added to phosphate-buffered saline (PBS; 50 mM, pH 6.5) following use of 5 mM ferric/ferro cyanide [Fe(CN)6]3−/4− and used as a redox couple to undertake electrochemical studies using a single-channel instrument (CS350; CorrTest, Wuhan, China) for a three-electrode system. Tyr/Pg-NiFe2O4/rGO/ITO, platinum (Pt), and silver/silver chloride (Ag/AgCl, 3.5 M KCl electrolyte solution) electrodes were applied as the working, counter, and reference electrode, respectively.
3.1 XRD analyses
XRD analysis of the hydrothermally synthesized Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite was carried out to confirm the phase and crystal-lattice system. After XRD (Cu Kα), 111, 002, 220, 311, 222, 400, 422, 511, 440, 620, 533, 622, 444, and 642 planes were obtained (Fig. 1A). We found that 2θ at 26.17° (002 plane) was like graphene oxide (Fig. 1A-a) and matched with JCPDS 75-1621; all other planes confirmed the formation of nickel ferrite (Fig. 1A-b) (JCPDS 86-2267). Fig. 1A-c shows a good match to confirm the formation of the Pg-NiFe2O4/rGO nanocomposite and exhibits a single-phase cubic spinel crystal.3 The diffraction peaks for Fe2O3 and NiO were absent, which suggested the formation of pure single-phase NiFe2O4. A FCC crystal system with space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m was noted. The Scherrer equation (D = 0.9λ/βcosθ) was used to calculate the crystallite size (D) using the highest peak intensity, where λ is the wavelength of the X-ray (Cu Kα 1.5418 Å), θ is Bragg's angle, and β is full-width at half maximum (FWHM). The obtained crystallite size was 29.8 nm for diffraction in the (311) plane. The lattice parameter (a) and density (d) of the nanocomposite were calculated using eqn (4) and (5), respectively, where h, k, and l are the Miller indices, a is the lattice parameter, d is the density of the X-ray, 8 molecules are present in each unit cell of a spinel lattice, M is molecular weight in g mol−1 of the spinel, and NA is the Avogadro number. The calculated values of the lattice parameter and density were 8.38 Å and 5.21 g cm−3, respectively.
m was noted. The Scherrer equation (D = 0.9λ/βcosθ) was used to calculate the crystallite size (D) using the highest peak intensity, where λ is the wavelength of the X-ray (Cu Kα 1.5418 Å), θ is Bragg's angle, and β is full-width at half maximum (FWHM). The obtained crystallite size was 29.8 nm for diffraction in the (311) plane. The lattice parameter (a) and density (d) of the nanocomposite were calculated using eqn (4) and (5), respectively, where h, k, and l are the Miller indices, a is the lattice parameter, d is the density of the X-ray, 8 molecules are present in each unit cell of a spinel lattice, M is molecular weight in g mol−1 of the spinel, and NA is the Avogadro number. The calculated values of the lattice parameter and density were 8.38 Å and 5.21 g cm−3, respectively.| | | a = λ(h2 + k2 + l2)1/2/2sinθ | (4) |
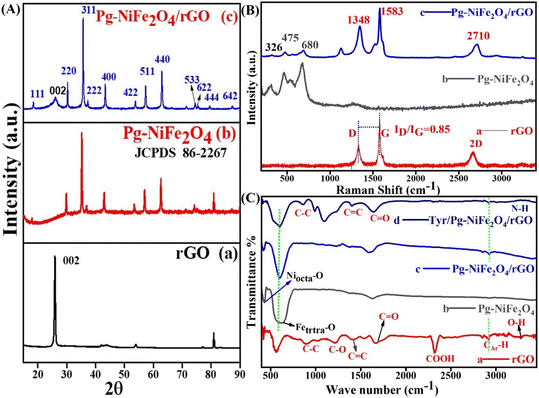 |
| | Fig. 1 (A) XRD spectra of (a) rGO, (b) Pg-NiFe2O4, and (c) Pg-NiFe2O4/rGO nanocomposite with their respective planes. (B) Raman spectra of (a) rGO, (b) Pg-NiFe2O4, and (c) Pg-NiFe2O4/rGO nanocomposite. (C) FTIR spectra of (a) rGO, (b) Pg-NiFe2O4, (c) Pg-NiFe2O4/rGO, and (d) Tyr/Pg-NiFe2O4/rGO, in transmittance mode. | |
3.2 Raman spectroscopy
Raman spectroscopy is a fast and nondestructive tool. It can provide direct electron–phonon interactions, as well as phase and structural (order/disorder) confirmations. The Raman spectra of rGO, Pg-NiFe2O4 and Pg-NiFe2O4/rGO (He–Ne laser beam at 633 nm,39 accumulation time = 7 s, acquisition time = 10 s, grating = 1800 grooves per mm, slit width = 100 μm) are shown in Fig. 1B. The Raman spectrum of rGO exhibited D (diamond) and G (graphite) bands at around 1348 cm−1 and 1583 cm−1 due to an out-of-plane breathing mode on sp2 atoms with A1g symmetry (local defects/disorder) and scattering of first-order in E2g phonons of Csp2 atoms in the Brillouin zone. A second-order two-phonon mode of the 2D band was observed at 2710 cm−1. The Csp2 atoms associated with vibrations of the G band in a 2D hexagonal lattice and 2D band provided evidence for the reduction of GO and formation of monolayer graphene-like structure40,41 The average size of sp2 domains was correlated with the intensity ratio (ID/IG) of D and G bands. However, the intensity, shape, and position of the D and G bands were dependent upon the number of layers of graphene. A ratio of ID/IG = 0.85 gave information about the degree of graphitization and defects present in the crystal lattice (Fig. 1B-a). Classical theory explains that an interaction between lattice vibrations and crystal structure appear during the synthesis of Pg-NiFe2O4, but some other phase formations, such as Fe3O4, α-Fe2O3, and γ-Fe2O3, are also possible. The scattering of Raman rays was used to differentiate the Pg-NiFe2O4 phase from another possible phase (like that found using XRD). The strongest peaks of the γ-Fe2O3 phase are at ∼1380 cm−1 and ∼1580 cm−1. However, no such peak was observed in this range42 (1300–1600 cm−1), which indicated the formation of a pure crystalline Pg-NiFe2O4 phase (Fig. 1B-b). The cubic inverse spinel structure of Pg-NiFe2O4 having a space group Oh7 (Fdm) and inverse spinel gave the vibrational mode predicted by the group theory in eqn (6) based on Raman (R), infrared (ir) active, and inactive (in) vibration. We found five (A1g + Eg + 3T2g) Raman-active modes and 4T1u IR-active modes. The peak at 675–610 cm−1 indicated the chemical nature of the divalent cation in the inverse spinel structure. Fig. 1B confirmed the formation of the pure crystalline Pg-NiFe2O4/rGO nanocomposite.| | | Γ(AB2O4) = A1g(R) + Eg(R) + 3T2g(R) + T1g(in) + 2A2u(in) + 2Eu(in) + 2T2u(in) + 4T1u(ir) | (6) |
3.3 Detection of functional groups by FTIR spectroscopy
FTIR spectroscopy was used for confirmation of the functional groups present in the hydrothermally synthesized nanocomposite. We used a range of detection of 400–3500 cm−1 for FTIR spectroscopy and assigned into two important regions (fingerprint and functional group). However, the peak observed in Pg-NiFe2O4 was below 750 cm−1; peaks at 430 cm−1 (Niocta–O bond) and 620 cm−1 (Fetetra–O bond) represented the stretching vibrations of the spinel cubic crystal in octahedral and tetrahedral lattices,43 respectively (Fig. 1C-b). The peak observed at 1595 cm−1 corresponded to O–H bending vibrations. However, the transmittance peaks at 1215 cm−1 and 1675 cm−1 indicated C–O alkoxy and epoxy groups,44 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl group. After that, the peaks at 900 cm−1 (C–C), 1420 cm−1 (CC), 2320 cm−1 (COOH), and 3380 cm−1 (N–H) were related to stretching vibrations in rGO and Tyr (Fig. 1C-a and d). The peak observed in the range of 2920–2980 cm−1 corresponded to CAr–H stretching of the aromatic ring present in rGO.
O carbonyl group. After that, the peaks at 900 cm−1 (C–C), 1420 cm−1 (CC), 2320 cm−1 (COOH), and 3380 cm−1 (N–H) were related to stretching vibrations in rGO and Tyr (Fig. 1C-a and d). The peak observed in the range of 2920–2980 cm−1 corresponded to CAr–H stretching of the aromatic ring present in rGO.
3.4 UV-visible spectra
UV-visible spectroscopy was used to analyze absorption in the absorbance range 200–800 nm. Fig. 2A shows the absorption spectra of rGO (265), Pg-NiFe2O4 (270), Pg-NiFe2O4/rGO (270), and Tyr/Pg-NiFe2O4/rGO (266) nanocomposites. The d–d transition of metal ions lead to a red (bathochromic) shift. The absorption peak of rGO (265 nm) corresponded to the π–π* transition, indicating that some functional groups on the surface of GO were removed and the conjugated structure was maintained.45 The band gap (Eg) of rGO, Pg-NiFe2O4, Pg-NiFe2O4/rGO, and Tyr/Pg-NiFe2O4/rGO absorption peaks was calculated using Tauc plots using eqn (7), where Eg is the band gap in eV, hν (1240 eV) is the photon energy, α is the absorbance coefficient, n is constant (n = 1/2 direct, 2 indirect, allowed, and 3/2 direct, 3 indirect forbidden transitions, respectively) for a direct band gap n = 1/2, and K is the proportionality constant. The graph was constructed using (αhν)2vs. hν to find the value of the band gap on the x-axis. The band gap (Eg on the X-axis) of rGO, Pg-NiFe2O4, Pg-NiFe2O4/rGO, and Tyr/Pg-NiFe2O4/rGO was 4.22, 2.74, 2.65, and 4.06 eV, respectively (Fig. 2B). The Pg-NiFe2O4/rGO nanocomposite had a lower band gap than Pg-NiFe2O4, which gave a faster electronic transition.
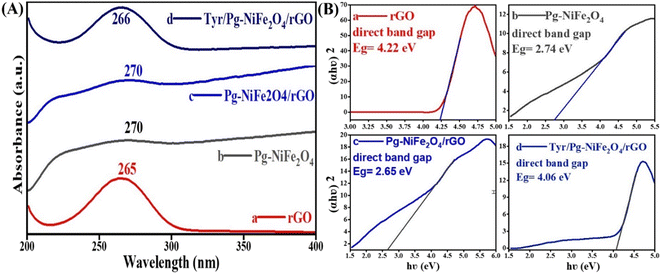 |
| | Fig. 2 (A) UV-visible absorbance spectra of (a) rGO, (b) Pg-NiFe2O4, (c) Pg-NiFe2O4/rGO, and (d) Tyr/Pg-NiFe2O4/rGO nanocomposites. (B) Band gap analysis by Tauc plots of (a) rGO, (b) Pg-NiFe2O4, (c) Pg-NiFe2O4/rGO, and (d) Tyr/Pg-NiFe2O4/rGO nanocomposites. | |
3.5 TEM, SEM, and EDS for elemental analysis
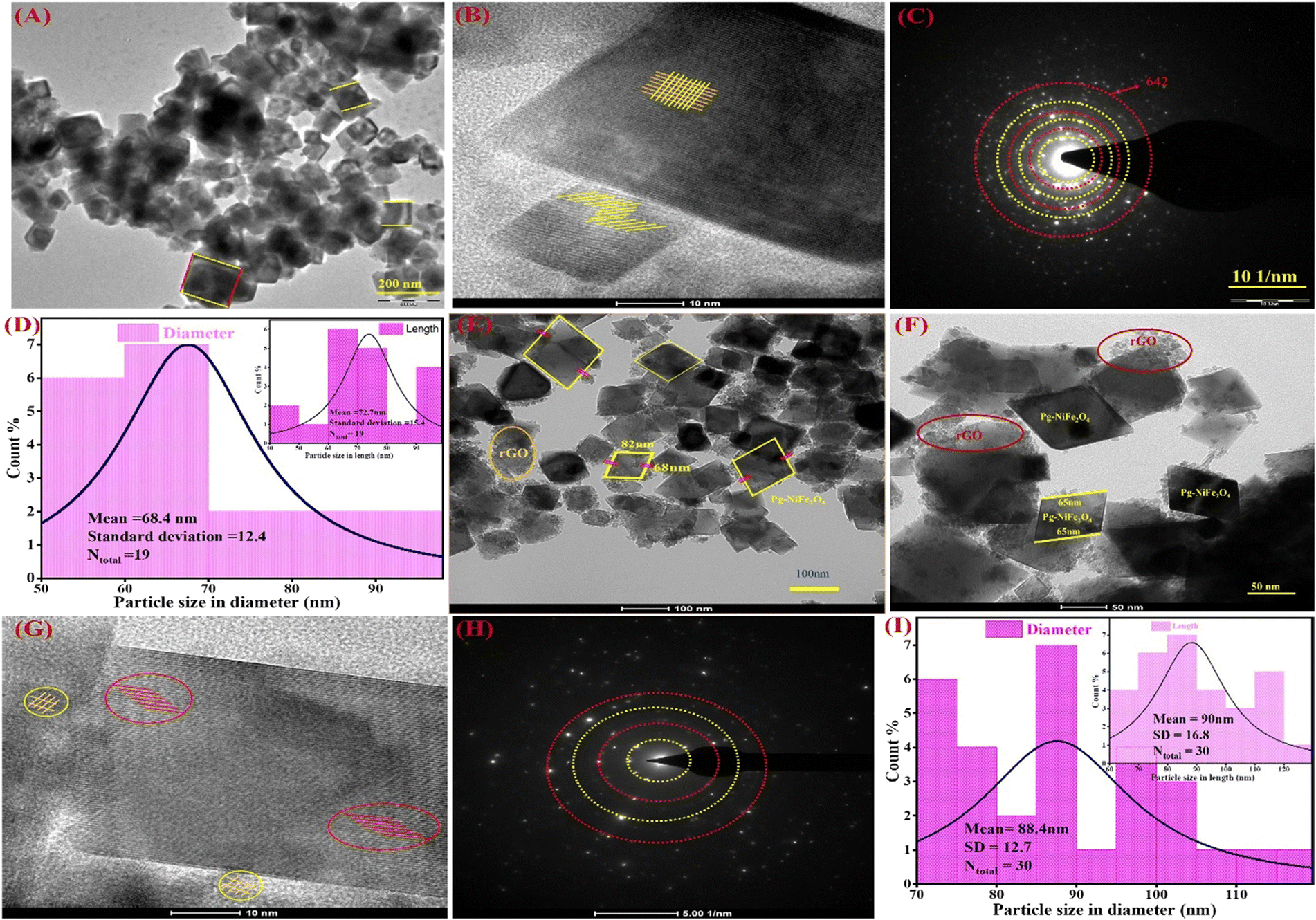
The internal surface morphology of the Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite was examined by TEM. The rhombus–parallelogram morphology of the calcinated NiFe2O4 and NiFe2O4/rGO nanocomposite is shown in Fig. 3. These parallelograms contained two opposite edges that were equal and parallel with the angle. The other two edges (at right angles to each other) were different. The high-resolution TEM (HR-TEM) image (Fig. 3B) showed lattice fringes in the Pg-NiFe2O4 phase distributed equally and also a selected area electron diffraction (SAED) pattern is shown in Fig. 3C. A particle-size histogram of Pg-NiFe2O4 with regard to diameter and length (Fig. 3D) was calculated to discover the ratio of diameter/length (Ntotal 30, 88.4 and 90 nm). TEM images of the NiFe2O4/rGO nanocomposite revealed an identical parallelogram morphology to that of NiFe2O4 (Fig. 3E and F). HR-TEM indicated the lattice fringes of Pg-NiFe2O4/rGO in two different phases of Ni and Fe metal (Fig. 3G). The SAED pattern (Fig. 3H) and particle size of Pg-NiFe2O4/rGO were calculated with regard to diameter and length (Fig. 3I), and were found to be 68.4 nm and 72.7 nm, respectively. Pg-NiFe2O4/rGO showed a uniform distribution of a parallelogram on rGO because Ni2+/Fe3+ interacted electrostatically with negatively charged rGO containing CO, C–O, and the OH group stimulated the uniform attachment of Ni2+/Fe3+ on the rGO surface. These homogeneous attachments enhanced the catalytic efficiency and inhibited the agglomeration of nanoparticles.46
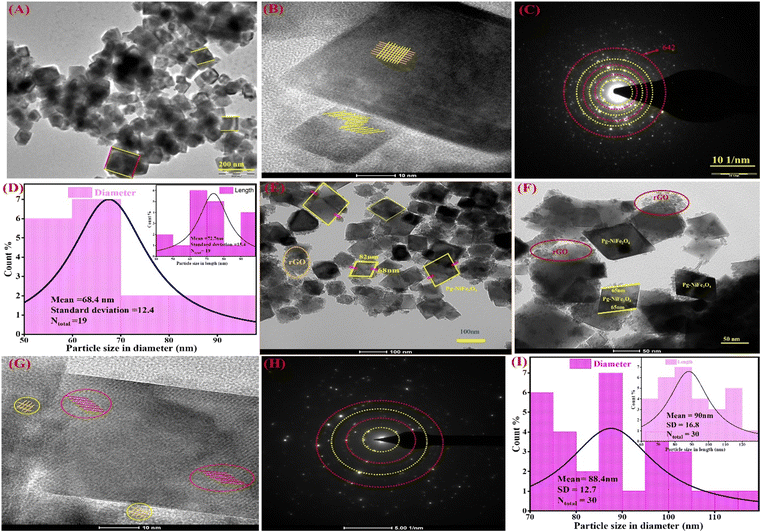 |
| | Fig. 3 TEM images of (A) Pg-NiFe2O4 and (B) HR-TEM images demonstrating fringes in a parallelogram. (C and D) SAED pattern and particle size (diameter and in-set is based on length) of Pg-NiFe2O4. (E and F) Parallelogram of NiFe2O4/rGO nanocomposite. (G) HR-TEM fringes of Pg-NiFe2O4/rGO, (H and I) SAED pattern and particle size of Pg-NiFe2O4/rGO with regard to diameter and in-set is based on length. | |
SEM was used for surface-morphology analysis of Pg-NiFe2O4 and Pg-NiFe2O4/rGO. These materials were evaluated at an acceleration voltage of 20 kV, and the source of the X-ray laser (at 3 nm) used for the secondary image was lanthanum hexaboride (LaB6). The surface of materials was coated with gold metal using a sputtering method to boost their electron emission, thereby resulting in the continuous flow of electrons and improving the quality of the image. Fig. 4A and B showed the unique parallelogram morphology in Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite to validate the result of TEM. Thus, the distribution of Pg-NiFe2O4 in the embedded surface of rGO provided a perfect interaction with a metal ion to enable uniform Pg distribution in the nanocomposite.
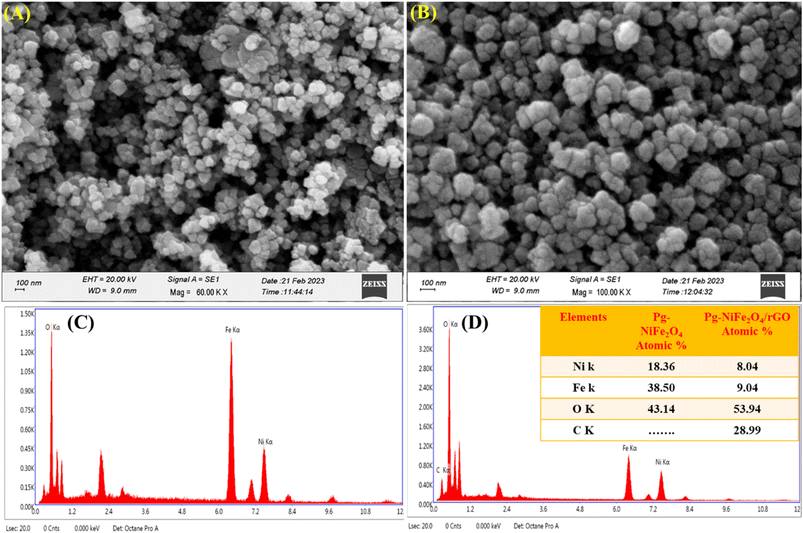 |
| | Fig. 4 SEM image of (A) Pg-NiFe2O4. (B) EDX image of Pg-NiFe2O4. (C) SEM image of Pg-NiFe2O4/rGO. (D) EDX image of Pg-NiFe2O4/rGO nanocomposite showing elemental composition (atomic%) in tabular form. | |
Detection of the elemental composition of Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite was done using EDX (Fig. 4C and D). Elemental composition is described in Table 1 with atomic percentages. Ni, Fe, O, and C elements were present in the Pg-NiFe2O4/rGO nanocomposite. The EDX spectrum exhibited an Ni/Fe atomic ratio of nearly 1:2, which indicated formation of the Pg-NiFe2O4 phase.
Table 1 Elemental composition of Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite
| Element |
Series |
Pg-NiFe2O4 |
Pg-NiFe2O4/rGO |
| Atomic% |
Atomic% |
| Fe (26) |
K-series |
38.50 |
9.04 |
| O (8) |
K-series |
43.14 |
53.94 |
| Ni (28) |
K-series |
18.36 |
8.04 |
| C (6) |
K-series |
— |
28.99 |
3.6 AFM
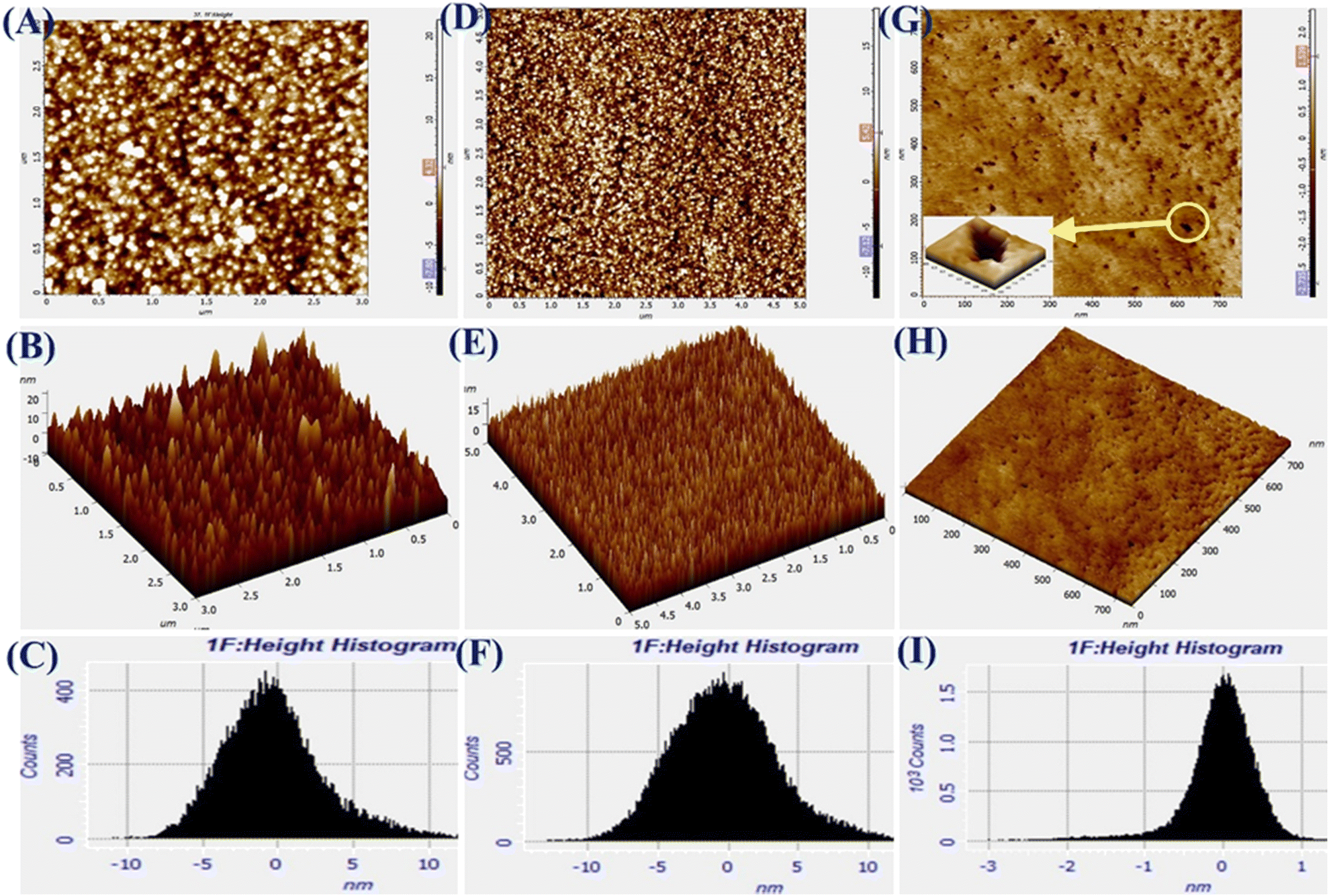
The AFM images of Pg-NiFe2O4, Pg-NiFe2O4/rGO, and Tyr/Pg-NiFe2O4/rGO bioelectrodes are shown in Fig. 5A–I. It reveals 2D and 3D images, and a height-distribution histogram of thin films for uniform and crack-free surfaces. Two important roughness parameters, average roughness (Ra) and root mean square roughness (Rq), were measured to explain the morphology of the thin film. Ra measures the height of average surface area roughness. Rq represents the standard deviation of surface height. Sq indicates the maximum area peak height (Table 2). After the immobilization of Tyr on the nanocomposite electrode, granules agglomerated due to a high charge density and particle–particle interaction. The corresponding Ra, Rq, and Sq values are shown in Table 2. The surface roughness decreased due to the adsorption of Tyr on the nanocomposite surface via strong interactions between the positive metal ion and oxygen atom of Tyr. The surface of Tyr/Pg-NiFe2O4/rGO has a vascular cavity that has an important role in the loading of biomolecules and is efficient during electroanalysis. The other two important parameters, skewness (Rsk) and kurtosis (Rku), were used to quantify surface morphological changes. The Rsk value was positive for Pg-NiFe2O4 and Pg-NiFe2O4/rGO, which indicated that peaks were predominant, with more surface peaks than valleys. A negative value of Rsk for Tyr/Pg-NiFe2O4/rGO indicated more valleys than peaks, and the surface was more planar (Table 2). The kurtosis (Rku) value was parallel to measurement of surface sharpness. We obtained an Rku value > 3 for Pg-NiFe2O4, Pg-NiFe2O4/rGO, and Tyr/Pg-NiFe2O4/rGO, which indicated more peaks than valleys on the surface47 (Table 2).
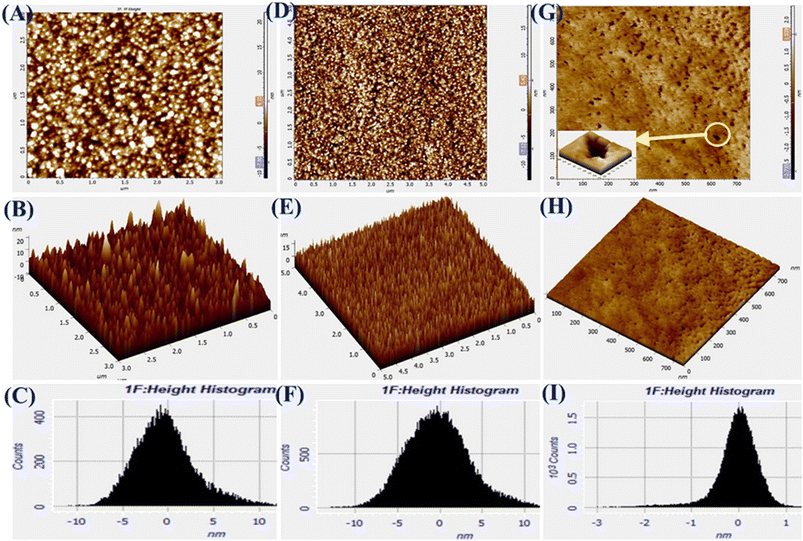 |
| | Fig. 5 A–C are AFM images denoting 2D, 3D, and the height histogram of Pg-NiFe2O4. D–F are AFM images denoting 2D, 3D, and the height histogram of Pg-NiFe2O4/rGO. G–I are AFM images denoting 2D, 3D, and the height histogram of Tyr/Pg-NiFe2O4/rGO. | |
Table 2 Different parameters in surface analysis using AFM
| Sample |
R
q (nm) |
R
a (nm) |
S
q (nm) |
R
ku
|
R
sk
|
| Pg-NiFe2O4 |
3.51 |
2.66 |
21.85 |
+4.68 |
+0.84 |
| Pg-NiFe2O4/rGO |
3.78 |
2.98 |
19.12 |
+3.28 |
+0.36 |
| Tyr/Pg-NiFe2O4/rGO |
0.43 |
0.31 |
2.41 |
+7.52 |
−1.30 |
3.7 XPS
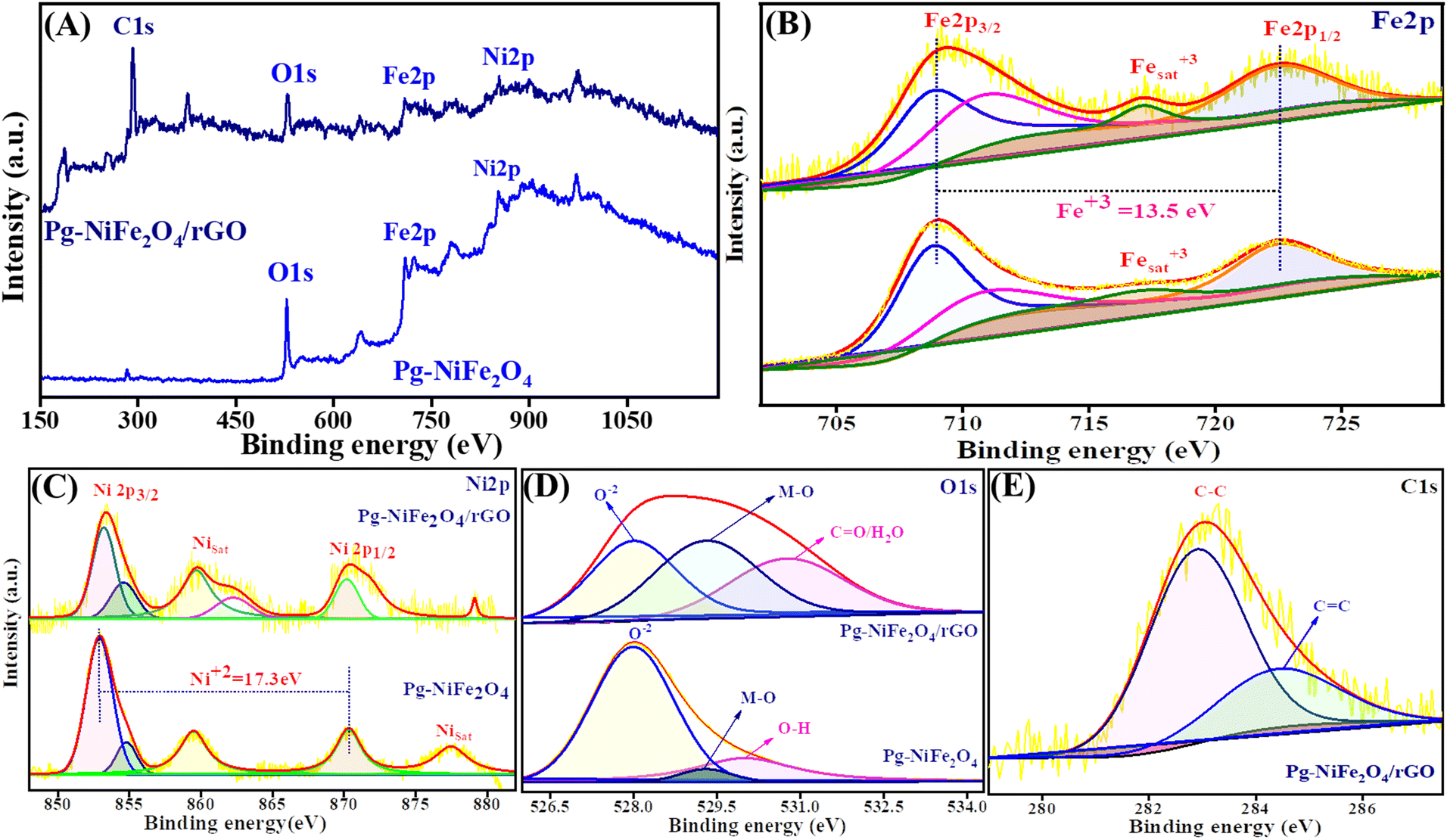
The oxidation state and composition of the Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite were investigated using XPS. Fig. 6A represents the survey spectra of the Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite to confirm the presence of Ni, Fe, O, and C elements (Fe 2p, Ni 2p, O 1s, C 1s). The presence of Fe in Pg-NiFe2O4 (Fig. 6B) in the Fe3+ oxidation state (13.5 eV difference between two energy states of Fe 2p) was indicated by peaks with high intensities at 709–711.3 eV (corresponding to Fe 2p3/2), 722.6 eV (corresponding to Fe 2p1/2), and a satellite peak at 717.1 eV.48,49 Furthermore, the two prominent peaks of Ni at 853.5–854.5 (Ni 2p3/2) and 870.3 (Ni 2p1/2) eV were equivalent to spin–orbit coupling, and other satellite peaks were observed at 859.6, 877.5, and 879.1 eV.50 These two peaks of Ni were confirmed to be the +2 oxidation state (17.3 eV) in cubic lattices (Fig. 6C). In addition, the strong metal–oxygen binding in the Pg-NiFe2O4 and Pg-nanocomposite caused the O 1s deconvoluted spectra to give peaks at 528 (O2−), 529.3 (metal–O) and 530.8 (O–H/CO) eV (Fig. 6D). The six O2− in octahedral voids and four O2− in tetrahedral voids were present in cubic spinel lattices. Fig. 6E, shows the deconvoluted C 1s peaks at 283 (C–C) and 284.5 (C–O) groups.48 The comparative interaction of Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite towards the binding energy elicited similar (±0.4 eV) results, which led to charge distribution at the interface. Thus, the XPS survey confirms the formation of Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite. The combined effect of these two components may be advantageous for achieving outstanding electrochemical activity.
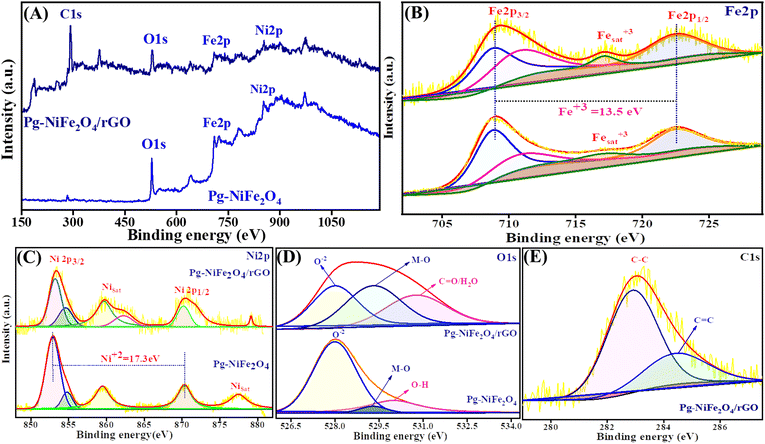 |
| | Fig. 6 XPS survey spectra of (A) Pg-NiFe2O4 and Pg-NiFe2O4/rGO. (B) Fe 2p deconvoluted spectra of XPS having different binding-energy states. (C) Ni 2p deconvoluted spectra of XPS having different binding-energy states. (D and E) O 1s and C 1s deconvoluted spectra of XPS having different binding-energy states. | |
3.8 BET analysis
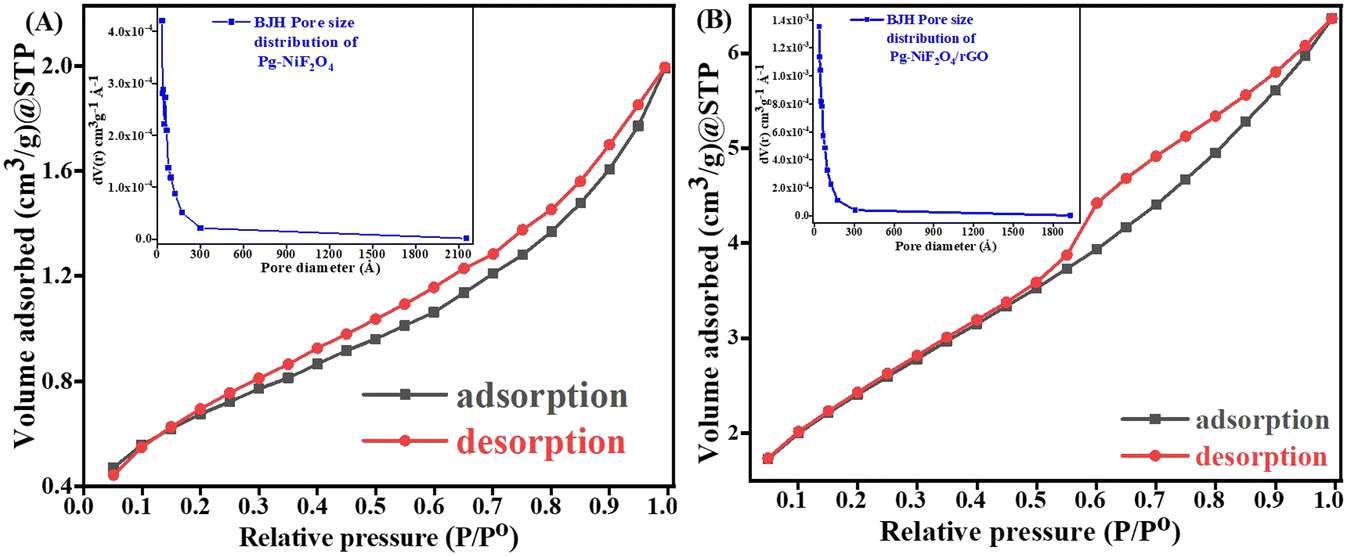
Fig. 7A and B represent the adsorption–desorption N2 isotherm curves of Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite for a specific surface area and pore-size distribution with pore volume. According to IUPAC, the characteristics of the adsorption isotherm of Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite exhibit type-IV hysteresis loop, which indicates the mesoporous nature of the material. The pore size of Pg-NiFe2O4 and Pg-NiFe2O4/rGO were calculated by the Barrett–Joyner–Halenda (BJH) method: diameter = 0–2300 Å. The value of the pore size we obtained was a radius of 17.04 and 24.45 Å, respectively. The specific surface area of Pg-NiFe2O4 and Pg-NiFe2O4/rGO was 16.35 m2 g−1 and 49.58 m2 g−1 according to the BET equation. The pore volume of Pg-NiFe2O4 and Pg-NiFe2O4/rGO was 0.016 and 0.043 cc g−1, respectively. The large surface area of the nanocomposite indicated higher loading of a biomolecule. A larger pore size, as seen in Pg-NiFe2O4/rGO (24.45 Å), enhanced the surface area available for biomolecule immobilization, thereby facilitating stronger binding of enzymes or biomolecules.51 These features lead to improved sensing capabilities and higher sensitivity due to more active sites for target–analyte interaction.
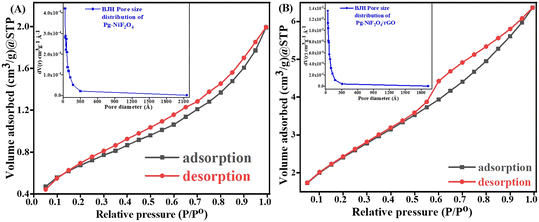 |
| | Fig. 7 A and B Specific surface area of Pg-NiFe2O4 and Pg-NiFe2O4/rGO based on BET calculation. Inset image shows the pore-size distribution. | |
4 Electrochemical analysis
4.1 Cyclic voltammetry (CV) and differential pulse voltammetry (DPV)
CV is one of the most important electrochemical methods used to detect neurochemicals, such as DA, serotonin, and epinephrine, undergoing redox reactions. A combination of a working electrode (Tyr/Pg-NiFe2O4/rGO/ITO), counter electrode (Pt), and reference electrode (Ag/AgCl in 3.5 M KCl) was used to detect these neurotransmitters. The reference electrode was used to measure and control the working potential. All electrochemical studies were carried out in the working-potential range (−0.5 to +1.5 V) and PBS (50 mM, 0.9% NaCl) with 5 mM [Fe2(CN)6]3−/4− used as a redox couple with a scan rate of 50 mV s−1. Fig. 8A is a cyclic voltammogram of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode. It showed the highest amperometric response (0.784 mA) compared with ITO (0.633 mA), Pg-NiFe2O4 (0.692 mA), and Pg-NiFe2O4/rGO (0.736 mA) due to a perfect electrostatic interaction in the bioelectrode and analyte (DA solution at 100 μM). After immobilization of the enzyme on the surface of the Pg-NiFe2O4/rGO/ITO electrode, there was a potential change in the surface charge and electrostatic interactions between the electrode and electrolyte. This could affect the electron-transfer kinetics and redox reactions of the electrode. If the enzyme immobilization increased the negative charge on the electrode surface, it could repel the negatively charged ions in the electrolytic solution and attract positively charged ions. This phenomenon could shift the electrode potential to a more positive value and more negative value in anodic and cathodic directions.52 A similar result was observed with DPV (Fig. 8B). The current response in DPV was 0.127 mA, 0.132 mA, 0.149 mA, and 0.183 mA for ITO, Pg-NiFe2O4/ITO, Pg-NiFe2O4/rGO/ITO, and Tyr/Pg-NiFe2O4/rGO/ITO bioelectrodes, respectively. In addition, Nyquist plots of the different fabricated electrodes were optimized by electrochemical impedance spectroscopy (EIS). The latter was carried out at frequency range of 100000 to 0.05 Hz, an AC amplitude of 10 mV, and point decade of 10. Fig. 8C shows the Nyquist plot between Z′ real value versus −Z′′ imaginary value to obtain a solution resistant (Rs) and charge transfer resistant (Rct) after applying equivalent circuit fitting on different electrodes. Rct values for ITO (3342 Ω), Pg-NiFe2O4/ITO (2625 Ω), Pg-NiFe2O4/rGO/ITO (2228 Ω), and Tyr/Pg-NiFe2O4/rGO/ITO (1974 Ω) bioelectrodes were obtained. We confirmed that the lowest Rct value (1974 Ω) in the fabricated electrode showed the highest electrical conductivity. These results were also validated by CV and DPV to show that the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was reliable for DA monitoring.
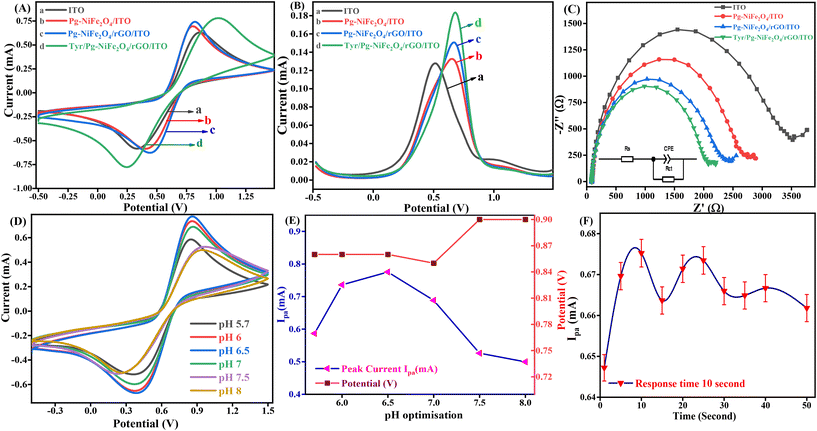 |
| | Fig. 8 Cyclic voltammogram of (A) ITO (a), Pg-NiFe2O4/ITO (b), Pg-NiFe2O4/rGO/ITO (c), and Tyr/Pg-NiFe2O4/rGO/ITO (d) electrodes. (B) DPV of ITO (a), Pg-NiFe2O4/ITO (b), Pg-NiFe2O4/rGO/ITO (c), and Tyr/Pg-NiFe2O4/rGO/ITO (d), (C) Nyquist plot (100000–0.05 Hz) of different fabricated electrodes. (D and E) Cyclic voltammograms showing optimisation at different pH values for the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode and optimisation with respect to Ipaversus pH versus potential (y-axis). (F) Response time of the bioelectrode with respect to DA. | |
4.2 pH optimization of Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode
An enzyme is a protein, which is affected by changes in pH. A very low or high pH diminishes the enzymatic activity due to changes in the shape and structure of the enzyme, and also affects the rate of the chemical reaction. The pH of a solution also affects the adsorption performance on the surface of an electrode, along with electrostatic interactions with biomolecules. An optimal pH is required for the maximum activity of an enzyme. The maximum amperometric response (0.775 mA) of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was obtained in pH range 5.7–8 (at pH 6.5) (Fig. 8D) in PBS after addition of DA solution (100 μM). Therefore, the optimum pH value of 6.5 was selected for amperometric detection of DA by the immobilized Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode.
4.3 Response time
The response time is an important parameter in biosensor development. We undertook a response-time study between 1 s and 50 s. The magnitude of the current increased for 1 s to 10 s, and then decreased up to 50 s (1.96%). The maximum amperometric response was obtained at 10 s (0.675 mA, 100%) (Fig. 8F). Therefore, a response time of 10 s was used for further electrochemical detection of DA.
4.4 Scan rate and kinetics
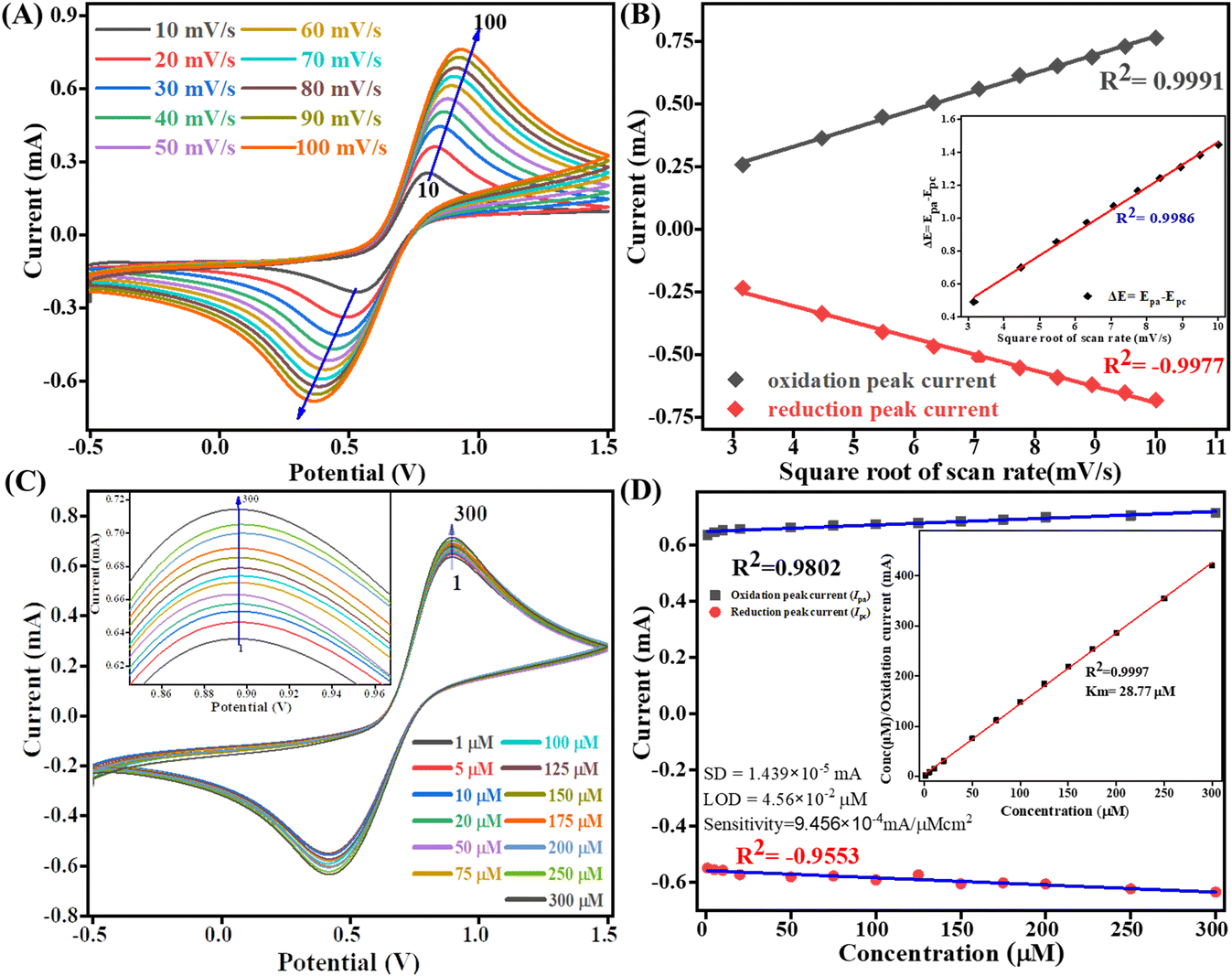
The kinetics study of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was done by varying the scan rate (10–100 mV s−1) during CV (Fig. 9A). The scan rate is an experimentally controlled process and depends on how fast the applied potential scans the interaction between the analyte and electrode. A faster scan rate decreases the size of the diffusion layer, so a higher current response is observed.53 In addition, the redox peak-current response of diffused species varies linearly with the square root of the scan rate (ν1/2), and shifting of the potential occurred (Fig. 9B). Oxidation and reduction peak potentials shift towards positive and negative values upon increasing the scan rate to confirm the electrochemical quasi-reversible redox process. Fig. 9B represents the peak shift of potential and also gives a linear relationship between peak-to-peak separation (ΔEp = Epa − Epc) and ν1/2. The value of the linear regression coefficient (R2) we obtained was 0.9987, which indicated improved electro-oxidation behavior. The diffusion coefficient (D) of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was investigated using the Randles–Sevcik (eqn (8)).54 The surface concentration of the Tyr/β-MnO2/RGO/ITO bioelectrode at 6.5 pH in PBS was calculated using eqn (9) based on the Brown–Anson model:55| | | Ip = (2.69 × 105)n3/2AD1/2ν1/2C | (8) |
where Ip is the peak current of the bioelectrode (Ipa and Ipc, anodic and cathodic peak current in V), n is electron stoichiometry or number of electrons involves (1), A is the bioelectrode surface area (0.25 cm2), D is the diffusion coefficient (cm2 s−1), ν is the scan rate in V (50 mV s−1 = 0.05 V s−1), C is the concentration of ferric/ferro [Fe2(CN)6]3−/4− in mol cm3 (5 mM = 0.000005 mol cm3), F is the Faraday constant (96500 C mol−1), I* is the electrode surface concentration (mol cm−2), R is the gas constant (8.314 J mol−1 K−1), and T (300 K) is the absolute room temperature. Thus, the calculated D value was 0.4674 cm2 s−1 and the I* of ferric/ferro [Fe(CN)6]3−/4− on the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode surface was 3.147 × 10−7 mol cm−2. The charge transfer rate constant (Ks) of the bioelectrode changed due to surface variation, and the value was calculated using eqn (10):56where m is the separation between peak-to-peak in V, n (1) is number of electron transfers, F is the Faraday constant (96500 C mol−1), R is the gas constant (8.314 J mol−1 K−1), ν (50 mV s−1) is the scan rate in V, and T (300 K) is the absolute room temperature. Thus, the Ks value of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was estimated to be 2.075 s−1. This increased Ks value on the surface of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode confirmed that the surface physiochemistry and electronic structure of the Pg-NiFe2O4/rGO/ITO nanocomposite improved the electron transfer rate arising from efficient catalytic activity of the nanocomposite.
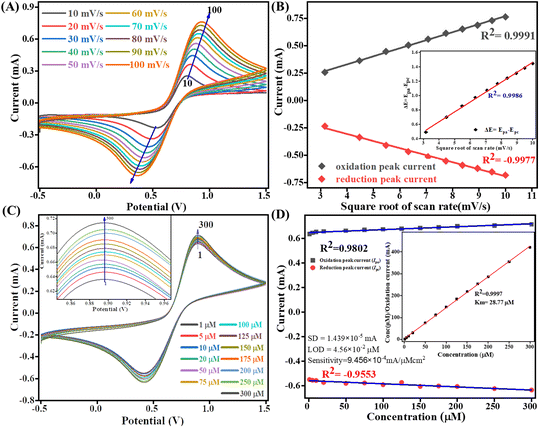 |
| | Fig. 9 Scan-rate analysis of (A) Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode at 10–100 mV s−1. (B) Linear-fit calibration curve of the square root of the scan rate (ν1/2) vs. peak current of the redox reaction. The inset image reveals the linear-fit curve between ν1/2 and ΔE (quasi-reversible nature of the bioelectrode). (C) Electro-oxidation of DA at a linear range of 1–300 μM. (D) Concentration linear-fit curve of DA at 1–300 μM, and the inset image shows determination of the Michaelis–Menten constant (Km). | |
4.5 Electro-oxidation of DA and the Michaelis–Menten constant
The electro-oxidation of the fabricated Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was investigated using a DA concentration of 1–300 μM in PBS (50 mM), 0.9% sodium chloride, at pH 6.5, containing [Fe(CN)6]3−/4− solution as a mediator (5 mM), with a scan rate of 50 mV s−1 using CV. The electro-oxidation mechanism of the DA biosensor is represented in Scheme 1. As shown in Fig. 9C, the oxidation potential of O-dopaquinone was 0.89 V. The oxidation peak current increased linearly with successive addition of DA due to the availability of high electron transfer in the enzymatic reaction. DA is converted to O-dopaquinone (oxidized form) by redox processes involving the enzyme Tyr, which has a vital role in stimulating and controlling this chemical reaction. Tyr uses dioxygen (O2) to complete the oxidation of the phenol group found in DA, resulting in O-dopaquinone being produced, and hydrogens removed from DA associate with oxygen to form water. O-Dopaquinone is formed when DA loses electrons and oxygen receives them in this reaction. The switching of electrons in this reaction is important in the redox process, which is a basic component of many biological reactions. The transfer of electrons from DA to O2 is involved in this oxidation reaction. Tyr functions as a catalyst, accelerating this transformation without being changed or consumed. Tyr remains intact and ready to participate in further reactions. Tyr can stimulate the oxidation of DA molecules regularly without becoming depleted or sustaining significant alterations, thereby maintaining neurotransmitter equilibrium in the body.
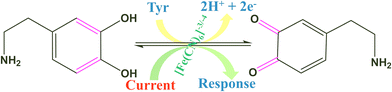 |
| | Scheme 1 Electrochemical oxidation of DA on the surface of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode. | |
A calibration graph between the electrochemical current vs. DA concentration was linear with an increasing concentration of DA up to 1–300 μM with R2 = 0.9802 at an oxidation potential of 0.89 V and R2 = 0.9553 at a reduction potential of −0.41 V (Fig. 9D). The sensitivity calculation was made using the equation limit of detection (LOD) = 3 × SD/sensitivity, where the standard deviation (SD) can be obtained from the calibration curve after doing a linear fit in the calibration curve: SD = 1.4390 × 10−5 mA. We obtained a high value of sensitivity 9.4569 × 10−4 mA μM−1 cm−2 (sensitivity = slope/area) due to a large surface area. The final obtained LOD was 4.564 × 10−2 μM based on a signal-to-noise ratio of 3. Therefore, this modified Tyr/Pg-NiFe2O4/rGO/ITO surface was used as a specific and efficient biosensor for DA detection.
The mechanism of action of the enzyme–substrate of a composite can be explained by the Michaelis–Menten constant (Km) using a Hanes–Woolf plot. The kinetic parameter Km was 28.77 μM and linear regression coefficient (R2) was 0.999, indicating that a low value of Km was responsible for the excellent affinity between the enzyme and Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode. A plot of the substrate concentration versus the substrate concentration/current is shown in the inset of Fig. 9D. Thus, a lower value of Km led to favorable enzyme activity and higher loading of the analyte on the surface of the modified Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode. The Km value is dependent upon the medium, conformational changes in enzyme structure, immobilization method, and the sensitivity of enzyme kinetics towards the environment.57 A comparative study of DA biosensors using different materials and sensing parameters is summarized in Table 3, and some merit discussion. Ali et al.58 developed barium titanate “nanocubes” as dual electrochemical sensors for DA monitoring on a glassy carbon electrode (GCE) in a linear range of 10 to 100 μM: the LOD was 0.35 μM. Lu et al.59 utilized a CoO nanoparticles/N-doped carbon sheets/reduced graphene oxide composites (CoO/N–CS–rGO) fabricated electrode on GCE to develop a hybrid nanozyme sensor for DA detection in human serum samples in the range 0.5 to 110 μM: the LOD was 0.15 μM. Liu et al.60 developed an electrochemical sensor based on the Fe2O3–NG/GCE electrode to optimize the DA concentration in the range 0.5 to 10 μM: the LOD was 0.080 μM in the standard sample and sample of human serum. Those data can be compared with our results with respect to electrochemical parameters such as linear range, LOD, and response time. Other developed sensors are compared and results mention in Table 3. Our synthesized Pg-NiFe2O4/rGO nanocomposite appeared to be an excellent material for efficient electrochemical sensing of DA.
Table 3 Comparison of the electrochemical estimation of DA with regard to linear range, limit of detection, sensitivity, response time, and Km, on fabricated electrode materials
| Modified electrode |
Method of sensing |
Range of detection (μM) |
LOD (μM) |
Sensitivity (mA μM−1 cm−2) |
Response time (second) |
Michaelis–Menten constant, Km (μM) |
Ref. |
| Tyr/Au–La2O3/ITO |
CV |
2–100 |
0.258 |
2.4806 × 10−4 |
30 |
5.649 |
61
|
| BaTiO3/GCE |
DPV |
10–100 |
0.35 |
— |
— |
— |
58
|
| N–rGO |
Electrochemical |
1–60 |
0.1 |
— |
— |
— |
62
|
| g-C3N4/GO/GCE |
Electrochemical |
0.03–30 |
0.0054 |
— |
— |
— |
63
|
| MnO2/NG/GCE |
Electrochemical |
0.1–100 |
0.039 |
— |
— |
— |
64
|
| Co–O/NCS–rGO |
DPV, CV |
0.5–110 |
0.15 |
— |
— |
— |
59
|
| Tyr/NiO/ITO |
CV |
2–100 |
1.04 |
6 × 10−5 |
45 |
— |
65
|
| PANI/TRGO-400 |
Electrochemical |
0.5–30 |
0.70 |
3 × 10−3 |
— |
— |
66
|
| rGO-500/SPCE |
CV |
0.5–20 |
1.11 |
— |
— |
— |
67
|
| Fe2O3–NG/GCE |
Voltammetric |
0.5–10 |
0.080 |
— |
— |
— |
60
|
| ZnO/ERGO/GCE |
LSV |
0.01–6 |
0.0036 |
0.028 |
— |
— |
68
|
| 6–80 |
| Tyr/Pg-NiFe2O4/rGO/ITO |
CV |
1–300 |
0.045 |
9.456 × 10−4 |
10 |
28.77 |
This work |
4.6 Shelf-life stability and reusability of the bioelectrode
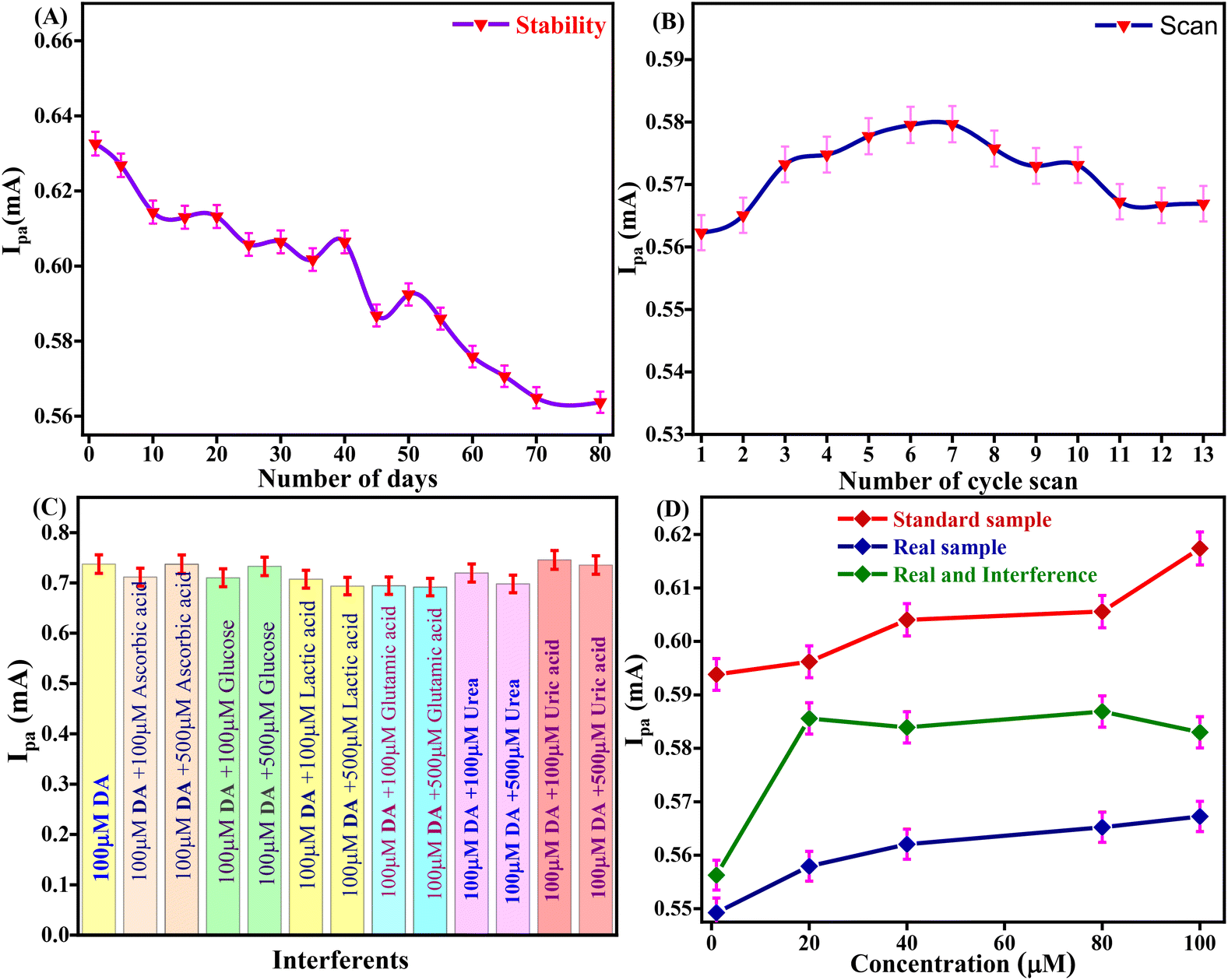
The storage stability of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode to retain enzyme activity was investigated from 1 to 80 days in DA (100 μM) and PBS with regular intervals of 5 days (Fig. 10A). After 40 days, the activity of the bioelectrode was up to 95.8% (RSD = 4.12%). After 80 days, the activity of the bioelectrode was 89.08% (RSD = 10.925%). The self-life stability was good up to 40 days with significant activity of the bioelectrode due to interactions between Pg-NiFe2O4/rGO and Tyr on the surface, whereas the ITO substrate ensured good adhesion and electrical properties. Thus, addition of Pg-NiFe2O4/rGO led to strong binding to the enzyme and the removal of immobilized enzyme was retained.57
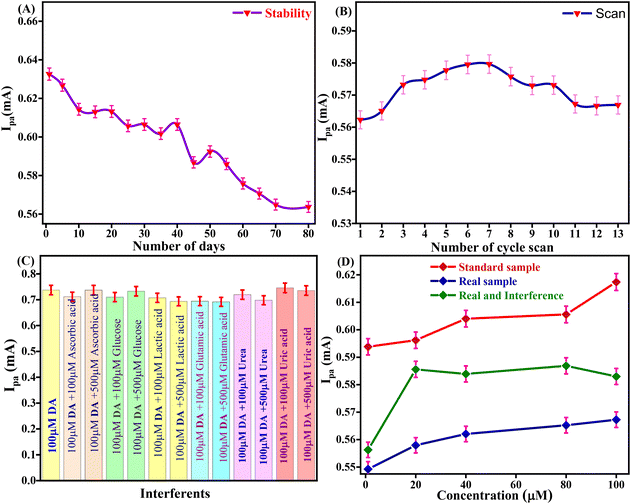 |
| | Fig. 10 (A and B) Stability and reusability of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode. (C) Interference study with DA. (D) Standard sample, real spiked sample, and real sample with interference being due to analytes of different concentration. | |
The Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode was used for repetitive measurement of CV signal response to ascertain the reusability in DA (100 μM) and PBS. The current response was measured for 13 scans. The current response increased from 1 to 6 scans, was constant for 6 to 7 scans (0.579 mA), and then decreased at 13 scans (0.566 mA) with RSD = 2.25% (Fig. 10B). Thus, the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode showed excellent reusability and retained enzyme activity towards DA sensing.
4.7 Interference and spiking a sample of fetal bovine serum
The selectivity of the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode-based biosensor was examined by its amperometric response in the presence of communal interferents such as ascorbic acid, glucose, lactic acid, glutamic acid, and urea using CV in PBS (50 mM) at pH 6.5 and 0.9% NaCl solution containing [Fe(CN)6]3−/4− (5 mM) as a mediator with a scan rate of 50 mV s−1 in the potential range +1.5 to −0.5 V. The changes in electrochemical current response in standard DA solution (100 μM) were measured in PBS with common interferents (Table 4). A bar graph (Fig. 10C) showed a slight decrement in Ipa (RSD < 6%) with the standard sample in the presence of interferents. Good agreement was also seen with the standard sample (addition of an error bar of 0.5% in the line graph). Thus, the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode had no major interference effect with other analytes present in a standard sample, which was acceptable (RSD < 6%) for fabricated biosensors.
Table 4 Effect of different interferents on the oxidation peak current and relative standard deviation (% RSD)
| Analyte |
Concentration (μM) |
I
pa (mA) |
% RSD |
| Dopamine (DA) |
100 |
0.73735 |
0 |
| DA + ascorbic acid (AA) |
100 + 100 |
0.71144 |
3.52 |
| DA + ascorbic acid (AA) |
100 + 500 |
0.73706 |
0.04 |
| DA + glucose (Glu) |
100 + 100 |
0.71016 |
3.69 |
| DA + glucose (Glu) |
100 + 500 |
0.73292 |
0.61 |
| DA + lactic acid (LA) |
100 + 100 |
0.70744 |
4.06 |
| DA + lactic acid (LA) |
100 + 500 |
0.69366 |
5.93 |
| DA + (+) glutamic acid (Gla) |
100 + 100 |
0.69435 |
5.84 |
| DA + (+) glutamic acid (Gla) |
100 + 500 |
0.69172 |
6.19 |
| DA + urea (U) |
100 + 100 |
0.71975 |
2.39 |
| DA + urea (U) |
100 + 500 |
0.69802 |
5.31 |
| DA + uric acid (UA) |
100 + 100 |
0.7457 |
1 |
| DA + uric acid (UA) |
100 + 500 |
0.7353 |
0.24 |
A spiked real sample was also investigated with the standard sample (Fig. 10D). A fetal bovine serum solution of DA (diluted 15 times with PBS at pH 6.5) was spiked with DA using the standard addition method and used for analyses. DA (1, 20, 40, 80, and 100 μM) was used to record the current response in standard, spiked serum, and interference samples. Table 5 shows that a real spiked sample with and without interferences achieved a recovery rate of 93%. However, the Tyr/Pg-NiFe2O4/rGO/ITO bioelectrode showed good agreement with the real spiked sample, and can be utilized for effective DA sensing in samples of blood and serum.
Table 5 Detection of the DA concentration in a sample of fetal bovine serum and serum with interferents
| Standard concentration of DA (μM) |
Determination of concentration of DA in serum samples (μM) |
Recovery (%) |
Determination of concentration of DA in serum samples with interferents (μM) |
Recovery (%) |
| 1 |
0.924 |
92.4 |
0.93 |
93.60 |
| 20 |
18.71 |
93.5 |
19.64 |
98.22 |
| 40 |
37.22 |
93 |
38.67 |
96.67 |
| 80 |
74.67 |
93.3 |
77.53 |
96.92 |
| 100 |
91.89 |
91.8 |
94.42 |
94.42 |
5 Conclusions
A low-temperature, hydrothermally synthesized Pg-NiFe2O4 and Pg-NiFe2O4/rGO nanocomposite by an in situ approach was obtained. That demonstrated unique Pg morphology, which was confirmed by HR-TEM. XRD, Raman spectroscopy, XPS, BET calculations, FTIR spectroscopy, and UV-visible spectroscopy. Further, the electrochemical analysis of a Pg morphology-based electrode was performed by CV and DPV, from this the kinetics of the material were evaluated by scan-rate analysis to calculate the surface concentration, diffusion coefficient, charge transfer rate constant, and quasi-reversible nature of the bioelectrode. While exploring the sensing performacne of DA from the fabricated bioelectrode the LOD of 4.56 × 10−2 μM and Km of 28.77 μM in the linear detection range (1–300 μM) of DA using the fabricated bioelectrode was obtained. This reveals that the developed sensor covered a wide range of detection and showed outstanding sensitivity towards DA in a spiked real sample (bovine serum albumin) with marginal changes in current response due to the presence of interferents. In addition, the developed biosensor showed marvelous recovery of ≥93% for real-sample analysis and was stable up to 40 days, underlining its robustness for practical applications. Thus, these obtained results from electrochemical studies demonstrates that the unique Pg-NiFe2O4/rGO nanocomposite material is an excellent candidate for electro-oxidation of DA and it can also be an excellent biosensing material for the detection of other analytes, that in turn proves that the prospects of this work are highly promising. Hence, the exceptional performance, broad linear range, and stability of this biosensor make it a strong candidate for integration into sustainable biosensing devices. By harnessing the capabilities of this unique material, future research can advance the development of reliable, cost-effective, and eco-friendly biosensors, contributing to improved healthcare, environmental monitoring, and analytical chemistry practices. This work sets the stage for innovative biosensing technologies with broader implications in various fields.
Author contributions
R. V.: data curation, investigation, visualization, and writing (original draft). K. R. B. S.: data curation, validation, visualization, and writing (review and editing of the original draft). R. V.: data curation, validation, visualization, and writing (original draft). J. S.: conceptualization, validation, project administration, supervision, and writing (review and editing of the original draft).
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported herein.
Acknowledgements
This work received no specific grant from public, commercial, or not-for-profit funding agencies. R. V. is thankful to his affiliated institution for providing constant financial support. K. R. B. S. is grateful to BHU (IoE grant project) for financial support throughout this work. R. V. acknowledges UGC New Delhi for giving a Dr. D. S. Kothari Postdoctoral Fellowship (F.4-2/2006 (BSR)/CH/18-19/0302). J. S. acknowledges BHU for providing a seed grant under MoE Govt. India, Institute of Eminence (Dev. Scheme No. 6031).
References
- Y. Wang, Y. Zhou, M. Han, Y. Xi, H. You, X. Hao, Z. Li, J. Zhou, D. Song, D. Wang and F. Gao, Small, 2019, 15, 1805435 CrossRef.
- B. Yan, W. Zhang, X. Qin, Y. Choi, G. Diao, X. Jin and Y. Piao, Chem. Eng. J., 2020, 400, 125895 CrossRef CAS.
- L. Sun, R. Zhang, Z. Wang, L. Ju, E. Cao and Y. Zhang, J. Magn. Magn. Mater., 2017, 421, 65–70 CrossRef CAS.
- K. Kombaiah, J. J. Vijaya, L. J. Kennedy and K. Kaviyarasu, Mater. Chem. Phys., 2019, 221, 11–28 CrossRef CAS.
- V. S. Kumbhar, A. D. Jagadale, N. M. Shinde and C. D. Lokhande, Appl. Surf. Sci., 2012, 259, 39–43 CrossRef CAS.
- P. Devi, C. Sharma, P. Kumar, M. Kumar, B. K. S. Bansod, M. K. Nayak and M. L. Singla, J. Hazard. Mater., 2017, 322, 85–94 CrossRef CAS.
- Y. Fang, S. Lin, F. Yang, J. Situ, S. Lin and Y. Luo, BioMed Res. Int., 2020, 2020, 1–12 Search PubMed.
- S. Li, N. Yang, L. Liao, Y. Luo, S. Wang, F. Cao, W. Zhou, D. Huang and H. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 37038–37045 CrossRef CAS.
- R. Devi, S. Gogoi, H. S. Dutta, M. Bordoloi, S. K. Sanghi and R. Khan, Nanoscale Adv., 2020, 2, 239–248 RSC.
- P. Salarizadeh, M. B. Askari, M. Seifi, S. M. Rozati and S. S. Eisazadeh, Mater. Sci. Semicond. Process., 2020, 114, 105078 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- H.-K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M.-H. Park, K. H. An, I. J. Kim, C.-W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- M. Carbone, L. Gorton and R. Antiochia, Electroanalysis, 2015, 27, 16–31 CrossRef CAS.
- Y. Guo, B. Wu, H. Liu, Y. Ma, Y. Yang, J. Zheng, G. Yu and Y. Liu, Adv. Mater., 2011, 23, 4626–4630 CrossRef CAS PubMed.
- G. Jiang, Z. Lin, C. Chen, L. Zhu, Q. Chang, N. Wang, W. Wei and H. Tang, Carbon, 2011, 49, 2693–2701 CrossRef CAS.
- R. Li, Y. Liu, L. Cheng, C. Yang and J. Zhang, Anal. Chem., 2014, 86, 9372–9375 CrossRef CAS PubMed.
- R. Sivasubramanian and P. Biji, Mater. Sci. Eng., B, 2016, 210, 10–18 CrossRef CAS.
- J. Liu, J. Tang and J. J. Gooding, J. Mater. Chem., 2012, 22, 12435 RSC.
- Z. Yang, Q. Lan, J. Li, J. Wu, Y. Tang and X. Hu, Biosens. Bioelectron., 2017, 89, 312–318 CrossRef CAS PubMed.
- Y. Chen, L. Chen, H. Bai and L. Li, J. Mater. Chem. A, 2013, 1, 1992–2001 RSC.
- A. T. E. Vilian, S. An, S. R. Choe, C. H. Kwak, Y. S. Huh, J. Lee and Y.-K. Han, Biosens. Bioelectron., 2016, 86, 122–128 CrossRef CAS PubMed.
- J. Jiang and X. Du, Nanoscale, 2014, 6, 11303–11309 RSC.
- T. Pradhan, H. S. Jung, J. H. Jang, T. W. Kim, C. Kang and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4684–4713 RSC.
- I. M. Taylor, E. M. Robbins, K. A. Catt, P. A. Cody, C. L. Happe and X. T. Cui, Biosens. Bioelectron., 2017, 89, 400–410 CrossRef CAS PubMed.
- L. Ma, T. Zhao, P. Zhang, M. Liu, H. Shi and W. Kang, Anal. Biochem., 2020, 593, 113594 CrossRef.
- D. Rithesh Raj, S. Prasanth, T. V. Vineeshkumar and C. Sudarsanakumar, Sens. Actuators, B, 2016, 224, 600–606 CrossRef CAS.
- F. Mamashli, A. A. Meratan, A. Ghasemi, N. Obeidi, B. Salmani, D. Atarod, M. Pirhaghi, F. Moosavi-Movahedi, M. Mohammad-Zaheri, M. B. Shahsavani, Z. Habibi-Kelishomi, B. Goliaei, M. Gholami and A. A. Saboury, ACS Chem. Neurosci., 2023, 14(5), 851–863 CrossRef CAS.
- G. Li, J. Wu, X. Qi, X. Wan, Y. Liu, Y. Chen and L. Xu, Mater. Today Chem., 2022, 26, 101043 CrossRef CAS.
- G. Li, X. Qi, G. Zhang, S. Wang, K. Li, J. Wu, X. Wan, Y. Liu and Q. Li, Microchem. J., 2022, 179, 107515 CrossRef CAS.
- G. Li, X. Wan, Y. Xia, D. Tuo, X. Qi, T. Wang, M. Mehmandoust, N. Erk, Q. He and Q. Li, ACS Appl. Nano Mater., 2023, 6, 17040–17052 CrossRef CAS.
- G. Li, X. Qi, J. Wu, L. Xu, X. Wan, Y. Liu, Y. Chen and Q. Li, J. Hazard. Mater., 2022, 436, 129107 CrossRef CAS.
- S. Alwarappan, G. Liu and C.-Z. Li, Nanomedicine, 2010, 6, 52–57 CrossRef CAS.
- A. Afkhami, H. Khoshsafar, H. Bagheri and T. Madrakian, Sens. Actuators, B, 2014, 203, 909–918 CrossRef CAS.
- A. A. Ensafi, B. Arashpour, B. Rezaei and A. R. Allafchian, Mater. Sci. Eng., C, 2014, 39, 78–85 CrossRef CAS PubMed.
- A. A. Ensafi, F. Saeid, B. Rezaei and A. R. Allafchian, Anal. Methods, 2014, 6, 6885–6892 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- S. Bae, S. W. Lee and Y. Takemura, Appl. Phys. Lett., 2006, 89, 252503 CrossRef.
- M. Wahadoszamen, A. Rahaman, N. M. R. Hoque, A. I. Talukder, K. M. Abedin and A. F. M. Y. Haider, J. Spectrosc., 2015, 2015, 1–8 CrossRef.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- H. Zhang and P. X. Feng, Carbon, 2010, 48, 359–364 CrossRef CAS.
- K. Maaz, A. Mumtaz, S. K. Hasanain and M. F. Bertino, J. Magn. Magn. Mater., 2010, 322, 2199–2202 CrossRef CAS.
- M. Srivastava, A. K. Ojha, S. Chaubey and A. Materny, J. Alloys Compd., 2009, 481, 515–519 CrossRef CAS.
- B. Mu, W. Zhang, S. Shao and A. Wang, Phys. Chem. Chem. Phys., 2014, 16, 7872 RSC.
- C. Zhu, S. Guo, Y. Fang and S. Dong, ACS Nano, 2010, 4, 2429–2437 CrossRef CAS PubMed.
- Q. Qiu, Y. Chen, J. Xue, J. Zhu, Y. Fu, G. He and H. Chen, Ceram. Int., 2017, 43, 2226–2232 CrossRef CAS.
- Z. Bazhan, F. E. Ghodsi and J. Mazloom, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 551 CrossRef.
- Z. Jiang, K. Chen, Y. Zhang, Y. Wang, F. Wang, G. Zhang and D. D. Dionysiou, Sep. Purif. Technol., 2020, 236, 116272 CrossRef CAS.
- T. Ge, Z. Jiang, L. Shen, J. Li, Z. Lu, Y. Zhang and F. Wang, Sep. Purif. Technol., 2021, 263, 118401 CrossRef CAS.
- Z. Chang, T. Li, G. Li and K. Wang, J. Mater. Sci.: Mater. Electron., 2019, 30, 600–608 CrossRef CAS.
- Y. Zhang, W. Cao, Y. Cai, J. Shu and M. Cao, Inorg. Chem. Front., 2019, 6, 961–968 RSC.
- S. Tajik, A. Lohrasbi-Nejad, P. Mohammadzadeh Jahani, M. B. Askari and H. Beitollahi, J. Mater. Sci.: Mater. Electron., 2022, 33, 2020–2030 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- J. Singh, A. Roychoudhury, M. Srivastava, V. Chaudhary, R. Prasanna, D. W. Lee, S. H. Lee and B. D. Malhotra, J. Phys. Chem. C, 2013, 117, 8491–8502 CrossRef CAS.
- J. Singh, M. Srivastava, A. Roychoudhury, D. W. Lee, S. H. Lee and B. D. Malhotra, J. Phys. Chem. B, 2013, 117, 141–152 CrossRef CAS.
- P. Malik, M. Srivastava, R. Verma, M. Kumar, D. Kumar and J. Singh, Mater. Sci. Eng., C, 2016, 58, 432–441 CrossRef CAS.
- J. Singh, M. Srivastava, P. Kalita and B. D. Malhotra, Process Biochem., 2012, 47, 2189–2198 CrossRef CAS.
- M. Ali, S. Sharma, R. Singh, K. Sharma, S. Majhi, D. Guin and C. S. P. Tripathi, J. Electrochem. Soc., 2022, 169, 067512 CrossRef CAS.
- N. Lu, Y. Liu, X. Yan, Z. Xu, Y. Xing, Y. Song, P. Zhao, M. Liu, Y. Gu, Z. Zhang and S. Zhai, ACS Appl. Nano Mater., 2022, 5, 11361–11370 CrossRef CAS.
- H. Liu, R. Xiong, P. Zhong, G. Li, J. Liu, J. Wu, Y. Liu and Q. He, New J. Chem., 2020, 44, 20797–20805 RSC.
- A. Srivastava, G. Mishra, J. Singh and M. D. Pandey, Mater. Lett., 2022, 308, 131231 CrossRef CAS.
- H. Zhang and S. Liu, J. Alloys Compd., 2020, 842, 155873 CrossRef CAS.
- Y. Xia, G. Li, Y. Zhu, Q. He and C. Hu, Microchem. J., 2023, 190, 108726 CrossRef CAS.
- Q. Li, Y. Xia, X. Wan, S. Yang, Z. Cai, Y. Ye and G. Li, Mater. Sci. Eng., C, 2020, 109, 110615 CrossRef CAS.
- A. Roychoudhury, S. Basu and S. K. Jha, Biosens. Bioelectron., 2016, 84, 72–81 CrossRef CAS PubMed.
- D. Minta, Z. González, S. Melendi-Espina and G. Gryglewicz, Surf. Interfaces, 2022, 28, 101606 CrossRef CAS.
- N. Gupta, G. Kaur, V. Sharma, R. Nagraik and M. Shandilya, J. Electroanal. Chem., 2022, 904, 115904 CrossRef CAS.
- F. Li, B. Ni, Y. Zheng, Y. Huang and G. Li, Surf. Interfaces, 2021, 26, 101375 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kshitij RB
Singh
a,
Kshitij RB
Singh