
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A thiomolybdate cluster for visible-light-driven hydrogen evolution: comparison of homogeneous and heterogeneous approaches†
Samar
Batool
 a,
Jasmin S.
Schubert
a,
Pablo
Ayala
a,
Hikaru
Saito
b,
Maria J.
Sampaio
cd,
Eliana S.
Da Silva‡
cd,
Cláudia G.
Silva
cd,
Joaquim L.
Faria
cd,
Dominik
Eder
a and
Alexey
Cherevan
*a
a,
Jasmin S.
Schubert
a,
Pablo
Ayala
a,
Hikaru
Saito
b,
Maria J.
Sampaio
cd,
Eliana S.
Da Silva‡
cd,
Cláudia G.
Silva
cd,
Joaquim L.
Faria
cd,
Dominik
Eder
a and
Alexey
Cherevan
*a
aTU Wien, Institute of Materials Chemistry, Getreidemarkt 9/BC/02, 1060, Vienna, Austria. E-mail: alexey.cherevan@tuwien.ac.at
bInstitute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasugakoen, Kasuga, Fukuoka 816-8580, Japan
cLSRE-LCM – Laboratory of Separation and Reaction Engineering – Laboratory of Catalysis and Materials, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal
dALiCE – Associate Laboratory in Chemical Engineering, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal
First published on 1st February 2024
Abstract
This study investigates the hydrogen evolution reaction (HER) efficiency of two photosystems incorporating an all-inorganic molecular thiomolybdate [Mo3S13]2− cluster as a HER catalyst. First, we delve into the performance of a homogeneous [Mo3S13]2−/[Ru(bpy)3]2+ (Mo3/Ru) dyad which demonstrates high turnover frequencies (TOFs) and apparent quantum yields (AQYs) at 445 nm approaching the level of 0.5%, yet its performance is marked by pronounced deactivation. In contrast, a heterogeneous approach involves anchoring [Mo3S13]2− onto graphitic carbon nitride (GCN) nanosheets through weak electrostatic association with its triazine/heptazine scaffold. [Mo3S13]2−/GCN (Mo3/GCN) displays effective H2 generation under visible light, with TOF metrics on par with those of its homogeneous analog. Although substantial leaching of [Mo3S13]2− species from the Mo3/GCN surface occurs, the remaining {Mo3}-based centers demonstrate impressive stability, leading to enduring HER performance, starkly distinguishing it from the homogeneous Mo3/Ru photosystem. Photoluminescence (PL) quenching experiments confirm that the performance of Mo3/GCN is not limited by the quality of the inorganic interface, but could be optimized by using higher surface area supports or a higher concentration of [Mo3S13]2− sites. Our findings showcase complexities underlying the evaluation and comparison of photosystems comprising well-defined catalytic centers and pave the way for developing analogous surface-supported (photo)catalysts with broad use in energy applications.
1. Introduction
Photocatalysis is often cited as one of the most sustainable and straightforward methods for producing solar fuels from renewable energy resources. Despite decades of research,1 however, only a limited number of efficient and stable photosystems for water splitting have been identified,2,3 indicating that the path towards developing high-performance photocatalysts is far from straightforward.4 As a result, the scientific community continues to search for a set of comprehensive design principles that can guide the synthesis of selective and stable photocatalysts, enabling the efficient conversion of solar energy into chemical energy.5–8Direct capture of sunlight energy and its use to facilitate the desired chemical reaction can be accomplished using either a homogeneous or heterogeneous photosystem that by definition bears pronounced differences in terms of their structure (molecules vs. solids), composition (mostly organic vs. mostly inorganic), solubility characteristics (molecular solutions vs. suspensions), underlying charge transfer and separation processes as well as the nature of catalytic sites. Many homogeneous photocatalytic systems show high turnover frequencies (TOFs); however – compared to their heterogeneous counterparts – often suffer from low turnover numbers (TONs) on account of photosensitizer degradation, catalyst self-aggregation, or formation of colloidal oxide species. Heterogeneous photocatalysts, on the other hand, benefit from adjustable absorption characteristics and superior stability under harsh redox conditions of the photocatalytic reaction but face complications associated with their poorly defined catalytic surface, which limit the degree of control over the processes of charge extraction and interfacial charge transfer.
In light of these challenges and prospects, a hybrid approach that combines the advantages of both homogeneous and heterogeneous photocatalytic systems emerges as a promising solution. Several important avenues for this combination have been explored by the community over the past few decades. One example involves single-metal-site and single-metal-atom catalysts which allow the creation of robust all-inorganic photosystems with unparalleled atom-utilization efficiency.9–11 Another strategy exploits stabilization of well-defined molecular catalysts on the surface of solid-state (photoactive) supports, such as in the case of the Co-based (Co4O4) water oxidation catalyst (WOC) recently embedded within the pores of a rigid coordination network,12 or in the field of surface organometallic chemistry which relies on the immobilization of complexes onto structurally controlled inorganic surfaces.13,14 Additionally, metal–organic frameworks (MOFs) can be regarded as a promising hybrid photosystem due to their ability to merge molecular units (ligands and metal nodes) into a crystalline solid-state material, thus incorporating the benefits of both molecular and inorganic approaches to (photo)catalysis.15 Despite these and many other combinations of molecular and solid-state photosystems that have been explored, only rare studies were able to directly compare the photocatalytic performance of similar photocatalytic systems under homogeneous and heterogeneous conditions.16 Nevertheless, by investigating the effects of molecular catalyst immobilization, one can gain insights into the factors that contribute to or restrict the performance of heterogenized catalysts. This type of investigation enables a more comprehensive understanding of the underlying mechanisms and provides valuable information for the design and optimization of highly efficient and stable hybrid photosystems.
Aiming to complement this knowledge gap, we turned our attention to an inorganic thiometalate cluster [Mo3S13]2−, which recently emerged as a promising noble-metal-free catalyst for the hydrogen evolution reaction (HER).17 This trinuclear cluster features all the advantages of a molecular catalyst – including the defined geometry, structure and composition – which together allow the shedding of light on its active sites and elucidate the reaction mechanisms.18–20 Over the past few years, examples of [Mo3S13]2− immobilization onto visible-light-active carbon nitride have been reported confirming the ability of surface-attached [Mo3S13]2− to act as a HER co-catalyst.21,22 Additionally, two recent studies reported that the encapsulation of [Mo3S13]2− clusters into porous heterogeneous scaffolds – such as covalent organic frameworks (COFs)23 and MOFs24 – yield stable photocatalytic systems, which further emphasizes the benefits of the heterogenization strategy. Most recently, our group reported surface-anchoring of [Mo3S13]2− on a carbon-free support, i.e. TiO2, which involved covalent binding and resulted in high and stable HER rates comparable to those of benchmark Pt/TiO2.25
This work sets out to provide a more comprehensive understanding of the benefits and limitations of heterogenization strategy. Specifically, we aim to directly compare the activity, stability, and other important performance indicators of surface-anchored [Mo3S13]2− clusters with their performance towards the HER under strictly homogeneous conditions. Our data reveal that despite [Mo3S13]2− being able to deliver excellent HER performance under homogeneous conditions, strong and rapid deactivation is imminent for this photosystem. In contrast to this, heterogenized [Mo3S13]2− shows much more stable HER performance with no apparent cluster degradation. Our photoluminescence spectroscopy quenching studies further compare redox pathways in both the photosystems and quantitatively compare their charge transfer kinetics. The data reveal that the performance of the heterogenized [Mo3S13]2− can be strongly limited by the degree and effectiveness of the electron–hole separation and that an increase in the [Mo3S13]2− loading or support surface area available for cluster immobilization may play a crucial role in unravelling full potential of this and other relevant thiomolybdate catalysts.20
2. Results and discussion
We start by describing two photosystems – a homogeneous and a heterogeneous – involving a thiomolybdate [Mo3S13]2− cluster as an HER catalyst. On one hand, we evaluate HER performance of the cluster under strictly homogeneous conditions – denoted here as Mo3/Ru – which involves the use of a state-of-the-art molecular dye, tris(bipyridine)ruthenium(II) ([Ru(bpy)3]2+), as a visible-light photosensitizer (Fig. 1a left).18,26 On the other hand, we explore the performance of the heterogenized [Mo3S13]2− cluster following its deposition on thermally exfoliated graphitic carbon nitride (GCN, details on synthesis are in the Experimental section). This photosystem is denoted here as Mo3/GCN. As shown in Fig. 1a (right), GCN sheets play the role of the light absorber (i.e. photosensitizer), while the [Mo3S13]2− clusters anchored on the surface act as a formal co-catalyst,21,22i.e. they extract the photoexcited charge carriers from the GCN bulk and promote the reaction of interest by reducing H+ to H2. Compared to our previous work in which [Mo3S13]2− was anchored onto a titania surface,25 the use of narrow-band gap GCN enables absorption of visible-light photons and thus allows a direct comparison of Mo3/GCN with the homogeneous visible-light-driven Mo3/Ru photosystem (Fig. 1b and Section 2 ESI†).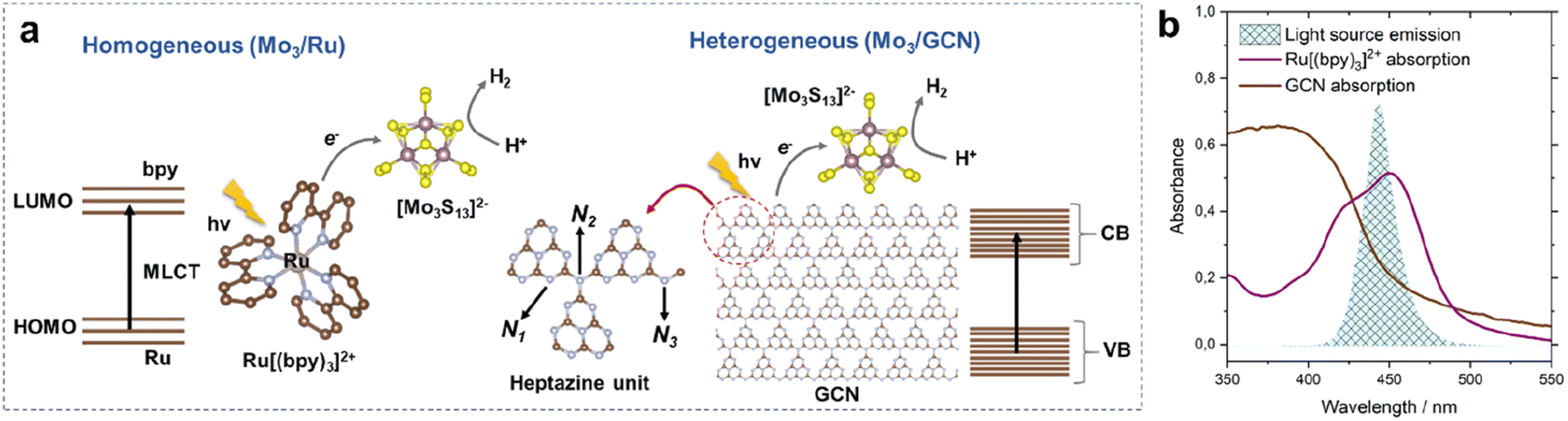 | ||
| Fig. 1 Photocatalytic HER mechanisms on (a) Mo3/Ru (homogeneous) and Mo3/GCN (heterogeneous) photosystems (brown: C; blue: N; purple: Mo; yellow: S) showing their main components as well as the photoexcitation and charge transfer pathways; LUMO and HOMO stand for the lowest unoccupied and the highest occupied molecular orbitals of [Ru(bpy)3]2+; CB and VB stand for the conduction and valence bands of GCN; N1, N2 and N3 correspond to the three types of N atoms building the heptazine unit of the GCN framework and (b) absorption spectra of [Ru(bpy)3]2+ (in methanol, 10−5 M) and GCN (via diffuse-reflectance spectroscopy, DRS, details in the Experimental section) used to construct the photosystems overlaid with the emission spectrum of a narrow-band LED lamp (center wavelength of 445 nm, details in the Experimental section) used in the photocatalytic studies. | ||
The heterogeneous Mo3/GCN will be first described from the point of view of its composition and structure. Following this, the photocatalytic HER results – in terms of activity, mechanism and stability – of Mo3/Ru and Mo3/GCN will be presented and compared aiming to reveal factors that control or limit their catalytic performance.
2.1 Heterogenized Mo3/GCN photosystem
Na2[Mo3S13] and graphitic carbon nitride (GCN) were synthesized according to previously established methods (see the Experimental section) and their purity and structure were confirmed using powder X-ray diffraction (XRD) and IR spectroscopy (Fig. S1†). Diffuse reflectance spectroscopy (DRS) of the as-obtained GCN nanosheets reveals pronounced absorption in the visible range (absorption tail beyond 500 nm) corresponding to an expected optical band gap of 2.75 eV (Fig. S2a and b†), which confirms the capability of the supporting GCN to absorb in the range of the molecular [Ru(bpy)3]2+ sensitizer.27 The [Mo3S13]2−/GCN composite (Mo3/GCN) was synthesized following a wet-impregnation route (for more details see the Experimental section). Compared to the DRS spectrum of bare GCN, the DRS spectra of Mo3/GCN show an additional broad band centered at 456 nm (Fig. S3†), which can be ascribed to the characteristic ligand-to-metal charge transfer transition of [Mo3S13]2− suggesting successful deposition of [Mo3S13]2− onto the GCN surface.28 Total reflection X-ray fluorescence spectroscopy (TXRF) further confirms the presence of Mo in the composite and allows the estimation of the real [Mo3S13]2− loading to be around 3.9 wt% (Table 1, details in the Experimental section). Interestingly, this value is significantly lower compared to the intended loading of 10 wt% and – in contrast to oxide-based supports25 – indicates a weaker nature of the interaction between organic GCN and [Mo3S13]2−. A similar actual-to-expected loading is also attained for a 1 wt% nominal loading value (Table 1), which further corroborates that the adsorption of [Mo3S13]2− on GCN is not governed by the presence of suitable adsorption sites on the GCN surface, but is rather defined by the adsorption/desorption equilibrium.| Composites | Nominal loadings [Mo3S13]2− (wt%) | Real loadings [Mo3S13]2− (wt%) |
|---|---|---|
| Mo3/GCN | 10 | 3.9 |
| 1 | 0.36 | |
| Mo3/H-GCN | 10 | 5.1 |
Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) spectra of pristine GCN (Fig. S4†) match well with those in the literature, confirming the formation of a heptazine-based scaffold (Fig. 1a, right).27 Notably, the results for Mo3/GCN composites show no vibrations corresponding to [Mo3S13]2−, as expected from the low cluster loadings. Therefore, scanning transmission electron microscopy (STEM) was used to provide evidence for the presence of [Mo3S13]2− on the GCN surface. Energy dispersive X-ray (EDX) maps acquired on the nanoscale (Fig. 2a and b) confirm chemical identity of the elements and reveal an even distribution of Mo and S over the entire surface of the GCN aggregates. High-resolution fast-Fourier transformed (FFT) image in Fig. 2c focuses on the edge of a typical GCN flake and shows the presence of multiple bright spots. Considering the high Z-contrast between Mo and C/N atoms, these spots likely correspond to heavy Mo atoms dispersed on GCN and their high density further indicates close proximity of the clusters. Interestingly, a closer look at Fig. 2c also reveals the formation of chain-like structures suggesting the possibility of partial cluster oligomerization, in line with MoSx-based nanostructuring observed on similar carbon-based surfaces.29
 | ||
| Fig. 2 STEM micrographs of the Mo3/GCN composite (a) high-resolution high-angle annular dark field (HAADF) STEM image of the Mo3/GCN composite showing an aggregate of GCN nanoflakes along with its (b) EDS-derived elemental mappings featuring the distribution of (i) C, (ii) Mo, (iii) N, and (iv) S elements, and (c) FFT-transformed magnified region of one of the GCN flake's edges (see selected area in (a)) showing collections of bright spots that correspond to the heavy Mo atoms. | ||
Surface-sensitive X-ray photoelectron spectroscopy (XPS) was further used to verify the structure of [Mo3S13]2− after attachment. The survey spectra confirm the presence of all elements expected from the Mo3/GCN composition (Fig. S5†). The detailed Mo 3d profile of Mo3/GCN in Fig. 3a only shows peaks corresponding to Mo4+ with an overall profile that resembles that of the free [Mo3S13]2− cluster. This indicates the preference for a trinuclear structure of the cluster cores and further suggests that neither oxidation nor decomposition of the central {Mo3} units of the cluster occurred upon anchoring. Importantly, we also observe a shift of the Mo 3d peaks maxima by 1.2 eV to higher binding energies, which is indicative of electronic interactions involving charge transfer from the cluster to the support, in line with the anionic nature of [Mo3S13]2−. The S 2p signal profile of Mo3/GCN undergoes a more significant change upon attachment, which suggests a restructuring of the S-containing ligands. A closer look at the deconvoluted profiles (Fig. 3b) indicates partial loss of bridging disulfides, which can be linked to the partial transformation of the clusters into more complex MoSx fragments, in line with cluster oligomerization observed in STEM.30 Detailed analysis of the N 1s edge (Fig. 3c) further allows evaluation of the nature of the cluster binding and the degree of [Mo3S13]2−/GCN interactions from the support point of view. In the case of the Mo3/GCN composite, we observe a strong drop in the major C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C component (N1) and a concurrent increase of the quaternary N–(C)3 signal (N2), which suggests that the cluster attachment causes the disruption of the GCN framework – specifically its heptazine/triazine units – in line with the electrostatic binding model (Fig. 1a, right). Overall, the data suggest successful heterogenization of high-density thiomolybdates on the surface of exfoliated GCN nanosheets and confirm their homogeneous deposition along with the intact nature of their {Mo3} cores.
N–C component (N1) and a concurrent increase of the quaternary N–(C)3 signal (N2), which suggests that the cluster attachment causes the disruption of the GCN framework – specifically its heptazine/triazine units – in line with the electrostatic binding model (Fig. 1a, right). Overall, the data suggest successful heterogenization of high-density thiomolybdates on the surface of exfoliated GCN nanosheets and confirm their homogeneous deposition along with the intact nature of their {Mo3} cores.
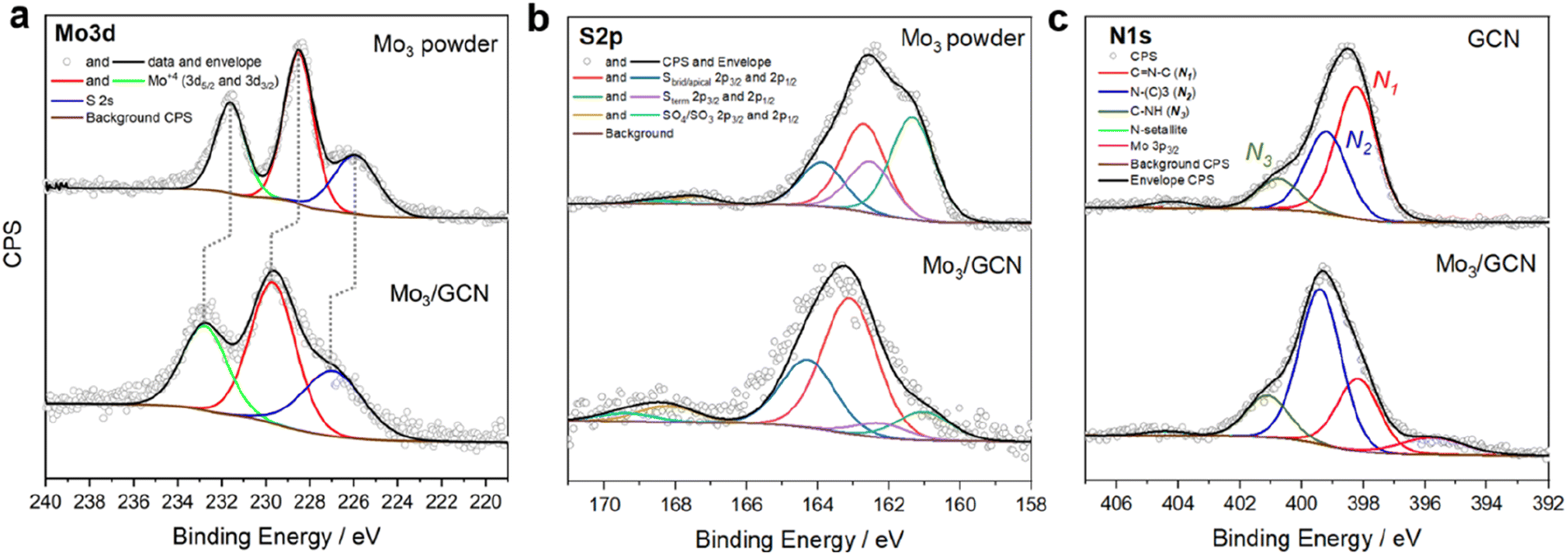 | ||
| Fig. 3 XPS spectra of the Mo3/GCN cluster. The [Mo3S13]2− clusters before (Mo3 powder) and after (Mo3/GCN) attachment to the GCN surface, (a) Mo 3d, (b) S 2p, and (c) N 1s (N1: pyridinic nitrogen; N2: quaternary nitrogen; N3: secondary amine), with corresponding fits. | ||
2.2 Optimization of HER conditions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (MeOH:H2O, 9:1) as a mixed solvent system. In parallel, Lei et al.26 evaluated HER performance of [Mo3S13]2− with [Ru(bpy)3]PF6 as a sensitizer, using 0.1 M of H2A as a sacrificial agent in an acetonitrile:water (ACN:H2O, 9:1) solvent mixture. Considering the relatively similar set of conditions reported in these studies, we first validated our Mo3/Ru photosystem by using both MeOH:H2O and CH3CN:H2O solvent mixtures with various sensitizer-to-catalyst concentration ratios: 65-to-1 (similar to conditions by Dave et al.18) and 13-to-1 (similar to conditions by Lei et al.) – details are in the Experimental section. Table S1† shows that the Mo3/Ru couple could generate H2 in both solvent systems with apparent quantum yield (AQY) values of up to 0.5%, whereas a 5-fold decrease in the [Mo3S13]2− concentration only resulted in a 2-fold drop in the H2 amount generated. This non-correlated behavior indicates that the HER performance of this homogeneous photosystem is rather limited by light capture efficiency i.e. photosensitization.
:water (MeOH:H2O, 9:1) as a mixed solvent system. In parallel, Lei et al.26 evaluated HER performance of [Mo3S13]2− with [Ru(bpy)3]PF6 as a sensitizer, using 0.1 M of H2A as a sacrificial agent in an acetonitrile:water (ACN:H2O, 9:1) solvent mixture. Considering the relatively similar set of conditions reported in these studies, we first validated our Mo3/Ru photosystem by using both MeOH:H2O and CH3CN:H2O solvent mixtures with various sensitizer-to-catalyst concentration ratios: 65-to-1 (similar to conditions by Dave et al.18) and 13-to-1 (similar to conditions by Lei et al.) – details are in the Experimental section. Table S1† shows that the Mo3/Ru couple could generate H2 in both solvent systems with apparent quantum yield (AQY) values of up to 0.5%, whereas a 5-fold decrease in the [Mo3S13]2− concentration only resulted in a 2-fold drop in the H2 amount generated. This non-correlated behavior indicates that the HER performance of this homogeneous photosystem is rather limited by light capture efficiency i.e. photosensitization.
Next, aiming for the ultimate comparison of homogeneous Mo3/Ru and heterogenized Mo3/GCN samples, apart from ascorbic acid previously employed as an electron donor, we also considered methanol (MeOH, being a representative alcohol) and triethanolamine (TEOA, being a representative tertiary amine) commonly used as sacrificial agents in the literature (details in Section 3 in the ESI†). Two important results can be highlighted. Firstly, since the use of the MeOH/H2O solvent mixture originally proposed by Dave et al. yielded much higher HER performance of the Mo3/Ru couple (4-fold increase), we aimed to verify if MeOH additionally acts as an electron and a proton donor.31 HER experiments in MeOH solutions with and without the addition of a sacrificial donor (H2A) in Fig. S6† show that no H2 could be generated in the absence of H2A, confirming that MeOH acts only as a solvent. Secondly, we observed that the replacement of H2A with TEOA yielded no H2 (see Table 2), which we relate to the inability of [Ru(bpy)3]2+ to oxidize the amine due to its low-lying redox potential. Based on these results, we continue the benchmarking of the Mo3/Ru performance obtained from methanolic H2A solutions.
| Phase | Catalyst | Sacrificial donor | Solvent | Photosensitizer | H2 (nmol) | TOF (min−1) |
|---|---|---|---|---|---|---|
| Homogeneous | [Mo3S13]2−50 μM | H2A 0.1 M | MeOH:H2O (9:1) |
[Ru(bpy)3]PF6 | 390 | 0.065 |
| TEOA 0.1 M | 0.645 mM | <0.1 | — | |||
| Heterogeneous | Mo3/GCN 10 wt% | H2A 0.1 M | H2O | None | 52 | 0.062 |
| TEOA 0.1 M | 197 | 0.237 | ||||
| Mo3/GCN 1 wt% | TEOA 0.1 M | 9 | 0.117 | |||
| Mo3/H-GCN 10 wt% | TEOA 0.1 M | 241 | 0.221 |
2.3 Comparison of both photosystems
These performance-related findings indicate that: (a) the number of electrons taking part in H+ reduction (i.e. the HER) is directly proportional to the amount of [Mo3S13]2− on the surface, suggesting that the overall HER performance can be improved by having more [Mo3S13]2− sites present. (b) The transfer of photoexcited electrons from GCN to [Mo3S13]2− is rapid with respect to H+ reduction (i.e. the HER) and is not improved when a stronger [Mo3S13]2− to GCN interaction/interface is created. (c) The effective scavenging of photoexcited holes can be seen as a performance-limiting factor, which needs to be addressed possibly by introducing other sacrificial agents or appropriate oxidation co-catalysts to aid in efficient charge extraction, separation and utilization.
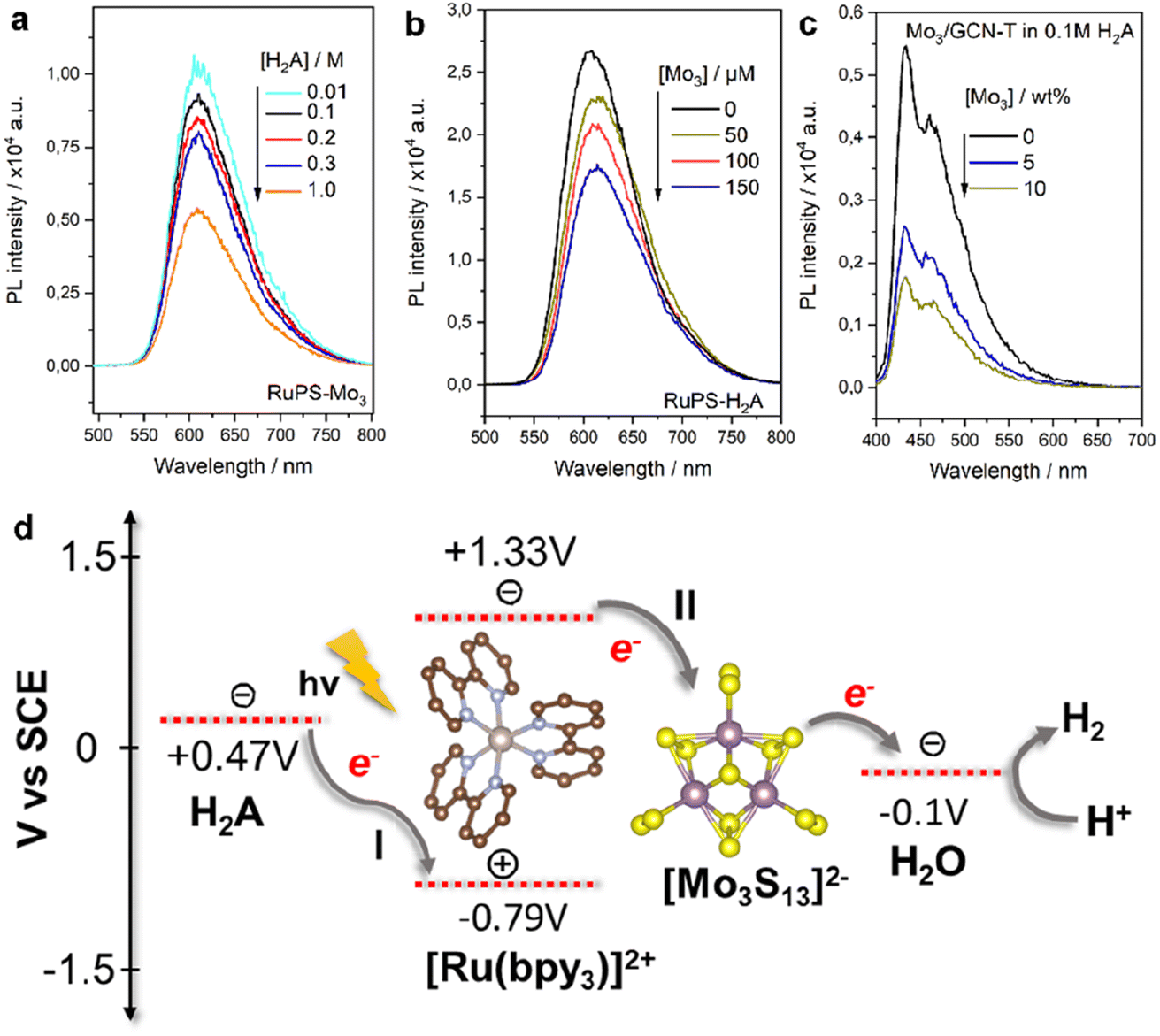 | ||
| Fig. 4 PL emission spectra of the Mo3/Ru system with varying (a) H2A and (b) [Mo3S13]2− concentrations and (c) Mo3/GCN system with varying [Mo3S13]2− loadings and (d) schematic of reductive (I) and oxidative (II) quenching mechanisms of the [Ru(bpy)3]2+ photosensitizer in the presence of the [Mo3S13]2− catalyst and H2A sacrificial donor. Energy levels are based on the relevant literature source.26 | ||
For Mo3/GCN, PL spectra of the composites feature two absorption maxima at around 440 and 480 nm that correspond to the π–π* and n–π* transitions within GCN (Fig. 4c).27 Compared to the emission of bare GCN, the deposition of [Mo3S13]2− leads to a strong PL quenching which can be associated with the facilitated charge separation that highlights the role of [Mo3S13]2− as a reductive co-catalyst capable of effective extraction of photoexcited electrons. Following the idea of concentration-dependent quenching studies of our Mo3/Ru, we also observed that heterogenized Mo3/GCN composites with different loadings of [Mo3S13]2− exhibit different emission intensities. As can be seen in Fig. 4c, increasing the cluster content from 5 wt% to 10 wt% (nominal values) results in more pronounced PL quenching indicating that charge transfer at the [Mo3S13]2−/GCN interface is further facilitated when a higher number of [Mo3S13]2− is present at the surface. Analyzing the Mo3/GCN quenching datasets based on three [Mo3S13]2− loading values – and assuming that the Stern–Volmer relationship can be applied to describe the kinetics of the GCN quenching – yields a quenching constant Kq,ox of around 4.1 × 107 M−1 s−1 (Fig. S8c†). This value is several orders of magnitude lower than the one obtained for the homogeneous Mo3/Ru case (1.8 × 1010 M−1 s−1), which is expected for the solid/liquid Mo3/GCN interface. In conjunction with the HER discussion above, these insights suggest that the use of photoactive supports with higher surface areas available for [Mo3S13]2− immobilization could lead to further optimization of the Mo3/GCN photosystem's performance.
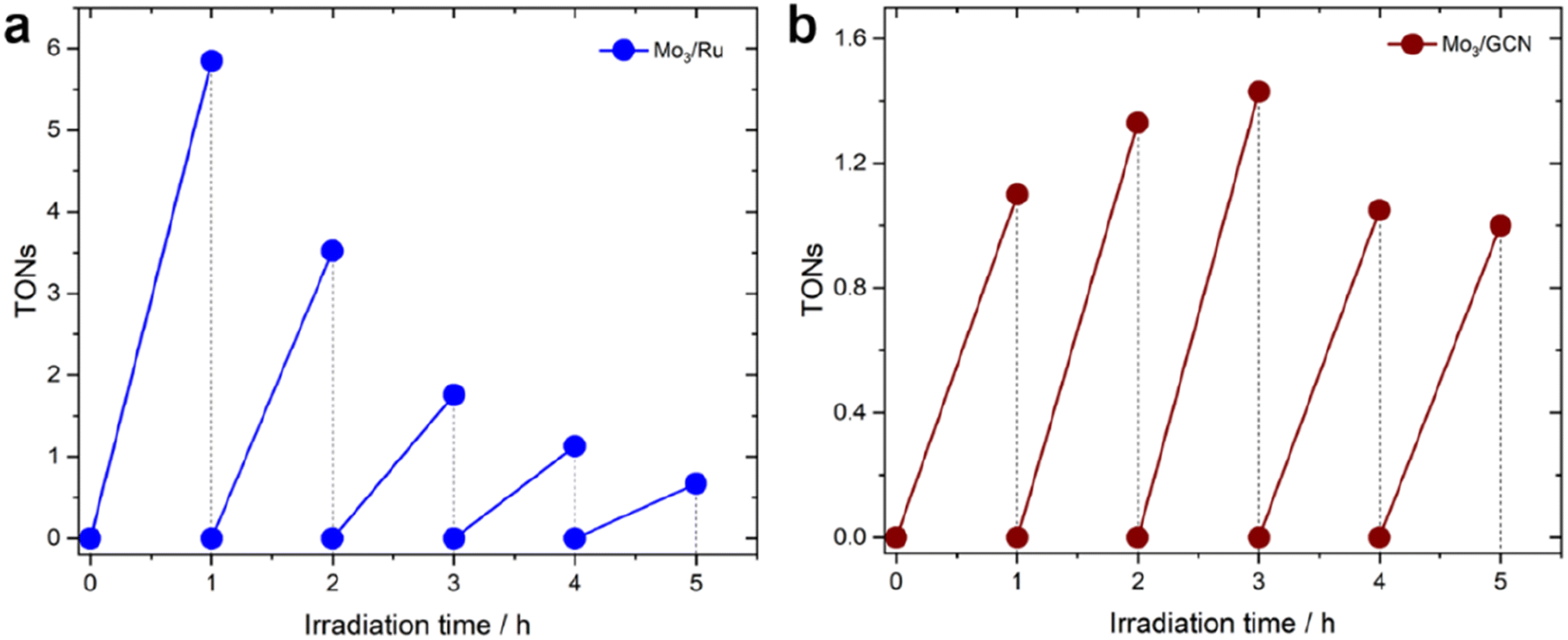 | ||
| Fig. 5 Long-term HER profiles (in H2A) of (a) Mo3/Ru system and (b) Mo3/GCN system. TONs for the Mo3/GCN system were calculated based on real loadings of [Mo3S13]2− clusters on GCN mentioned in Table 1. The experiments were conducted using an LED 445 nm lamp (see Methods section 4.3 for more details). | ||
In contrast to this, the heterogenized Mo3/GCN (Fig. 5b) shows no pronounced deactivation, but rather stable performance – despite with lower average TOFs – over the time course of 5 hours of illumination. XPS of the Mo3/GCN composites recovered after the HER run indicate that partial loss (dissolution/leaching) of the clusters takes place during the reaction, which is also suggested by the recovered CN–C contribution of GCN observed in the N 1s edge (see Fig. S10† for more details). This result is further in agreement with elemental analyses using TXRF (Table S2†), which indicates that only 50% of the original [Mo3S13]2− species are present in Mo3/GCN after the HER. In view of the relatively stable HER performance observed for the Mo3/GCN composites (Fig. 5b), however, we can propose that this partial leaching of the [Mo3S13]2− takes place at the initial stages of the reaction and does not affect long-term HER performance. In fact, detailed Mo 3d and S 2p XPS profiles look qualitatively similar to those of the as-prepared Mo3/GCN composite (Fig. S10†), suggesting that no transformation of the surface-bound Mo3-species takes place. These remaining [Mo3S13]2− clusters (1.7 wt% compared to the original 3.9 wt% loading) likely constitute stable and active catalytic sites for the HER that are able to effectively extract the electrons photogenerated in the GCN support and drive stable H2 generation.
3. Conclusions
Here, we compared HER performances of homogeneous and heterogeneous photosystems involving an all-inorganic molecular [Mo3S13]2− cluster as an HER site. On one hand, we explored a prototypical homogeneous Mo3/Ru couple in which visible light excitation leads to a rapid oxidative quenching of *[Ru(bpy)3]2+ (PS*) by the anionic [Mo3S13]2−. When combined with a H2A hole scavenger, this photosystem delivered high TOFs and AQY445 values up to 0.5%; it however suffered from rapid photo-degradation. On the other hand, we demonstrated that [Mo3S13]2− can be immobilized onto the surface of GCN by relying on electrostatic interactions with its triazine/heptazine framework. We show that this heterogenized Mo3/GCN photosystem could generate H2 under visible light with TOF values close to those measured for its homogeneous counterpart. Despite significant leaching of the [Mo3S13]2− species from the surface of Mo3/GCN, the rest of the {Mo3}-based centers remain structurally intact. They are able to deliver stable long-term HER performance, which is in strong contrast to that of Mo3/Ru, which showed photodegradation after 1 hour of photocatalytic reaction. Mechanistic and photoluminescence quenching studies further demonstrated that the performance of Mo3/GCN may still be largely limited by the extent of the Mo3/GCN interface and the efficiency of electron–hole separation. These results provide valuable insights into the challenges in activity evaluation and comparison for photosystems that involve well-defined catalytic centers (be it clusters or single-metal-atoms or sites), which will help develop similar surface-supported (photo)catalysts for various energy applications.4. Experimental section
4.1 Chemicals
The chemicals used for the synthesis were obtained from commercial suppliers and include ammonium molybdate tetrahydrate ((NH4)6[Mo7O24]·4H2O, Sigma-Aldrich, 99.98% trace metals basis), hydroxylamine hydrochloride (NH2OH·HCl, Sigma-Aldrich, 98.0%, ACS Reagent), ammonium sulfide solution ((NH4)2Sx, Sigma-Aldrich, 20 wt% in H2O), carbon disulfide (CS2, Sigma-Aldrich, 99.9%), sodium chloride (NaCl, Carl Roth), sodium hydroxide (NaOH, Carl Roth) and dicyandiamide (C2H4N4, 99%, Sigma-Aldrich). The solvents used for the synthesis were deionized water, ethanol (EtOH, from Chem-Lab NV), HPLC-gradient grade methanol (MeOH, from VWR), diethyl ether (from Sigma-Aldrich), and N,N-dimethylformamide (DMF, from Acros Organics, 99.8%, extra dry over the molecular sieve, acroseal).4.2 Synthetic procedures
The protonation of GCN was carried out following a procedure already reported34 wherein GCN was dispersed in 10 mL of 37% HCl solution and stirred for 4 hours at room temperature. The solution was then filtered and washed with water until neutral pH was achieved. The as-modified sample denoted as H-GCN was dried in air at 105 °C overnight.
4.3 Methods
UV-vis spectroscopy was performed on a Jasco V670 UV-vis spectrometer. The samples were prepared in methanol and aqueous methanol (1:1 vol.) solution with a concentration of 0.05 mM; UV-vis spectra were recorded in absorbance mode. Absorption spectra of powdered samples were measured by solid-state via diffuse-reflectance spectroscopy (DRS) using MgSO4 and GCN as references.
ATR-FTIR spectra of the samples were recorded via a PerkinElmer FTIR Spectral UATR-TWO with a Spectrum Two Universal ATR (Single Reflection Diamond) instrument. Powdered samples were directly loaded onto the sample holder and the spectra were recorded in the region of 4000–400 wavenumbers (cm−1). Raman measurements were performed with a WITec alpha 300 RSA+ Raman microscope equipped with a 488 nm excitation laser (532 nm) maintaining the laser intensity at 5 mW.
The quantitative elemental analysis of the samples was performed with X-ray photoelectron spectroscopy (XPS) using a custom-built SPECS XPS-spectrometer equipped with a monochromatized Al-Kα X-ray source and a hemispherical WAL-150 analyzer (acceptance angle: 60°). To improve the sensitivity of the measurements, Mo3/TiO2 samples were prepared and investigated in the form of thin-films (see Additional methods in the ESI†). This was followed by wet impregnation of the Na2[Mo3S13] clusters from methanolic solutions. For a single XPS measurement, pass energies of 100 eV and 30 eV and energy resolutions of 1 eV and 100 meV were used for survey and detailed spectra, respectively (excitation energy: 1486.6 eV, beam energy and spot size: 70 W onto 400 μm, angle: 51° to sample surface normal, base pressure: 5 × 10−10 mbar, and pressure during measurements: 2 × 10−9 mbar). Data analysis was performed using CASA XPS software, employing transmission corrections (as per the instrument vendor's specifications), Shirley backgrounds and Scofield sensitivity factors. Charge correction was applied so the adventitious carbon peak (C–C peak) was shifted to 284.8 eV binding energy (BE). All content values shown are in units of relative atomic percent (at%), where the detection limit in survey measurements usually lies around 0.1–1 at%, depending on the element.
Quantitative determination of the [Mo3S13]2− cluster loadings was performed by X-ray fluorescence spectroscopy in total reflection geometry (TXRF) using an ATOMIKA 8030C X-ray fluorescence analyzer (Atomika Instruments GmbH, Oberschleissheim, Munich, Germany). The X-ray tube was employed at 50 kV and 47 mA and the selected excitation source was the continuous spectrum of tungsten monochromatized at 35 keV. The samples were excited for 100 s and a Si(Li)-detector was used for X-ray acquisition. The samples were prepared by suspending 1 mg in 1 mL H2O for 10 minutes (c = 1 mg mL−1). 10 μL of a 1000 ppm Yttrium internal standard (for quantification) were added to the suspension, which was subsequently vortexed for 1 min. 5 μL of the suspension with the internal standard were drop cast onto a clean quartz reflector, which was then dried for 5 minutes on a hot plate. After this time, the residue was sealed with 5 μL of a 1% PVA solution and dried for another 5 minutes on a hot plate. The absolute amounts of Mo with respect to Y were then quantified based on the calibration curve and the proportion of Mo (K-line) and Y peak (K-line) areas. This calculation yielded real [Mo3S13]2− loading values presented in Table 1, considering the undisturbed stoichiometry of Mo to S.
Scanning electron microscopy (SEM) images were acquired using an FEI Quanta 250 FEG scanning electron microscope to obtain visual information on the morphology of the samples. Typically, an acceleration voltage of 10 kV and secondary electron detection mode were used.
High-angle annular dark field (HAADF) STEM imaging and energy dispersive X-ray spectroscopy (EDS) were performed by using a Titan Cubed G2 60-300 (TEM/STEM, FEI Co., now Thermo Fisher Scientific) operated at 300 kV. This microscope has an aberration corrector for STEM (DCOR, CEOS), four-quadrant windowless super-X SDD (silicon drift detector) system. The probe current was ∼60 pA for STEM observation as well as EDS. The convergence semi-angle of the electron probe was 18 mrad. The typical probe diameter was less than 0.1 nm. Forward scattered electrons in an angular range from 38 to 184 mrad were detected using a HAADF detector for STEM imaging.
Powder X-ray diffraction (XRD) of (NH4)2[Mo3S13] and Na2[Mo3S13] was performed using an XPERT II: PANalytical XPert Pro MPD (Θ–Θ diffractometer) for the ex situ experiments. The sample was placed on a Si sample holder and irradiated with a Cu X-ray source (8.04 keV, 1.5406 Å). The signals were then acquired with Bragg–Brentano Θ/Θ-diffractometer geometry ranging from 5° to 80° degrees using a semiconductor X'Celerator (2.1°) detector. XRD analysis of GCN powder was carried out on a PANalytical X'Pert MPD equipped with an X'Celerator detector and a secondary monochromator (Cu Kα λ = 0.154 nm, 50 kV, 40 mA; data recorded at a 0.017° step size, 100 s per step).
Steady state photoluminescence (PL) measurements were performed using a PicoQuant FluoTime 300 spectrophotometer. A Xe arc lamp (300 W power) was the excitation source, coupled with a double-grating monochromator. The detection system was composed of a PMA Hybrid 07 detector along with a high-resolution double monochromator. The PL properties of homogeneous solutions were observed using 445 nm excitation wavelength whereas 370 nm light excitation was used to probe the PL spectra of heterogeneous catalysts suspended in water; the concentration of each reaction component was set to mimic those from the corresponding HER experiments. The Mo3/GCN samples were prepared by dispersion of the composite in water (0.5 mg mL−1), sonication for 30 min, and centrifugation for 30 min. The supernatant solution was diluted with water and 0.1 M ascorbic acid. The data were collected and later fitted using EasyTau2 software. The Stern–Volmer treatment was applied to extract the bimolecular rate constant based on the [Ru(bpy)3]2+ lifetime of 198.58 ns measured at 615 nm.
4.4 Photocatalytic experiments
The visible-light-driven hydrogen evolution experiments were carried out using a 5 mL batch reactor equipped with a monochromatic LED light source (445 ± 13 nm, Thorlabs SOLIS). For the experiments in the homogeneous phase, the reactor was filled with a 2 mL of reaction mixture which comprises 1:1 MeOH/H2O containing [Ru(bpy)3]2+ (bpy stands for 2,2′-bipyridine) as a photosensitizer (0.645 mM), L-ascorbic acid (H2A) as a proton donor (0.1 M), and the corresponding catalyst Na2[Mo3S13]·H2O (50 μM). For the heterogeneous hydrogen evolution experiments the reactor was filled with 2 mL of water containing 0.5 mg mL−1 of the Mo3/GCN photocatalyst and 0.1 M sacrificial donor (0.1 M ascorbic acid (H2A) and triethanolamine (TEOA), details in Fig. S7 and S8d†). Exposure to ambient light was minimized during the solution mixture preparation and transfer to the reactor. The reaction volume was purged with Ar (15 mL min−1) for 10 min to ensure the removal of headspace and dissolved oxygen prior to the start of the reaction. The temperature of the reactor was maintained at 15 °C with a water-cooling system. The reaction mixture was stirred at 600 rpm. The H2 produced was monitored by sampling the reactor headspace (200 μL) and analyzing its composition via gas chromatography (Shimadzu GC 2030) equipped with a barrier ionization discharge detector and a Micropacked-ST column using helium as a carrier gas. Injections were performed with an interval of 30 minutes. Calibration was performed using a range of H2 in argon gas mixtures. A set of blank experiments in the absence of [Mo3S13]2− was conducted otherwise mimicking the conditions of both the homogeneous and heterogeneous HER tests resulting in negligible (<0.1 nmol) amounts of H2 generated.
4.5 TON, TOF, and AQY calculation
The H2 concentrations in ppm (derived from the chromatograms) were converted to μmol and turnover numbers (TONs – expressed per [Mo3S13]2− species) based on reactor parameters and the ideal gas equation. Initial turnover frequencies (TOFs) were calculated after 10 minutes of illumination (in most of the cases a close to linear H2 evolution trend within the first 60 minutes of the HER was observed). The calculation of the apparent quantum yield (AQY) values considered the ratio between the number of reacted electrons and the number of photons absorbed by the reaction solution.39,40 The latter was extracted using a power meter PM100D (Thorlabs) by measuring photon flux at the reactor position (12 mW cm−2). To estimate the number of photons absorbed by the reaction solution we first measured intensity of light that reached the detector after passing through the reactor filled with pure solvent and then measured intensity of light that reached the detector after passing through the reactor filled with the Mo3/Ru photocatalyst dissolved in the solution. The difference between the values indicated the amount of light that has been trapped by Mo3/Ru.Author contributions
Conceptualization, SB and AC; methodology, SB and AC; formal analysis, SB, JS, and PA; investigation, SB, JS, PA, HS, MJS and ESS; resources, DE; data curation, SB; writing: original draft preparation, SB; writing: review and editing, SB, JLF and AC; visualization, SB; supervision, CGS, JLF, DE and AC; project administration, AC; funding acquisition, CGS, JLF and AC. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded in whole, or in part, by the Austrian Science Fund (FWF) (grant number P32801-N and Cluster of Excellence MECS). The authors would like to acknowledge the support by national funds through FCT/MCTES (PIDDAC): LSRE-LCM, UIDB/50020/2020 (DOI: https://doi.org/10.54499/UIDB/50020/2020) and UIDP/50020/2020 (DOI: https://doi.org/10.54499/UIDP/50020/2020); and ALiCE, LA/P/0045/2020 (DOI: https://doi.org/10.54499/LA/P/0045/2020). MJS acknowledges FCT funding under the Scientific Employment Stimulus – Institutional Call (CEECINST/00010/2021). Open Access was funded by the Austrian Science Fund (FWF). The authors would like to acknowledge the facilities of Technische Universität Wien (TU Wien) for technical support and fruitful discussions: the X-Ray Center; Analytical Instrumentation Center and Electron Microscopy Center (USTEM). The authors would like to further acknowledge Dr Sreejith P. Nandan for his support of XRD and PL measurements, Stephen Nagaraju Myakala for his support in sample preparation and Dr Dogukan H. Apaydin for his support with FTIR measurements.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- H. Lyu, T. Hisatomi, Y. Goto, M. Yoshida, T. Higashi, M. Katayama, T. Takata, T. Minegishi, H. Nishiyama, T. Yamada, Y. Sakata, K. Asakura and K. Domen, Chem. Sci., 2019, 10, 3196–3201 RSC.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- D. Bahnemann, P. Robertson, C. Wang, W. Choi, H. Daly, M. Danish, H. de Lasa, S. Escobedo, C. Hardacre, T. H. Jeon, B. Kim, H. Kisch, W. Li, M. Long, M. Muneer, N. Skillen and J. Zhang, J. Phys. Energy, 2023, 5, 012004 CrossRef.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- R. Huang, X. Li, W. Gao, X. Zhang, S. Liang and M. Luo, RSC Adv., 2021, 11, 14844–14861 RSC.
- S. Subudhi, D. Rath and K. M. Parida, Catal. Sci. Technol., 2018, 8, 679–696 RSC.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y.-T. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CrossRef CAS PubMed.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- P. Ayala, A. Giesriegl, S. P. Nandan, S. N. Myakala, P. Wobrauschek and A. Cherevan, Catalysts, 2021, 11, 417 CrossRef CAS.
- A. I. Nguyen, K. M. Van Allsburg, M. W. Terban, M. Bajdich, J. Oktawiec, J. Amtawong, M. S. Ziegler, J. P. Dombrowski, K. V. Lakshmi, W. S. Drisdell, J. Yano, S. J. L. Billinge and T. D. Tilley, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11630–11639 CrossRef CAS PubMed.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- G. Jeantelot, M. Qureshi, M. Harb, S. Ould-Chikh, D. H. Anjum, E. Abou-Hamad, A. Aguilar-Tapia, J.-L. Hazemann, K. Takanabe and J.-M. Basset, Phys. Chem. Chem. Phys., 2019, 21, 24429–24440 RSC.
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- S. P. Nandan, N. I. Gumerova, J. S. Schubert, H. Saito, A. Rompel, A. Cherevan and D. Eder, ACS Mater. Au, 2022, 2, 505–515 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- M. Dave, A. Rajagopal, M. Damm-Ruttensperger, B. Schwarz, F. Nägele, L. Daccache, D. Fantauzzi, T. Jacob and C. Streb, Sustainable Energy Fuels, 2018, 2, 1020–1026 RSC.
- J. McAllister, N. A. G. Bandeira, J. C. McGlynn, A. Y. Ganin, Y.-F. Song, C. Bo and H. N. Miras, Nat. Commun., 2019, 10, 370 CrossRef CAS PubMed.
- S. Batool, M. Langer, S. N. Myakala, M. Heiland, D. Eder, C. Streb and A. Cherevan, Adv. Mater., 2023, 2305730 Search PubMed.
- F. Guo, Y. Hou, A. M. Asiri and X. Wang, Chem. Commun., 2017, 53, 13221–13224 RSC.
- A. Rajagopal, E. Akbarzadeh, C. Li, D. Mitoraj, I. Krivtsov, C. Adler, T. Diemant, J. Biskupek, U. Kaiser, C. Im, M. Heiland, T. Jacob, C. Streb, B. Dietzek and R. Beranek, Sustainable Energy Fuels, 2020, 4, 6085–6095 RSC.
- Y.-J. Cheng, R. Wang, S. Wang, X.-J. Xi, L.-F. Ma and S.-Q. Zang, Chem. Commun., 2018, 54, 13563–13566 RSC.
- T. N. Nguyen, S. Kampouri, B. Valizadeh, W. Luo, D. Ongari, O. M. Planes, A. Züttel, B. Smit and K. C. Stylianou, ACS Appl. Mater. Interfaces, 2018, 10, 30035–30039 CrossRef CAS PubMed.
- S. Batool, S. P. Nandan, S. N. Myakala, A. Rajagopal, J. S. Schubert, P. Ayala, S. Naghdi, H. Saito, J. Bernardi, C. Streb, A. Cherevan and D. Eder, ACS Catal., 2022, 12, 6641–6650 CrossRef CAS PubMed.
- Y. Lei, M. Yang, J. Hou, F. Wang, E. Cui, C. Kong and S. Min, Chem. Commun., 2018, 54, 603–606 RSC.
- E. S. Da Silva, N. M. M. Moura, A. Coutinho, G. Dražić, B. M. S. Teixeira, N. A. Sobolev, C. G. Silva, M. G. P. M. S. Neves, M. Prieto and J. L. Faria, ChemSusChem, 2018, 11, 2681–2694 CrossRef CAS PubMed.
- A. Müller, R. Jostes, W. Jaegermann and R. Bhattacharyya, Inorg. Chim. Acta, 1980, 41, 259–263 CrossRef.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef CAS PubMed.
- E. Ronge, S. Hildebrandt, M.-L. Grutza, H. Klein, P. Kurz and C. Jooss, Catalysts, 2020, 10, 856 CrossRef CAS.
- A. S. Hainer, J. S. Hodgins, V. Sandre, M. Vallieres, A. E. Lanterna and J. C. Scaiano, ACS Energy Lett., 2018, 3, 542–545 CrossRef CAS.
- V. Kumaravel, M. D. Imam, A. Badreldin, R. K. Chava, J. Y. Do, M. Kang and A. Abdel-Wahab, Catalysts, 2019, 9, 276 CrossRef CAS.
- J. Kröger, A. Jiménez-Solano, G. Savasci, P. Rovó, I. Moudrakovski, K. Küster, H. Schlomberg, H. A. Vignolo-González, V. Duppel, L. Grunenberg, C. B. Dayan, M. Sitti, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2021, 11, 2003016 CrossRef.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50–51 CrossRef CAS PubMed.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- A. Müller, S. Pohl, M. Dartmann, J. P. Cohen, J. M. Bennett and R. M. Kirchner, Z. Naturforsch. B, 1979, 34, 434–436 CrossRef.
- V. P. Fedin, J. Czyzniewska, R. Prins and T. Weber, Appl. Catal. Gen., 2001, 213, 123–132 CrossRef CAS.
- M. J. Sampaio, A. R. L. Ribeiro, C. M. R. Ribeiro, R. A. Borges, M. F. Pedrosa, A. M. T. Silva, C. G. Silva and J. L. Faria, Chem. Eng. J., 2023, 459, 141617 CrossRef CAS.
- A. Cherevan, P. Gebhardt, A. Kunzmann, R. D. Costa and D. Eder, ACS Appl. Energy Mater., 2018, 1, 1259–1267 CrossRef CAS.
- M. Qureshi and K. Takanabe, Chem. Mater., 2017, 29, 158–167 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01658g |
| ‡ Current affiliation: Department de Química, Unitat de Química Inorgánica, Universitát Autónoma de Barcelona, Bellaterra, 08193 Barcelona, Spain. |
| This journal is © The Royal Society of Chemistry 2024 |