
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous synthesis of ruthenium nanoparticles with tuneable sizes using ruthenium nitrosyl nitrate precursor†
Joseph
El-Kadi
 ,
Eugenio
Fenoaltea Pieche
,
Seung Woo
Ko
and
Laura
Torrente-Murciano
*
,
Eugenio
Fenoaltea Pieche
,
Seung Woo
Ko
and
Laura
Torrente-Murciano
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, CB3 0AS, Cambridge, UK. E-mail: lt416@cam.ac.uk
First published on 6th February 2024
Abstract
This paper presents a novel approach for the synthesis of ruthenium nanoparticles via the reduction of ruthenium nitrosyl nitrate with sodium borohydride in flow 3D helical reactors in the absence of capping ligands. Manipulating the pH-speciation of the ruthenium precursor and the fluid dynamics of the flow system allows for the synthesis of small nanoparticles and the tuning of average size with narrow size distributions (2–4 ± 0.5 nm). A mechanism is proposed for the NP synthesis involving the formation of a stable ruthenium nitrite complex from the ruthenium nitrosyl nitrate precursor in the presence of sodium hydroxide, which avoids unwanted metal oxide hydrolysis or precipitation. In contrast, more conventional metal precursors such as chlorides or nitrates easily hydrolyse under basic conditions forming metal oxides or precipitates. We also demonstrate the need of achieving fast mixing of reactants (<50 ms) to enable a homogeneous nucleation under such fast reduction kinetics. This work is a demonstration of the need of combining reaction chemistry and engineering approaches on the synthesis of nanomaterials.
Introduction
Ruthenium (Ru) is a transition and platinum group metal with a range of applications mainly in the electronics and catalysis sectors.1 It is commonly used as an alloying agent to strengthen titanium, platinum and palladium to prepare wear-resistant electrical contacts.2 In heterogeneous catalysis, ruthenium is gaining substantial attention for the synthesis3 and decomposition4–6 of ammonia, due to its interaction with nitrogen and ammonia being optimum according to the Sabatier principle. Indeed, ruthenium nanoparticles were at the centre of the so-called second generation of ammonia synthesis catalysts, however, they have only been deployed in isolated commercial cases due to its high cost and hydrogen poisoning at low conversion levels.7 In homogeneous catalysis, ruthenium and its complexes display activity for various reactions including acetic acid synthesis, water splitting, hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds, formic acid synthesis, alcohol dehydration, and oxidation reactions.8 Currently, the scarcity and high price of ruthenium, a rare metal, limits its widespread commercial application. Manipulating and maximising the nanoscale properties of ruthenium has the potential to increase its industrial relevance, particularly for the catalysis of structurally sensitive reactions, where activity strongly depends on metal nanoparticle size within a certain range, usually below 10 nm.9
C and CO bonds, formic acid synthesis, alcohol dehydration, and oxidation reactions.8 Currently, the scarcity and high price of ruthenium, a rare metal, limits its widespread commercial application. Manipulating and maximising the nanoscale properties of ruthenium has the potential to increase its industrial relevance, particularly for the catalysis of structurally sensitive reactions, where activity strongly depends on metal nanoparticle size within a certain range, usually below 10 nm.9
Traditional synthesis of heterogeneous catalysts relies on wetness preparation methods due to their simplicity and low cost10 where nanoparticles are synthesised directly on the surface of the support. The resulting size of the nanoparticles depends on a range of factors interacting in a complex manner, such as choice of metal precursor, pore volume and surface area of the support, support hydrophobicity and solvent, drying and calcination method. An alternative method is the synthesis of colloidal ruthenium nanoparticles and their post-synthesis deposition. A number of studies have demonstrated the synthesis of Ru NPs in a batch fashion, generally conducted via i. chemical reduction of a ruthenium salt with a borohydride agent11–15 or with a polyol (glycols or diols),14,16–21 and ii. organometallic decomposition of an organometallic ruthenium complex.22–33 However, past studies rely on the use of stabilisers and/or capping ligands to avoid agglomeration and control the size of Ru NPs,20 with potential detrimental effects on their final catalytic activity due to the blockage of active sites.28,34 For example, Simakova et al.34 reported the blocking of catalytic Ru active sites for the hydrogenation of arbinose and galactose due to the polyvinylpyrrolidone (PVP) stabiliser used in the polyol synthesis of the Ru NP colloids, compared to catalysts prepared by conventional incipient wetness impregnation. Jansat et al.28 also reported that the most active Ru NPs for catalysing the hydrogenation of unsaturated substrates were synthesised without amino stabilisers via organometallic decomposition.
An alternative and attractive approach for the negation of capping ligands is the synthesis of metal nanoparticles in flow microreactors, where laminar flow prevents their agglomeration under laminar flow. Our group has recently demonstrated the ligand-free flow synthesis of silver and gold nanoparticles with tuneable diameters and narrow size distribution in micro-flow devices through the chemical reduction of metal chlorides and nitrate precursors.35–37 However, this strategy is strongly dependent on the relative kinetics of nucleation and growth of the metal precursor and it is yet to be tailored to Ru NPs.
Alyami et al.38 demonstrated an interesting example of Ru NP flow synthesis with the average Ru NP size changing from 2.9 ± 0.5 nm with Ru(acac) precursor versus 4.8 ± 0.5 nm with a different RuCl3 precursor, however, oleylamine was used as a stabiliser in an organic solvent (toluene) under harsh conditions of 30 bar and 160 °C. The only example of ligand-free synthesis of Ru NPs was reported by Hu et al.39 whereby 1.5 nm sized NPs were prepared via the polyol method with ethylene glycol and the addition of sodium hydroxide (NaOH). In this case, it is likely that NaOH electrostatically stabilised the Ru NPs during the synthesis, as suggested by other ligand-assisted Ru NP synthesis studies13,21 which have use acids or bases to induce an environment of electrostatic repulsion between colloidal NPs. Hu et al.39 did not vary the average size of Ru, likely due to the limitations of batch synthesis to vary size while maintaining a narrow size distribution in the absence of capping ligands.
In this work, we present a new method to prepare colloidal ruthenium nanoparticles with precise sizes using a continuous/flow synthesis strategy in the absence of capping ligands. The Ru NPs are synthesised in a 3D microscale coiled flow inverter reactor (CFIR) via the reduction of ruthenium nitrosyl nitrate with sodium borohydride, producing smaller average sizes than in batch. The pH speciation of the metal precursor is found to be just as important as the kinetics of nucleation and growth. A mechanism is proposed for the NP synthesis involving the formation of a stable ruthenium nitrite complex in the presence of sodium hydroxide, which avoids unwanted metal oxide hydrolysis or precipitation. Manipulating the fluid dynamics of the flow system allows for the tuning of average size with narrow size distributions from 2.1 ± 0.1 nm to 2.9 ± 0.5 nm to 3.9 ± 0.5 nm. This demonstrates a departure from traditional Ru NP synthesis methods and presents a strategy to isolate the parameter of nanoparticle size for size-effect catalytic investigations.
Experimental section
Materials
Ruthenium nitrosyl nitrate solution (Ru(NO)(NO3)3, 1.5 w/v% in dilute nitric acid), alkaline sodium borohydride solution (NaBH4, ∼12 wt% in 14 M sodium hydroxide), powdered sodium borohydride (NaBH4, ≥98.0%), sodium hydroxide solution (NaOH, 0.1 M, 95–100%), powdered sodium hydroxide (NaOH, 97%), polyvinylpyrrolidone powder (PVP, average Mr 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and bovine serum albumin powder (BSA, lyophilized, ≥96%) were purchased from Sigma Aldrich. Nitric acid solution (HNO3, 1 M) was purchased from Fisher Scientific. All chemicals were used without further purifications within 3 months of purchase to ensure fresh solutions free of degradation. Ultra-high-purified 18.2 MΩ cm MilliQ water was used in preparing all solutions and washing.
000), and bovine serum albumin powder (BSA, lyophilized, ≥96%) were purchased from Sigma Aldrich. Nitric acid solution (HNO3, 1 M) was purchased from Fisher Scientific. All chemicals were used without further purifications within 3 months of purchase to ensure fresh solutions free of degradation. Ultra-high-purified 18.2 MΩ cm MilliQ water was used in preparing all solutions and washing.
Synthesis of colloidal Ru NPs
Ruthenium nanoparticles (Ru NPs) were synthesised via wet reduction of ruthenium precursors (Ru(NO)(NO3)3 and RuCl3) using NaBH4 as a reducing agent. In a typical flow synthesis, 2.5 mM Ru(NO)(NO3)3 and 6.25 mM of alkaline NaBH4 aqueous solutions were each fed at 115 mL h−1 by a Chemyx Fusion 6000 syringe pump through a continuous coiled flow inverter reactor (CFIR) (Fig. 1).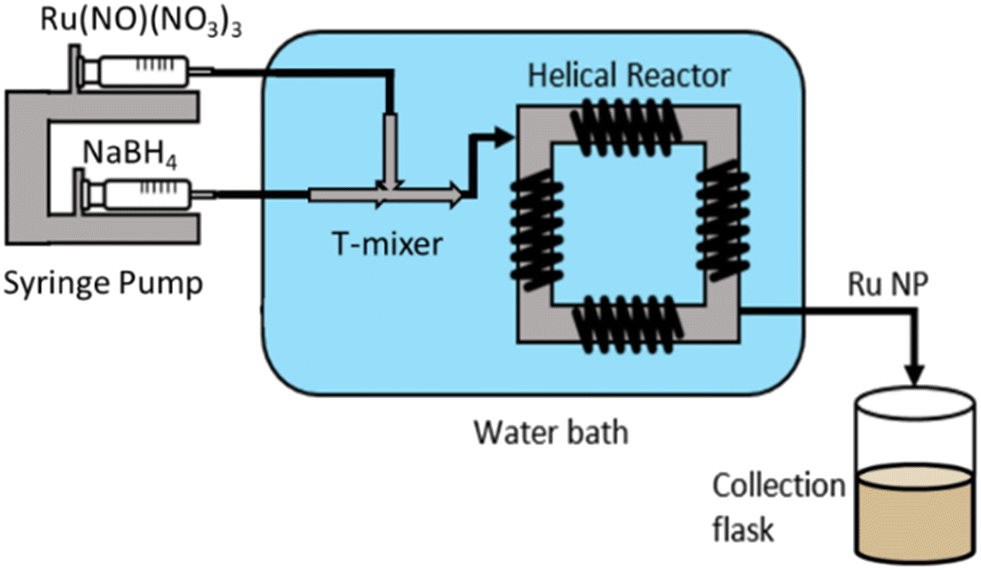 | ||
| Fig. 1 Ru NP continuous reactor set up: two syringe pumps control flowrates of Ru(NO)(NO3)3 and NaBH4 solutions. Reactants were mixed with a T-mixer in crossflow configuration and the resulting solution passes through the coiled flow inverter reactor (CFIR). | ||
The precursors entered a 0.02-inch T-mixer in a crossflow configuration, where the Ru-containing stream entered the system perpendicular to the resulting direction of flow. The resulting solution passed through the CFIR, consisting of 3.16 m of perfluoro alkoxy (PFA) tubing (0.02-inch inner diameter and 1 cm outer diameter) coiled around a 3D printed support (printed using a Formlabs Form 2 3D stereolithographic printer with a high temperature resin and 0.1 mm resolution). Both the mixer and the reactor were immersed in a water bath maintained at 25 °C. Steady-state conditions were assumed after 2 min of operation, after which samples were collected. For post-synthesis characterisation, 5 mL of Ru NPs were then collected in a glass vial containing 0.56 mL of 1 M HNO3 solution (final HNO3 concentration of 0.1 M) to instantaneously decompose any excess of NaBH4 to prevent any possible further reaction, and to provide electrostatic stabilisation.
For comparison, a batch synthesis method was used whereby the same precursor solutions were simultaneously added into a round bottle flask maintained at 25 °C with a 500 rpm magnetic stirrer. The solutions reacted during 10 s (emulating the residence time in the standard flow synthesis) before the addition of nitric acid to quench the reaction (final HNO3 concentration of 0.1 M). In an initial batch synthesis screening of different molar ratios of NaBH4:Ru, 2 mL of each precursor solution was ejected simultaneously from pipettes into a glass vial at room temperature. The pH values of the precursors were measured, and the pH values of mixed solutions were measured 10 s after mixing, after which UV-vis spectrums were obtained for the solutions that did not display immediate large agglomeration.
Characterisation
pH measurements were carried out with a Fisherbrand™ accumet™ XL150 pH Benchtop Meter with a Tris-compatible double junction refillable glass-body pH electrode. Synthesised Ru NP suspensions were analysed by Dynamic Light Scattering (DLS) to estimate average size and zeta potential (ZP) to assess colloidal stability using a Malvern Zetasizer Nano S90 instrument equipped with a 633 nm laser. Ultraviolet-visible (UV-vis) spectrometry was conducted with an Agilent Cary-60 UV-vis spectrometer. Transmission Electron Microscopy (TEM) was conducted with a Talos, FEI microscope (200 kV), with samples prepared by drop-casting 3 μl of colloidal Ru NP suspension onto a carbon-coated copper TEM grid (400 mesh, Agar Scientific) and drying naturally for 30 min. Particle size distribution histograms were constructed using ImageJ open-source software using a minimum of 200 nanoparticles from at least 3 TEM images taken from different mesh sections of the TEM grid. Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) was used to measure the concentration of Ru in solution using a Thermo-Fisher Nexion 2000-S machine.Results and discussion
Ruthenium nanoparticles were synthesised in a flow reactor by reduction of Ru(NO)(NO3)3 with NaBH4 to produce Ru(0) as shown by eqn (1).40| 8Ru3+ + 3BH4− + 12H2O → 8Ru + 3B(OH)4− + 24H+ | (1) |
A total flowrate of 230 mL min−1 (equal flowrate for both inlet streams) results on a Reynolds number of 179 and a Dean number of 40, confirming laminar flow and the formation of Dean vortexes respectively. An initial reaction concentration of Ru(NO)(NO3)3 and NaBH4 of 1.25 mM and 3.13 mM were used respectively, the latter containing 0.03 M NaOH. As a result, the pH during the synthesis is 9.5, avoiding the fast deposition of NaBH4 by hydrolysis under non-alkaline conditions (eqn (2)).43
| NaBH4 + 4H2O → H3BO3 + NaOH + 4H2 | (2) |
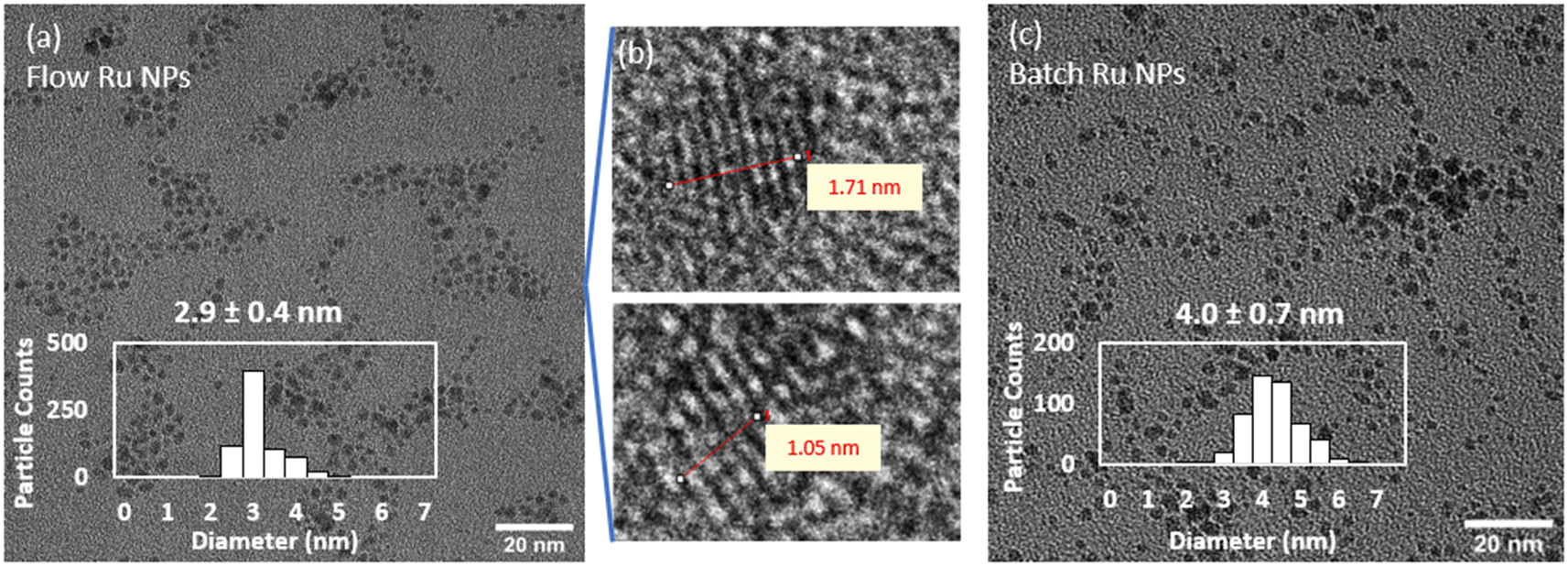 | ||
| Fig. 2 Representative TEM images of (a) flow synthesis of Ru NPs collected in HNO3 (b) lattice fringes from flow-synthesised Ru NPs showing spacing of 0.21 nm corresponding to hexagonal close-packed Ru(0), and (c) comparative batch synthesis. Conditions: 1.25 mM Ru(NO)(NO3)3 and 3.13 mM NaBH4, pH upon mixing: 9.5, pH after HNO3 quench: 1.3, 25 °C. | ||
For comparison purposes, a batch synthesis of Ru NPs was carried out under the same conditions by quickly mixing the Ru- and the NaBH4-containing streams together in a round bottom flask reactor. Similarly, the reaction was quenched after 10 s by adding a HNO3 solution (0.1 M final concentration). A representative picture of the resulting Ru NPs is shown in Fig. 2c, showing slightly larger sizes and a broader size distribution (4.0 ± 0.7 nm) than the flow-synthesised counterpart. As demonstrated below, these differences are associated to the enhanced mixing rate achieved in flow reactors in comparison to batch ones.
As mentioned above, the pH during the flow synthesis was 9.5. This value was a result of mixing the Ru(NO)(NO3)3 aqueous solution with a pH of 1.2 (as received before diluting to the desired concentration) and the alkaline NaBH4 aqueous solution with pH of 15 (as received). Nanoparticle synthesis at different pH conditions resulted in either heavily agglomerated particles or no measurable conversion of Ru, which prompted us to study the effect of the precursors speciation under different pH conditions, an aspect previously overlooked in the literature.
Speciation of Ru(NO)(NO3)3 at different pH values
The speciation of Ru and NaBH4 are both dependent on pH. Ru3+ is thermodynamically stable in very acidic pH values below 1.8, above which it can hydrolyse forming Ru(OH)3.49 Hence, Ru(III) salt solutions are often stored under acidic conditions to maintain stability. Although there exists pH speciation studies for Ru3+ from RuCl3,49 the pH-speciation of Ru(NO)(NO3)3 has not been documented to the best of our knowledge. Ru(NO)(NO3)3 contains a nitrosyl (NO) group that is π back bonded to Ru.50 This is such a stable bond that the Ru–NO(III) group often behave like a single element.51 Indeed, we believe that the nitrosyl group (NO) is responsible of the small Ru NP observed as its bond to the Ru atom stabilises it against agglomeration during the nanoparticle synthesis. To prove this theory, palladium nanoparticles were synthesised using a counterpart K2[Pd(NO)(NO2)4(NO3)] leading to 4.9 ± 0.9 nm particles in a batch synthesis (Fig. S2†), comparable sizes to the ones achieved with Ru(NO)(NO3)3 precursor in batch (Fig. 2c).To investigate the strength of the nitrosyl ligand in the Ru(NO)(NO3)3, its hydrolysis was investigated under different pH values from 3.0 (after dilution to 2.5 mM) to 12.6 using NaOH. No colour change was observed across all pH values, nor the presence of particles by DLS. UV-vis measurements in Fig. S3a† reveal the appearance of an absorbance shoulder at ∼380–400 nm on the precursor spectrum, which increases in magnitude as pH increases, indicating changes in the structure of the Ru(NO)(NO3)3 complex. The nitrate (NO3−) groups can be easily replaced by OH− ligands upon addition of NaOH, however, there is not a complete Ru hydrolysis to form oxide particles or precipitates. This likely suggests the formation of a new complex as pH increases, whereby the stable nitrosyl (NO) group is hydrolysed into a nitrite group, as reported for other alkyl, amino, and cyano ruthenium nitrosyl complexes.50–52 Such partial hydrolysis of Ru(NO)(NO3)3 in basic conditions is partially reversible as the new UV-vis absorption shoulder at ∼380–400 nm partially relaxes over 24 hours (Fig. S3b†) although it does not fully return to its initial spectrum. Visually, a change in the colour of the solution is not apparent to the naked eye even for the most alkaline Ru(NO)(NO3)3 solution (Fig. S3c†). These observations are opposite to those for RuCl3 (ref. 53) or other metal nitrates54 which hydrolyse and form metal oxides particles or precipitates in basic pH conditions.
To investigate the effect of the reducibility of Ru(NO)(NO3)3 speciation at different pH values, the pH of 2.5 mM Ru(NO)(NO3)3 precursor solution was first varied between 1.8 to 12 and subsequently 6.25 mM NaBH4 solution (aqueous solutions of powdered NaBH4 in the absence of NaOH) was added (note that these conditions are similar to those in the flow synthesis above). Observations are summarised in Table 1. The most alkaline Ru(NO)(NO3)3 solution (pH 12) experienced no colour change within 20 min of NaBH4 addition with DLS results showing poor repeatability, substantially low count rates, and unstable raw correlogram plots due to low Ru conversion. However, agglomerates were observed after 20 h, suggesting that under alkaline conditions, the reduction reaction is very slow. Similar observations took place on the other side of the pH spectrum. The most acidic Ru(NO)(NO3)3 solution (pH 1.8) experienced no subsequent colour change after 20 min of NaBH4 addition and showed unstable DLS results, also indicating low Ru conversion. No agglomerates or colour change were observed even the following day, likely because the very acidic conditions instantly hydrolytically decomposed the NaBH4 solution, preventing any further reduction, as well as the high stability of Ru(NO)(NO3)3 under these conditions as previously discussed.
| Starting pH of Ru precursor solution (pHRu) | DLS results 30 min after mixing | Visual observations | |||
|---|---|---|---|---|---|
| Average size by number (nm) | Standard deviation (nm) | Mean count rate (kcps) | Indication of reduction | ||
| 1.8 | 81 | 49 | 4 | No colour change | |
| 3.3 | 4400 | 550 | 79 | ✓ | Immediate colour change and agglomeration |
| 5.4 | 240 | 13 | 420 | ✓ | Immediate colour change |
| 6.3 | 110 | 6 | 260 | ✓ | Colour change within 20 min |
| 8.4 | 59 | 1 | 180 | ✓ | Colour change within 20 min |
| 9.4 | 120 | 1 | 370 | ✓ | Colour change within 20 min |
| 9.7 | 80 | 0.2 | 330 | ✓ | Colour change within 20 min |
| 12 | 140 | 20 | 3 | No colour change, but agglomerates after 20 h | |
The Ru(NO)(NO3)3 solutions at pH values between 6.3–9.7, experienced a delay of ∼20 min for the colour change after NaBH4 addition with formation of relatively large particles (<150 nm) according to DLS. These suspensions showed repeatable DLS measurements across 3 scans (Fig. S4†) with stable raw correlogram plots, indicating the formation of stable NPs. On the other hand, the Ru(NO)(NO3)3 solutions with acidic pH values (3.3 and 5.4) showed immediate colour changes from light yellow to brown-grey upon NaBH4 addition, indicating faster reduction of the Ru precursor under these acidic conditions.11 However, agglomerates observable with a naked eye formed rapidly from the Ru solution of pH 3.3 (Fig. S4†).
These results suggest that the partial hydrolysis of Ru(NO)(NO3)3 with NaOH under basic conditions form Ru complexes with a slow reduction by NaBH4. However, NaOH is still needed to prevent the immediate hydrolytic decomposition of NaBH4 in acidic conditions. Therefore, a compromise in the NaOH concentration is required to achieve fast reduction for the formation of small NPs.
For comparison purposes, RuCl3, a more common precursor in the literature,11,13–15 was used for the synthesis of Ru NP colloids via NaBH4 reduction. The reduction of 2 mM RuCl3via dropwise addition of NaBH4 solution, in the absence of ligands, led to significant precipitation as soon as the pH rose above 5 (Fig. S5†), in contrast to previous observations.13 Attempts to maintain the pH of 2 mM RuCl3 under acidic conditions (through HCl addition) to prevent the aforementioned precipitation were also unsuccessful, resulting in initially large DLS sizes and significant agglomeration after 24 h (Table S2†).
Batch synthesis screening of NaBH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Ru molar ratios
:Ru molar ratios
The precursor concentrations used in the flow synthesis were selected after conducting an initial 6 point-screening in batch mixing conditions to investigate the effect of NaBH4:Ru molar ratios. The Ru(NO)(NO3)3 concentration was kept constant at 1.25 mM and the concentration of NaBH4 (prepared from NaBH4 in 14 M NaOH solution, with an NaBH4:NaOH ratio of 4.4) was varied to study NaBH4:Ru molar ratios in range of 1.5–3 – a sensitive region identified by preliminary tests. The reduction and colour change of the Ru precursor was followed with UV-vis spectroscopy. The formation of any agglomerates visible by the naked eye was recorded. Initial and final pH's upon mixing the precursors were measured. Results are summarised in Fig. 3 and Table 2.
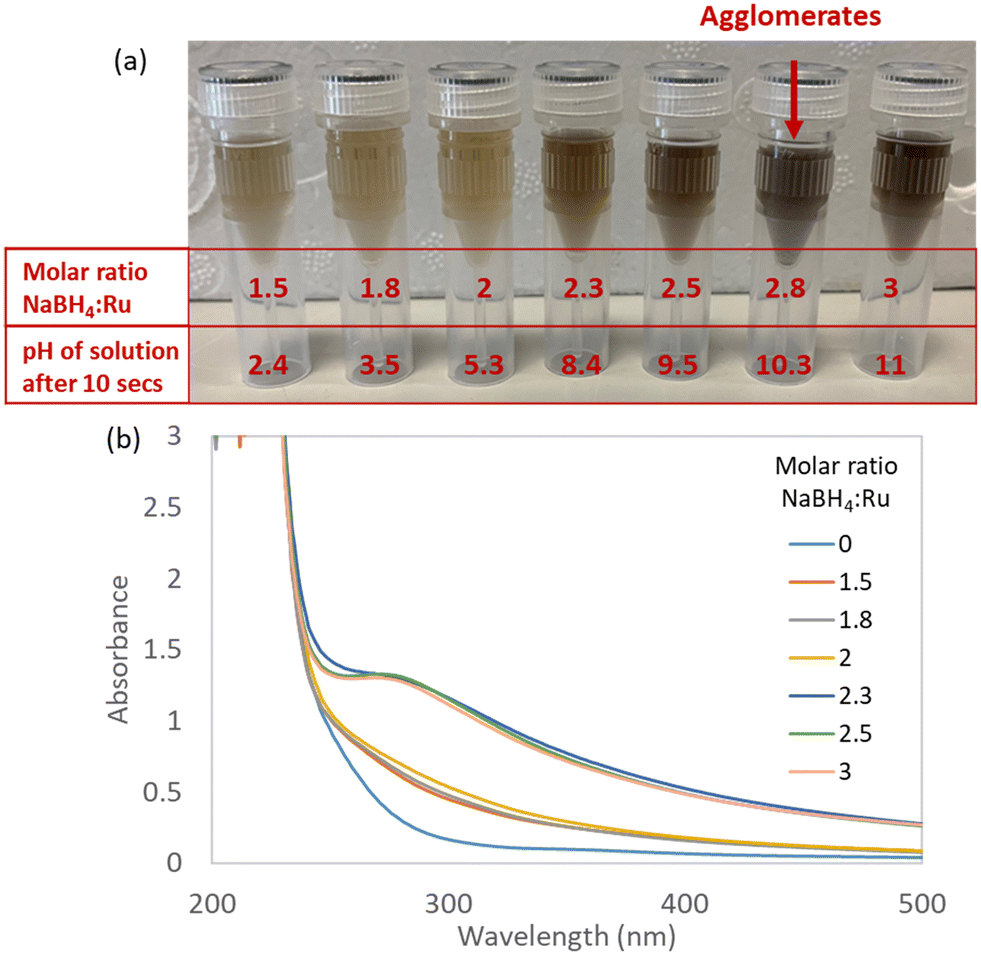 | ||
| Fig. 3 Effect of NaBH4:Ru ratio with NaOH present on Ru NP formation. Solutions of 1.25 mM Ru(NO)(NO3)3 and NaBH4 (prepared from NaBH4 in 14 M NaOH with an NaBH4:NaOH ratio of 4.4), mixed at room temperature in various NaBH4:Ru molar ratios from 0 to 3, with 0 corresponding to only water addition in the absence of NaBH4 (a) image of mixed solutions after 30 min of conducting experiments with pH measured after 10 s of mixing. (b) UV-vis absorbance spectra for mixed solutions after pH measurements. | ||
:Ru molar ratio with NaOH present
| Before mixing | Initially after mixing | pH of solution 10 s after NaBH4 addition | Molar ratio of NaBH4:Ru |
Visible agglomeration after 30 min (Y/N) | ||||
|---|---|---|---|---|---|---|---|---|
| Ru(NO)(NO3)3 solution | NaBH4 solution | [Ru] mM | [NaBH4] mM | |||||
| [Ru] mM | pH | [NaBH4] mM | pH | |||||
| 2.5 | 1.8 | 3.8 | 12.3 | 1.25 | 1.9 | 2.4 | 1.5 | N |
| 4.4 | 12.2 | 2.2 | 3.5 | 1.8 | N | |||
| 5.0 | 12.4 | 2.5 | 5.3 | 2 | N | |||
| 5.6 | 12.5 | 2.8 | 8.4 | 2.3 | N | |||
| 6.3 | 12.5 | 3.1 | 9.5 | 2.5 | N | |||
| 6.9 | 12.6 | 3.2 | 10.3 | 2.8 | Y | |||
| 7.5 | 12.6 | 3.8 | 11.0 | 3 | N | |||
At low NaBH4:Ru ratios of 1.5, 1.8 and 2, minor colour changes are observed which indicate incomplete reduction of the Ru precursor (Fig. 3). This is likely due to the instant hydrolytic decomposition of the NaBH4 reducing agent, as indicated by the acidic final pHs for these molar ratios of 2.4, 3.5 and 5.3, respectively (Table 2). At medium NaBH4:Ru ratios of 2.3 and 2.5, a colour change from light yellow to brown-grey (Fig. 3) is observed within seconds of mixing, indicating reduction of Ru(III) to Ru(0).11 No agglomerates are observed with the naked eye. The corresponding UV-vis spectra for NaBH4:Ru ratios of 2.3 and 2.5 show a shoulder between 280–300 nm, characteristic of OH– anions stabilising Ru NPs18,21 – which agrees with the final pH becoming basic at exactly these ratios (Table 2).
At the highest NaBH4:Ru ratios of 2.8 and 3, immediate large agglomerates are observed for the ratio of 2.8 but not for the ratio of 3 (Fig. 3). For the highest NaBH4:Ru ratio of 3, the reduction of the Ru precursor is likely inhibited by the formation of the stable Ru nitrite complex under the basic conditions (final pH of 11.0, Table 2), as discussed in the previous section on the pH speciation of the Ru precursor. This reveals that the Ru complex formation occurs faster than reduction at these conditions.
Based on these initial screenings, a NaBH4:Ru molar ratio of 2.5 is selected for flow synthesis to achieve sufficient conversion of the Ru precursor while avoiding the formation of agglomerates or the stable Ru nitrite complex. Both NaBH4:Ru molar ratios of 2.3 and 2.5 are deemed suitable, however, 2.5 is selected based on ensuring that there is an excess of NaBH4 to effectively reduce the stable Ru nitrite complex which forms in the presence of NaOH. This ratio of 2.5 is substantially larger than the stoichiometric ratio of NaBH4:Ru of 0.375 for the reduction of Ru3+.
Transport phenomena (mixing) effects
The relatively fast reduction of Ru(NO)(NO3)3 by NaBH4 at a pH of 9.5 and the differences in Ru particle size in batch and flow reactors indicate that mixing time (i.e. the time required to achieve homogeneous mixing between both reactant streams) has a similar scale to the reaction time, an aspect usually overlooked in material synthesis.55 In these cases, a fast-mixing rate is critical to trigger a homogeneous nucleation of Ru NP in the whole reactor volume leading to narrow size distributions.56 Flow reactors are particularly suited to evaluate transport phenomena effects as their fluid dynamics are well-defined and can be manipulated easily by simply varying their design and operating conditions. To understand these effects, Ru NPs were synthesised in flow systems under the same conditions (1.25 mM Ru(NO)(NO3)3 and 3.13 mM NaBH4, pH upon mixing: 9.5, 25 °C), but with varying reactor design parameters and operating conditions as summarised in Table 3, alongside resulting Ru particle size and distribution (TEM). Further DLS and zeta potential characterisation is provided in Table S3.† The disparity between the DLS and TEM results in Table S3† are because the DLS measurements have a low limit of particles size of ∼10 nm.57 DLS measures the hydrodynamic sizes of particles, being particularly sensitive to the presence of large particles (a sixth-power relationship).44,58 Hence, DLS for these sub 4 nm Ru NPs should only be used qualitatively, as an initial indication of the presence of a NP colloidal suspension before conducting TEM to obtain more accurate quantitative results.| Experiment | Total flowrate (mL h−1) | Residence time (s) | Reactor detailsa | TEM results | ||||
|---|---|---|---|---|---|---|---|---|
| T-mixer | Helix diameter (cm) | Reynolds number | Dean number | Average size (nm) | Standard deviation (nm) | |||
| a Reactor length: 3.16 m, reactor internal diameter: 0.02 inch, temperature of water bath: 25 °C. | ||||||||
| 1 | 230 | 10 | 0.02′′ | 1 | 179 | 40 | 2.9 | 0.5 |
| 2 | 115 | 20 | 0.02′′ | 1 | 90 | 20 | 3.9 | 0.5 |
| 3 | 20 | 115 | 0.02′′ | 1 | 16 | 4 | 4.0 | 1.4 |
| 4 | 230 | 10 | 0.01′′ | 1 | 179 | 40 | 2.1 | 0.3 |
| 5 | 230 | 10 | 0.05′′ | 1 | 179 | 40 | 3.2 | 0.4 |
| 6 | 230 | 10 | 0.02′′ | 10 | 179 | 13 | 3.0 | 0.5 |
In each size control experiment, the Ru NPs were electronically stabilised in the presence of HNO3 in the receiving outlet vial (0.1 M final concentration). The acidic conditions also quenched the reaction by rapidly decomposing any NaBH4 remaining in the solution. Keeping constant the flow system configuration and decreasing the total precursors flowrate from 230 to 115 ml h−1 leads to an increase in particle size from 2.9 ± 0.4 to 3.9 ± 0.5 (experiments 1 and 2, Table 3, Fig. 4). Further decrease of the total flowrate to 20 ml h−1 does not lead to further increases of sizes but instead a considerably broader size distribution, 4.0 ± 1.4 nm (experiment 3, Table 3, Fig. 4). This relative upper limit of ∼4 nm agrees with other observations in the literature using NaBH4 as a strong reducing agent.11,13–15 Increasing total flowrate has two convoluted effects on mixing. On one hand, it increases the early mixing in the T-mixer promoted by a higher level of engulfment of the streams.59 On the other hand, it also promotes a higher level of mixing in the reactor itself by increasing the magnitude of the Dean vortexes formed in the coiled flow inverter reactor. Dean vortexes consist of the rotation of the fluid enhancing the radial mixing. They are created by the centripetal force experienced by fluids flowing inside curved reactor. The dimensional Dean number  provides a quantification of the difference forces acting in the fluid and represent the magnitude of the vortices.41 As the Dean number is directly proportional to the Reynolds number, an increase of the flowrate increases the magnitude of the vortices and thus, the mixing in the reactor (within laminar flow).
provides a quantification of the difference forces acting in the fluid and represent the magnitude of the vortices.41 As the Dean number is directly proportional to the Reynolds number, an increase of the flowrate increases the magnitude of the vortices and thus, the mixing in the reactor (within laminar flow).
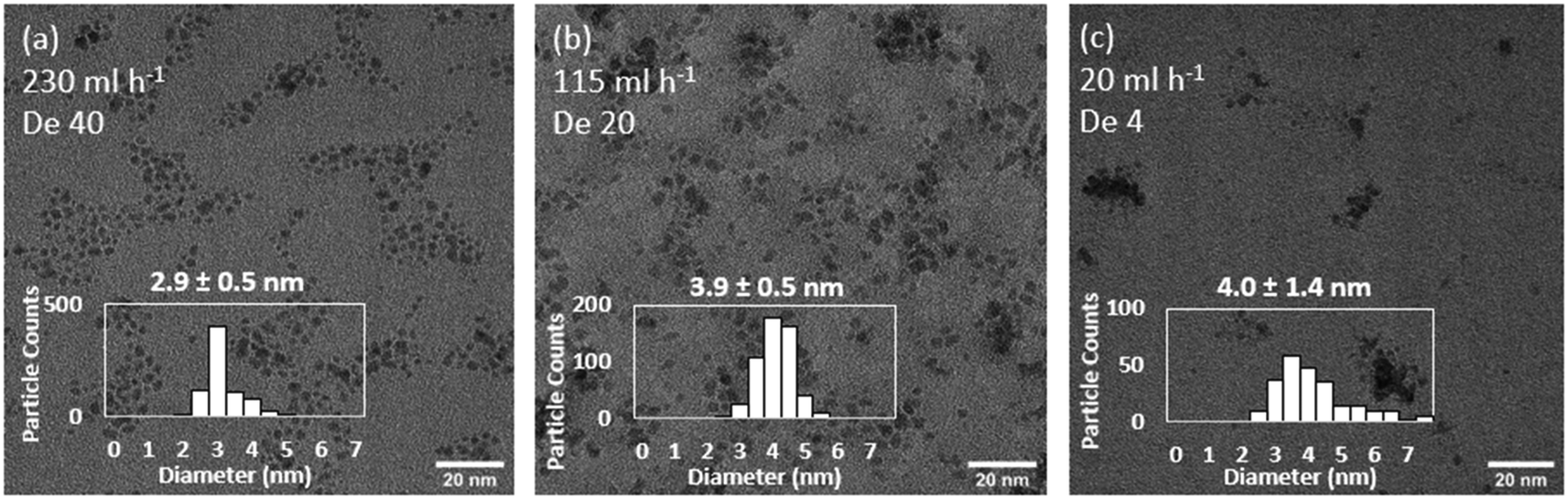 | ||
| Fig. 4 Effect of total flowrate of (a) 230 ml h−1, (b) 115 ml h−1 and (c) 20 ml h−1 on the size and distribution of continuously synthesised Ru NPs. Representative TEM images. Experiments 1–3, Table 3. | ||
To decouple the mixing effects in the T-mixer and the reactor, the total flowrate (230 mL h−1) and reactor configuration (CFIR) was fixed, while the internal diameter of the T-mixer was varied between 0.01 and 0.05′′ (experiments 1, 4 and 5, Table 3, Fig. 5). The Ru NPs sizes increased from 2.1 to 2.9 and 3.1 nm as the T-mixer internal diameter increased from 0.01 to 0.02 to 0.05′′ respectively. In all cases, a similar size distribution of ±0.3–0.4 nm was measured (Fig. 5). As the fluid dynamics in the reactor is the same in the three experiments, with constant Reynolds and Dean numbers (179 and 40 respectively), any differences in size can only be attributed to the early mixing in the T-mixer, within the first 50 ms. As a result, it can be concluded the faster the mixing rate (i.e. rate at which homogeneous mixing is achieved), the faster the average nucleation rate which leads to a higher number of nuclei and consequently a smaller resulting size of Ru NPs. Similar conclusions have been confirmed using computational fluid dynamic simulations for gold nanoparticles37 and perovskite nanocrystals.60
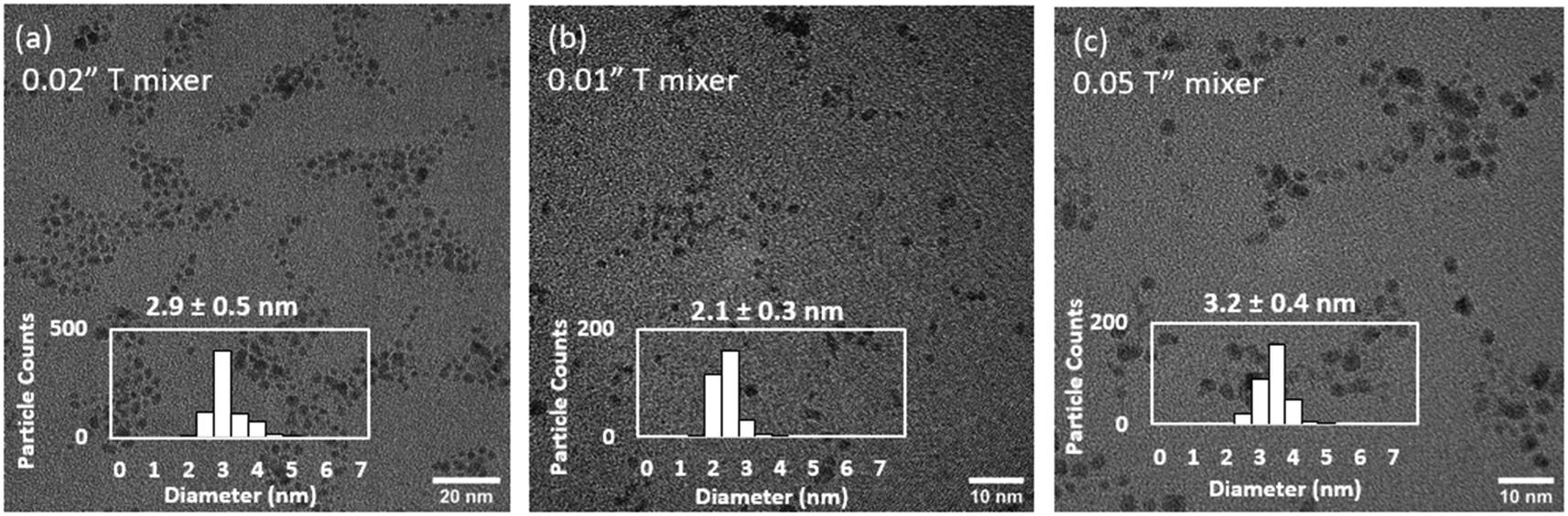 | ||
| Fig. 5 Effect of internal diameter of T-mixer of (a) 0.02′′ (b) 0.01′′ and (c) 0.05′′ on determining early mixing rate (within 50 ms) on the size and distribution of continuously synthesised Ru NPs. Representative TEM images. Experiments 1, 4, 5, Table 3. | ||
On the other hand, keeping constant the flowrate (230 mL h−1) and T-mixer (0.02′′) but varying the curvature of the coiled reactor from 1 cm to 10 cm (experiments 1 and 6, Table 3) led to almost negligible differences in the resulting particle size and distribution (2.9 ± 0.4 nm and 3.0 ± 0.5 nm respectively, Fig. S6†) despite the considerable differences in Dean number (40 and 13 respectively) and thus, radial mixing in the reactor. It is important to note that the flow inversions were omitted in the latter experiment to further minimise any radial mixing. The high early mixing rate promoted at high Reynolds number in the T-mixer likely leads to a homogeneous distribution of nuclei across the reaction volume promoting homogeneous growth in the reactor, independent on any further passive mixing during this stage (>50 ms). Further comparison of experiments 2 and 3 (Table 3) where the synthesis of Ru NPs were carried out in the same flow set-up but with decreasing flowrates (58 and 10 mL h−1 leading to 3.9 ± 0.5 nm and 4.0 ± 1.4 nm respectively) demonstrates that in order to achieve a homogeneous growth in the reactor and thus, narrow size distribution, it is critical to have previously achieved a high level of mixing during the nucleation stage. It must be noted that due to the absence of stabilisers, there is also the possibility that the inner walls of the reactor function as sites for heterogeneous nucleation of the NPs. Further research is needed to investigate this synthetic avenue.
Conclusions
A novel flow synthesis method has been developed to produce electrostatically stable small and tuneable (2–4 nm) Ru nanoparticles with narrow size distributions in the absence of capping ligands. The use of Ru(NO)(NO3)3 as a ruthenium precursor avoids the formation of aggregates during the synthesis due to the formation of a stable intermediate ruthenium nitrite (NO2−) from the nitrosyl group (NO) in the presence of NaOH. An understanding of the speciation of this ruthenium precursor as a function of pH reveals that a balance in NaOH concentration is required. An NaOH deficit results in the fast degradation of the sodium borohydride reducing agent while an NaOH excess slows down the reduction. The synthesis of small and narrow size dispersed nanoparticles under such fast reduction kinetics requires a fast early mixing (<50 ms) to enable a fast and homogeneous nucleation across the reaction volume. Manipulating such early mixing (e.g. by varying the mixer configuration for the inlet streams) leads to size tuneability between 2 and 4 nm. Homogeneous growth is also required to achieved narrow size distributions, but this can happen either in the mixer or throughout the whole synthesis time.Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.Acknowledgements
The authors greatly acknowledge the financial support from UK Engineering and Physical Science and Research Council (grant numbers EP/L020432/2 and EP/V025759/1) and the Department of Chemical Engineering and Biotechnology at the University of Cambridge for JEK's PhD scholarship.References
- Royal Society of Chemistry, Ruthenium, https://www.rsc.org/periodic-table/element/44/ruthenium, (accessed 19/04/ 2023) Search PubMed.
- Thomas Jefferson National Accelerator Facility, The element ruthenium, https://education.jlab.org/itselemental/ele044.html, (accessed 19/04/ 2023) Search PubMed.
- C. Smith and L. Torrente-Murciano, Adv. Energy Mater., 2021, 11, 2003845 CrossRef CAS.
- T. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- A. K. Hill and L. Torrente-Murciano, Appl. Catal., B, 2015, 172, 129–135 CrossRef.
- Z. G. Hu, J. Mahin, S. Datta, T. E. Bell and L. Torrente-Murciano, Top. Catal., 2019, 62, 1169–1177 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- M. R. Axet and K. Philippot, Chem. Rev., 2020, 120, 1085–1145 CrossRef CAS PubMed.
- M. P. C. van Etten, B. Zijlstra, E. J. M. Hensen and I. A. W. Filot, ACS Catal., 2021, 11, 8484–8492 CrossRef CAS PubMed.
- J. R. A. Sietsma, A. Jos van Dillen, P. E. de Jongh and K. P. de Jong, in Studies in Surface Science and Catalysis, ed. E. M. Gaigneaux, M. Devillers, D. E. De Vos, S. Hermans, P. A. Jacobs, J. A. Martens and P. Ruiz, Elsevier, 2006, vol. 162, pp. 95–102 Search PubMed.
- R. Herbois, S. Noël, B. Léger, S. Tilloy, S. Menuel, A. Addad, B. Martel, A. Ponchel and E. Monflier, Green Chem., 2015, 17, 2444–2454 RSC.
- T. Tsukatani and H. Fujihara, Langmuir, 2005, 21, 12093–12095 CrossRef CAS PubMed.
- J. Yang, J. Y. Lee, T. C. Deivaraj and H.-P. Too, J. Colloid Interface Sci., 2004, 271, 308–312 CrossRef CAS PubMed.
- J. Yang, T. C. Deivaraj, H.-P. Too and J. Y. Lee, Langmuir, 2004, 20, 4241–4245 CrossRef CAS PubMed.
- W. Yu, M. Liu, H. Liu, X. Ma and Z. Liu, J. Colloid Interface Sci., 1998, 208, 439–444 CrossRef CAS PubMed.
- Y. Chen, K. Y. Liew and J. Li, Mater. Lett., 2008, 62, 1018–1021 CrossRef CAS.
- N. Chakroune, G. Viau, S. Ammar, L. Poul, D. Veautier, M. M. Chehimi, C. Mangeney, F. Villain and F. Fiévet, Langmuir, 2005, 21, 6788–6796 CrossRef CAS PubMed.
- R. Harpeness, Z. Peng, X. Liu, V. G. Pol, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2005, 287, 678–684 CrossRef CAS PubMed.
- G. Viau, R. Brayner, L. Poul, N. Chakroune, E. Lacaze, F. Fiévet-Vincent and F. Fiévet, Chem. Mater., 2003, 15, 486–494 CrossRef CAS.
- X. Yan, H. Liu and K. Y. Liew, J. Mater. Chem., 2001, 11, 3387–3391 RSC.
- Y. Wang, J. Ren, K. Deng, L. Gui and Y. Tang, Chem. Mater., 2000, 12, 1622–1627 CrossRef CAS.
- G. Salas, C. C. Santini, K. Philippot, V. Collière, B. Chaudret, B. Fenet and P. F. Fazzini, Dalton Trans., 2011, 40, 4660–4668 RSC.
- P. S. Campbell, C. C. Santini, D. Bouchu, B. Fenet, K. Philippot, B. Chaudret, A. A. H. Pádua and Y. Chauvin, Phys. Chem. Chem. Phys., 2010, 12, 4217–4223 RSC.
- G. Salas, A. Podgoršek, P. S. Campbell, C. C. Santini, A. A. H. Pádua, M. F. Costa Gomes, K. Philippot, B. Chaudret and M. Turmine, Phys. Chem. Chem. Phys., 2011, 13, 13527–13536 RSC.
- T. Gutel, C. C. Santini, K. Philippot, A. Padua, K. Pelzer, B. Chaudret, Y. Chauvin and J.-M. Basset, J. Mater. Chem., 2009, 19, 3624–3631 RSC.
- I. Favier, S. Massou, E. Teuma, K. Philippot, B. Chaudret and M. Gómez, Chem. Commun., 2008, 3296–3298 RSC.
- T. Gutel, J. Garcia-Antõn, K. Pelzer, K. Philippot, C. C. Santini, Y. Chauvin, B. Chaudret and J.-M. Basset, J. Mater. Chem., 2007, 17, 3290–3292 RSC.
- S. Jansat, D. Picurelli, K. Pelzer, K. Philippot, M. Gómez, G. Muller, P. Lecante and B. Chaudret, New J. Chem., 2006, 30, 115–122 RSC.
- K. Pelzer, K. Philippot and B. Chaudret, Z. Phys. Chem., 2003, 217, 1539–1548 CrossRef CAS.
- K. Pelzer, O. Vidoni, K. Philippot, B. Chaudret and V. Collière, Adv. Funct. Mater., 2003, 13, 118–126 CrossRef CAS.
- C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante and M.-J. Casanove, J. Am. Chem. Soc., 2001, 123, 7584–7593 CrossRef CAS PubMed.
- O. Vidoni, K. Philippot, C. Amiens, B. Chaudret, O. Balmes, J. O. Malm, J. O. Bovin, F. Senocq and M. J. Casanove, Angew. Chem., Int. Ed., 1999, 38, 3736–3738 CrossRef CAS.
- P. Lara, K. Philippot and B. Chaudret, ChemCatChem, 2013, 5, 28–45 CrossRef CAS.
- I. L. Simakova, Y. S. Demidova, E. V. Murzina, A. Aho and D. Y. Murzin, Catal. Lett., 2016, 146, 1291–1299 CrossRef CAS.
- K. J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267–276 RSC.
- B. Pinho and L. Torrente-Murciano, Adv. Energy Mater., 2021, 11, 2100918 CrossRef CAS.
- Y. Gao, PhD Thesis, University of Cambridge, 2021 Search PubMed.
- N. M. Alyami, A. P. LaGrow, D. H. Anjum, C. Guan, X. Miao, L. Sinatra, D.-J. Yuan, O. F. Mohammed, K.-W. Huang and O. M. Bakr, Cryst. Growth Des., 2018, 18, 1509–1516 CrossRef CAS.
- X. C. Hu, X. P. Fu, W. W. Wang, X. Wang, K. Wu, R. Si, C. Ma, C. J. Jia and C. H. Yan, Appl. Catal., B, 2020, 268, 118424 CrossRef CAS.
- J. Y. Lee, J. Yang, T. C. Deivaraj and H.-P. Too, J. Colloid Interface Sci., 2003, 268, 77–80 CrossRef CAS PubMed.
- K. J. Wu, G. M. D. Bohan and L. Torrente-Murciano, React. Chem. Eng., 2017, 2, 116–128 RSC.
- R. Baber, L. Mazzei, N. T. K. Thanh and A. Gavriilidis, Nanoscale, 2017, 9, 14149–14161 RSC.
- S. Chen and K. Kimura, Langmuir, 1999, 15, 1075–1082 CrossRef CAS.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- V. R. Patel and Y. K. Agrawal, J. Adv. Pharm. Technol. Res., 2011, 2, 81–87 CrossRef CAS PubMed.
- S. Agarwal and J. N. Ganguli, RSC Adv., 2014, 4, 11893–11898 RSC.
- X. Zhou, T. Wu, B. Hu, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2009, 306, 143–148 CrossRef CAS.
- Q. Zhang, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, Nat. Commun., 2018, 9, 510 CrossRef PubMed.
- I. Povar and O. Spinu, J. Electrochem. Sci. Eng., 2016, 6, 135–143 CrossRef CAS.
- T. W. Hayton, P. Legzdins and W. B. Sharp, Chem. Rev., 2002, 102, 935–992 CrossRef CAS PubMed.
- A. Leroy and J. C. Morris, J. Inorg. Nucl. Chem., 1971, 33, 3437–3453 CrossRef CAS.
- J. L. Walsh, R. M. Bullock and T. J. Meyer, Inorg. Chem., 1980, 19, 865–869 CrossRef CAS.
- I. Povar and O. Spin, J. Electrochem. Sci. Eng., 2016, 6, 145–153 CrossRef CAS.
- J.-P. Jolivet, M. Henry and J. Livage, Metal oxide chemistry and synthesis: from solution to solid state, Wiley, Chichester, 2000 Search PubMed.
- B. Pinho, K. W. Zhang, R. L. Z. Hoye and L. Torrente-Murciano, Adv. Opt. Mater., 2022, 10, 2200524 CrossRef CAS.
- C. Casado, B. Pinho, J. Marugán and L. Torrente-Murciano, J. Chem. Eng., 2024, 479, 147684 CrossRef CAS.
- E. Tomaszewska, K. Soliwoda, K. Kadziola, B. Tkacz-Szczesna, G. Celichowski, M. Cichomski, W. Szmaja and J. Grobelny, J. Nanomater., 2013, 2013, 313081 Search PubMed.
- nanoComposix, NanoComposix's guide to dynamic light scattering measurement and analysis v1.4, https://docplayer.net/45227932-Nanocomposix-s-guide-to-dynamic-light-scattering-measurement-and-analysis.html, (accessed online 11/02/ 2023) Search PubMed.
- Y. Gao, D. F. Zhu, Y. Han and L. Torrente-Murciano, J. Flow Chem., 2021, 11, 589–598 CrossRef CAS.
- K. Zhang, Y. Gao, B. Pinho, R. L. Z. Hoye, S. D. Stranks and L. Torrente-Murciano, Chem. Eng. J., 2023, 451, 138752 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00585b |
| This journal is © The Royal Society of Chemistry 2024 |