
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetics of dehydrogenation of n-heptane over GaPt supported catalytically active liquid metal solutions (SCALMS)†
Oshin
Sebastian‡
a,
Asem
Al-Shaibani‡
 a,
Nicola
Taccardi
a,
Marco
Haumann
*ab and
Peter
Wasserscheid
*ac
a,
Nicola
Taccardi
a,
Marco
Haumann
*ab and
Peter
Wasserscheid
*ac
aFriedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Lehrstuhl für Chemische Reaktionstechnik (CRT), Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: marco.haumann@fau.de; peter.wasserscheid@fau.de
bResearch Centre for Synthesis and Catalysis, Department of Chemistry, University of Johannesburg, P.O. Box 524, Auckland Park 2006, South Africa
cForschungszentrum Jülich GmbH, Helmholtz-Institute Erlangen-Nürnberg for Renewable Energy (IEK 11), Egerlandstraße 3, 91058 Erlangen, Germany
First published on 30th January 2024
Abstract
The concept of Supported Catalytically Active Liquid Metal Solutions (SCALMS) was explored for the catalytic dehydrogenation of n-heptane. For this purpose, a GaPt on alumina (Ga84Pt/Al2O3) was compared with a Pt on alumina catalyst at different reaction temperatures and feed compositions. While the observed activation energies with both catalysts for the overall n-heptane depletion rate were similar with both catalysts, the SCALMS systems provides a lower activation energy for the desired dehydrogenation path and significantly higher activation energies for the undesired aromatization and cracking reaction. Thus, the SCALMS catalyst under investigation shows technically interesting features, in particular at high temperature operation. The partial pressure variation revealed an effective reaction order of around 0.7 for n-heptane for both catalysts, while the effective order for hydrogen was 0.35 for Pt/Al2O3 and almost zero for SCALMS.
Introduction
Dehydrogenation of n-paraffins with chain length >C6 is a promising method to the respective linear olefins,1 which have many applications in, e.g., fine and commodity chemicals as well as polymers.2 For example, C8–C10 olefins are converted in hydroformylation processes with syngas (H2 and CO) into plasticizer alcohols, while C10–C19 alkenes are utilized for the synthesis of biodegradable detergents.1,3 In 2022, the C4–C20 olefin market reached ∼9 billion US$ and is expected to reach ∼12 billion US$ by 2028.4Dehydrogenation of alkanes, being an endothermic equilibrium reaction, is preferred at high temperatures. Furthermore, low pressures are favorable to shift the equilibrium toward the product side. For longer-chain alkanes, several product isomers can be formed depending on the position of the double bond. In Scheme 1, this is illustrated for heptane dehydrogenation yielding five different heptene products. Out of the heptene isomers, 1-heptene has the lowest stability at typical dehydrogenation conditions of >673 K and quickly transforms into geometric isomers such as trans-2-heptene and trans-3-heptene.5 The equilibrium conversion of n-heptane at 723 K and 0.1 MPa is around 19.1% and yields 7.8% trans-2-heptene and 5.9% trans-3-heptene as major isomers, with 1-heptene being formed with only 1.2% (see Fig. S4 in ESI† for more information). The conversion of n-heptane can be increased to >50% when the reaction temperature is increased to >773 K. However, since the energy required to break the C–C bond (245 kJ mol−1) is much smaller than for breaking the C–H bond (365 kJ mol−1),1 the rate of thermal cracking reactions is accelerated at higher temperatures6,7 and the selectivity towards the desired olefins is reduced at such elevated temperatures.8
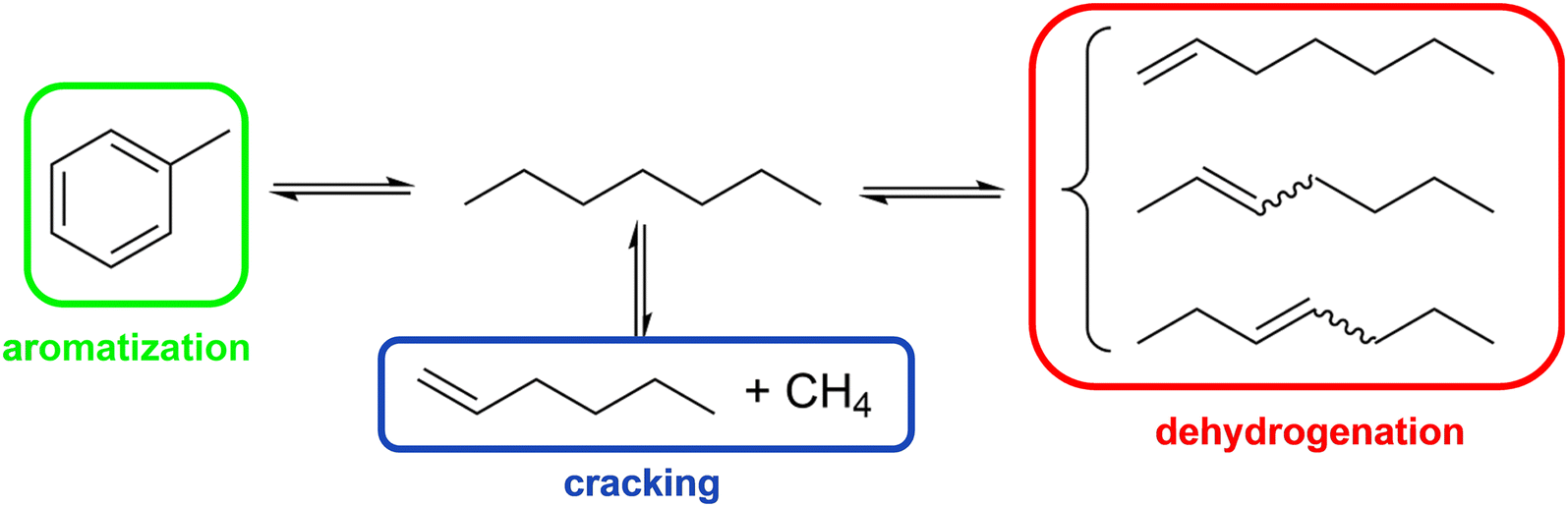 | ||
| Scheme 1 Possible reaction routes under typical n-heptane dehydrogenation conditions and resulting major products. | ||
It is widely accepted that with a typical dehydrogenation catalysts, such as Pt/Al2O3, the metal center promotes the dehydrogenation reactions, while the acidic support serves as site for isomerization.9 Hence, during the dehydrogenation of n-heptane using Pt/Al2O3, there is a multitude of competing reactions at play. The combination of reactions results in a broad spectrum of products. Direct dehydrogenation leads to the formation of C7n-olefins, hydroisomerization leads to the formation of C7iso-paraffins, dehydrocyclization results in C7 naphthenes, and hydrogenolysis/cracking leads to cracked products with less than 7 carbon atoms (the literature reported products in each of these families are shown in Table S2 in the ESI†).10,11
Platinum-based catalysts have been employed for catalytic reforming since the late 1940s. In the early 1960s, Sinfelt, Rohrer, and Hurwitz12,13 studied platinum on acidic aluminum oxide support using n-heptane and methylcyclopentane. Their work highlighted what they described as the characteristic effect of hydrogen.12 These authors showed that the amount of hydrogen in the feed has a critical effect on the n-heptane dehydrocyclization rate and selectivity. While too low hydrogen partial pressures lead to reduced activity due to coke formation, very high hydrogen partial pressures disfavor the intended dehydrogenation equilibrium for thermodynamic reasons. Sinfelt14 also demonstrated the acidic support to be the location of the isomerization reaction following a migration of alkenes from the metal sites. Furthermore, Rohrer and Sinflet15 revealed the effect of hydrogen in cleaning the metal surface from coke precursors.15 Later, in 1987, Bond16 and co-workers confirmed that Pt metal sites promote hydrogenation, dehydrogenation, and hydrogenolysis, while the acidic sites on Al2O3 support promote isomerization reactions. Based on this insight into the bifunctional nature of reforming catalysts, Liu17 and co-workers established in 2002 a kinetic model based on a lumped reaction network for n-heptane reforming on Pt–Re/Al2O3. Similarly, a comprehensive kinetic model for n-heptane reforming on Pt/Al2O3 catalyst was prepared by Olafadehan18 and co-workers in 2008. The rival kinetic models were discriminated using experimental data from Susu and Adenusi,6,18 and the best-fitting kinetic model indicated that the surface reaction of adsorbed iso-heptane to adsorbed methylcyclohexane is the rate-determining step for n-heptane reforming.18
In contrast to reforming, there is little kinetic information on the direct dehydrogenation of n-heptane to n-heptenes available in the literature. Recently, the group of Jess investigated the dehydrogenation of n-octane using a Pt/Al2O3 catalyst with modifiers (Sn, K, Ce).19 The formation of mono-olefins, di-olefins as well as aromatics was found, and a Langmuir–Hinshelwood mechanism was established, assuming the surface reaction to be the rate-determining step (RDS) with re-adsorption of formed products inhibiting the overall dehydrogenation.
A summary of the kinetic parameters reported in the literature for the conversion of n-heptane using Pt-based catalysts is provided in Table S1 in the ESI.† These kinetic parameters vary largely since they were determined under different operating conditions and with varying catalyst systems. These variations make the comparison between the reported parameters difficult. However, general trends can be identified. In particular, the activation energy for the overall reforming reactions is typically lower for monometallic Pt/Al2O3 catalysts (approx. 30–80 kJ mol−1) compared to bimetallic Pt–Sn/Al2O3 (approx. 110–200 kJ mol−1). Additionally, the activation energy for cracking reactions is higher than the one for isomerization and aromatization. The reaction order with respect to hydrogen is negative at low partial pressures but becomes positive at higher pressures.
To increase the selectivity towards linear olefins, the side reactions during alkane dehydrogenation must be restricted. This can be achieved by carefully designing the active sites. One way to achieve such selective active sites is to downsize the active metal nanoparticles to a “single atom” level (Fig. 1).20–26
 | ||
| Fig. 1 a) SEM-EDX mapping of GaPt located on the outer surface of Al2O3 support. b) Schematic representation of GaPt SCALMS catalysis at the gas-alloy interface. | ||
Recently, some of us have demonstrated that atomically dispersed single-atom sites can be stabilized under harsh conditions with the help of Supported Catalytically Active Liquid Metal Solutions (SCALMS).26 In SCALMS, the single atom sites of catalytically active metals (e.g., Pd,26 Rh,27 Pt,28,29 Ni,30etc.) are created by diluting them with large excess amount of low melting metal like gallium (Tmelt < 303 K). In these alloys, the active metal concentration is in the range of a few at%. These alloy droplets are dispersed onto a porous support, and under reaction conditions, they become liquid, thereby creating a dynamic liquid gas-alloy interface.31 The liquid nature of these alloy under reaction conditions has been proven by means of spectroscopy and microscopy.26–31 The unique properties of isolated Pt atoms in Ga matrix such as resistance to poison by CO have been well investigated by Bauer et al.32 using operando DRIFTS.32 More recently, we have studied effect of the synthesis parameters of GaPt SCALMS via ultrasonication on the rate of the galvanic displacement, thus on the morphology of the GaPt nanocomposites, and on the catalytic performance of the resulting materials in n-heptane dehydrogenation.33
Under reaction conditions, dopant active metal atoms exist as isolated homogeneous sites in the much less reactive liquid metal matrix and are considered to be analogous to single-atom alloys (SAAs).24 The properties of SCALMS are linked to the unique dynamic interface and modified electronic properties of isolated active metal atoms in the Ga matrix,26 which enhances its activity. In addition, the presence of uniform active sites makes SCALMS systems highly selective.26,31 GaPt SCALMS were shown to enhance the selectivity towards the desired products and reduce side reactions in propane28 and methylcyclohexane29 dehydrogenation.
In this contribution, we present the dehydrogenation of n-heptane using SCALMS systems. We determined the apparent reaction kinetics and reveal differences in the reaction kinetics of GaPt/Al2O3 SCALMS systems compared to Pt/Al2O3.
Experimental
All chemicals were used as received. Al2O3 support (Brockmann activity I, pH: 7.0 ± 0.5, particle size: 50–150 μm, Sigma Aldrich), gallium nuggets (size: 3 mm, purity: 99.999%, Materion), chloroplatinic acid hexahydrate (Pt basis: ≥37.50%, Sigma-Aldrich), and anhydrous propan-2-ol (purity: ≥99.8%, water content max.: 0.003%, VWR Chemicals) were used for catalyst preparation. The feedstock n-heptane (≥99%) was purchased from Honeywell.Catalysts preparation
GaPt/Al2O3 SCALMS was prepared using a top-down method described previously.28 Here, millimetre-sized Ga-nuggets were dispersed using ultrasonication into Ga nanodroplets (100–300 nm on average) in propan-2-ol. This dispersion was deposited onto an Al2O3 support by slowly evaporating the solvent under vacuum at 313 K using a rotary evaporator. To remove the solvent completely, the solid was calcined at 773 K under ambient conditions to produce Ga-decorated Al2O3. Pt was introduced into Ga-decorated Al2O3via partial galvanic displacement in a solution of hexachloroplatinic acid. The desired Ga to Pt atomic ratio was obtained by adjusting the amount of stock solution added. The solid obtained was then calcined at 773 K under air. Calcination resulted in a thin gallium oxide skin which, after the material has been placed into the reactor, was removed by pre-treatment with hydrogen (723 K, atmospheric pressure, 50 mLN min−1 H2 flow, 120 min).Pt/Al2O3 catalyst was prepared via the classical wet impregnation method. Al2O3 support was suspended in propan-2-ol, and the required amount of Pt precursor (hexachloroplatinic acid) was added. The solvent was then slowly evaporated under vacuum at 313 K using a rotary evaporator. The solid obtained was then calcined for 2 hours at 773 K under in air to produce the Pt/Al2O3 material that was placed in the catalytic reactor and pre-treated under hydrogen atmosphere.
Catalysts characterization
The chemical compositions of the prepared catalysts (see Table S3†) were determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES, Ciros CCD instrument, Spectro Analytical Instruments GmbH). The analysis method applied digesting the catalyst in a solution of hydrochloric acid, nitric acid, and hydrofluoric acid in volumetric ratios of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.
:1:1.
The surface area and pore size of the catalyst materials were determined by nitrogen sorption (QUADRASORB™ v5.06) at 77.35 K. Multipoint BET surface area was determined at a relative pressure range of 0.05 to 0.35. The coefficient of determination R2 was ensured to be above 0.999 for all samples. CO-temperature programmed desorption (CO-TPD) experiments were carried out on a Micromeritics Autochem II 2920 Chemisorb. Density functional theory (DFT) was employed to determine the pore size distribution (PSD).
Experimental set-up
Catalytic dehydrogenation of n-heptane was carried out in a fixed-bed tubular reactor. A continuous flow laboratory mini-plant was used for this purpose (see Fig. S1 and S2 in the ESI†). The catalyst material (2.5 g), prepared as described before, was placed in the isothermal zone of the tubular reactor (material: Inconel®, inner diameter: 10 mm, length: 500 mm) between two quartz wool beds. The tubular reactor was positioned inside an electrically heated tubular split furnace (Carbolite). Prior to each reaction, the catalyst was pre-treated under 50 mLN min−1 H2 (purity: 99.99%, Linde Gas) at 723 K for 2 hours. For SCALMS systems, this step minimizes the oxide skin of the supported Ga–Pt droplets formed during calcination. Following this pre-treatment, the reactor was flushed with 100 mLN min−1 He (purity: 99.996%, Linde Gas) for 30 minutes and allowed to cool down or heated up to the reaction temperature (683–743 K). Once the temperature stabilized, the reactor was sealed under the inert atmosphere, and the flow of reactants was started through the by-pass. Liquid n-heptane (0.062 g min−1) was evaporated in a stream of H2 and He. After stabilization, the reaction was started by closing the by-pass line and diverting the flow of the reactants to the reactor.The composition of the product stream was analyzed by gas chromatography (GC) using an Agilent 7820A GC equipped with a Restek Rtx®-100-DHA column (100 m, 0.25 mm ID, 0.5 μm coating) coupled with a Restek Rtx®-5-PONA tuning column (5 m, 0.25 mm ID, 1.0 μm coating) and a flame ionization detector (FID). Since the relative response factors of all the hydrocarbons were close to unity,34 the mole fractions of all components i (xi) were calculated directly from the peak areas. Further details, including products detected, their classification, and sample chromatogram, are provided in the ESI† (Fig. S3 and Table S2).
Kinetic measurements
The kinetics measurements were performed in a temperature range between 683 to 743 K. The n-heptane reaction order was determined between 25 and 100 mbar at an interval of 50 mbar while keeping H2 partial pressure (200 and 800 hPa) and temperature (703 K) constant. The reaction order of H2 was determined between 200 and 800 hPa at an interval of 200 hPa while keeping the n-heptane partial pressure (100 hPa) and temperature constant (703 K).The conversion of n-heptane Xheptane, the individual selectivities Si, and the product yields Yi were calculated according to eqn (1)–(3).
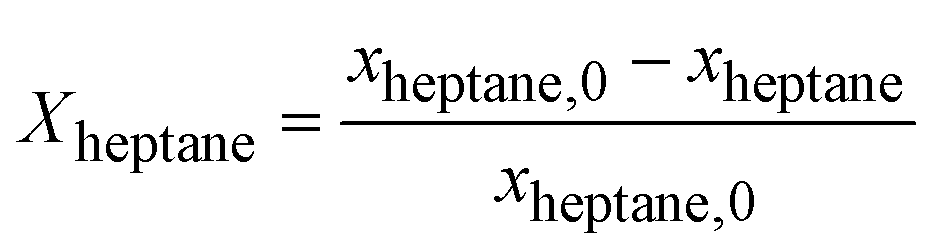 | (1) |
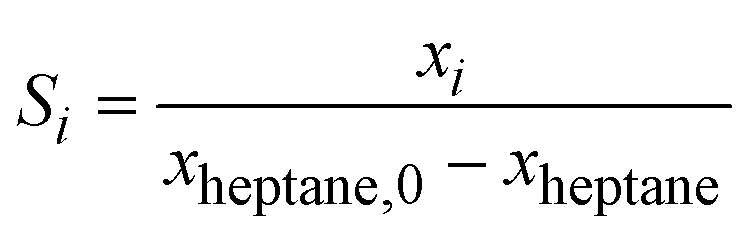 | (2) |
| Yi = Xheptane × Si | (3) |
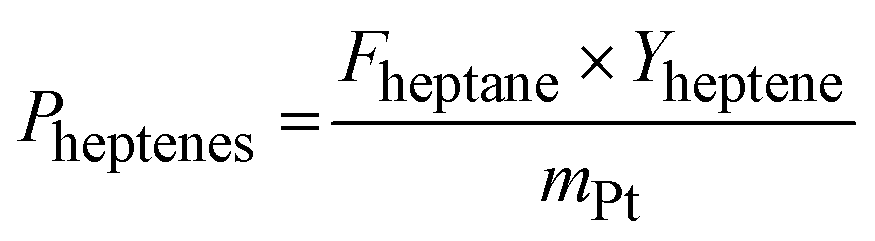 | (4) |
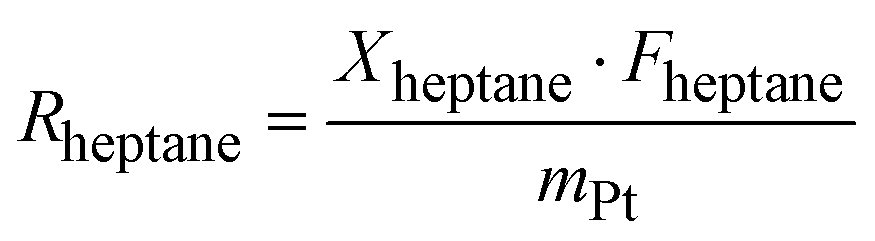 | (5) |
| Rheptane = k × p(H2)n × p(heptane)m | (6) |
Combining eqn (5) and (6) and linearizing the resulting equation yields eqn (7), from which the effective activation energy is obtained using an Arrhenius plot. Note that the initial rates have been used here.
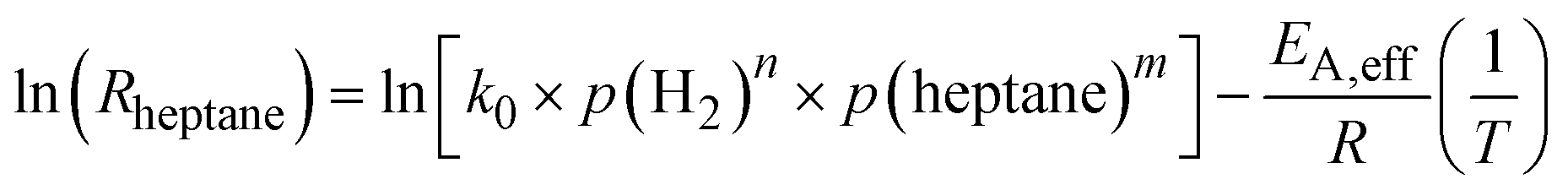 | (7) |
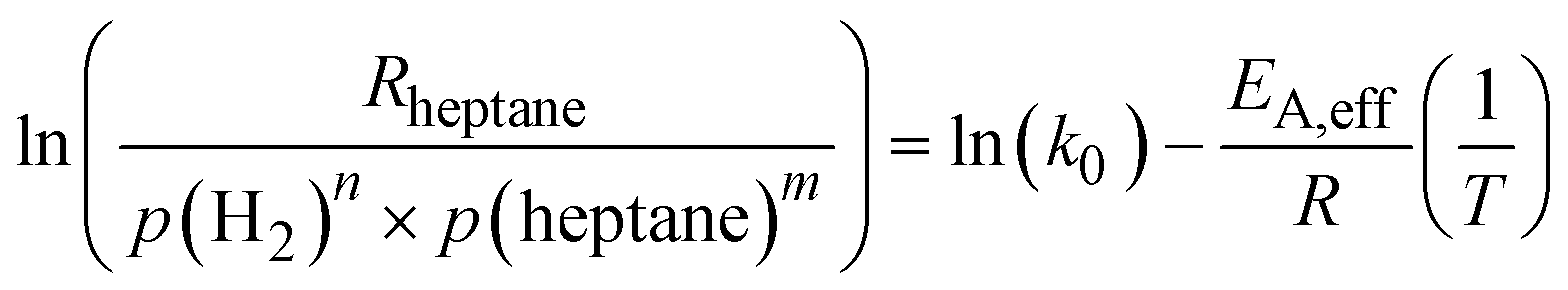 | (8) |
Results and discussion
Table 1 summarizes the textural characterization of the applied bare support and the two prepared catalysts. The bare Al2O3 support and both catalysts show very similar textural characterization (surface area, pore volume, and pore size). This indicates that neither the preparation of Pt/Al2O3 catalyst via wet impregnation nor the SCALMS synthesis via ultrasonication followed by galvanic displacement changed the textural properties of the support significantly. The dispersion of deposited Pt and GaPt was calculated from CO-TPD studies (see Table S8 in ESI† for details) and was found to be 1.8% for Pt/Al2O3 and 2.4% in case of GaPt SCALMS.| Material | S BET | V Pore | D Pore |
|---|---|---|---|
| m2 g−1 | cm3 g−1 | nm | |
| Al2O3 | 145.0 | 0.30 | 7.9 |
| Pt/Al2O3 | 145.7 | 0.30 | 7.4 |
| Ga84Pt/Al2O3 | 130.0 | 0.28 | 8.3 |
Prior to the start of our kinetic experiments, the blind activity of the reactor, the bare Al2O3 support, and of Ga supported on Al2O3 were tested. None of these blank experiments showed significant conversion of n-heptane (see Fig. S5 and S6 in ESI†).
Next, the performance and reproducibility of the n-heptane dehydrogenation reaction with the two Pt-based catalyst under investigation were evaluated. Fresh catalysts (Pt/Al2O3 and Ga84Pt/Al2O3 SCALMS) were tested at 703 K and atmospheric pressure. Fig. 2 reveals the surprising finding that both catalysts perform in a similar range in activity and n-heptenes selectivity. Still some important differences are notable.
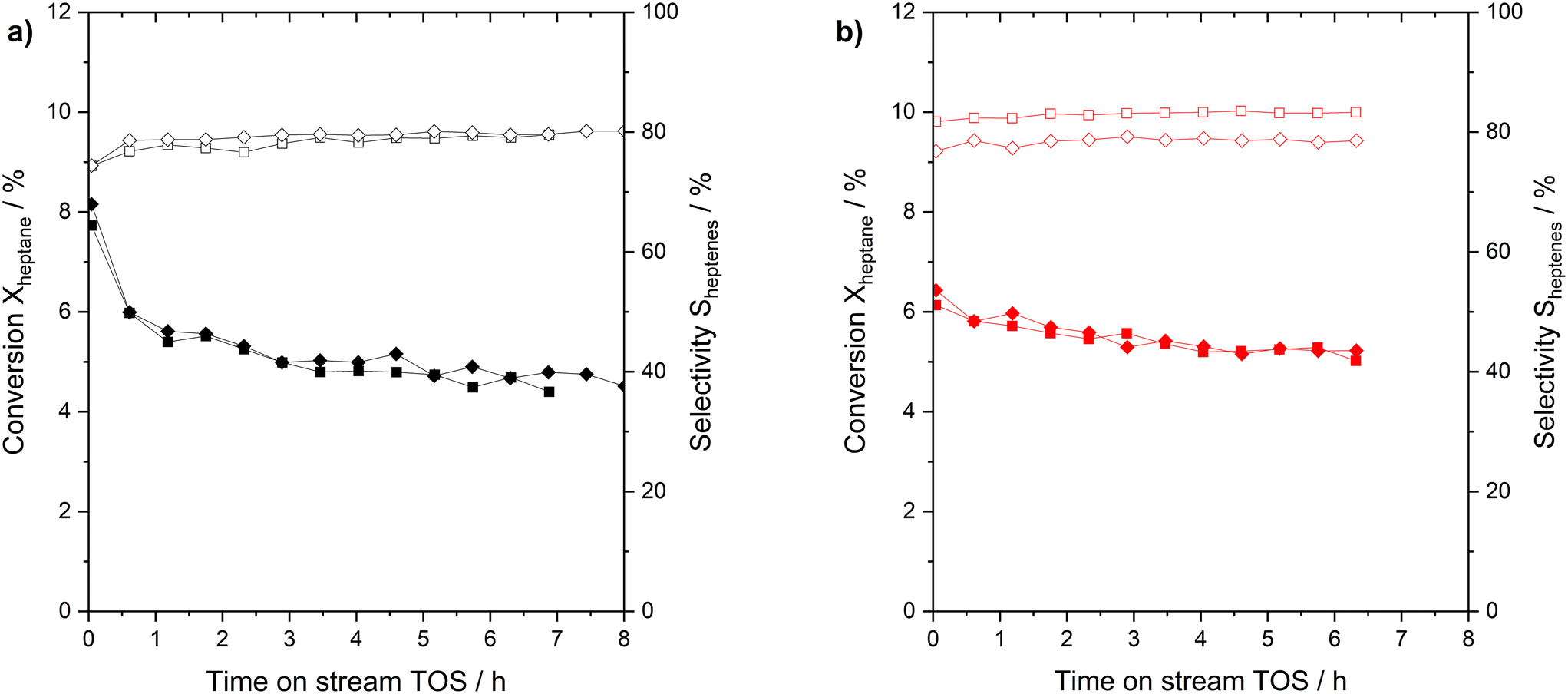 | ||
| Fig. 2 Performance and reproducibility studies for n-heptane dehydrogenation using a) Pt/Al2O3 (black symbols) and b) Ga84Pt/Al2O3 (red symbols). Conversion of n-heptane is indicated by filled symbols, and selectivity for n-heptenes is shown in open symbols. Pre-reduction conditions: 723 K, atmospheric pressure, 50 mLNmin−1 H2 flow, 120 min. Reaction conditions: 703 K, atmospheric pressure, 2.5 mL catalyst bed volume, 150 mLN min−1 total flow, 100 hPa n-heptane, 200 hPa H2, and 700 hPa He. | ||
Both runs with the Pt/Al2O3 catalyst gave initial conversions slightly above 8% which declined rapidly during the initial runtime and reached around 5% conversion after 7 h time on stream. The SCALMS catalyst showed a slightly lower initial activity with conversions of around 6%. It showed, however, a lower degree of deactivation over the 7 h time-on-stream so that the final conversion in the experiment was found even slightly higher than for the Pt/Al2O3 catalyst. Deactivation of solid catalysts in alkane dehydrogenation is mainly caused by coking at vicinal sites.35 In SCALMS systems, we attributed the much lower degree of coking to the support surface, in this case alumina.36 The SCALMS catalysts showed a very stable selectivity over time. Regarding the calculation of the Pt-based productivity (see Fig. S10 in ESI† for details) a value of 50 gheptenes gPt−1 h−1 was calculated for Pt/Al2O3, while the SCALMS system reaches 80 gheptenes gPt−1 h−1. The increase is indicative of the high utilization of the dissolved Pt atoms in the dynamic liquid alloy. No post run analysis of the catalysts was made, but we reported in previous studies that the nature of SCALMS catalysts remains, with e.g., Pt being associated to Ga even after harsh conditions of propane dehydrogenation.28
Our kinetic experiments targeted first the aspect of a potential influence of internal or external diffusion limitation on the observed overall performances of the catalysts under investigation. With the Pt/Al2O3 catalyst at 743 K and a constant volume flow of 150 mLN min−1, a variation of the catalyst pellet size from an average of 150 μm to 350 μm was performed and did not reveal any change in the overall rate (see Fig. S7 in ESI†). Therefore, pore diffusion limitation can be excluded for this catalyst under the applied conditions. To study the effect of external mass transfer limitation, the volumetric flow rate through the reactor was varied at constant residence time by adjusting the catalyst bed volume. The rate of depletion was not affected above 120 mLN min−1 (see Fig. S8 and S9 in ESI†). Consequently, film diffusion effects can be excluded for the Pt/Al2O3 catalyst under feedstock flow conditions above 120 mLN min−1. In a similar manner, we also excluded potential diffusion effects for the Pt/Al2O3 catalyst at 743 K, which was the upper limit of our temperature experiments.
For the applied SCALMS catalysts, it has been shown in previous work that most of the GaPt droplets produced during preparation are about one order of magnitude bigger than the support pores. Thus, the biggest part of these droplets is located at the outer surface of the support material.37 Thus, no significant pore diffusion limitation is expected. In-line with the similar activity range compared to the Pt/Al2O3 catalyst, we found that also for the SCALMS material under investigation the internal and external diffusion effects can be excluded for using alumina pellets with a particle size between 50–150 μm and a total volumetric flow rate of 150 mLN min−1.
Fig. 3 shows the logarithmic plot of n-heptane depletion rate as a function of hydrogen partial pressure for both Pt/Al2O3 and the SCALMS material. From the conversion data (see Fig. 2) the data points of Fig. 3 have been determined by averaging the first three consecutive data points (see ESI† and Fig. S13 and S14 for details). Noteworthy, the rate is higher for Ga84Pt/Al2O3 at any hydrogen partial pressure than for the Pt/Al2O3 catalyst. The hydrogen reaction order for SCALMS was determined to be 0.06, while Pt/Al2O3 resulted in a higher value of 0.35. It is generally agreed that the presence of hydrogen helps in keeping the active Pt sites free from coking by hydrogenation of olefinic species adsorbed on the catalyst surface.15,38,39 It seems that this effect is not as pronounced for SCALMS, assumingly because the highly dynamic liquid–gas interface in SCALMS is less affected by coking.26,28,32 This effect is confirmed here by the low dependency of the reaction rate on the hydrogen partial pressure in the reactor.
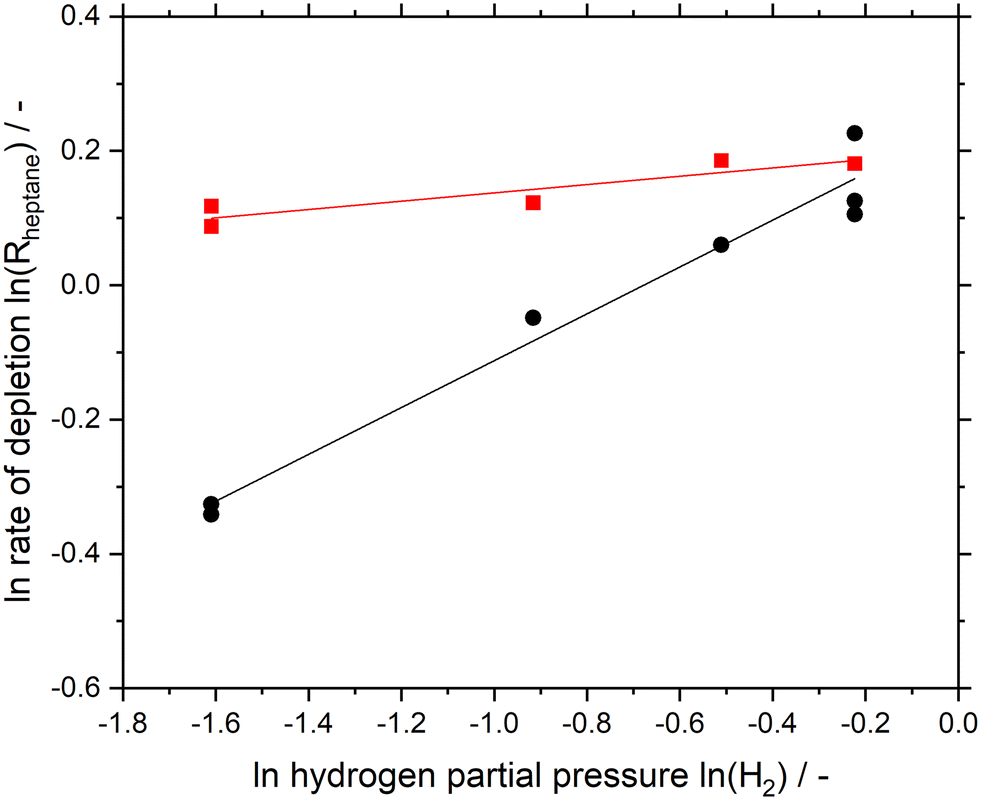 | ||
| Fig. 3 Logarithmic plot of rate of depletion for n-heptane dehydrogenation as a function of hydrogen partial pressure at 100 mbar constant n-heptane partial pressure for Ga84Pt/Al2O3 SCALMS (red squares) and Pt/Al2O3 (black circles). Pre-reduction conditions: 723 K, atmospheric pressure, 50 mLNmin−1 H2 flow, 120 min. Reaction conditions: 703 K, atmospheric pressure, 2.5 mL catalyst bed volume, 150 mlN min−1 total flow, 100 hPa n-heptane, 200–800 hPa H2, and 100–700 hPa He. Note: each data point represents an average of three consecutive measurements, excluding the very first measurement point (see Fig. S13 and S14† for details). | ||
Looking at the selectivity of n-heptenes at different H2 partial pressures (see Fig. 4 and Table S4†), it appears that for both Pt-based catalysts under investigation higher hydrogen partial pressure lowers the n-heptene yield from 80 to 75%. For the SCALMS system the selectivities are by few percent points higher. For the Pt/Al2O3 catalyst, the selectivity loss with higher partial pressure of H2 is slightly more pronounces than with the SCALMS system. All selectivity comparison results have been elaborated at a conversion level of 6%.
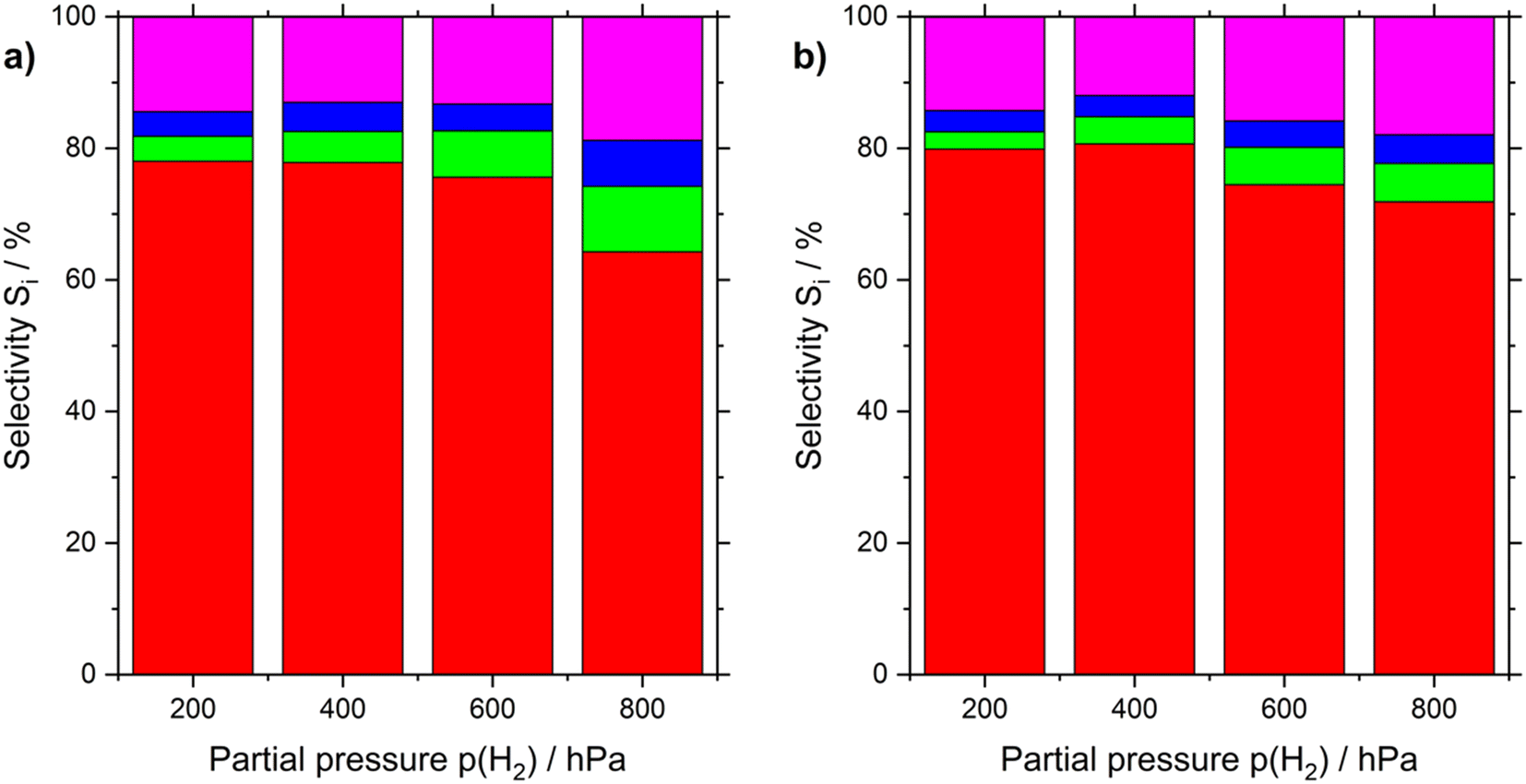 | ||
| Fig. 4 Effect of H2 partial pressure on product distribution in n-heptane dehydrogenation using a) Pt/Al2O3 and b) Ga84Pt/Al2O3 SCALMS. Selectivity of n-heptenes (red), toluene (green), cracking (blue) and other (pink) products are shown. Pre-reduction conditions: 723 K, atmospheric pressure, 50 mLN min−1 H2 flow, 120 min. Reaction conditions: 703 K, atmospheric pressure, 2.5 mL catalyst bed volume, 150 mlN min−1 total flow, 100 hPa n-heptane, 200–800 hPa H2, and 100–700 hPa He. | ||
The reaction orders of n-heptane were determined both at 200 and 800 hPa partial pressure of hydrogen (see Fig. S11 in ESI†). In both cases, the conversion decreased as the n-heptane partial pressure increased (see Tables S5 and S6 in ESI†). Both catalysts revealed similar reaction orders around 0.64 (for 200 hPa H2) and 0.75 (for 800 hPa H2). The by-product formation for both catalysts followed similar trends, with toluene and cracking product yields decreasing at higher partial pressure of n-heptane.
The effective activation energies EA,eff for n-heptane dehydrogenation on Pt/Al2O3, and the Ga84Pt/Al2O3 SCALMS system under investigation were determined in the temperature range between 683 and 743 K using 800 hPa partial pressure of H2. As shown in Fig. 5, both catalysts showed very similar activation energies of around 68 kJ mol−1. Note, that literature values for the activation energy in alkane dehydrogenation vary significantly. While for monometallic catalysts such as Pt/Al2O3, values in the range of 30–80 kJ mol−1 were reported, bimetallic systems such as PtSn/Al2O3 showed much higher values > 110 kJ mol−1. The activation energy for Pt/Al2O3 (68 ± 12 kJ mol−1) found in this study indicates the absence of severe internal and external transport limitations and agrees with our studies to exclude these effects (see ESI† for details). The similar value for the SCALMS catalyst also indicates the absence of transport effects.
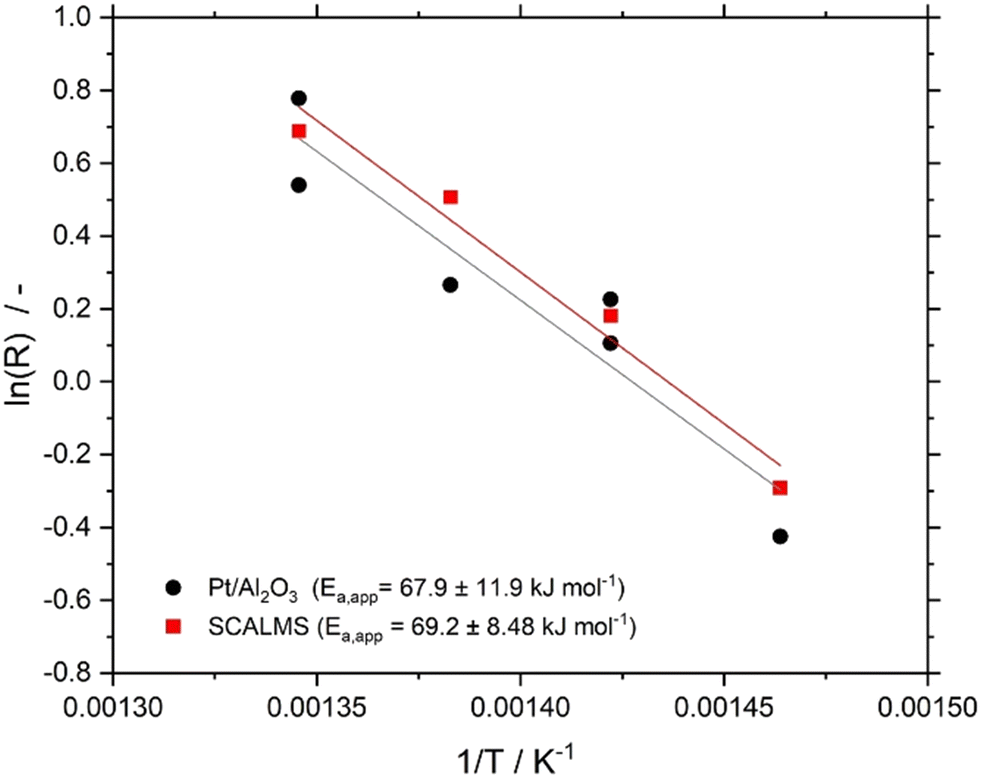 | ||
| Fig. 5 Arrhenius plots for n-heptane dehydrogenation reaction using Ga84Pt/Al2O3 SCALMS (red squares) and Pt/Al2O3 (black circles). Pre-reduction conditions: 723 K, atmospheric pressure, 50 mLN min−1 H2 flow, 120 min. Reaction conditions: 683–743 K, atmospheric pressure, 2.5 mL catalyst bed volume, 150 mlN min−1 total flow, 100 hPa n-heptane, 800 hPa H2, and 100 hPa He. | ||
Between 683 and 723 K, both catalysts yielded a similar product distribution. However, a remarkable difference in selectivity is found for the two catalysts under investigation above 723 K (see Fig. 6 and Table S7 in ESI†). While the Pt/Al2O3 catalyst turns to a significant extent into a cracking and aromatization catalyst, the supported liquid metal alloy system keeps its preference for alkane dehydrogenation to the desired olefinic products. The respective activation energies are summarized in Table 2.
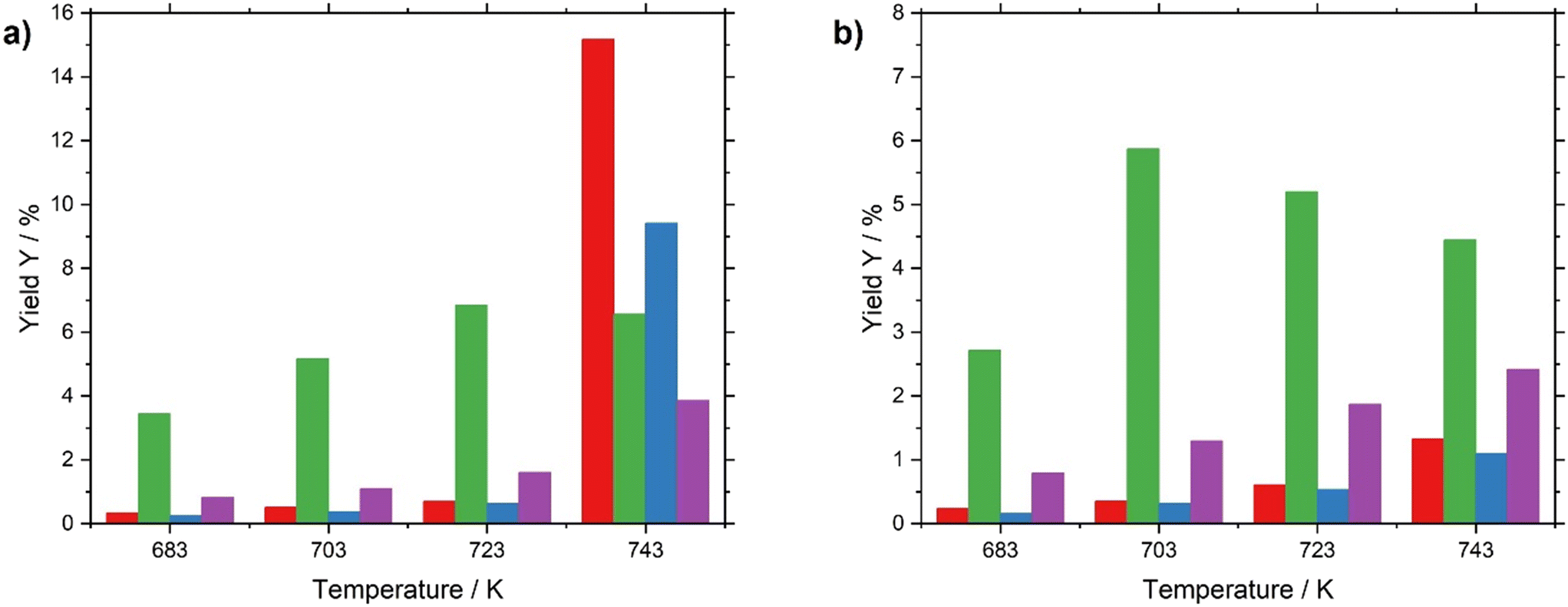 | ||
| Fig. 6 Effect of temperature on product distribution in n-heptane dehydrogenation using a) Pt/Al2O3 and b) Ga84Pt/Al2O3 SCALMS. Yields of cracking (red), n-heptenes (green), toluene (blue), and other (purple) products are shown. Pre-reduction conditions: 723 K, atmospheric pressure, 50 mLNmin−1 H2 flow, 120 min. Reaction conditions: 683–743 K, atmospheric pressure, 2.5 mL catalyst bed volume, 150 mlN min–1 total flow, 100 mbar n-heptane, 800 mbar H2, and 100 mbar He. | ||
| Reaction | Activation energy/kJ mol−1 | |
|---|---|---|
| Pt/Al2O3 | Ga84Pt/Al2O3 | |
| Total heptane depletion | 67.9 | 69.2 |
| Dehydrogenation | 65.0 | 49.5 |
| Aromatization | 96.3 | 137.4 |
| Cracking | 56.5 | 111.1 |
As shown in the table, these selectivity differences are well reflected in the individual activation of the different reaction products. While both the Pt on alumina and the SCALMS material show very similar overall activation energies, the one for n-heptenes formation is significantly lower for the SCALMS system and the ones for aromatization and cracking are significantly lower for the Pt on alumina catalyst. The value for the Pt on alumina catalyst for the dehydrogenation reaction of 65.0 kJ mol−1 is in good agreement with the recently published data for n-octane dehydrogenation (57.3 kJ mol−1) from the Jess group using a similar, promoted catalyst.19
We interpret the differences in reaction selectivities found at high temperature between Pt on alumina and the SCALMS material as result of the Pt atoms being atomically dissolved in the Ga matrix. Consequently, side-reaction requiring larger ensembles of Pt atoms40 are disfavored. Selectivity effects through reduction of Pt ensemble sizes were experimentally proven by De Jongste and co-workers for a PtCu system.41 Reducing the Pt ensemble size down to a single-atom level was reported to be beneficial to suppress coking and yielded higher selectivity.25,42
Following the determination of the reaction orders and the activation energy, the pre-exponential factor was determined from eqn (8). The reaction orders for n-heptane were similar at the two different hydrogen partial pressures, hence an average value of 0.7 was used. The pre-exponential factors for SCALMS and Pt/Al2O3 were found to be 6.04 × 105 and 8.04 × 105 molheptane gPt−1 h−1 bar−(n+m), respectively (see Table 3 for details). To validate the power law model, a comparison of the reaction rate observed experimentally, and the reaction rate calculated using the power law model with the parameters as determined was made (see ESI† for details). The parity plot of Rcalcvs. Rmodel reveals good agreement with all but two data points being inside the ±10% envelope.
These parameters were further improved by employing a numerical evaluation using a minimization algorithm provided by the MATLAB software (see Fig. S12 in ESI† for more details). The so-optimized parameters showed an improvement of about 3% for both catalyst systems in the parity plot (see Fig. 7). The results indicated that for the SCALMS catalyst, a zero-order in H2 gave a better fit for the experimental data, as summarized in Table 3.
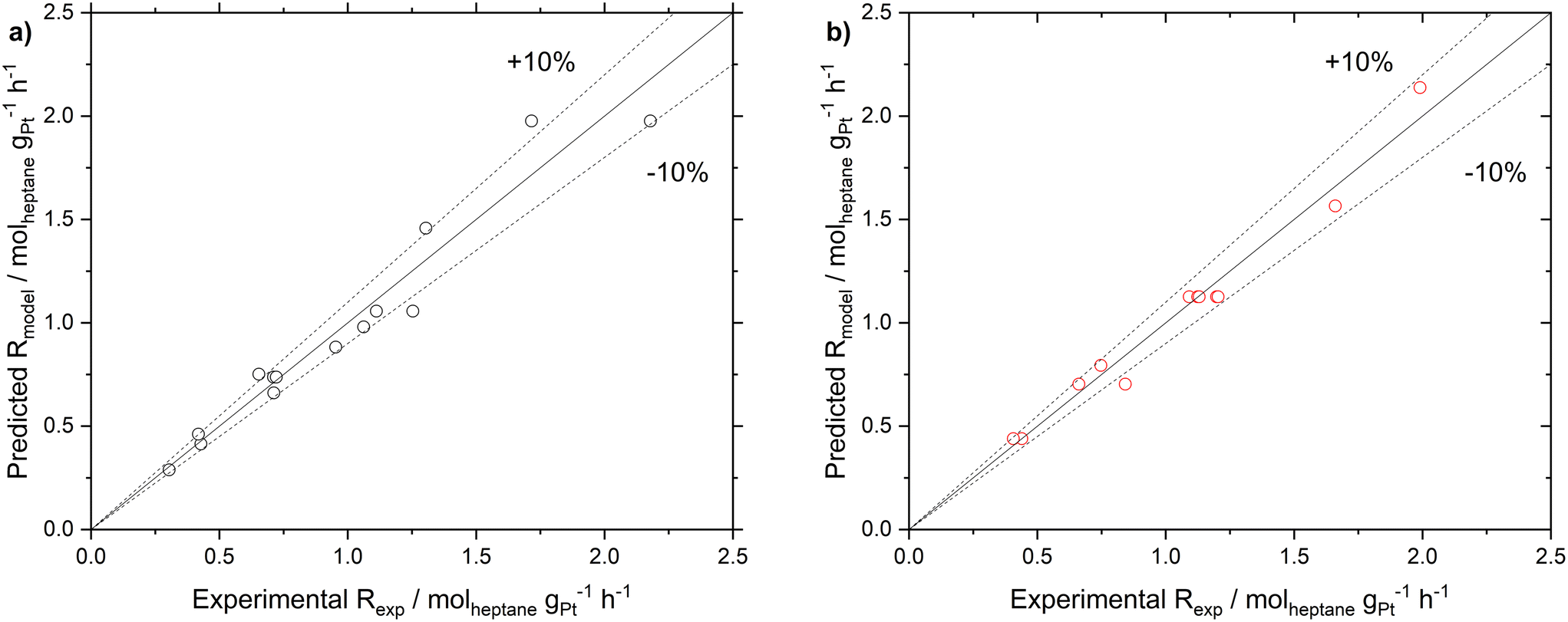 | ||
| Fig. 7 Parity graph of the calculated reaction rate (Rmodel) and the experimental reaction rate (Rcalc) in n-heptane dehydrogenation using a) Pt/Al2O3 and b) Ga84Pt/Al2O3 SCALMS catalysts. Fitted parameters used for Pt/Al2O3: EA,app = 68.1 kJ mol−1, H2 reaction order = 0.26, n-heptane reaction order = 0.68, and frequency factor = 6.04 × 105. Average error = 8.47%). Fitted parameters used for Ga84Pt/Al2O3 SCALMS: (EA,app = 69.7 kJ mol−1, H2 reaction order = 0, n-heptane reaction order = 0.68, and frequency factor = 8.04 × 105. Average error = 5.55%). | ||
Conclusion
In the present work, we have extended the application of Supported Catalytically Active Metal Solutions (SCALMS) to the dehydrogenation of intermediate-chain length alkanes. In detail, we studied the dehydrogenation of n-heptane to n-heptenes. A classical Pt/Al2O3 catalyst system was compared to a supported catalytically active liquid metal solution (SCALMS) system of the composition Ga84Pt/Al2O3. Both catalysts gave reproducible results in the continuous gas-phase dehydrogenation of n-heptane. Conversion and selectivities at 703 K were comparable with approx. 6% initial conversion and 80% selectivity to heptenes at the applied reaction conditions. Both catalysts deactivated slightly during the first six hours of the experiment with lower initial deactivation rate found for the SALMS system.Since the amount of precious Pt was significantly lower in the case of SCALMS, the resulting Pt-based productivity was higher with the Ga84Pt/Al2O3 system by almost 50%, showcasing the efficient utilization of precious metals in supported liquid metal catalysis. The kinetic parameters determined for the Pt on alumina catalyst were found to be in good agreement with the literature. For the SCALMS system, a significantly lower effective activation energy of 50 kJ mol−1 was obtained for the dehydrogenation, while the undesired side-reactions cracking, and aromatization showed much higher barriers than for the Pt on alumina catalyst. The reaction order for hydrogen was found to be zero for SCALMS and 0.35 for Pt/Al2O3, while the one for n-heptane was around 0.7 for both catalysts. The resulting kinetic data were used in a power rate model and gave reliable predictions of catalyst performance within a ±10% confidence interval. The use of SCALMS catalysts in high-temperature applications offers some benefits compared to standard heterogeneous catalysts. Given the higher dynamics at the gas–liquid alloy interface, the SCALMS material benefits from better utilization of Pt and of a lower tendency for deactivation by coke formation.
Author contributions
Oshin Sebastian – investigation and visualization; Asem Al-Shaibani – investigation and formal analysis; Nicola Taccardi – validation and conceptualization; Marco Haumann – conceptualization and writing; Peter Wasserscheid – project administration and writing.Conflicts of interest
There are no conflicts to report.Acknowledgements
Financial support from the European Research Council is gratefully acknowledged (Project 786475: Engineering of Supported Catalytically Active Liquid Metal Solutions). We also acknowledge support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 431791331 – SFB 1452 (CLINT).References
- K. Griesbaum, et al., in Ullmann's Encyclopedia of Industrial Chemistry, 2000, pp. 133–189 Search PubMed.
- K. Das and A. Kumar, in Advances in Organometallic Chemistry, 2019, vol. 72, pp. 1–57 Search PubMed.
- G. Lappin, Alpha Olefins Applications Handbook, CRC Press, 1st edn, 1989 Search PubMed.
- Linear Alpha Olefins Market: Global Industry Trends, Share, Size, Growth, Opportunity and Forecast 2022–2027. (2022), https://www.imarcgroup.com, (2022).
- N. Yu, J. Long, H. Zhou, A. Ma and Z. Dai, Simulation of the Reaction Mechanism of Dehydrogenation of n-Heptane to Produce Olefins, Shiyou Xuebao, 2016, 32, 437–443, DOI:10.3969/j.issn.1001-8719.2016.03.001.
- D. S. Aribike and A. A. Susu, Kinetics and mechanism of the thermal cracking of n-heptane, Thermochim. Acta, 1988, 127, 247–258, DOI:10.1016/0040-6031(88)87501-4.
- K. K. Pant and D. Kunzru, Pyrolysis of n-heptane: kinetics and modeling, J. Anal. Appl. Pyrolysis, 1996, 36, 103–123, DOI:10.1016/0165-2370(95)00925-6.
- D. Sanfilippo and P. N. Rylander, in Ullmann's Encyclopedia of Industrial Chemistry, 2009, pp. 451–471 Search PubMed.
- B. H. Davis, Alkane dehydrocyclization mechanism, Catal. Today, 1999, 53, 443–516, DOI:10.1016/S0920-5861(99)00136-4.
- O. Said-Aizpuru, et al., Non-Monotonous Product Distribution Dependence on Pt/γ-Al2O3-Cl Catalysts Formulation in n-Heptane Reforming, ChemCatChem, 2020, 12, 2262–2270, DOI:10.1002/cctc.201902260.
- M. R. Usman and F. M. Alotaibi, Unified kinetics of n-heptane hydroisomerisation over various Pt/zeolite catalysts, Prog. React. Kinet. Mech., 2016, 41, 177–192, DOI:10.3184/146867816x14646899008350.
- J. C. Rohrer, H. Hurwitz and J. H. Sinfelt, Kinetics of the catalytic dehydrocyclization of n-heptane, J. Phys. Chem., 1961, 65, 1458–1460, DOI:10.1021/j100826a513.
- J. H. Sinfelt, H. Hurwitz and J. C. Rohrer, Role of dehydrogenation activity in the catalytic isomerization and dehydrocyclization of hydrocarbons, J. Catal., 1962, 1, 481–483, DOI:10.1016/0021-9517(62)90097-0.
- J. H. Sinfelt, Bifunctional Catalysis, 1964 Search PubMed.
- J. C. Rohrer and J. H. Sinfelt, Interaction of hydrocarbons with Pt-Al2O3 in the presence of hydrogen and helium, J. Phys. Chem., 1962, 66, 1193–1194, DOI:10.1021/j100812a507.
- G. C. Bond, Metal-Catalysed Reactions of Hydrocarbons, Springer, 2005, p. 666 Search PubMed.
- K. Liu, S. C. Fung, T. C. Ho and D. S. Rumschitzki, Heptane Reforming over Pt–Re/Al2O3: Reaction Network, Kinetics, and Apparent Selective Catalyst Deactivation, J. Catal., 2002, 206, 188–201, DOI:10.1006/jcat.2001.3485.
- O. A. Olafadehan, A. A. Susu and A. Jaiyeola, Mechanistic Kinetic Models for n-Heptane Reforming on Platinum/Alumina Catalyst, Pet. Sci. Technol., 2008, 26, 1459–1480, DOI:10.1080/10916460701675157.
- T. Prucker, A. Lamberty, J. Thiessen, M. König and A. Jess, Kinetics of dehydrogenation of n-octane on a promoted Pt-catalyst, Chem. Eng. J., 2022, 442(2), 136233, DOI:10.1016/j.cej.2022.136233.
- B. Qiao, et al., Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3, 634–641, DOI:10.1038/nchem.1095.
- M. Flytzani-Stephanopoulos, Supported metal catalysts at the single-atom limit – A viewpoint, Chin. J. Catal., 2017, 38, 1432–1442, DOI:10.1016/s1872-2067(17)62886-9.
- J. M. Thomas, R. Raja and D. W. Lewis, Single-site heterogeneous catalysts, Angew. Chem., Int. Ed., 2005, 44, 6456–6482, DOI:10.1002/anie.200462473.
- E. J. Peterson, et al., Low-temperature carbon monoxide oxidation catalysed by regenerable atomically dispersed palladium on alumina, Nat. Commun., 2014, 5, 4885, DOI:10.1038/ncomms5885.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Single-Atom Alloy Catalysis, Chem. Rev., 2020, 120, 12044–12088, DOI:10.1021/acs.chemrev.0c00078.
- M. D. Marcinkowski, et al., Pt/Cu single-atom alloys as coke-resistant catalysts for efficient C-H activation, Nat. Chem., 2018, 10, 325–332, DOI:10.1038/nchem.2915.
- N. Taccardi, et al., Gallium-rich Pd-Ga phases as supported liquid metal catalysts, Nat. Chem., 2017, 9, 862–867, DOI:10.1038/nchem.2822.
- N. Raman, et al., Highly Effective Propane Dehydrogenation Using Ga-Rh Supported Catalytically Active Liquid Metal Solutions, ACS Catal., 2019, 9, 9499–9507, DOI:10.1021/acscatal.9b02459.
- N. Raman, et al., GaPt Supported Catalytically Active Liquid Metal Solution Catalysis for Propane Dehydrogenation – Support Influence and Coking Studies, ACS Catal., 2021, 11, 13423–13433, DOI:10.1021/acscatal.1c01924.
- O. Sebastian, et al., Stable and Selective Dehydrogenation of Methylcyclohexane using Supported Catalytically Active Liquid Metal Solutions – Ga52Pt/SiO2 SCALMS, ChemCatChem, 2020, 12, 4533–4537, DOI:10.1002/cctc.202000671.
- A. Søgaard, A. L. de Oliveira, N. Taccardi, M. Haumann and P. Wasserscheid, Ga-Ni supported catalytically active liquid metal solutions (SCALMS) for selective ethylene oligomerization, Catal. Sci. Technol., 2021, 11, 7535–7539, 10.1039/d1cy01146d.
- G. Rupprechter, Supported liquid metal catalysts: Popping up to the surface, Nat. Chem., 2017, 9, 833–834, DOI:10.1038/nchem.2849.
- T. Bauer, et al., Operando DRIFTS and DFT Study of Propane Dehydrogenation over Solid- and Liquid-Supported Ga(x)Pt(y) Catalysts, ACS Catal., 2019, 9, 2842–2853, DOI:10.1021/acscatal.8b04578.
- O. Sebastian, et al., Ga–Pt supported catalytically active liquid metal solutions (SCALMS) prepared by ultrasonication – influence of synthesis conditions on n-heptane dehydrogenation performance, Catal. Sci. Technol., 2023, 13, 4435–4450, 10.1039/d3cy00356f.
- W. A. Dietz, Response Factors for Gas Chromatographic Analyses, J. Chromatogr. Sci., 1967, 5, 68–71, DOI:10.1093/CHROMSCI/5.2.68.
- B. K. Vu, et al. Electronic density enrichment of Pt catalysts by coke in the propane dehydrogenation, Korean J. Chem. Eng., 2010, 28, 383–387, DOI:10.1007/s11814-010-0363-8.
- M. Wolf, et al., Capturing spatially resolved kinetic data and coking of Ga-Pt supported catalytically active liquid metal solutions during propane dehydrogenation in situ, Faraday Discuss., 2021, 229, 359–377, 10.1039/d0fd00010h.
- N. Raman, et al., Top-down vs. bottom-up synthesis of Ga based supported catalytically active liquid metal solutions (SCALMS) for the dehydrogenation of isobutane, Chem. Eng. J., 2023, 475, 146081, DOI:10.1016/j.cej.2023.146081.
- K. Foger and H. Jaeger, The effect of chlorine treatment on the dispersion of platinum metal particles supported on silica and γ-alumina, J. Catal., 1985, 92, 64–78, DOI:10.1016/0021-9517(85)90237-4.
- S. M. Davis, F. Zaera and G. A. Somorjai, The reactivity and composition of strongly adsorbed carbonaceous deposits on platinum. Model of the working hydrocarbon conversion catalyst, J. Catal., 1982, 77, 439–459, DOI:10.1016/0021-9517(82)90185-3.
- C. T. Campbell, et al. Probing ensemble effects in surface reactions. 1. Site-size requirements for the dehydrogenation of cyclic hydrocarbons on platinum(111) revealed by bismuth site blocking, J. Phys. Chem., 1989, 93, 806–814, DOI:10.1021/j100339a056.
- H. C. D. Jongste and V. Ponec, On the Reforming Reactions of Hydrocarbons on Platinum-Copper Alloys, Stud. Surf. Sci. Catal., 1981, 7(A), 186, DOI:10.1016/S0167-2991(09)60270-6.
- X. H. Li, D. Ma and B. A. O. Xinhe, Dispersion of Pt Catalysts Supported on Activated Carbon and Their Catalytic Performance in Methylcyclohexane Dehydrogenation, Chin. J. Catal., 2008, 29, 259–263, DOI:10.1016/s1872-2067(08)60027-3.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00490b |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |