
Reactivity control of nitrate-incorporating octadecavanadates by changing the oxidation state and metal substitution†
Isshin
Yoshida
a,
Yuji
Kikukawa
 *a,
Ryoji
Mitsuhashi
b and
Yoshihito
Hayashi
a
*a,
Ryoji
Mitsuhashi
b and
Yoshihito
Hayashi
a
aDepartment of Chemistry, Graduate School of Natural Science and Technology, Kanazawa University, Kakuma, Kanazawa 920-1192, Japan. E-mail: kikukawa@se.kanazawa-u.ac.jp
bInstitute of Liberal Arts and Science, Kanazawa University, Kakuma, Kanazawa 920-1192, Japan
First published on 1st May 2024
Abstract
Clarification and control of the active sites at the atomic/molecular level are important to develop nanocatalysts. The catalytic performance of two oxidation states of nitrate-incorporating octadecavanadates, [V18O46(NO3)]5− (V18) and [V18O46(NO3)]4− (V18ox), and a copper-substituted one, [Cu2V16O44(NO3)]5− (Cu2V16), in selective oxidation was investigated. Both V18 and V18ox possessed the same double-helical structures and one of two tetravalent vanadium sites of V18 was oxidized in V18ox. The comparison of the mobility of the incorporated nitrate reveals that tetravalent vanadium centres show stronger interaction with the incorporated anions than pentavalent ones. The oxidation reaction with V18ox proceeded more smoothly with tert-BuOOH as an oxidant than that with V18. The reactivity and selectivity of the oxidation of 2-cyclohexen-1-ol were different among the derivatives. V18ox showed the higher reactivity with 72% selectivity to epoxide. With V18, reactivity was lower but higher selectivity to epoxide was achieved. In the presence of Cu2V16, 2-cyclohexen-1-one was selectively obtained with 81% selectivity. The order of the reactivity for cyclooctene was V18ox, V18 and Cu2V16. These results shows that the cap part of the double-helix acts as the active site. Even though the vanadium–oxygen species exhibit the same structures, the catalytic properties can be controlled by changing the valence of vanadium and metal substitution.
Introduction
Vanadium oxide V2O5 is one of the well-known practical oxidation catalysts. Development of vanadium-based catalysts is still one of the attractive fields.1–3 To develop vanadium-based catalysts, the rational understanding of the active sites at the atomic/molecular level is required. Deep understanding of the molecular catalysts is one of the important approaches, because of their tuneable properties.4,5 Vanadium oxide is dissolved in basic solutions to form well-defined molecular structures called polyoxovanadates.6,7 Since the principal properties of vanadium–oxygen species are different in different coordination geometries, the preparation of polyoxovanadates with the appropriate coordination geometry is important. The dominant coordination geometries of vanadium–oxygen species vary with the pH value in the aqueous medium, such as octahedral VO6-based decavanadate and tetrahedral VO4-based metavanadate in acidic and basic solution, respectively.8 Solid V2O5 exhibits a layered structure, and each sheet is composed of the square pyramidal geometry VO5. Once VO5 units are condensed to form sphere structures, an anion is stabilized at the centre of the sphere. The anion-centred polyoxovanadate structures depend on the size and shape of the centred anion. Small anion templates give relatively small polyoxovanadates, such as [V7O19F]3−, [V11O29F2]4− and [V12O32Cl]5−, and large templates give large poloxovanadates, such as [V18O44(N3)]10−, [V18O42(SO4)]8−, and [HV22O54(X)]6− (X = SCN, ClO4).9–14 Their properties, such as redox potential, stability and reactivity, depend on their structures. Some polyoxovanadates show catalytic performance for several oxidation reactions such as oxidation of alcohols, alkenes, and sulfides.15,16 There are several chemical environments of vanadium atoms in a polyoxovanadate even considering the symmetry. In addition, most spherical polyoxovanadates possess both V4+ and V5+ and some of them are disordered.17–19 Therefore, it is difficult to accurately determine the active vanadium sites and the valence of the vanadium centres in a polyoxovanadate. Local structure control among the related polyoxovanadates leads to understanding of the reactivity of the specific sites.20Nitrate-incorporating octadecavanadate [V18O46(NO3)]5− (V18) possesses helicity.21 The original procedure for synthesis of V18 involved the oxidation of dark purple decavanadate [V10O26]4− in nitromethane in the presence of nitrate. Recently, the Streb group reported a new precursor by heating yellow decavanadate [H3V10O28]3− and reaction with nitrate to yield V18.22 Generally, the structures of polyoxovanadates vary under slightly different synthetic conditions. The fact that the V18 structure is obtained with the different procedures means that V18 can be considered to be one of the stable polyoxovanadates. The charge of V18 is 5−, showing the presence of two V4+ ions. From the bond valence sum (BVS) values, the V4+ sites are localized on the top and bottom parts of the helix. From the cyclic voltammetry, V18 shows multi-step reversible redox properties. The stability, the localized valence, and the redox properties led us to investigate the effect of the valence of the vanadium centre of the cap parts on the oxidation catalytic properties of the V18 helix. In addition, we recently reported a derivative of V18, dicopper-substituted polyoxovanadate (Cu2V16) and its catalytic activity for alcohol oxidation (Fig. 1).23 In this work, preparation of the oxidized species of V18 (V18ox) and reactivity of the local vanadium site are investigated with a comparison of the catalytic performance among the derivatives.
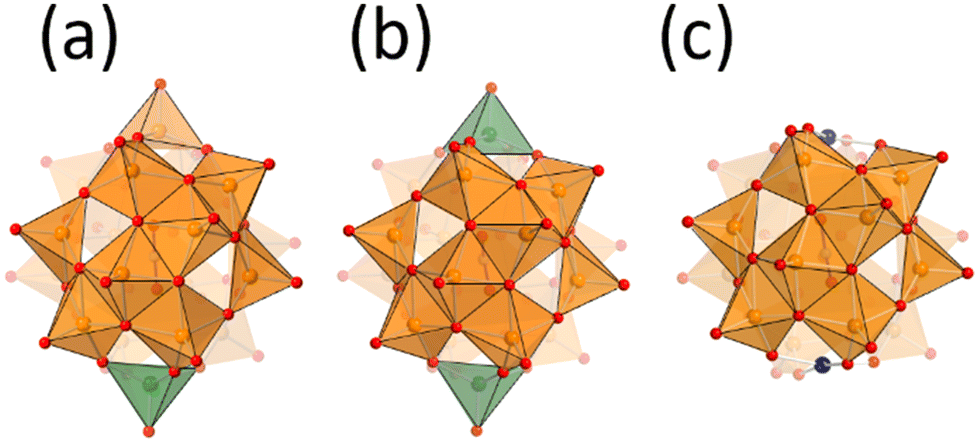 | ||
| Fig. 1 Anion structures of (a) V18ox, (b) V18, and (c) Cu2V16. Orange and green square pyramids represent V5+O5 and V4+O5. Red and blue spheres represent oxygen and copper atoms. The deep and pale colours of the V5+O5 units represent front and back sides. Nitrate locates at the centre of the clusters. | ||
Experimental
Materials
All reagents were obtained from commercial suppliers and were used without further purification unless otherwise noted. {n-Bu4N}5[V18O46(NO3)] (V18) and {n-Bu4N}5[Cu2V16O44(NO3)] (CuV16) were synthesized following the literature methods.21,23Synthesis of oxidized octadecavanadate, [V18O46(NO3)]4− (V18ox)
{n-Bu4N}4[V10O26] (640 mg, 340 mmol) was dissolved in 20 mL of acetonitrile. After the addition of 60% HNO3 (99 μL, 1.3 mmol), the solution was refluxed for 3.5 h. Then the solution was filtrated to remove the insoluble materials. To the filtrate, diethyl ether (ca. 80 mL) was added, and the solution was left to stand overnight at 0 °C. A brown powder (430 mg) was obtained by filtration (86% yield based on vanadium). Elemental analysis calcd for {n-Bu4N}4[V18O46(NO3)]: C 28.63%, H 5.41%, N 2.61%, V 34.15; found: C 27.90%, H 5.26%, N 2.57%, V 34.43%. Crystalline samples suitable for single crystal X-ray analysis were obtained from the mixed solvent of acetonitrile and diethyl ether in the presence of 4 equiv. of {Et4N}BF4 with respect to V18ox. CCDC 2341040 contains the supplementary crystallographic data for V18ox (Table S1†).Results and discussion
Synthesis and characterization of oxidized octadecavanadate
The cyclic voltammogram of V18 shows two peaks in the positive region with respect to the open circuit potential (Fig. S1†). The controlled potential electrolysis at 0.65 V were carried out. The solution colour changed from green to brown (Fig. S2†). In the UV/vis spectra, the intensity of the band around 600–1500 nm due to the intervalence charge transfer (IVCT) of the obtained solution decreased by half in comparison with that of V18, showing the 1 electron oxidation of V18 (Fig. 2). By addition of ethyl acetate into the solution, brown crystals were obtained. Although the obtained crystallographic data was not enough good, it was indicated that the polyoxovanadate structure of the brown crystals is identical to that of the original V18. The preparation of the 2 electron oxidized species was also attempted by the controlled potential electrolysis at higher potential. However, only the 1 electron oxidized species were isolated probably because the 2 electron oxidized species were easily reduced and/or decomposed. Among polyoxovanadates formed from VO5 square pyramids, fully oxidized ones possess lacunary sites, as far as we know.24–28 This fact may be related to the stability of the fully oxidized species. | ||
| Fig. 2 UV/vis spectra of (a) V18 and (b) V18ox prepared by chemical reaction. The red line is the spectrum after the controlled potential electrolysis and dilution with acetonitrile. The concentration of the polyoxovanadates was 0.2 mM in acetonitrile. | ||
With reference to the electrochemical investigation, synthesis of the oxidized V18 by a chemical reaction was investigated. In the previous report, V18 was synthesized in nitroethane. During the reaction, the synthetic solution colour changed from purple to brown and finally to green, indicating that the reaction contains oxidation and reduction processes of the polyoxovanadate.21 Without addition of NO3−, V18 was also obtained by the reaction of [V10O26]4− and tert-BuOOH (TBHP) in nitromethane at 85 °C for 2 days. Its IR spectrum showed the typical peaks at 1342 and 1359 cm−1 due to the incorporated NO3− in V18, suggesting that nitromethane is oxidized to form NO3− during the reaction (Fig. S3†). Through the experimental facts, we made the hypothesis that the over oxidized product was reduced by the nitroalkane in the previous report, and that without the nitroalkane, oxidized products are prepared. To obtain the oxidized products, we selected acetonitrile as the solvent. As we expected, the brown colour of the synthetic solution was maintained. Upon the addition of diethyl ether, a brown product was obtained.
Single crystals suitable for X-ray crystallographic analysis were obtained from the mixed solvent of acetonitrile and ethyl acetate in the presence of 4 equiv. of {Et4N}BF4 (Fig. 1 and Table S1†). Two tetra-n-butylammonium (TBA) and two tetraethylammonium (TEA) cations per one polyoxovanadate were observed. The total number of cations was one less than for the original V18. The anion was isostructural with the original V18. These results showed the formation of one-electron-oxidized products (V18ox). The double helical structure was capped by two vertex-sharing VO5 units. The distances between the capping vanadium atoms of V18ox and V18 were 8.49 and 8.55 Å, respectively. The angles of the inserted nitrate triangles from the line between the capping vanadium atoms of V18ox and V18 are 5° and 12°, respectively (Fig. S4†). These results show the inside volume of the sphere of the V18ox is smaller than that of V18. The bond lengths between the capping vanadium atoms, V1 and V2, and the surrounding oxygen atoms in V18ox are 1.851(3), 1.873(3), and 1.584(4) Å, and 1.904(3), 1.925(3), and 1.597(4) Å, respectively. The bond valence sum values of V1 and V2 are 5.22 and 4.23, respectively, showing that one of the capped vanadium atoms is oxidized.29,30
Thermogravimetry analysis of TBA salts of V18ox and V18 showed no weight loss under 200 °C except for that of hydrated water (Fig. S5†), suggesting their stability below 200 °C. The IR spectrum of V18ox synthesized by the chemical reaction was the same as that obtained by the electrochemical method. It shows the typical split peaks at 1342 and 1360 cm−1 due to the incorporated nitrate (Fig. S6†). The bond length differences of the cap parts led to the different intensities of each peak. The temperature-dependent IR spectra of TBA salts of V18 and V18ox were measured under vacuum conditions (Fig. 3 and Fig. S7†). Although no shifts of the peaks due to nitrate in V18 were observed even at 200 °C, the peak at 1342 cm−1 due to nitrate in V18ox at room temperature was shifted to 1346 cm−1 at 200 °C. After cooling the original peak position was retrieved. With increasing the temperature, the spectra in the region between 700 and 900 cm−1 due to the polyoxovanadate frameworks were slightly changed, and the original spectra were recovered after cooling. These results indicated the higher mobility of the incorporated nitrate in V18ox with the distortion of the polyoxovanadate framework than that in V18. Recently we reported the mobility of the incorporated nitrate in Cu2V16 below room temperature.18 Although the inside cavity of Cu2V16 was smaller than that of V18, the incorporated nitrate of Cu2V16 showed smoother mobility. Among the derivatives, the helical body was identical. Therefore, the mobility of the nitrate strongly depended on the cap parts. Tetravalent vanadium centres showed stronger interactions with the incorporated anion than pentavalent ones.
 | ||
| Fig. 3 Temperature-dependent IR spectra of V18ox (a) at 200 °C, (b) after cooling from 200 °C to room temperature, and (c) at room temperature under vacuum conditions before heating. The red line represents the peak at 1346 cm−1. | ||
The UV/vis spectrum of V18ox showed a halved intensity of the band around 600–900 nm due to IVCT of V18ox in comparison with that of V18, matching the V18ox obtained by the electrochemical method (Fig. 2). Upon addition of {n-Bu4N}I into the solution of V18ox, the intensity in the IVCT region increased and in the presence of 2.25 equivalents of {n-Bu4N}I with respect to V18ox, the intensity at 900 nm was identical to that of V18, showing the reduction of V18ox to give V18 (Fig. S8†).
Oxidation catalysis
To clarify the effect of the one-electron oxidation of V18, oxidation of sulfides was investigated (Table 1). 5.5 M decane solution of TBHP was used as the oxidant, since V18 and V18ox were stable in the presence of 200 equiv. of TBHP maintaining their oxidation states (Fig. S9†). Without catalysts, thioanisole oxidation hardly proceeded. Thioanisole oxidation with 0.5 mol% V18ox required 10 min to give 91% and 1% yields of methyl phenyl sulfoxide and methyl phenyl sulfone, respectively. With V18 for 10 min, a 26% total yield was obtained. V18 required 60 min to achieve a 91% total yield and 98% selectivity to sulfoxide. The reactivity of V18ox is higher than those of the precursors, [V10O26]4– and [NO3]−. After the reaction, addition of an excess amount of diethyl ether gave the solid catalysts. From the IR spectra, V18 and V18ox maintained their structures (Fig. S10†). The copper-substituted derivative, Cu2V16 showed a lower and slightly higher performance than V18ox and V18, respectively. Since V18 and the derivative possessed the same helical body, the pentavalent vanadium cap part of V18ox promoted the reaction more efficiently. The capping square pyramid parts of the clusters were bridged only by the vertex-sharing oxygen atoms, and the square pyramids of the body parts were connected by the corner-sharing oxygen atoms. Therefore, the relatively sterically open cap parts allowed the access of the oxidants and/or substrates.| Entry | Substrate | Catalyst | Time/min | Sulfoxide yielda/% | Sulfone yielda/% |
|---|---|---|---|---|---|
| The typical catalytic reaction conditions: substrate (1 mmol), TBA salts of catalysts (5 μmol), 5.5 M decane solution of TBHP (1 mmol), acetonitrile (2 mL), internal standard (0.2 mmol), 32 °C, 800 rpm with a Teflon-coated magnetic stirrer bar, under Ar. Yields were determined by GC with the internal standard method and/or 1H NMR (see the ESI†). The reaction was carried out in a screw-top test tube.a Yields were determined by GC with naphthalene as an internal standard.b Yields were determined by GC with naphthalene as an internal standard and 1H NMR.c Yields were determined by 1H NMR. | |||||
| 1 |
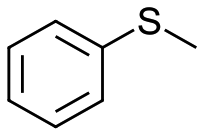
|
V18ox | 10 | 91 | 1 |
| 2 | V18 | 10 | 25 | 1 | |
| 3 | 60 | 89 | 2 | ||
| 4 | [V10O26]4− | 60 | 47 | 9 | |
| 5 | [NO3]− | 60 | 19 | 3 | |
| 6 | Cu2V16 | 10 | 34 | 2 | |
| 7 | 60 | 88 | 2 | ||
| 8 | — | 60 | 8 | — | |
| 9b |
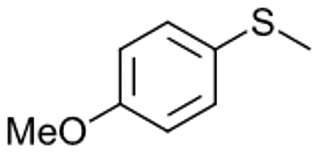
|
V18ox | 2 | 89 | 1 |
| 10b | V18 | 2 | 3 | — | |
| 11b | V18 | 120 | 91 | 2 | |
| 12 |

|
V18ox | 5 | 82 | 9 |
| 13 | V18 | 5 | 9 | — | |
| 14 | 120 | 86 | 5 | ||
| 15b |
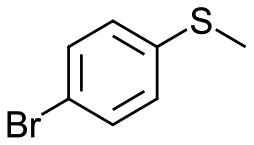
|
V18ox | 10 | 76 | 1 |
| 16b | V18 | 10 | 10 | — | |
| 17b | 120 | 76 | 5 | ||
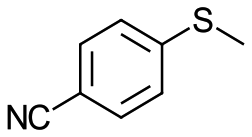
| 18c |
|
V18ox | 15 | 93 | 3 |
| 19c | V18 | 120 | 93 | 3 | |
| 20c |
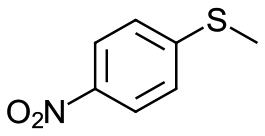
|
V18ox | 15 | 97 | 1 |
| 21c | V18 | 120 | 90 | 2 | |
| 22 |

|
V18ox | 3 | 73 | 7 |
| 23 | V18 | 3 | 31 | 4 | |
| 24 | 10 | 83 | 4 | ||
| 25 |
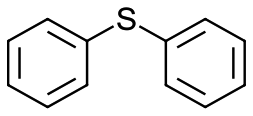
|
V18ox | 10 | 93 | 2 |
| 26 | V18 | 10 | 64 | 1 | |
| 27 | 15 | 90 | 3 | ||
| 28 |

|
V18ox | 5 | 94 | 3 |
| 29 | V18 | 5 | 18 | 1 | |
| 30 | 20 | 81 | 3 | ||
| 31 |

|
V18ox | 5 | 98 | 1 |
| 32 | V18 | 5 | 33 | 8 | |
| 33 | 20 | 94 | 2 | ||
In the presence of V18 and V18ox, the oxidation of various kinds of sulfides with TBHP proceeded to give the corresponding sulfoxides and sulfones. In all cases, the reaction with V18ox proceeded more efficiently than that with V18. Aromatic sulfides with electron-donating groups required short reaction times. Phenyl vinyl sulfide gave the corresponding sulfoxide and sulfone with retention of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The small difference in the reactivity of diphenyl sulfide between V18 and V18ox indicates that the steric hinderance of the two phenyl groups prevents the substrate from approaching the active site of the cap part of V18ox. The oxidation of cyclic and linear aliphatic sulfides also proceeded to afford the corresponding sulfoxides.
C double bond. The small difference in the reactivity of diphenyl sulfide between V18 and V18ox indicates that the steric hinderance of the two phenyl groups prevents the substrate from approaching the active site of the cap part of V18ox. The oxidation of cyclic and linear aliphatic sulfides also proceeded to afford the corresponding sulfoxides.
Next, oxidation of alcohols was investigated. For the oxidation of 1-phenyl ethanol, the yield of acetophenone after 24 h with V18ox was 50% (Scheme 1). Cu2V16 showed a higher catalytic performance than V18ox and the reaction hardly proceeded in the presence of V18. The order of the catalytic activity for the alcohol oxidation was different from that of sulfide oxidation.
 | ||
| Scheme 1 Oxidation of 1-phenyl ethanol. Reaction time was 24 h. Naphthalene was used as an internal standard. The typical catalytic reaction conditions: substrate (1 mmol), TBA salts of catalysts (5 μmol), 5.5 M decane solution of TBHP (1 mmol), acetonitrile (2 mL), internal standard (0.2 mmol), 32 °C, 800 rpm with a Teflon-coated magnetic stirrer bar, under Ar. Yields were determined by GC with the internal standard method and/or 1H NMR (see the ESI†). The reaction was carried out in a screw-top test tube. | ||
Oxidation of 2-cyclohexen-1-ol, a kind of allylic alcohol possessing two functional groups (CC, and OH) which can be oxidized, was investigated (Scheme 2). With V18ox, the total yield of the products reached 76% in 24 h. The ketone to epoxide selectivity was 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :72. Although the total yield with V18 was lower under the same conditions, the selectivity toward epoxidation was higher than that with V18ox. In the presence of Cu2V16, selective alcohol oxidation proceeded. With the immobilization of metal cations into the polyoxometalate frameworks, the preference of the selectivity changed dramatically.31,32
:72. Although the total yield with V18 was lower under the same conditions, the selectivity toward epoxidation was higher than that with V18ox. In the presence of Cu2V16, selective alcohol oxidation proceeded. With the immobilization of metal cations into the polyoxometalate frameworks, the preference of the selectivity changed dramatically.31,32
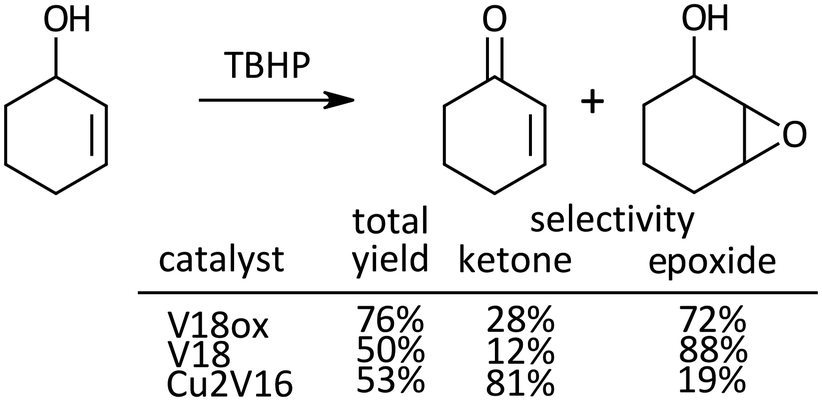 | ||
| Scheme 2 Oxidation of 2-cyclohexen-1-ol. Reaction time was 24 h. Chlorobenzene was used as an internal standard. The typical catalytic reaction conditions: substrate (1 mmol), TBA salts of catalysts (5 μmol), 5.5 M decane solution of TBHP (1 mmol), acetonitrile (2 mL), internal standard (0.2 mmol), 32 °C, 800 rpm with a Teflon-coated magnetic stirrer bar, under Ar. Yields were determined by GC with the internal standard method and/or 1H NMR (see the ESI†). The reaction was carried out in a screw-top test tube. | ||
In the case of the epoxidation of cyclooctene, the order of the reactivity was V18ox, V18 and Cu2V16 (Scheme 3).
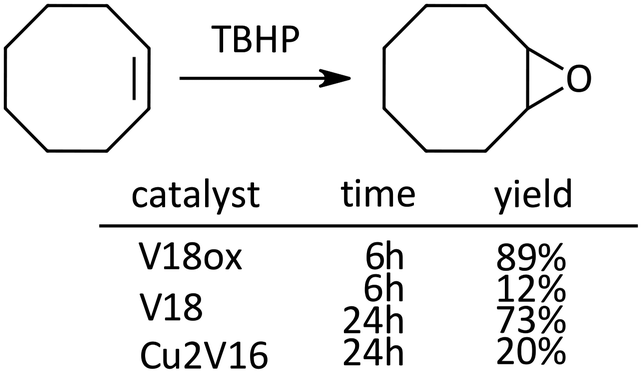 | ||
| Scheme 3 Epoxidation of cyclooctene. Naphthalene was used as an internal standard. The typical catalytic reaction conditions: substrate (1 mmol), TBA salts of catalysts (5 μmol), 5.5 M decane solution of TBHP (1 mmol), acetonitrile (2 mL), internal standard (0.2 mmol), 32 °C, 800 rpm with a Teflon-coated magnetic stirrer bar, under Ar. Yields were determined by GC with the internal standard method and/or 1H NMR (see the ESI†). The reaction was carried out in a screw-top test tube. | ||
The comparison of oxidation catalytic activity among the derivatives revealed that the changing of the oxidation state or the constituent metal cation provided control of the reactivity.
Conclusions
By the avoidance of reductive conditions, the one-electron-oxidized species was successfully obtained. Almost all reported sphere type polyoxovanadates with an anion at the centre adopted the mixed valence states of V4+ and V5+. Through monitoring the mobility of the incorporated nitrate, we found that tetravalent vanadium centres contributed to the interaction with the incorporated anion to form thermodynamically (meta)stable polyoxovanadate frameworks for the first time, as far as we know. The different catalytic performances for sulfide oxidation, alcohol oxidation and epoxidation among V18ox, V18 and Cu2V16 revealed that the reactions proceed not on the common helical body part, but the cap parts. The pentavalent VO5 cap parts showed high reactivity with low selectivity. The tetravalent VO5 cap parts showed low reactivity with high selectivity for the epoxidation. By substituting the cap part with copper, the reactivity and selectivity to alcohol oxidation increased. Polyoxovanadates exhibit structural versatility with various chemical environments of vanadium atoms in each cluster. Although the detailed discussion of the reaction mechanism including the detection of the real active species is still challenging, the comparison among the related derivatives is one of the promising ways to discuss the active sites.Author contributions
Conceptualization, funding acquisition, Y. K.; characterization, R. M.; other experimental investigation, I. Y.; data correction, Y. K. and I. Y.; supervision, Y. H. All authors have contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Imoto at Kanazawa University for his preliminary experiments. This research was partly supported by JSPS KAKENHI Grant Number JP23H04621 and JP23K04776, JSPS Core-to-Core program, and JST FOREST Program Grant Number JPMJFR2023720466.References
- Y. Inomata, H. Kubota, S. Hata, E. Kiyonaga, K. Morita, K. Yoshida, N. Sakaguchi, T. Toyao, K. Shimizu, S. Ishikawa, W. Ueda, M. Satake and T. Murayama, Nat. Commun., 2021, 12, 557 CrossRef CAS PubMed.
- B. Liu, Z. Zheng, Y. Liu, M. Zhang, Y. Wang, Y. Wan and K. Yan, J. Energy Chem., 2023, 78, 412 CrossRef CAS.
- Y. Liu, Y. Wang, B. Liu, M. Amer and K. Yan, Acta Phys. -Chim. Sin., 2023, 39, 2205028 Search PubMed.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456 CrossRef CAS PubMed.
- Z. Li, Y. Yan, M. Liu and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218261120 CrossRef CAS PubMed.
- M. Anjass, G. A. Lowe and C. Streb, Angew. Chem., Int. Ed., 2021, 60, 7522 CrossRef CAS PubMed.
- S. Chakraborty, B. E. Petel, E. Schreiber and E. M. Matson, Nanoscale Adv., 2021, 3, 1293 RSC.
- C. F. Baes and R. E. Mesmer, The hydrolysis of cations, John Wiley, New York, 1976 Search PubMed.
- Y. Kikukawa, T. Yokoyama, S. Kashio and Y. Hayashi, J. Inorg. Biochem., 2015, 147, 221 CrossRef CAS PubMed.
- K. Kastner, J. T. Margraf, T. Clark and C. Streb, Chem. – Eur. J., 2014, 20, 12269 CrossRef CAS PubMed.
- L. Roubatis, N. C. Anastasiadis, C. Paratriantafyllopoulou, E. Moushi, A. J. Tasiopoulos, S. C. Karlabounas, P. G. Veltsistas, S. P. Perlepes and A. M. Evangelou, Inorg. Chem. Commun., 2016, 69, 85 CrossRef CAS.
- A. Müller and J. Döring, Z. Anorg. Allg. Chem., 1991, 595, 251 CrossRef.
- A. Notario-Estévez, P. Kozłowski, O. Linnenberg, C. de Graaf, X. López and K. Y. Monakhov, Phys. Chem. Chem. Phys., 2018, 20, 17847 RSC.
- A. Müller, E. Krickemeyer, M. Penk, R. Rohlfing, A. Armatage and H. Bögge, Angew. Chem., Int. Ed. Engl., 1991, 30, 1674 CrossRef.
- K. Wang, Q. Xu, P. Ma, C. Zhang, J. Wang and J. Niu, CrystEngComm, 2018, 20, 6273 RSC.
- K. Wang, Y. Niu, D. Zhao, Y. Zhao, P. Ma, D. Zhang, J. Wang and J. Niu, Inorg. Chem., 2017, 56, 14053 CrossRef CAS PubMed.
- C.-D. Zhang, S. X. Liu, B. Gao, C.-Y. Sun, L.-H. Xie, M. Yu and J. Peng, Polyhedron, 2007, 26, 1514 CrossRef CAS.
- A. Mïller, R. Sessoli, E. Krickemeryer, H. Bögge, J. Meyer, D. Gatteschi, L. Pardi, J. Westphal, K. Hovemeier, R. Rohlfing, J. Döring, F. Hellweg, C. Beugholt and M. Schmidtmann, Inorg. Chem., 1997, 36, 5239 CrossRef.
- M. I. Khan, E. Yohannes and R. J. Doedens, Angew. Chem., Int. Ed., 1999, 38, 1291 CrossRef.
- Y. Kikukawa, Y. Sakamoto, H. Hirasawa, Y. Kurimoto, H. Iwai and Y. Hayashi, Catal. Sci. Technol., 2022, 12, 2438 RSC.
- Y. Koyama, Y. Hayashi and K. Isobe, Chem. Lett., 2008, 37, 578 CrossRef CAS.
- M. Anjass, G. A. Lowe and C. Streb, Angew. Chem., Int. Ed., 2021, 60, 7522 CrossRef CAS PubMed.
- I. Yoshida, R. Mitsuhashi, Y. Kikukawa and Y. Hayashi, Inorganics, 2024, 12, 61 CrossRef CAS.
- J. M. Cameron, G. N. Newton, C. Busche, D. Long, H. Oshio and L. Cronin, Chem. Commun., 2013, 49, 3395 RSC.
- K. Okaya, T. Kobayashi, Y. Koyama, Y. Hayashi and K. Isobe, Eur. J. Inorg. Chem., 2009, 2009, 5156 CrossRef.
- T. Kobayashi, S. Kuwajima, T. Kurata and Y. Hayashi, Inorg. Chim. Acta, 2014, 420, 69 CrossRef CAS.
- S. Kuwajima, Y. Arai, H. Kitajima, Y. Kikukawa and Y. Hayashi, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2018, 47, 1295 CrossRef PubMed.
- Y. Kikukawa, K. Seto, S. Uchida, S. Kuwajima and Y. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 16051 CrossRef CAS PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- W. Liu and H. H. Thorp, Inorg. Chem., 1993, 32, 4102 CrossRef CAS.
- M. Carraro, B. S. Bassil, A. Sorarú, S. Berardi, A. Suchopar, U. Kortz and M. Bonchio, Chem. Commun., 2013, 49, 7914 RSC.
- Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2010, 49, 6096 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2341040. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr01243g |
| This journal is © The Royal Society of Chemistry 2024 |