
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering high-valence metal-enriched cobalt oxyhydroxide catalysts for an enhanced OER under near-neutral pH conditions†
Ruijing
Dong
ab,
Jiajian
Gao
b,
Truong-Giang
Vo
 b,
Shibo
Xi
b,
Choon Wee
Kee
b,
Xun
Cao
b,
Wei
Chu
*a and
Yan
Liu
*b
b,
Shibo
Xi
b,
Choon Wee
Kee
b,
Xun
Cao
b,
Wei
Chu
*a and
Yan
Liu
*b
aSchool of Chemical Engineering, Sichuan University, No. 24 South Section 1, Yihuan Road, Chengdu, 610065, China. E-mail: chuwei1965@scu.edu.cn
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology, and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore. E-mail: liu_yan@isce2.a-star.edu.sg
First published on 3rd June 2024
Abstract
Understanding water splitting in pH-neutral media has important implications for hydrogen production from seawater. Despite their significance, electrochemical water oxidation and reduction in neutral electrolytes still face great challenges. This study focuses on designing efficient electrocatalysts capable of promoting the oxygen evolution reaction (OER) in neutral media by incorporating high-valence elements into transition-metal hydroxides. The as-prepared and optimized two-dimensional Mo–Co(OH)2 nanosheets, which undergo operando transformation into oxyhydroxide active species, demonstrated an overpotential of 550 mV at 10 mA cm−2 with a Tafel slope of 110.1 mV dec−1 in 0.5 M KHCO3. In situ X-ray absorption spectroscopy revealed that the incorporation of high-valence elements facilitates the generation of CoOOH active sites at low potential and enhances electron transfer kinetics by altering the electronic environment of the Co center. This study offers new insights for developing more efficient OER electrocatalysts and provides fresh ideas for seawater utilization through the study of the reaction mechanism of the near-neutral-pH OER.
1. Introduction
Water splitting plays an important role in generating green hydrogen as a CO2-free sustainable fuel that can transform the global energy architecture in the context of a low-carbon economy.1–3 Electricity-driven water splitting is regarded as an optimal process for hydrogen energy production owing to its minimal environmental impact and lack of significant requirements for large-scale, high-risk equipment. Electrolysis of water involves two concurrent semi-reactions: the hydrogen evolution reaction (HER) involving the reduction of water at the cathode and the oxygen evolution reaction (OER) involving the oxidation of water at the anode.4,5 However, the slow reaction kinetics of the four-electron transfer mechanism of the OER, as opposed to the two-electron transfer of the HER, limits the wide application of water electrolysis.6 Transition-metal oxides (TMOs) and their hydroxides have been widely studied as catalytic materials to improve OER performance owing to the advantages of their lower cost than noble metals, magnificent conductivity and stability. For example, Hao et al. studied the OER mechanism on NiFe oxyhydroxides using a molecular probe and demonstrated that the chemical O–O coupling step serves as the rate-determining step.7 Yang et al. developed a novel Co-based NiCoFe LDH, which exhibited excellent OER performance with an overpotential of 233 mV.8 Subsequently, Vo et al. reported a transition-metal-based material, namely Ni–Fe hybrid films, which reached a low overpotential of 316 mV at a current density of 10 mA cm−2.9 McCrory et al. reported the benchmarking protocol of TMO catalysts using CoOx, CoPi, CoFeOx, NiOx, and NiCeOx, which showed high OER performance in alkaline solutions with overpotentials between 0.35 and 0.43 V.10Further, to improve the intrinsic activity of transition metal-based catalysts (TMCs), doping high-valence metals (HVMs) into TMCs appears as an effective approach allowing control over the local electronic structure and improving the oxidation states of the catalytic active centers of the TMCs.11 Meanwhile, HVMs also synergize with TMCs and participate in the OER reaction as dual active sites. For example, Li et al. demonstrated that the Ce-induced reinforcement of Co–O covalencies led to a remarkable enhancement in the OER performance, achieving an ultra-low overpotential of 261 mV in 0.1 M KOH.12 Meanwhile, Zhao et al. observed a dual-phase structural transition in bimetallic NiCo-based nanocrystals during the OER process, resulting in abundant oxygen vacancies and high oxidation states, which accounted for the high OER activity.13 Tang et al. observed the potential-dependent formation of OOH* intermediates at chromium (Cr) sites by confining high-valence Cr into a Co oxyhydroxide lattice.14 Moreover, molybdenum (Mo) played a similar role to Cr, as observed in Mo–CoOOH nanoframes, which exhibited the lowest overpotential of 400 mV and high durability for over 120 hours.15 These reports indicate that high-valence metals can adjust the microstructure and electronic configuration of transition metals when HVMs are embedded into the TMC crystal lattice. Additionally, high-valence metals can also serve as adsorption sites for oxygen-containing intermediates, such as *OOH, *O, and *OH, thereby further improving the catalytic activity of OER. However, the application of HVMs to enhance TMC catalysts with high intrinsic activity in near-neutral-pH OER has rarely been reported.
Recently, OER in pH-neutral electrolytes has received growing attention.16–18 One perspective is that neutral dielectrics are necessary for microbial electrocatalysis systems because most microorganisms can only survive under near-neutral pH conditions.19 Unlike strong acid and alkali solutions, neutral conditions are more environmentally friendly and sustainable. However, despite the environmental advantages, there are challenges associated with OER in near-neutral-pH electrolytes. The low solubility of reactants in the neutral electrolyte limits the OER reaction kinetics, which poses a hurdle to achieving efficient OER.20 Additionally, the overall reaction pathway for OER in alkaline and neutral solutions differs, indicating potentially different rate-determining steps.21 Even though the OER pathways in alkaline media (eqn (1)) and neutral media (eqn (2)) are well-established, various perspectives exist in the literature regarding the rate-demining steps. For example, Niu et al. reported that the formation of OOH* is the rate-determining step, which is accelerated by weakening the adsorption of O on RuO2/(Co, Mn)3O4.22 In parallel, Zhong et al. explored O2 production over a cobalt catalyst, revealing that the turnover-limiting chemical step was preceded by a one-electron, one-proton equilibrium in the pH 7.0 electrolyte.23 Therefore, understanding the OER reaction mechanism under neutral conditions is important for designing catalysts.24 Additionally, there is also a pressing need to design an efficient HVO-enhanced TMC with high intrinsic activity under pH-neutral and understand its reaction mechanism in near-neutral-pH media to unlock its full potential in sustainable energy applications using seawater.
| * + 4OH− → 2H2O + O2 + 4e− | (1) |
| * + 2H2O → 4H+ + O2 + 4e− | (2) |
In this work, a series of high-valence metal elements, including Cr (+6), Mo (+6), and W (+6), were used to modify layered Co(OH)2, and the OER activity of these catalysts in near-neutral-pH media was investigated. The optimized two-dimensional Mo–Co(OH)2 nanosheets were utilized as the model catalyst to investigate the underlying contributions of the high-valence elements incorporated into Co(OH)2 through in situ X-ray absorption analysis and DFT theoretical simulation. The results indicate that the incorporation of Mo beneficially modulated the electronic structure of the Co metal hydroxides, facilitating the operando formation of CoOOH as the active species in the OER process. Moreover, compared with Co(OH)2, the Co–O bond length was shortened in Mo–Co(OH)2, promoting deprotonation and increasing the valence of Co. This study elucidates the dynamic evolution of active sites and offers guidance for boosting OER performance by incorporating appropriate high-valence metals into transition-metal hydroxides.
2. Experimental section
2.1 Chemicals
Sodium molybdate dihydrate (Na2MoO4·2H2O), potassium dichromate (K2Cr2O7), sodium tungstate dihydrate (Na2WO4·2H2O), cobalt nitrate hexahydrate (CoN2O6·6H2O), potassium bicarbonate (KHCO3), 2-methylimidazide (MIM) and Nafion (20 wt%) were bought from Sigma Aldrich. Hexadecyltrimethylammonium bromide (CTAB) was purchased from Alfa Aesar. Ethanol was purchased from VWR CHEMICALS.2.2 Synthesis of 2D nanosheets
Two-dimensional (2D) high-valence-metal-modified Co(OH)2 nanosheets were synthesized using zeolitic imidazolate frameworks (ZIF67) as the cobalt precursor.15 In brief, 236 mg of CoN2O6·6H2O and 6.4 mg of CTAB were fully dissolved in 8 mL of deionized water. Subsequently, the solution was swiftly added to 56 mL of an aqueous MIM solution (3 g) and stirred at ambient temperature for 20 minutes to yield ZIF-67. The centrifuged solid ZIF-67 product was dispersed in ethanol, followed by the addition of an aqueous Na2MoO4 (280 mg) solution as the Mo source. Thereafter, the mixtures were refluxed at 85 °C for 2 h in an oil bath. Centrifugal separation was used to collect the formed Mo–Co(OH)2 and dried at 60 °C after washing several times with ethanol. The synthetic procedure of W–Co(OH)2 and Cr–Co(OH)2 was similar to that of Mo–Co(OH)2, except that Na2WO4 (272 mg) and KCrO7 (160 mg) were used instead of Na2MoO4.2.3 Characterization of 2D nanosheets
The crystalline structure of all samples was determined using Powder X-ray diffraction (XRD) analysis conducted on a Bruker D8 Advance X-ray diffractometer using Cu-Kα radiation (λ = 1.54178 Å) and Bragg–Brentano geometry in the 2θ range of 5–85°. Elemental composition analysis was conducted via inductively coupled plasma-optical emission spectroscopy (ICP-OES) by employing an Agilent VISTA-MPX instrument. X-ray photoelectron spectroscopy equipped with an Mg-Kα source (XPS, XSAM800 XPS) was executed to analyze the oxidation state of the Co species, and all data were calibrated using the 284.8 eV peak of carbon 1s peak. The cubic morphology of ZIF-67 and sheet morphology of the prepared samples were evaluated by field-emission scanning electron microscopy (FESEM) using a JSM-6700F instrument. High-resolution transmission electron microscopy coupled with energy-dispersive X-ray spectroscopy (HRTEM/EDX JEM-2100F, 200 kV) was employed for examining the lattice structures and elemental mappings of all samples. The specific surface area of the prepared samples was determined via N2 adsorption/desorption isotherms, which were calculated using the Brunauer–Emmett–Teller equation (Micromeritics BET ASAP 2420). The X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses of the Co K-edge in the samples were conducted in the transmission mode at the XAFCA beamline of the Singapore Synchrotron Light Source (SSLS). The SSLS storage ring was operated at E = 700 MeV and Imax = 200 mA. Additionally, energy calibration was achieved using a Co foil as the reference.2.4 In situ X-ray absorption spectroscopy (XAS)
The in situ Co K-edge XAS of Co(OH)2 and Mo-Co(OH)2 was performed at the XAFCA beamline of SSLS in the fluorescence mode. Briefly, the in situ spectroscopic experiments were conducted using a three-electrode electrochemical Teflon cell operating under a sensitive fluorescence mode. A carbon paper bearing catalyst droplets was affixed to the cell, which featured Ag/AgCl (saturated KCl solution) and a Pt wire as the reference and counter electrodes, respectively. A 0.5 M KHCO3 solution was filled in the cell as the electrolyte. During the in situ measurements, a range of potentials was applied to the working electrode, which was linked to an electrochemical workstation (Corr Test).2.5 Oxygen evolution reaction measurements
The OER electrochemical tests were performed using an electrochemical workstation (CHI 760E) in which a 0.5 M KHCO3 solution (pH = 8.5) served as the electrolyte, a Pt mesh was employed as the counter electrode, and Ag/AgCl (saturated KCl solution, ERHE = EAg/AgCl + 0.059 × pH + 0.197) was used as the reference electrode. The working electrode made of glassy carbon with a geometric area of 0.196 cm2 was loaded with various catalysts. Normally, to achieve a uniform ink, 2 mg of the electrocatalyst and 6.25 μL of Nafion were added to 1 mL of ethanol, followed by ultrasonic dispersion. Subsequently, 20 μL of the ink was deposited onto the surface of a glassy carbon electrode by drop-casting and dried under an infrared lamp. The electrochemical measurements were performed using a rotating disk electrode (RDE) at 25 °C, a scan rate of 5 mV s−1 and 1600 rpm. Prior to the linear sweep voltammetry (LSV) test, the catalysts underwent repeated sweeps from 1.2 V to 1.9 V (vs. RHE) in a 0.5 M KHCO3 solution until a stable voltammogram curve was achieved.The OER performance was investigated based on LSV curves, which were obtained by using automatic iR (i, current; R, resistance) correction (100%) on the workstation. Electrochemical impedance spectroscopy (EIS) was conducted in a frequency range spanning from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz to 0.01 Hz while maintaining a potential of 1.8 V (vs. RHE). The double-layer capacitance (Cdl) was calculated using cyclic voltammetry (CV) under different scan rates from 5 to 25 mV s−1. In the stability test of Co(OH)2 and Mo–Co(OH)2 using chronoamperometry, the potential was maintained at 10 mA cm−2, as determined from the LSV profile.
000 Hz to 0.01 Hz while maintaining a potential of 1.8 V (vs. RHE). The double-layer capacitance (Cdl) was calculated using cyclic voltammetry (CV) under different scan rates from 5 to 25 mV s−1. In the stability test of Co(OH)2 and Mo–Co(OH)2 using chronoamperometry, the potential was maintained at 10 mA cm−2, as determined from the LSV profile.
2.6 Theoretical simulations
Theoretical simulations were conducted using density-functional theory (DFT) analysis. Spin-polarized calculations were executed using version 6.4.2 of the Vienna Ab initio Simulation Package (VASP) by employing projector-augmented-wave pseudopotentials.25–28 The Perdew–Burke–Ernzerhof generalized gradient approximation functional was selected as the density functional theory (DFT) functional. The high precision mode (PREC = accurate) was utilized with a plane-wave energy cut-off of 800 eV and an augmentation charge cut-off of 1000 eV. The convergence criterion for electronic energy was set to 10−8 eV. A Gaussian smearing of 0.05 width was applied in self-consistent field calculations. Brillouin zone integration was achieved by sampling a Gamma-centered k-point mesh with a spacing of 0.5 Å−1, which was generated automatically using the “KSPACING” keyword. Dispersion interactions were considered using the D3 method with Becke–Johnson damping.29 For ionic relaxation, the magnitude of all the forces acting on all atoms is less than 0.01 eV Å−1. Numerical integration was done via the finite differences approach. The width of displacement of each ion was set to 0.01 Å. Thermochemical corrections with the Gibbs free energy were calculated using VASPKIT.303. Results and discussion
3.1 Synthesis and characterization of two-dimensional (2D) HVM-modified Co(OH)2 nanosheets
2D nanosheets of high-valence-metal-modified Co(OH)2 were synthesized by introducing HVMs into cobalt hydroxides by using ZIF-67 cubes as the growth template. With the addition of a Mo source into the ZIF-67 dispersion, subsequent heating and refluxing, the ZIF-67 cube was transformed into Co(OH)2 double-layered hydroxides. Concurrently, MoO42− intercalated between the layers of Co(OH)2, resulting in the formation of nanosheets denoted as Mo–Co(OH)2. Similarly, Cr–Co(OH)2 and W–Co(OH)2 were prepared using a Cr source and a W source instead of Mo, respectively. Fig. 1(a) shows the SEM images of ZIF-67, revealing their nanocubic morphology with an average size of 300 nm. The XRD pattern of ZIF-67 is indicated in Fig. 1(b), with peaks at 2θ values 7°, 10.2°, 12.5°, 34.5°, 14.5°, 16°, 18°, 22°, 24.3°, 26.5°, corresponding to the ZIF-67 planes (011), (002), (112), (022), (013), (222), (114), (223), and (134), respectively. The XRD pattern of HVM-Co(OH)2 2D nanosheets is shown in Fig. 1(c), and all significant peaks correspond to α-Co(OH)2, which has a larger distance between layers than that in β-Co(OH)2. The larger layer spacing enables convenient mass transfer and HVM ion insertion.31 The 2θ peaks at 11.6°, 23.2°, 33.5°, 34.5°, and 60° correspond to the (003), (006), (012), (100), and (110) planes of cobalt hydroxides (JCPDS no. 46-0605).32 There was no striking distinction in the crystal structure with the introduction of different high valence metals, which demonstrates the preservation of the α-Co(OH)2 structure. Fig. 1(d)–(f) displays the nanosheet-shaped morphology of Mo–Co(OH)2, Cr–Co(OH)2, and W–Co(OH)2, respectively, as observed via high-resolution TEM (HRTEM). No separate particles of HVMs were observed, further suggesting the preservation of the original structure of α-Co(OH)2. The results also illustrate the versatility and wide applicability of the synthetic method due to its simplicity. The elemental mapping is depicted in Fig. 1(g), and Fig. S2† illustrates that Co and Mo (or Cr or W) elements were evenly dispersed on the nanosheets, proposing the successful incorporation of HVMs into Co(OH)2.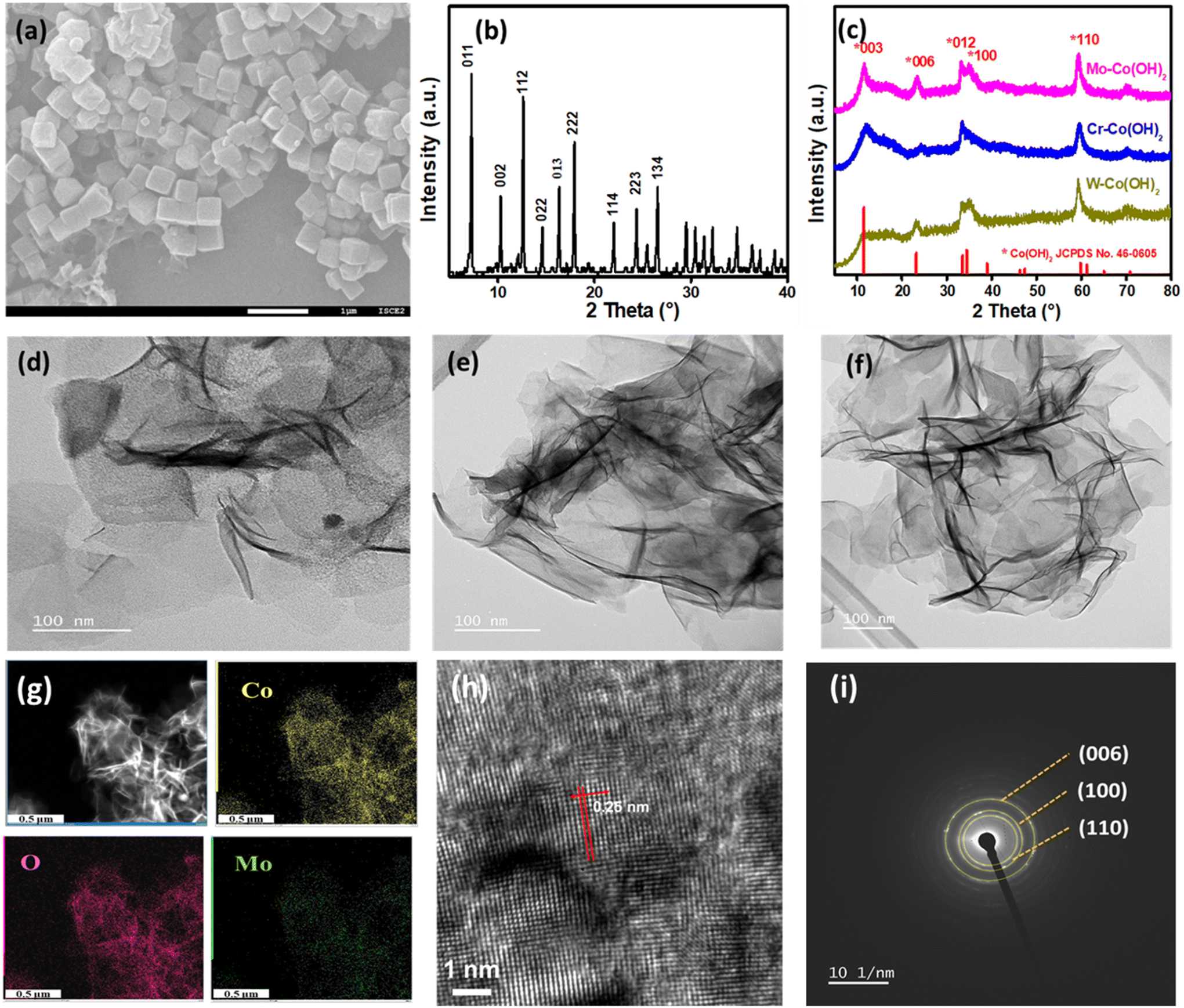 | ||
| Fig. 1 (a) SEM image and (b) XRD pattern of the ZIF-67 template. (c) XRD patterns of Mo–Co(OH)2, Cr–Co(OH)2 and W–Co(OH)2. (d–f) TEM images of Mo–Co(OH)2, Cr–Co(OH)2 and W–Co(OH)2 2D nanosheets. (g) TEM mapping images of Mo–Co(OH)2. (h) High-resolution TEM (HRTEM) image of Mo–Co(OH)2 and (i) selected-area electron diffraction pattern of Mo–Co(OH)2. | ||
Measured from the HRTEM image of Mo–Co(OH)2 (Fig. 1h), the lattice spacing of 0.25 nm reveals the existence of the (110) plane of crystalline Co(OH)2. Additionally, the selected area electron diffraction pattern depicted in Fig. 1(i) shows a good polycrystalline structure, with the (006), (100), and (110) diffraction planes of Co(OH)2 aligning closely with those observed in the XRD pattern.33 The specific surface areas of Mo–Co(OH)2, Cr–Co(OH)2 and W–Co(OH)2, as measured using the N2 adsorption/desorption isotherms (Fig. S3†), were 137.1 m2 g−1, 234.8 m2 g−1, and 172.1 m2 g−1, respectively, implying that catalysts with high specific surface areas could be effectively obtained through the simple hydrolysis method.
3.2 Electrocatalytic oxygen evolution performance of HVM-modified Co(OH)2
The OER performance was investigated using LSV in a 0.5 M KHCO3 solution (pH 8.5). Fig. 2(a) shows that the electrocatalytic ability of HVM-modified Co(OH)2 is significantly superior to that of unmodified Co(OH)2, indicating the enhancing effect of HVM on the OER. Among the samples tested, Mo–Co(OH)2 emerged as the most effective OER catalyst, exhibiting the highest current density at a given overpotential. Specifically, to achieve a current density of 10 mA cm−2, Mo–Co(OH)2 gave an overpotential of 550 mV. This value is notably lower compared with Cr–Co(OH)2 (570 mV) and W–Co(OH)2 (640 mV). Except for the pure Co(OH)2 sample, the anodic wave corresponding to the transformation of Co(OH)2 to CoOOH at the activity sites during the OER reaction was clearly observed in the LSV curve.34,35 The introduction of HVMs allowed the generation of CoOOH at a lower voltage. The results demonstrate that HVMs promote charge transfer, thereby facilitating the efficient removal of protons in the initial step of the OER process because of the strong covalency of the M–O interaction.36 It is noteworthy that the excessively strong electronic interaction between the HVM and Co could impede the oxidation of Co2+ to a certain extent, as evidenced by the positive shift in the anodic wave of Co2+/Co3+ from Cr–Co(OH)2 to W–Co(OH)2. In general, the electrocatalytic ability of Co(OH)2 was promoted by the incorporation of HVMs.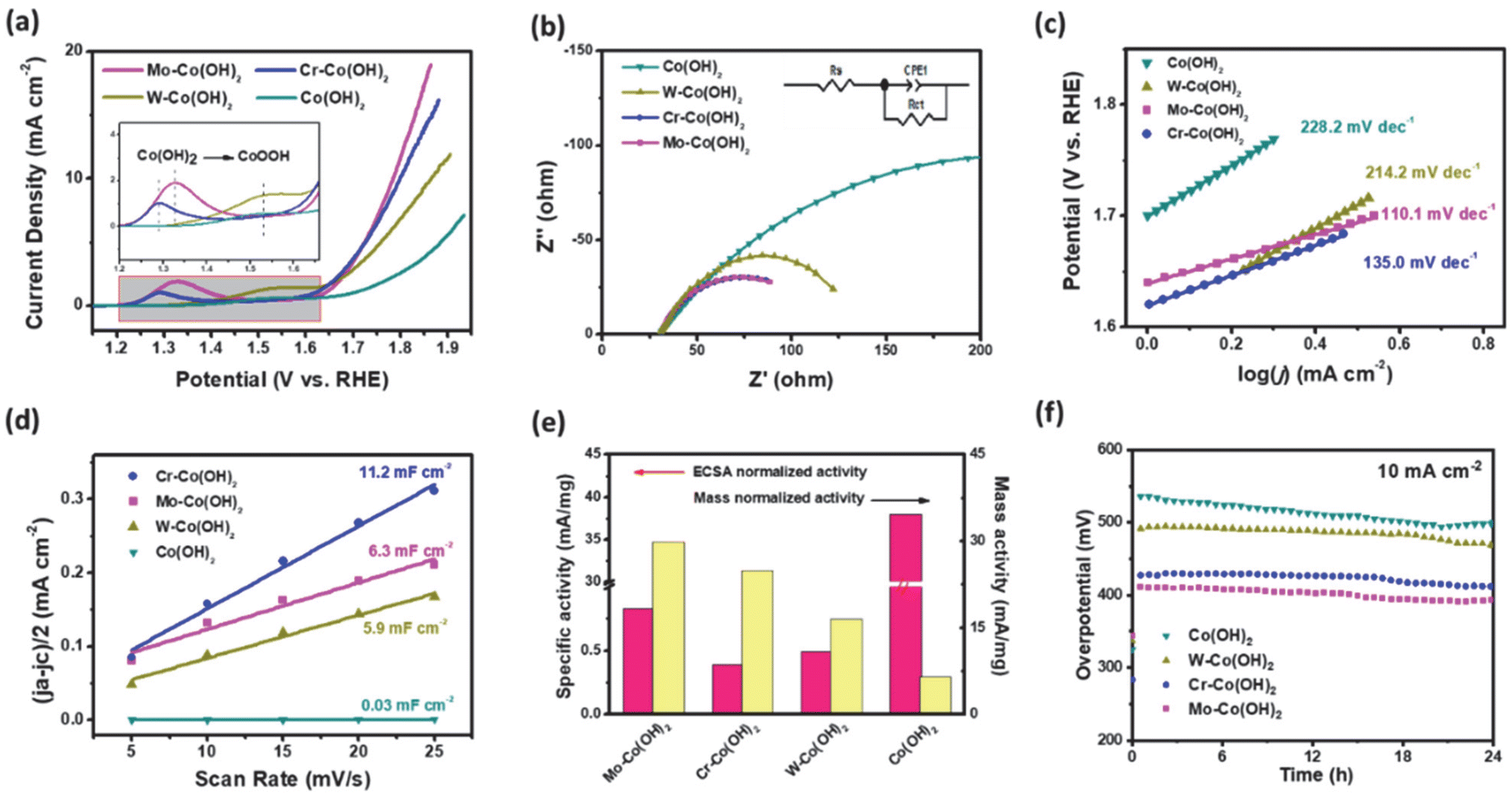 | ||
| Fig. 2 (a) iR-corrected polarization curves of HVM-modified Co(OH)2 during the OER. (b) Nyquist plots of the catalysts, and the inset shows the equivalent circuit mode. (c) Tafel plots of the different catalysts. (d) Double-layer capacitance plots of the different catalysts. (e) Specific activity normalized to ECSA and mass and (f) stability test of Co(OH)2 and HVM-modified Co(OH)2 under 10 mA cm−2. | ||
Electrochemical Impedance Spectroscopy (EIS) was carried out to investigate the charge-transfer processes and electrode kinetics. The corresponding Nyquist plots are depicted in Fig. 2(b), with the inset showcasing the equivalent circuit used for the analysis. The solution resistance (Rs), which includes the electrode resistance, of the as-prepared catalysts were close to each other, implying the similar conductivity of Mo–Co(OH)2, Cr–Co(OH)2 and W–Co(OH)2. The fitted charge transfer resistances (Rct) were 118.96 Ω for Mo–Co(OH)2, 112.61 Ω for Cr–Co(OH)2, and 136.2 Ω for W–Co(OH)2, which are lower than that of Co(OH)2 (389 Ω). This suggests that compared with pure Co(OH)2, the incorporation of high-valence elements into Co(OH)2 resulted in faster electron transfer kinetics. The optimized electronic structure and charge conductivity benefit the enhancement of OER performance.37 As seen in Fig. 2(c), the Tafel slope value of Mo–Co(OH)2 was 110.1 mV dec−1, demonstrating faster kinetics in Mo–Co(OH)2 than Cr–Co(OH)2 (135.0 mV dec−1), W–Co(OH)2 (214.2 mV dec−1) and Co(OH)2 (228.2 mV dec−1). The electrochemically active surface area (ECSA) was calculated using eqn (3) for all samples; the double-layer capacitance (Cdl) was acquired from cyclic voltammetry (CV) tests at different scan rates (Fig. S4†), and the specific capacitance (Cs) was 40 μF cm−2.38,39
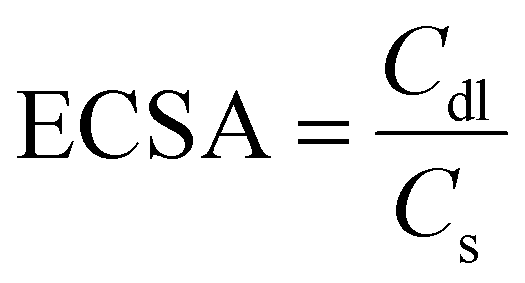 | (3) |
As shown in Fig. 2(d), there were distinct differences between different catalysts; the Cdl of Cr–Co(OH)2 was the maximum at 11.2 mF cm−2 compared with 6.3 mF cm−2 of Mo–Co(OH)2 and 5.9 mF cm−2 of W–Co(OH)2. The results propose that all HVM-modified Co(OH)2 could provide more active sites and possess higher ECSA (Table S1†). As shown in Fig. 2(e), the ECSA-normalized activity and mass-normalized activity revealed that Mo–Co(OH)2 had the greatest specific activity. Stability testing at 10 mA cm−2 is depicted in Fig. 2f. There was no significant decrease in the activity of Co(OH)2 and all HVM-modified Co(OH)2 samples within 24 hours. Among them, Mo–Co(OH)2 presented the lowest overpotential of 390 mV at a current density of 10 mA cm−2. Therefore, Mo–Co(OH)2 was chosen as the representative HVM-enhanced cobalt oxyhydroxide to investigate the microstructures and understand the catalytic mechanism in the following experiments.
3.3 Surface chemistry and local environment of the Mo–CoOOH nanosheets
The element valence and chemical content of Mo–Co(OH)2 and Co(OH)2 were analyzed using X-ray photoelectron spectroscopy (XPS). As shown in Fig. 3(a), the fitted peaks at 781.4 eV and 796.9 eV correspond to the 2p3/2 and 2p1/2 orbitals of Co2+; there was no distinct difference in the Co chemical state between pristine and HVM-modified Co(OH)2.40 However, compared with pristine Mo–Co(OH)2, the binding energy of Co2+ was shifted to a higher potential by around 0.4 eV after OER, and remained at 780.35 eV and 795.8 eV corresponding to the 2p3/2 and 2p1/2 orbitals of Co3+ in Mo–Co(OH)2 after the OER process. These results suggest that high-valence Mo can promote the oxidation of the Co species due to a strong covalent interaction between Mo and Co.41 The XPS peak-fitting analysis substantiated the Mo-facilitated increase in the valence state of the Co species, thereby promoting the formation of CoOOH as active sites for OER at a relatively lower potential. This result is consistent with the observed OER performance (Fig. 2). The electronic environment of the catalytic active center is optimized by the introduced HVMs, thus improving the intrinsic activity of the cobalt-based catalysts. As shown in Fig. S6,† the 2D nanosheet morphology of HVM-modified Co(OH)2 was well-maintained after the OER process.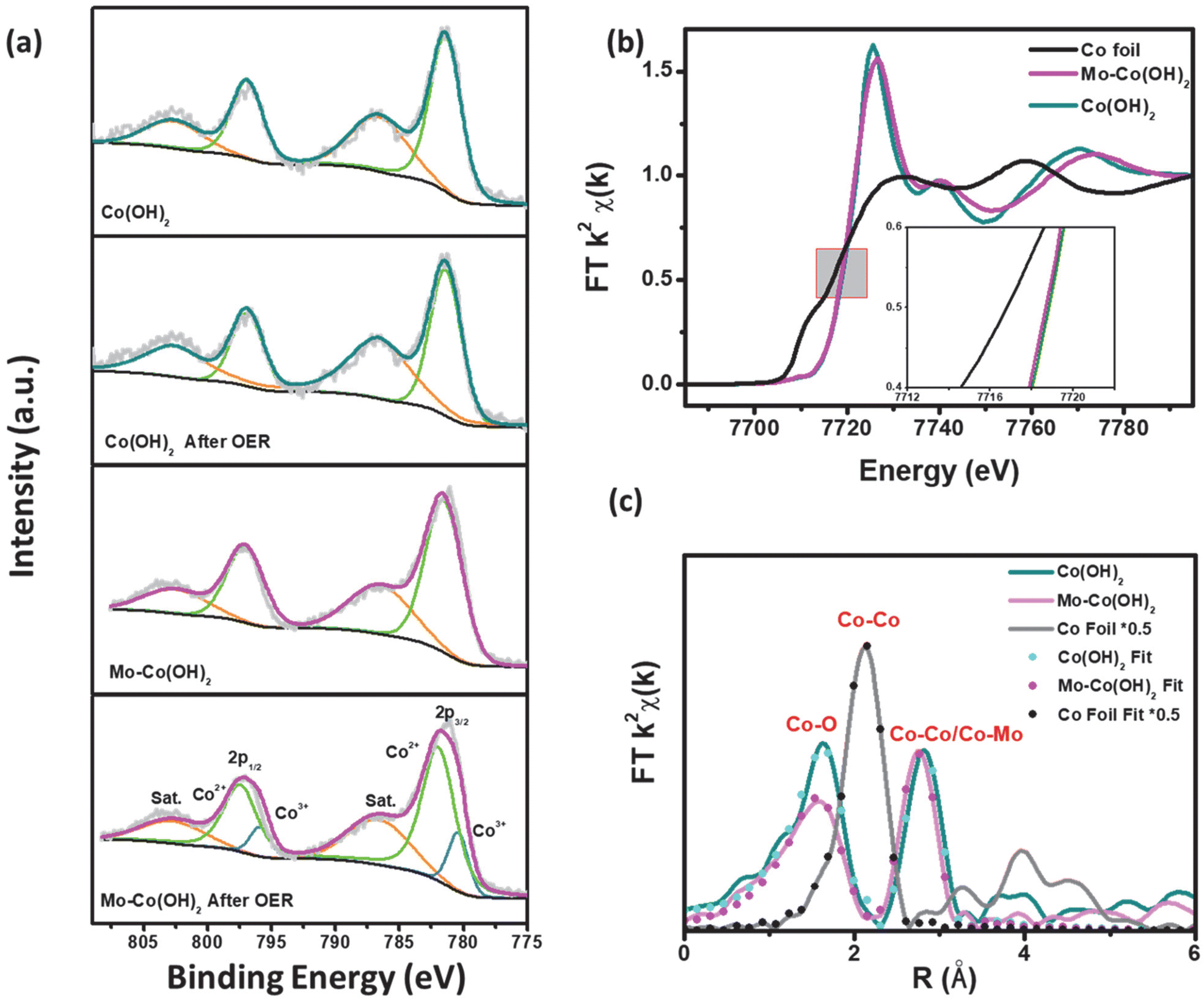 | ||
| Fig. 3 (a) Co 2p XPS absorption spectra of Mo–Co(OH)2 and Co(OH)2, (b) Co K-edge XAFS absorption spectra of Mo–Co(OH)2, Co(OH)2 and Co foil and (c) k2-weighted Fourier transform spectra of the Co K-edge EXAFS absorption spectra. | ||
To further investigate the impact of HVMs on the coordination environment of TMCs, X-ray absorption fine structure (XAFS) spectroscopy was conducted for Mo–Co(OH)2 and Co(OH)2. As seen in Fig. 3(b), the X-ray near-edge absorption fine structure (XANES) analysis showed a similar absorption energy of 7718 eV at the Co K-edge, which demonstrated the identical initial chemical states of the Co species in Mo–Co(OH)2 and Co(OH)2, which is consistent with the conclusion drawn from XPS analyses. Compared with Co(OH)2, the white line of the Co K-edge in Mo–Co(OH)2 was slightly weaker, indicating greater 4p orbital splitting of Co.42 This phenomenon causes changes in the spin state or symmetry properties of Co with the incorporation of high-valence Mo.43
As shown in Fig. 3(c), i.e. the Fourier transform (FT) of Co K-edge extended XAFS (EXAFS) of the Co foil showed a dominant peak of Co–Co at 2.49 Å. Co(OH)2 exhibited the Co–O peak at 2.09 Å and the Co–Co peak at 3.19 Å, which shifted to lower R values in Mo–Co(OH)2 (i.e. the Co–O peak was at 2.06 Å and the Co–Co/Mo peak at 3.13 Å). Simulation of the quantitative structural parameters of Co atoms in Mo–Co(OH)2 by least-squares EXAFS curve fitting exemplified that the Co–O coordination number was about 5.1 in Mo–Co(OH)2, which is distinct from the standard coordination number of 6 in Co(OH)2 (Table S2†), suggesting the existence of cationic vacancy.44
3.4 In situ/operando XAS studies for understanding the catalytic mechanism
An operando XAS analysis was performed to elucidate the potential catalytic mechanism by investigating the dynamic changes in the oxidation state of Co under OER-relevant potentials. Fig. 4(a) and (d) display the operando XANES spectra at the Co K-edge of Co(OH)2 and Mo–Co(OH)2 recorded at different applied potentials. From open-circuit potential (OCP) to 1.5 V, the Co absorption edge shifted to higher energy, showing an increase in the Co oxidation state. The weakened and broadened white line peak of the Co K-edge in Mo–Co(OH)2 with increased potential suggests molecular orbital formation due to the hybridization of Mo d orbital with the Co p orbital.36 Of the wavelet transforms of the k2-weighted EXAFS (WTEXAFS) signals of the Co K-edges of Co(OH)2 (Fig. 4b) and Mo–Co(OH)2 (Fig. 4e) at 1.4 V, the intensity of Co–M in Mo–Co(OH)2 was stronger than that of Co(OH)2, suggesting that atomic Mo was successfully incorporated in Co(OH)2. Moreover, between the WTEXAFS signals of the Co K-edges of Co(OH)2 (Fig. 4c) and Mo–Co(OH)2 (Fig. 4f) at 1.7 V, the intensity of Co–O in Mo–Co(OH)2 was stronger than that of Co(OH)2, further demonstrating that the introduction of Mo promoted the oxidation of the Co species, which is consistent with findings from the XPS analysis. Fig. 4(g) presents a schematic of the operando oxidation process of Co(OH)2 to CoOOH during the OER. It suggests that Co(OH)2 undergoes the first deprotonation step at a higher voltage (1.7 V) compared with Mo-enhanced Co(OH)2 (1.5 V). According to the above analysis, the radius of Co–O decreases, and the electronic environment of the Co center is regulated by the incorporation of Mo, thereby resulting in the better OER intrinsic activity of Mo–Co(OH)2.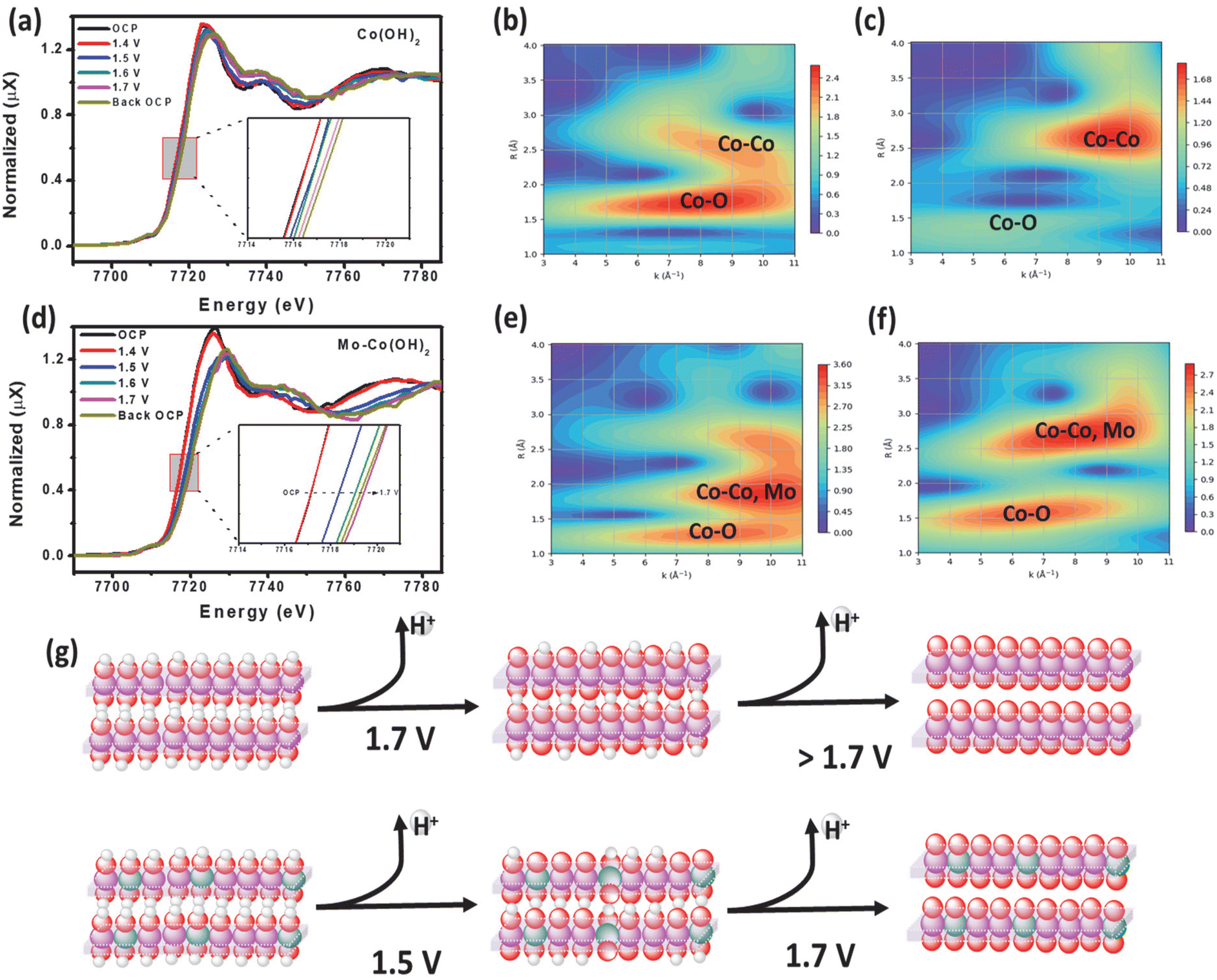 | ||
| Fig. 4 (a) Operando XAFS Co K-edge absorption spectra of Co(OH)2, wavelet transformation of the k2-weighted Co K-edge absorption EXAFS signals of Co(OH)2 at (b) 1.4 V and (c) 1.7 V, (d) operando XAFS Co K-edge absorption spectra of Mo–Co(OH)2, (e) wavelet transforms of the k2-weighted Co K-edge absorption EXAFS signals of Mo–Co(OH)2 at (e) 1.4 V and (f) 1.7 V. (g) Schematic of the oxidation process of Co(OH)2 to CoOOH during the OER: O (red), H (white), Co (purple), and Mo(green). | ||
3.5 Mechanism analysis
The theoretical OER mechanism analysis was carried out using Density Functional Theory (DFT) calculations. The thermodynamics of the OER was investigated according to the following electrochemical steps:| * + H2O → *OH + H+ + e− | (4) |
| *OH → *O + H+ + e− | (5) |
| *O + H2O → *OOH + H+ + e− | (6) |
| *OOH → O2* + H+ + e− | (7) |
| O2* → O2 (g) + * | (8) |
The Computational Hydrogen Electrode Model was utilized to compute the free energy diagram for the OER reaction (Fig. 5a).45,46 The fully oxidized form of Mo–CoOOH – Mo–CoO2 – was used as the model, as reported by Tang et al.15 However, the ratio of Mo to Co used in our model was 1 to 8 to closely reflect the stoichiometry determined from the ICP-OES results. Consistent with the result of Tang et al., the adsorption of OH does not take place if ionic relaxation is initiated with the OH on top of the Co atoms, but it occurs on the Mo atom. The limiting potential of Mo–CoOO in the neutral OER is +1.57 V. The probable rate-determining step of the OER is *O + OH to *OOH with a barrier of at least 0.34 V.
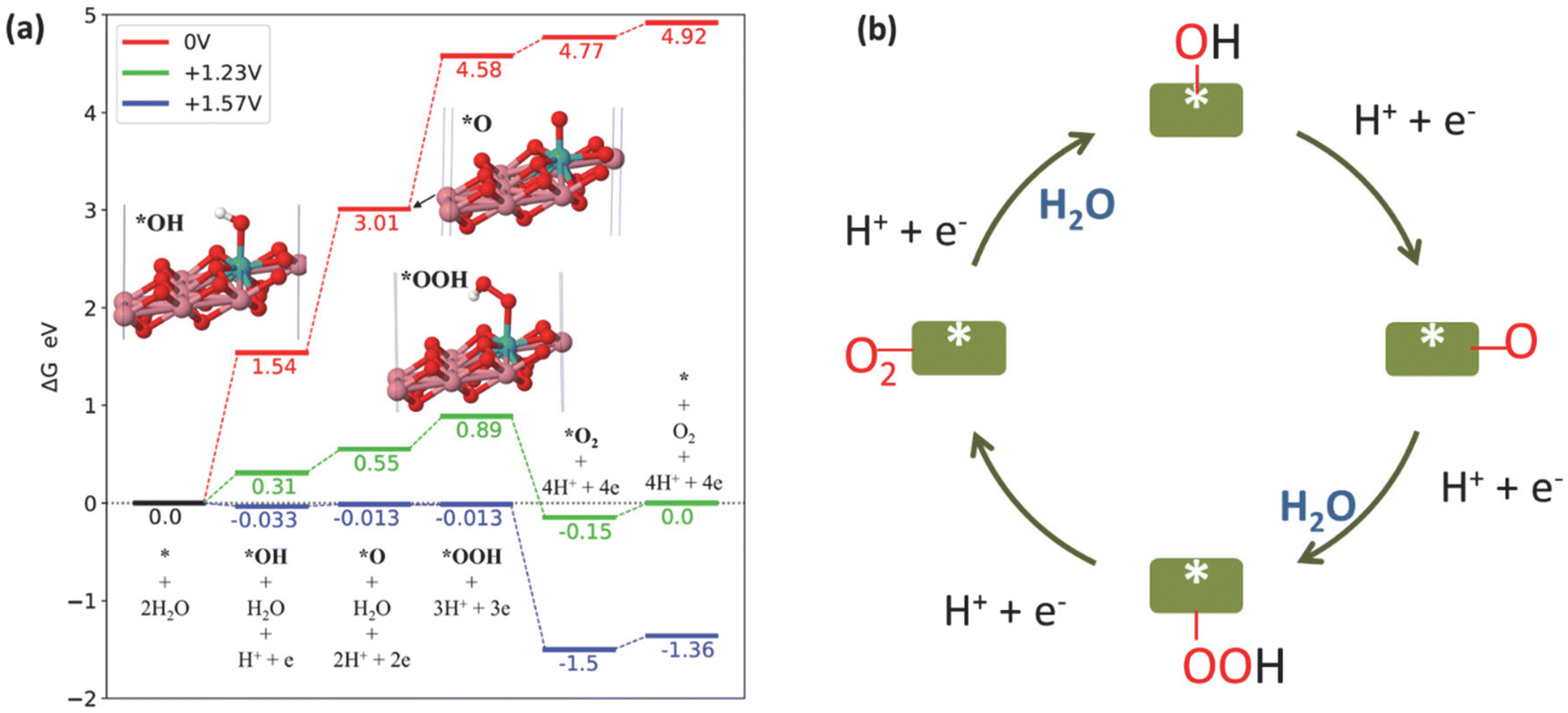 | ||
| Fig. 5 (a) Free energy diagram of the OER catalysed by Mo–CoO2 at different electrode potentials: 0 V (red), +1.23 V (green) and +1.57 V (blue). (b) Schematic of the OER mechanism on Mo–Co(OH)2. | ||
We observed a similar barrier energy of at least 0.31 V for *OOH formation and *OH formation (eqn (4)), while the barrier energy for *O formation (eqn (5)) was at least 0.24 V, demonstrating that the first three steps limited the OER in terms of thermodynamics. According to the Tafel slope investigation shown in Fig. 2(c), *OH formation (eqn (4)) extremely limits OER kinetics, resulting in a low reaction rate. Overall, the kinetics and thermodynamics analyses suggest that the rate-determining step of OER on HVMs-enhanced CoOOH in pH-neutral electrolytes is the adsorption and dissociation of H2O, which leads to *OH formation.
Fig. 5(b) shows the schematic of the OER pathway on Mo–Co(OH)2 according to the traditional adsorbate evolution mechanism. According to the four-electron transfer process of the OER, molecular H2O is adsorbed on the catalyst surface, initially forming *OH with the removal of one proton and one electron. Further, the deprotonation of *OH generates *O, which combines with another H2O to form the intermediate *OOH. The final step is the removal of the proton and electron of *OOH along with the desorption of O2. The adsorbate evolution mechanism indicates that the O atoms of O2 originate from H2O rather than the lattice O of the catalysts.47,48
4. Conclusion
In summary, a comprehensive investigation of the development of a series of high-valence-element-modified Co(OH)2 two-dimensional nanosheets and their OER electrocatalytic performance was successfully conducted. Among the investigated samples, Mo–Co(OH)2 emerged as the most efficient OER catalyst in near-neutral-pH media. Subsequent in situ XAS analysis revealed that incorporating HVMs was beneficial for the operando generation of CoOOH active sites at a low overpotential during the OER process. In near-neutral-pH media, there are two aspects to be considered for further improving their catalytic performance. One is to promote the adsorption and dissociation of H2O, while the other is to enhance the formation of the *OOH and *O intermediates. These findings provide new insights for developing more efficient OER electrocatalysts under near-neutral-pH conditions and understanding active species evolution, besides providing new ideas for seawater utilization toward green hydrogen production.Author contributions
Ruijing Dong: conceptualization, investigation and manuscript preparation. Jiajian Gao: results discussion and manuscript revision. Truong-Giang Vo: electrochemical testing discussion, analysis and manuscript revision. Shibo Xi: XAS analysis and data interpretation; Choon Wee Kee: DFT simulation and mechanism discussion. Xun Cao: HRTEM analysis and data interpretation; Wei Chu: supervision, manuscript revision. Yan Liu: supervision, formal analysis, manuscript revision, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could appear to influence the work reported in this paper.Acknowledgements
The authors appreciate the Chinese Scholarship Council (CSC) for providing the Ph. D. scholarship and ISCE2 (A-STAR, Singapore) for the financial support. This research is supported by the National Research Foundation, Singapore, and PUB, Singapore's National Water Agency under its Competitive Funding for Water Research (CWR-2101-0029). The computational work for this article was partially performed using resources of the National Supercomputing Centre, Singapore (https://www.nscc.sg). This work was also supported by the A*STAR Computational Resource Centre through its high-performance computing facilities. Authors gratefully acknowledge Ms Wang Zhan April, Ms. Zheng Jia E and Mr Yeo Wen Cong (ISCE2, A*STAR, Singapore) for their assistance with XPS, ICP and BET tests, respectively.References
- P. Hota, A. Das and D. K. Maiti, Int. J. Hydrogen Energy, 2023, 48, 523–541 CrossRef CAS.
- J. Guo, Y. Zhang, A. Zavabeti, K. Chen, Y. Guo, G. Hu, X. Fan and G. K. Li, Nat. Commun., 2022, 13, 5046 CrossRef CAS PubMed.
- D. E. H. J. Gernaat, H. S. de Boer, V. Daioglou, S. G. Yalew, C. Müller and D. P. van Vuuren, Nat. Clim. Change, 2021, 11, 119–125 CrossRef.
- Y. P. Zhu, C. Guo, Y. Zheng and S. Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- Y. Hao, Y. Li, J. Wu, L. Meng, J. Wang, C. Jia, T. Liu, X. Yang, Z. P. Liu and M. Gong, J. Am. Chem. Soc., 2021, 143, 1493–1502 CrossRef CAS PubMed.
- Q. Yang, T. Li, Z. Lu, X. Sun and J. Liu, Nanoscale, 2014, 6, 11789–11794 RSC.
- T. G. Vo, S. D. S. Hidalgo and C. Y. Chiang, Dalton Trans., 2019, 48, 14748–14757 RSC.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. Wang, T. Zhai, Y. Wu, T. Zhou, B. Zhou, C. Shang and Z. Guo, Adv. Sci., 2023, 10, e2301706 CrossRef PubMed.
- M. Li, X. Wang, K. Liu, H. Sun, D. Sun, K. Huang, Y. Tang, W. Xing, H. Li and G. Fu, Adv. Mater., 2023, 35, e2302462 CrossRef PubMed.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- L. Tang, M. Xia, S. Cao, X. Bo, S. Zhang, Y. Zhang, X. Liu, L. Zhang, L. Yu and D. Deng, Nano Energy, 2022, 101, 107562 CrossRef CAS.
- L. Tang, L. Yu, C. Ma, Y. Song, Y. Tu, Y. Zhang, X. Bo and D. Deng, J. Mater. Chem. A, 2022, 10, 6242–6250 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- L. Q. Zhou, C. Ling, H. Zhou, X. Wang, J. Liao, G. K. Reddy, L. Deng, T. C. Peck, R. Zhang, M. S. Whittingham, C. Wang, C. W. Chu, Y. Yao and H. Jia, Nat. Commun., 2019, 10, 4081 CrossRef PubMed.
- L. Zhang, L. Wang, Y. Wen, F. Ni, B. Zhang and H. Peng, Adv. Mater., 2020, 32, e2002297 CrossRef PubMed.
- C. Liu, B. C. Colon, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed.
- K. Xu, H. Cheng, L. Liu, H. Lv, X. Wu, C. Wu and Y. Xie, Nano Lett., 2017, 17, 578–583 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- S. Niu, X.-P. Kong, S. Li, Y. Zhang, J. Wu, W. Zhao and P. Xu, Appl. Catal., B, 2021, 297, 120442 CrossRef CAS.
- D. K. Zhong and D. R. Gamelin, J. Am. Chem. Soc., 2010, 132, 4202–4207 CrossRef CAS PubMed.
- J. Gao, H. Tao and B. Liu, Adv. Mater., 2021, 33, e2003786 CrossRef PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- N. P. Dileep, T. V. Vineesh, P. V. Sarma, M. V. Chalil, C. S. Prasad and M. M. Shaijumon, ACS Appl. Energy Mater., 2020, 3, 1461–1467 CrossRef CAS.
- R. Jia, M. Xia, L. Tang, L. Yu, Y. Yang, Y. Zhang, X. Bo, S. Zhou, Y. Tu and D. Deng, ACS Catal., 2022, 12, 13513–13522 CrossRef CAS.
- J. Zhao, Z. Huang, J. Zeng, M. Deng, G. Yin, X. Liao and J. Gu, J. Inorg. Organomet. Polym. Mater., 2011, 22, 492–499 CrossRef.
- T.-G. Vo, P.-Y. Ho and C.-Y. Chiang, Appl. Catal., B, 2022, 300, 120723 CrossRef CAS.
- S. Wang, Q. Jiang, S. Ju, C. S. Hsu, H. M. Chen, D. Zhang and F. Song, Nat. Commun., 2022, 13, 6650 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- A. Moysiadou, S. Lee, C. S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. D. Lou, Adv. Mater., 2020, 32, e2002235 CrossRef PubMed.
- X.-Z. Song, N. Zhang, F. Liu, Z.-H. Wang, W.-Y. Zhu, G.-Z. Zhang, Z.-Y. Niu, X.-F. Wang and Z. Tan, J. Alloys Compd., 2021, 873, 159776 CrossRef.
- X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y. W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Nature, 2022, 611, 702–708 CrossRef CAS PubMed.
- W. H. Lee, M. H. Han, Y. J. Ko, B. K. Min, K. H. Chae and H. S. Oh, Nat. Commun., 2022, 13, 605 CrossRef CAS PubMed.
- Y. J. Wu, J. Yang, T. X. Tu, W. Q. Li, P. F. Zhang, Y. Zhou, J. F. Li, J. T. Li and S. G. Sun, Angew. Chem., Int. Ed., 2021, 60, 26829–26836 CrossRef CAS PubMed.
- X. Zhao, Z. H. Levell, S. Yu and Y. Liu, Chem. Rev., 2022, 122, 10675–10709 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Gao, C.-Q. Xu, S.-F. Hung, W. Liu, W. Cai, Z. Zeng, C. Jia, H. M. Chen, H. Xiao, J. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2019, 141, 3014–3023 CrossRef CAS PubMed.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01168f |
| This journal is © The Royal Society of Chemistry 2024 |