
Ru doping and interface engineering synergistically boost the electrocatalytic performance of a WP/WP2 nanosheet array for an efficient hydrogen evolution reaction†
Zhichang
Hu
,
Zhizhong
Xiao
,
Wei
Wei
,
Jian
Yang
,
Xiaoyu
Huang
,
Qingcheng
Lu
,
Sundaram
Chandrasekaran
,
Huidan
Lu
 * and
Yongping
Liu
*
* and
Yongping
Liu
*
Guangxi Key Laboratory of Electrochemical and Magneto-Chemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, P. R. China. E-mail: lhuidangl@163.com; liuyp624@163.com
First published on 3rd June 2024
Abstract
The surface electronic structure and morphology of catalysts have a crucial impact on the electrocatalytic hydrogen evolution reaction performance. This work reports on the fabrication of a Ru-doped WP/WP2 heterojunction nanosheet array electrode via a one-step phosphating treatment of a Ru-doped WO3 precursor. Benefitting from the large electrochemical active surface of nanosheet arrays, rich WP/WP2 heterojunction interface, and trace Ru atom doping, the catalyst has a fairly low overpotential of 58.0 mV at 10 mA cm−2 and a Tafel slope of 50.71 mV dec−1 in acid solution toward the electrocatalytic HER. Further, theoretical calculations unveil that Ru atom doping and interface effect synergistically optimized the electronic structure of the catalyst and hence weakened the adsorption capacity of the catalyst surface toward hydrogen (H), which lowered the Gibbs free energy (ΔGH*) and consequently effectively improved the HER performance. This work may open new avenues for developing advanced nanoarray electrodes with efficient electrochemical energy conversion.
Introduction
Hydrogen (H2) is considered one of the ideal energy carriers most likely to replace traditional fossil energy because of its high energy density and clean and renewable characteristics.1,2 Electrocatalytic water splitting provides an effective H2 production method,3 driven by the electric energy converted from clean and renewable energy, such as wind or solar energy.4,5 To produce H2 through electrocatalytic water splitting on a large scale, it is essential to obtain suitable catalyst materials with high catalytic performance and economical properties.6 However, multi-active catalysts for electrocatalytic hydrogen evolution are mainly Pt series catalysts with scarce resources and high cost, significantly restricting their wide application.7Transition-metal phosphides (TMPs) show numerous fascinating physical and chemical features, including high electronic conductivity, luminescence, thermoelectric, and catalytic characteristics, resulting from the unique characteristics of phosphorus atoms.8–10 Tungsten phosphide is a suitable catalyst for electrocatalytic hydrogenation, whose catalytic activity is higher than other tungsten compounds such as carbides, sulfides, and nitrides.11 It has been stated in several reports that tungsten phosphide is highly active for electrocatalytic reactions and has low production costs.12–15 However, bare WP or WP2 show unsatisfactory HER overpotential. Thus, several groups used various methods to improve the HER activity. Wu et al.16 prepared a coral-like WP nanorod array (C-WP/W), which had low overpotential (only 109 mV at 10 mA cm−2) and low Tafel slope (79.8 mV dec−1). The excellent electrocatalytic performance was attributed to its surface profile characterized by high structural stability and specific surface area. Liu et al.2 synthesized an Ni-doped WP2 nanosheet array, which achieved an overpotential of 109 mV (at a current density of 10 mA cm−2) and a Tafel slope of 65 mV dec−1. The high efficiency of the HER activity was attributed to its surface morphology and the improvement of the internal electronic structure of WP2via Ni doping. Thus, the fundamental catalytic advantages of phosphorus in WP is likely to arise from modifying the nature of tungsten.
In recent years, the construction of the heterojunction structure has been proven to be one of the effective ways to prepare high-efficiency electrocatalysts.17,18 Abundant heterojunction interfaces are obtained through fine interface regulation, which leads to more catalytically active sites. Meanwhile, the charge transfer between the heterojunction interfaces can change the electronic structure of the two phases, thereby adjusting the hydrogen adsorption energy.19,20 Chen21 obtained a unique eutectoid WC/W2C heterostructure by the calcination of two-dimensional organic–inorganic tungsten precursors. It represented excellent HER activity at a current density of 10 mA cm−2; the overpotential is only 75 mV, and the lower Tafel slope is 59 mV dec−1. Li22 constructed a novel heterostructure composed of WP and Ni2P (WP/Ni2P P@NPC) as an efficient HER electrocatalyst. The catalyst had significant acid electrocatalytic activity for HER, with an overpotential of 157 mV and a current density of 10 mA cm−2. However, the HER performance of the catalyst currently obtained is still unable to achieve the level of commercial Pt/C. It is worth noting that Ru has been explored as a promising substitute for Pt electrocatalyst.23 Ru is a noble metal, and the cost is relatively less than Pt, and the low-level use of Ru will greatly reduce the production cost. In this regard, Chen,24 Liu,25 Du26 used low-level dopings of Ru on NiCoP, FeCoP, WC/WP, respectively, and they realized Pt-like HER. However, Ru with WP/WP2 heterostructure is unexplored in energy conversion and storage. Therefore, it is worth trying and feasible to dope the appropriate proportional Ru into tungsten phosphide.
In this work, we prepared an Ru-doped WP/WP2 nanosheet heterojunction array on carbon cloth (Ru-WP/WP2 NH/CC) by the one-step phosphating treatment of the Ru-doped WO3 (Ru-WO3/CC) precursor. The large electrochemical active surface of nanosheet arrays’ morphology, tightly bounded WP/WP2 heterojunction, and doping of Ru atom synergistically enhance the performance of electrocatalytic hydrogen evolution. This heterostructure exhibits Pt-like HER performance with a low overpotential of ∼58.0 mV at a current density of 10 mA cm−2 and a small Tafel slope value of ∼47.98 mV dec−1 in acidic electrolytes. We have discovered the phenomenon of local electronic enrichment in the heterogeneous structure by density functional theory (DFT). The doping of Ru optimizes the electronic system of WP/WP2, which rationally weakens the adsorption capacity of the catalyst surface to hydrogen (H). Thus, Ru-WP/WP2 NH/CC exhibits the appropriate adsorption of Gibbs free energy (ΔGH*), and the effective HER performance is improved.
Experimental
Materials
Carbon cloth (CC, W0S1009) was purchased from Carbon Energy Technology Co., Ltd. Anhydrous tungsten hexachloride (WCl6, 99.9%) and sodium hypophosphite monohydrate (NaH2PO2·H2O, AR) were purchased from Mclean Biochemical Technology Co., Ltd. Anhydrous ethanol (CH3CH2OH, AR) and acetone (CH3COCH3, AR) were purchased from Guangdong Guanghua Technology Co., Ltd. Oxalic acid dihydrate (C2H2O4·2H2O, AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. Anhydrous ruthenium trichloride (RuCl3, AR), platinum–carbon catalyst (20 wt% Pt/C), ruthenium oxide catalyst (RuO2, 98%), and Nafion reagent (5 wt%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. All chemicals were used directly without any further purification.Synthesis of WO3 and the Ru-WO3 NS/CC precursor
The precursor Ru-WO3 NS/CC was prepared by a simple solvothermal method. First, CC was put vertically into a 100 mL Teflon-lined stainless-steel autoclave, 50 mL nitric acid solution (volume ratio Vwater![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Vnitricacid = 20:30 mL) was added, and subsequently it was sent into an oven for hydrophilic treatment at 100 °C for 10 h. After cooling, it was taken out and fully cleaned with deionized water, followed by ultrasonic washing with acetone, ethanol, and deionized water for 30 min, and dried in an oven at 60 °C for 12 h later use. A 100 mL Teflon-lined stainless-steel autoclave was prepared and the cleaned CC was placed vertically into it for later use. 40 mL of anhydrous ethanol was added to the Teflon-lined stainless-steel autoclave, and 0.3 g WCl6, 4.7 mg RuCl3, and 0.6 g of oxalate were dissolved in 40 mL of anhydrous ethanol. To eliminate the influence of air and dissolved oxygen, argon gas was used to remove the air for about 20–30 s, and the Teflon-autoclave was covered quickly. Then, the Teflon-lined stainless-steel autoclave was preheated in an oven at 60 °C for 30 min and subsequently reacted at 180 °C for 10 h. After cooling at room temperature, the sample was taken out, the surface was washed with anhydrous ethanol and dried. Finally, the sample was annealed at 500 °C for 2 h to obtain 3% Ru-WO3 NS/CC. In addition, the doping amount of Ru in WO3 nanosheets was controlled by changing the amount of RuCl3 added during precursor synthesis. Samples with different Ru:W mole ratios of 0%, 0.1%, 0.5%, 1%, 3%, 5% and 7% were synthesized for concentration gradient optimization experiments.
:Vnitricacid = 20:30 mL) was added, and subsequently it was sent into an oven for hydrophilic treatment at 100 °C for 10 h. After cooling, it was taken out and fully cleaned with deionized water, followed by ultrasonic washing with acetone, ethanol, and deionized water for 30 min, and dried in an oven at 60 °C for 12 h later use. A 100 mL Teflon-lined stainless-steel autoclave was prepared and the cleaned CC was placed vertically into it for later use. 40 mL of anhydrous ethanol was added to the Teflon-lined stainless-steel autoclave, and 0.3 g WCl6, 4.7 mg RuCl3, and 0.6 g of oxalate were dissolved in 40 mL of anhydrous ethanol. To eliminate the influence of air and dissolved oxygen, argon gas was used to remove the air for about 20–30 s, and the Teflon-autoclave was covered quickly. Then, the Teflon-lined stainless-steel autoclave was preheated in an oven at 60 °C for 30 min and subsequently reacted at 180 °C for 10 h. After cooling at room temperature, the sample was taken out, the surface was washed with anhydrous ethanol and dried. Finally, the sample was annealed at 500 °C for 2 h to obtain 3% Ru-WO3 NS/CC. In addition, the doping amount of Ru in WO3 nanosheets was controlled by changing the amount of RuCl3 added during precursor synthesis. Samples with different Ru:W mole ratios of 0%, 0.1%, 0.5%, 1%, 3%, 5% and 7% were synthesized for concentration gradient optimization experiments.
Synthesis of Ru-WP, Ru-WP2 NS/CC, WP/WP2, and Ru-WP/WP2 NH/CC
In a typical synthesis of Ru-WP/WP2 NS/CC, the precursor was phosphatized via an in situ solid-state reaction. Generally, phosphating treatment was carried out in a tubular furnace with two-temperature controlled heating zones. The porcelain boat loaded with 4.0 g NaH2PO2·H2O was placed in the upstream heating zone and heated to 300 °C at a rate of 2.2 °C min−1 and maintained for 2 h. On the other hand, the precursor sample placed in the lower half heating zone was heated to 700 °C at a ramping rate of 5 °C min−1; this temperature was maintained for 2 h. Ar gas flows at a rate of 20 s.c.c.m. (standard-state cubic centimeter per minute) as a protective atmosphere. After the reaction was completed and cooled to room temperature, the cathode electrocatalyst product Ru-WP/WP2 NS/CC was obtained. By changing the phosphating temperature and keeping the other conditions the same, different products could be obtained. A phosphating temperature of 650 °C yields Ru-WP2 NS/CC and 800 °C yields Ru-WP NS/CC.Physical characterization
The PANalytical X'Pert3 diffractometer (Cu target, λ = 1.54056 Å) was used to identify the crystal structure of the prepared samples. The surface morphology and microstructure of catalyst materials were characterized by a SU5000 thermal field emission scanning electron microscope (SEM) and a JEM-2100F field emission transmission electron microscope (TEM). The surface chemical states and elemental chemical environments of the materials were analyzed by an ESCALAB 250Xi X-ray photoelectron spectrometer (XPS) with a monochromatic AlKα (1486.6 eV) line source.Electrochemical measurements
All electrochemical analyses were performed in a three-electrode cell for testing using an electrochemical workstation (CHI 760E, CH Instruments Inc.). The acid electrolyte was 0.5 M H2SO4 for all the tests, a graphite rod was used as the counter electrode, Ag/AgCl (sat. KCl) electrode was used as the reference electrode, and a 1 × 1 cm2 sample was used as the working electrode. During the HER test, the electrolyte was purged with N2 gas for 30 min to eliminate the air interference. For the comparison study, the benchmark Pt/C (20 wt%) and RuO2 catalysts were prepared on a 1 × 1 cm2-sized CC by a drop-casting method. In brief, 10 mg Pt/C (RuO2) catalyst was dispersed in a solution containing 230 μL of anhydrous ethanol, 250 μL of deionized water, and 20 μL of 5% Nafion. Ultrasonic treatment was conducted for ∼30 min to form a homogeneous catalyst ink. Then, 25 μL (∼0.5 mg cm−2 loading, consistent with the average load of the catalyst samples grown on CC) catalyst ink drops were cast on the pre-treated CC and dried at room temperature. The cathodic linear scanning voltammetry (LSV) measurement scan range was 0.2 V to −0.6 V (vs. RHE), and the scan rate was 5 mV s−1. To eliminate the effect of the electrolyte ohmic resistance, iR correction ∼95% was utilized. Finally, the measured potential was converted relative to RHE by the Nernst (eqn (1)).| E (vs. RHE) = E (vs. Ag/AgCl) + 0.197 V + (0.0591 × pH). | (1) |
Electrochemical impedance spectroscopy (EIS) was performed to characterize the electrolyte resistance and capacitance of the electrocatalyst. The double-layer capacitance (Cdl) of the catalyst was evaluated by cyclic voltammetry (CV) tests at different scanning rates (10–100 mV s−1). The electrochemical stability of the catalyst was assessed by conducting 2000 continuous CV cycles at a scanning rate of 100 mV s−1, followed by LSV tests. A continuous and stable overpotential with a current density of 10 mA cm−2 for 86000 s was conducted to test the electrochemical stabilities of the electrocatalyst.
Computational methods
Density functional theory (DFT) was calculated using the CASTEP module of Material Studio 8.0. Based on XRD analysis and previous simulation reports,2,11,14,27 we constructed two crystal faces with low surface energy, WP(011) and WP2(100), while the Layer tool was used to build the WP/WP2 heterostructure model. Furthermore, to construct the Ru-doped WP/WP2 system, one W atom in the WP/WP2 system was replaced by one Ru atom. For the initial cell structure optimization calculation of each model, 3 × 3 × 3 Monkhorst–Pack grid k-point was used to optimize the structure. In comparison, the adsorption H single-point energy and electronic structure were calculated by the 3 × 3 × 1 Monkhorst–Pack grid k-point. The Perdew–Burke–Ernzerhof (PBE) method of the Generalized Gradient Approximation (GGA) functional describes the electron exchange–correlation energy. Ultra-soft pseudopotentials were used to describe the electronic structure and interaction of the model more accurately. At the same time, the DFT-D of the Grimme method was applied for dispersion correction, spin polarization, and Broyden–Fletcher–Goldfarb–Shanno (BFGS) were set as the minimization algorithms for all calculations. The adsorption free energy of H on the catalyst surface ΔGH* can be calculated by the following formula (2).28,29| ΔGH* = ΔEH* + ΔZPE − TΔS | (2) |
| ΔEH* = EH*+surface − Esurface − 1/2EH2 | (3) |
Results and discussion
The synthesis process of the nanosheet heterostructure catalyst material is shown in Fig. 1. Ru-WP/WP2 NH/CC was synthesized by a two-step method: Ru-WO3 was grown on CC by the solvothermal method, and then Ru-WP/WP2 NH/CC was synthesized by in situ solid-phase phosphating. | ||
| Fig. 1 Schematic illustration of the fabrication of Ru-WP/WP2 NH/CC. | ||
Fig. 2(a, b) and Fig. S1† show the SEM images of Ru-WO3 on CC at different magnification rates. The WO3 nanosheets with about 30 nm thickness were vertically grown and interlaced with each other on the carbon cloth. After in situ phosphating at 700 °C for 2 h, as shown in Fig. 2(c, d) and Fig. S2,† the heterostructure of Ru-WP/WP2 nanosheets maintained the basic nanosheet morphology. It is worth noting that many zigzag nanoparticles appeared on the edge of the nanosheet, and some micropores appeared in the interior of the nanosheets, which were beneficial for exposing more active sites to facilitate the catalytic hydrogen evolution reaction. EDS mappings were used to identify the elements in the catalyst. Fig. 2(e–h) and Fig. S3† show the composition and distribution of features in Ru-WP/WP2 NH/CC, indicating that W, P, and trace Ru (0.5 at%) elements are uniformly distributed in the catalyst. As shown in Fig. 2(i) and (j), the HR-TEM images of the Ru-WP/WP2 NH/CC samples showed clear lattice fringes with a lattice spacing of 0.421 and 0.651 nm, corresponding to the (101) and (001) crystal planes of WP and WP2, respectively, and showed obvious nanosheet heterostructure interface, indicating that the WP/WP2 nanosheet heterostructure was successfully synthesized. In addition, the electron diffraction points of the heterojunction interface region of Ru-WP/WP2 NH/CC can be obtained from the TEM characterization results, such as the illustration shown in Fig. 2(g). The spots represent the crystal planes of WP and WP2, respectively, indicating that it is a heterojunction with two phases simultaneously.
 | ||
| Fig. 2 SEM images of (a and b) Ru-WO3 NH/CC and (c and d) WP/WP2 NH/CC samples with different magnifications. (e–h) EDS elemental mapping analysis of P, W, and Ru in the Ru-WP/WP2 NH/CC sample. (i and j) HR-TEM images of Ru-WP/WP2 NH/CC. | ||
Fig. 3 shows the X-ray diffraction (XRD) pattern and X-ray photoelectron spectroscopy (XPS) of the samples. As can be seen from Fig. 3(a), the diffraction peaks located at 20.97, 26.11, 31.10, 36.10, 43.95, and 46.60 were indexed to monoclinic WP2 (JCPDS no. 76-2365), and the peaks appearing at 21.06, 28.66, 31.04, 42.86, 43.20, 44.58, and 46.50 were attributed to orthorhombic WP (JCPDS no. 29-1364). These peaks were all observed in WP/WP2 NH/CC, confirming that the heterojunction is composed of two substrates, namely, monoclinic WP2 and orthorhombic WP. The broad peaks with 2θ of 26° and 43° were attributed to carbon cloth (Fig. S4c†).32 The XRD pattern of the heterojunction doped with Ru is shown in Fig. 3(d). The diffraction peaks of doped Ru are entirely consistent with the diffraction peaks of the undoped one, indicating that a trace amount of Ru doping has little effect on the lattice parameters of WP and WP2. Notably, as shown in Fig. S4(a),† products with different structures can be obtained by controlling different phosphating temperatures. At 550 and 600 °C, the mixed-phase of WO3 and WP2 is obtained, and when the temperature rises to 650 °C, the monoclinic WP2 phase is obtained and continues to increase to 800 °C; only the WP single phase is obtained. While phosphating at 700–750 °C, the result is the mixed-phase of WP and WP2, namely, the WP/WP2 nanosheet heterostructure.
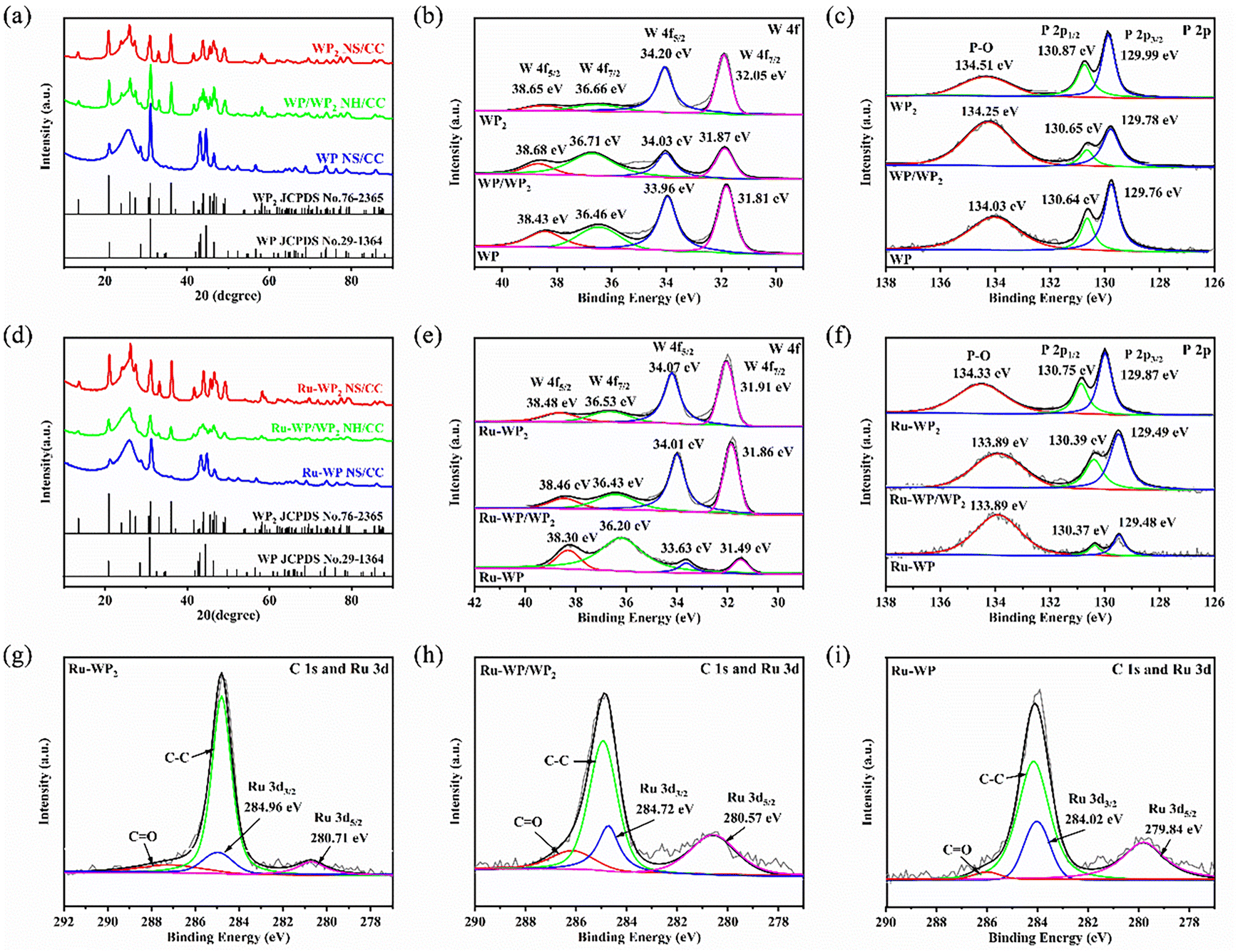 | ||
| Fig. 3 (a) XRD patterns of WP2 and WP/WP2 WP nanosheet arrays. XPS spectra of (b) W 4f and (c) P 2p of WP2 and WP/WP2 WP nanosheet arrays. (d) XRD patterns of Ru-WP2, Ru-WP/WP2 and Ru-WP nanosheet arrays. XPS spectra of (e) W 4f, (f) P 2p, (g–i) C 1s and Ru 3d of Ru-WP2, Ru-WP/WP2 and Ru-WP nanosheet arrays. | ||
The bonding composition and chemical environment of chemical elements on the surface of the samples can be further analyzed by XPS. From the XPS core-level spectra measurement in the ESI Fig. S5,† it can be seen that there are Ru, W, and P elements in the Ru-WP/WP2 catalyst. As shown in Fig. 3(e), the WP/WP2 nanosheet heterostructure doped with Ru shows the binding energies (BEs) of W 4f5/2 (34.01 eV) and W 4f7/2 (31.86 eV) assigned to W–P in WP2 (WP).33,34 Meanwhile, the deconvoluted peaks at 38.46 eV and 36.43 eV can be associated with the binding peaks of the 4f5/2 and 4f7/2 spectra, respectively, caused by the unavoidable air oxidation on the surface of the nanosheet heterostructure, which is very similar to the previous report.35,36Fig. 3(f) shows the P 2p energy spectrum, which produces two peaks at BEs of 130.39 eV and 129.49 eV corresponding to P 2p1/2 and P 2p3/2 satellite features generated by the WP bond in WP2 (WP), respectively.37 In addition, the large and broad peak of BEs at 133.89 eV is the P–O bond caused by the oxidation of P atoms on the surface of the nanosheet material exposed to air.38 As shown in Fig. 3(g–i), the deconvoluted Ru 3d spectra of Ru-WP/WP2 NH/CC gave rise to the peaks at 284.72 and 280.57 eV due to the Ru 3d3/2 and Ru 3d5/2 states of metallic Ru4+ and Ru0, respectively.7,39,40 By comparing the XPS measurement spectrum of Fig. 3(b, c) and (e, f) with the catalyst doped with a low content of Ru, the binding energy of W and P moves slightly to a lower binding energy, which indicates that the part of the electrons of Ru are transferred to W or P, and a small number of electrons are trapped by W and P.41 It may be that the strong electron action of Ru promotes the excitation of active sites in the catalyst, which is beneficial to the HER process. In addition, with the increase in the phosphating temperature, the catalyst will change from WP2 to WP/WP2 and finally to WP. The chemical valence of W and Ru becomes lower, and the binding energy decreases in XPS analysis.42,43
To further prove the effects of the heterostructure and metal Ru doping on the catalyst's performance, we evaluated the HER activity of the samples in an acidic medium in a three-electrode device. As shown in the polarization curves of Fig. 4(a), the Ru-WP/WP2 NH/CC synthesized under the optimal conditions suggested better electrocatalytic hydrogen evolution performance than the bare counterparts, Ru-doped single-phase WP or WP2, and the polarization curve showed a linear sagging trend similar to that of the Pt/C catalyst. The overpotential is only 58.0 mV at a current density of 10 mA cm−2, and a high current density of 100 mA cm−2 and 300 mA cm−2 can be achieved when the overpotential is 113.7 mV and 166.3 mV, respectively. The lower overpotential means that the reaction activation energy barrier of the HER process is smaller, which is more dominant in the thermodynamic conversion efficiency. The overpotentials of Ru-WP NS/CC and Ru-WP2 NS/CC were 131.2 mV and 89.1 mV at a current density of 10 mA cm−2, respectively. Tafel slope is an important parameter for evaluating the reaction kinetics in the HER process.44 As shown in Fig. 4(b), the Ru-WP/WP2 NH/CC sample exhibits a small Tafel slope (47.98 mV dec−1) close to that of the Pt/C catalyst (31.91 mV dec−1), which represents efficient HER activation reaction rate and fast electron transport kinetics, manifesting that the rate-determining step of the catalyst is possibly determined by the Volmer–Heyrovsky pathway.45 Ru-WP NS/CC (60.54 mV dec−1) and Ru-WP2 NS/CC (59.47 mV dec−1), Ru-WP/WP2 nanosheet heterostructures still maintain fascinating electrocatalytic hydrogen evolution properties, suggesting the importance of the heterostructure structure. In addition, we optimized the Ru concentration gradient and phosphating temperature gradient of synthetic catalyst materials. From the concentration gradient optimization (ESI Table S1†), it can be known that when Ru is added to the preparation precursor, Ru:W is 0.1%, 0.5%, 1%, 3%, 5%, and 7%, and the overpotential of Ru-WP/WP2 NH/CC is 184.7 mV, 180.7 mV, 131.1 mV, 58.0 mV, 94.7 mV, and 100.4 mV, respectively, at a current density of 10 mA cm−2. This shows that the optimal molar ratio of Ru:W is about 3%. At the same time, the temperature gradient optimization (Table S2†) shows that the performance of the catalyst material obtained at 700 °C is the best. The lower temperature cannot completely phosphatize to form a WO3/WP2 mixed-phase catalyst. At the same time, the too high temperature will destroy the surface morphology of the original tungsten trioxide catalyst nanosheet array and form relatively low HER activities of WP. Table S3† summarizes the reports of similar catalysts in recent years. In contrast, Ru-WP/WP2 NH/CC still exposes relatively pre-eminent HER behavior.
 | ||
| Fig. 4 Evaluation of the electrocatalytic hydrogen evolution activity of the catalyst in 0.5 M H2SO4. (a) Linear sweep voltammetry curves of the catalysts at a scanning rate of 5 mV s−1. (b) Tafel slope plots. (c) Nyquist fitting curves. (d) HER durability test. Polarization curve after 2000 cyclic voltammetry scans. (e) Cyclic voltammetry curves at different scanning rates (10–100 mV s−1). (f) The double-layer capacitance of the catalyst was calculated on the basis of the cyclic voltammetry curve. | ||
The charge transfer resistance (Rct) of the sample was analyzed by electrochemical impedance spectroscopy (EIS), such as the Nyquist diagram of Fig. 4(c), and the kinetic process of the polarization reaction was further studied. The equivalent circuit model is shown in the illustration of Fig. 4(c), where Rs and Rct are the electrolyte resistance and charge transfer resistance of the working electrode, respectively, and the CPE is generated by the constant phase element of the working electrode. Under the applied bias voltage corresponding to 10 mA cm−2 current density, compared with single-phase Ru-doped WP or WP2, the Rct of Ru-WP/WP2 NH/CC is only 2.20 Ω (Rct of Ru-WP NS/CC is 4.22 Ω and Rct of Ru-WP2 NS/CC is 5.20 Ω), which shows higher charge transfer efficiency and electron transfer kinetics for HER. We further studied the charge transfer resistance of the catalyst materials with different Ru doping amounts and phosphating temperatures, as shown in Fig. S9(a) and (b).† It was proved that an appropriate concentration of Ru doping, the heterostructure can effectively reduce the charge transfer resistance. The stability of Ru-WP/WP2 NH/CC is also studied by long-term durability. As shown in Fig. 4(d), the polarization curves of the catalyst display no noticeable change before and after 2000 cycles of continuous CV tests, which shows the excellent electrochemical stability of Ru-WP/WP2 NH/CC. As shown in Fig. S10,† we tested the XRD pattern of the catalyst before and after HER and found that only the crystal peak value decreased. Fig. S11† shows the time-dependent current density curve of 3% Ru-WP/WP2, and the catalyst maintained a constant current density of 10 mA cm−2 catalytic activity for at least 86000 s, implying that it has high stability in acidic electrolyte. For comparison, we provide stability tests for RuO2,46,47 as shown in Fig. S12.† In order to evaluate the electrochemically activity surface area (ECSA) of the catalyst, we collected the CV curve with the scanning rate of 10–100 mV s−1 and estimated the electrical double-layer capacitance (Cdl) by taking the non-Faraday region. Fig. 4(f) shows the double layer capacitance fitting line of the catalyst. Ru-WP/WP2 NH/CC has a potential of 0.21 V (vs. RHE) corresponding to the current density; the calculated Cdl is 100.68 mF cm−2, while the Cdl of Ru-WP NS/CC and Ru-WP2 NS/CC is 45.14 mF cm−2 and 61.51 mF cm−2, respectively. We further studied the TOF curves of different catalysts, which are shown in Fig. S13.† The results show that the Ru-WP/WP2 nanosheet heterostructure has an excellent electrochemically activity surface area, which is higher than that of the Ru-WP and Ru-WP2 nanosheet arrays. This is attributed to the WP/WP2 heterostructure composite phase having a rich phase interface than single-phase WP or WP2, which provides more new active sites resulting from the interfacial effect. At the same time, Ru atoms doping into the host lattice reduced the energy barrier of the HER reaction and improved the electrochemical activity. In additional, the nanosheet heterostructure itself has a wide specific surface area, which enables the fast mass and ions transfer during the reactions.
The Gibbs free energy of hydrogen adsorption (ΔGH*) is one of the important indexes to evaluate the HER performance. When ΔGH* is close to 0, it is considered the ideal electrocatalyst for HER.48,49 We used density functional theory (DFT) to evaluate the ΔGH* and electronic structure changes of the Ru-doped WP/WP2 nanosheet heterostructure. Fig. 5(a) shows the models of Ru-WP/WP2 employed in the calculation after geometry optimization. The Gibbs free energy of the catalyst at different hydrogen adsorption sites was evaluated. According to the calculation results and comparison data in Table S4,† the representative metal W site with H adsorption energy closer to 0 was finally selected as the hydrogen adsorption site. Moreover, Fig. S15 and S17† were demonstrated as the hydrogen adsorption models of Ru-WP/WP2 and WP/WP2 heterostructure after structural optimization. Fig. S19, S21, S23, and S25† exhibit the hydrogen adsorption models of Ru-WP2, WP2, Ru-WP, and WP, respectively, after structural optimization. It is worth noting that the unilateral WP and WP2 models in the heterostructure structure are only inconsistent with the WP and WP2 models in the number of extended supercells. In addition, we compared the effects of different replacement sites of Ru in the heterostructure structure on the Gibbs free energy of hydrogen adsorption, and detailed data are summarized in ESI Table S5.† As Fig. 5(b) shows the Gibbs free energy diagrams of Ru-WP/WP2, WP/WP2, Ru-WP2, WP2, Ru-WP and WP models, depicting the free energy barrier of H adsorption and desorption on the catalyst surface. As shown in Fig. 5(b) and Table S4,† the ΔGH* of Pt(111) is only −0.09 eV, which is very close to the ideal ΔGH* = 0 eV, indicating that it has excellent HER performance. Similarly, the ΔGH* value of the double-site Ru-doped double W-site adsorption (i.e., both the WP side and the WP2 side of the heterostructure are Ru-doped, and H is adsorbed on W) is very low −0.06 eV, which is more consistent with the ideal Gibbs free energy of hydrogen evolution. Also, the ΔGH* of Ru-WP and Ru-WP2 are −0.71 eV and −0.10 eV, respectively. It has been proved that the construction of WP/WP2 heterostructure can reduce the free energy barrier and make the ΔGH* value closer to zero.
 | ||
| Fig. 5 (a) Views of the structure model for Ru-WP/WP2. (b) Gibbs free energy diagram was calculated for different catalyst materials. (c and d) d-Orbital Skewness Density (d-DOS) diagrams of W atoms at the adsorption site. The black dotted line (0 eV) indicates the position of the Fermi level, and the other dotted lines represent the d-band center corresponding to each electrocatalyst material. (e) Ru-WP/WP2 and (f) WP/WP2 of charge density difference diagram. Among them, red represents electron accumulation, and blue represents electron consumption. Yellow is the Ru atom, green is the W atom, and pink is the P atom. The red dashed box shows the heterostructure interface and interface atoms. | ||
In addition, the ΔGH* value of H adsorption on pure WP/WP2 double W sites is −0.272 eV, which indicates that metal Ru doping can also balance the competition between H adsorption and desorption processes, making the ΔGH* value closer to zero, which will be a very favorable condition for HER. Generally speaking, a very high ΔGH* value indicates that the adsorption capacity of H is relatively weak, which makes it difficult to adsorb H on the surface of the catalyst, which is not conducive to the occurrence of HER, while a very low ΔGH* value means that H will be tightly adsorbed on the surface of the catalyst, which seriously limits the desorption of H and the precipitation of H2,50 suggesting that Ru-WP/WP2 NH/CC has outstanding HER activity. According to the d-band theory, the d-band center (εd) can effectively describe the binding strength between the adsorbed H and the catalyst surface.51 As shown in the partial density of states (d-DOS) diagram of the d orbital of the W atom at the adsorption site in Fig. 5(c) and (d), the d band center of Ru-WP/WP2 is farther away from the Fermi level than Ru-WP, Ru-WP2, and WP/WP2. In general, a more negative band center weakens the binding between the adsorbent (H) and the catalyst's surface. For Ru-WP/WP2, it is beneficial to the desorption of H and the precipitation of H2. It also endows it with a ΔGH* closer to the theoretical optimal value of 0. The distribution of local electrons and the difference in the electronic structure can be understood by calculating the differential charge density of each catalyst model. As can be seen from the differential charge density diagram in Fig. 5(e) and (f), compared with WP/WP2, there is a strong electron consumption behavior around the yellow Ru atom of Ru-WP/WP2 NH/CC, indicating that Ru has lost electrons, which is consistent with the previous XPS test and the analysis results. The electron accumulation behavior of W atoms between the heterostructure interfaces occurs, which means that electrons transfer from Ru to heterostructure interfaces and gather around W atoms, which increases their electron density. Also, the P atom near Ru shows a state of partial electron aggregation, which may be related to Ru's strong electron dissipation behavior. In addition, the differential charge densities of monomer WP and monomer WP2 and WP/WP2 heterostructures are compared, and they are shown in Fig. S20, S22, S24, and S25.† When single-phase WP and WP2 combine into heterostructures, except for the interface atoms, the phenomenon of local loss of electrons around other metal atoms (Ru or W) no longer exists but is replaced by a darker blue, which means that the behavior of dispersed electrons is more obvious, indicating that there is a significant modulation behavior in the local charge distribution of the heterostructure interface. These results show that there is an ideal electronic structure in the heterostructure of the WP/WP2 nanosheet, and after metal Ru doping, Ru as an electron donor provides more electrons to fill the interface, the heterostructure maintains a relatively electron-enriched state, and the adsorption capacity of H on the surface of the catalyst is reasonably regulated. Thus, a ΔGH* value close to zero is obtained, which effectively reduces the energy barrier of the HER process. This is also the performance of Ru-WP/WP2 NH/CC with high HER activity.
Conclusions
In conclusion, through the strategy of in situ phosphating, a Ru-doped WP/WP2 nanosheet heterostructure array (Ru-WP/WP2 NH/CC) was grown on the surface of carbon cloth for the first time. As the electrocatalyst of a cathode, Ru-WP/WP2 NH/CC shows excellent HER activities. In the acid electrolyte of 0.5 M H2SO4, the current density can reach 10 mA cm−2 when the overpotential is only 58.0 mV, and the corresponding low Tafel slope is only 47.98 mV dec−1. The HER activities are attributed to the fact that the heterostructure of the catalyst nanosheet replicates the fascinating nanosheet morphology of the precursor and forms many closely bonded interfaces; in addition, the doping of trace Ru atoms improves the inherent electronic structure and obtains more HER active sites. The DFT calculation further shows that the WP/WP2 of the heterostructure structure shows a favorable heterostructure interface. The doping of Ru optimizes the original electronic structure, which maintains the electron-enriched state of the heterostructure, weakens the binding ability between the adsorbent H and the catalyst surface, makes the hydrogen adsorption Gibbs free energy closer to the thermal median value 0, and enhances the HER activity. This work proposes a new strategy to improve the activity for electrocatalytic HER. It provides a valuable insight into the construction of reasonable heterostructure nanowires and the use of other atoms to regulate the electronic structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22062005, 22165005, U20A20128, 52004076) and the Guangxi Science Fund for Distinguished Young Scholars (2019GXNSFFA245016).References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- W. Liu, P. Geng, S. Li, W. Liu, D. Fan, H. Lu, Z. Lu and Y. Liu, J. Energy Chem., 2021, 55, 17–24 CrossRef CAS.
- H. Li, M. Hu, L. Zhang, L. Huo, P. Jing, B. Liu, R. Gao, J. Zhang and B. Liu, Adv. Funct. Mater., 2020, 30, 2003198 CrossRef CAS.
- J. Lin, Y. Liu, Y. Liu, C. Huang, W. Liu, X. Mi, D. Fan, F. Fan, H. Lu and X. Chen, ChemSusChem, 2019, 12, 961–967 CrossRef CAS PubMed.
- L. Shang, Y. Zhao, X.-Y. Kong, R. Shi, G. I. N. Waterhouse, L. Wen and T. Zhang, Nano Energy, 2020, 78, 105375 CrossRef CAS.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y. R. Lu, T. S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- Y. Xu, S. Yin, C. Li, K. Deng, H. Xue, X. Li, H. Wang and L. Wang, J. Mater. Chem. A, 2018, 6, 1376–1381 RSC.
- H. Wang, Y. Guo, Q. Mao, H. Yu, K. Deng, Z. Wang, X. Li, Y. Xu and L. Wang, Nanoscale, 2023, 15, 7765–7771 RSC.
- H. Ma, W. Yan, Y. Yu, L. Deng, Z. Hong, L. Song and L. Li, Nanoscale, 2023, 15, 1357–1364 RSC.
- Y. C. Huang, Z. Y. Hu, L. A. Huang, Z. P. Wang, Z. P. Lin, S. J. Shen, W. W. Zhong and J. Q. Pan, Nanoscale, 2023, 15, 3550–3559 RSC.
- F. Meng, Y. Yu, D. Sun, L. Li, S. Lin, L. Huang, W. Chu, S. Ma and B. Xu, Appl. Surf. Sci., 2021, 546, 148926 CrossRef CAS.
- X. Lv, X. Li, C. Yang, X. Ding, Y. Zhang, Y. Z. Zheng, S. Li, X. Sun and X. Tao, Adv. Funct. Mater., 2020, 30, 1910830 CrossRef CAS.
- L. Yu, I. K. Mishra, Y. Xie, H. Zhou, J. Sun, J. Zhou, Y. Ni, D. Luo, F. Yu, Y. Yu, S. Chen and Z. Ren, Nano Energy, 2018, 53, 492–500 CrossRef CAS.
- W. Liu, P. Geng, S. Li, R. Zhu, W. Liu, H. Lu, S. Chandrasekaran, Y. Pang, D. Fan and Y. Liu, Int. J. Hydrogen Energy, 2020, 45, 28576–28585 CrossRef CAS.
- Z. Pu, X. Ya, I. S. Amiinu, Z. Tu, X. Liu, W. Li and S. Mu, J. Mater. Chem. A, 2016, 4, 15327–15332 RSC.
- L. Wu, Z. Pu, Z. Tu, I. S. Amiinu, S. Liu, P. Wang and S. Mu, Chem. Eng. J., 2017, 327, 705–712 CrossRef CAS.
- Q. Liu, Z. Xue, B. Jia, Q. Liu, K. Liu, Y. Lin, M. Liu, Y. Li and G. Li, Small, 2020, 16, e2002482 CrossRef PubMed.
- R. Zhang, G. Wang, Z. Wei, X. Teng, J. Wang, J. Miao, Y. Wang, F. Yang, X. Zhu, C. Chen, E. Zhou, W. Hu and X. Sun, J. Mater. Chem. A, 2021, 9, 1221–1229 RSC.
- Y. Du, H. Zhao, W. Wang, Y. Yang, M. Wang, S. Li, Y. Liu and L. Wang, Dalton Trans., 2021, 50, 391–397 RSC.
- J. Wu, R. Zhao, H. Xiang, C. Yang, W. Zhong, C. Zhang, Q. Zhang, X. Li and N. Yang, Appl. Catal., B, 2021, 292, 120200 CrossRef CAS.
- Z. Chen, W. Gong, S. Cong, Z. Wang, G. Song, T. Pan, X. Tang, J. Chen, W. Lu and Z. Zhao, Nano Energy, 2020, 68, 104335 CrossRef CAS.
- T. Li, H. J. Chen, Z. H. Chen, Z. X. Xue, X. H. Zhang and B. Hui, ACS Sustainable Chem. Eng., 2023, 11, 14549–14558 CrossRef CAS.
- P. Li, W. Li, Y. Huang, J. Li, Q. Huang, S. Zhao and S. Tian, Nanoscale, 2022, 14, 6258–6267 RSC.
- D. Chen, R. Lu, Z. Pu, J. Zhu, H.-W. Li, F. Liu, S. Hu, X. Luo, J. Wu, Y. Zhao and S. Mu, Appl. Catal., B, 2020, 279, 119396 CrossRef CAS.
- H. Liu, X. Li, L. Ge, C. Peng, L. Zhu, W. Zou, J. Chen, Q. Wu, Y. Zhang, H. Huang, J. Wang, Z. Cheng, Z. Fu and Y. Lu, Catal. Sci. Technol., 2020, 10, 8314–8324 RSC.
- H. Du, Z. Du, T. Wang, S. He, K. Yang, K. Wang, L. Xie, W. Ai and W. Huang, Sci. China Mater., 2022, 65, 967–973 CrossRef CAS.
- M. Pi, T. Wu, D. Zhang, S. Chen and S. Wang, Electrochim. Acta, 2016, 216, 304–311 CrossRef CAS.
- S. Chandrasekaran, P. Zhang, F. Peng, C. Bowen, J. Huo and L. Deng, J. Mater. Chem. A, 2019, 7, 6161–6172 RSC.
- D. Zhao, K. Sun, W. C. Cheong, L. Zheng, C. Zhang, S. Liu, X. Cao, K. Wu, Y. Pan, Z. Zhuang, B. Hu, D. Wang, Q. Peng, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 8982–8990 CrossRef CAS PubMed.
- F. Yang, Y. Zhao, Y. Du, Y. Chen, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2018, 8, 1703489 CrossRef.
- Q. Zhou, Z. Shen, C. Zhu, J. Li, Z. Ding, P. Wang, F. Pan, Z. Zhang, H. Ma, S. Wang and H. Zhang, Adv. Mater., 2018, 30, e1800140 CrossRef PubMed.
- H. Yu, C. Zhu, K. Zhang, Y. Chen, C. Li, P. Gao, P. Yang and Q. Ouyang, J. Mater. Chem. A, 2014, 2, 4551–4557 RSC.
- M. Pi, T. Wu, D. Zhang, S. Chen and S. Wang, Electrochim. Acta, 2016, 216, 304–311 CrossRef CAS.
- M. Pi, W. Guo, T. Wu, X. Wang, D. Zhang, S. Wang and S. Chen, J. Power Sources, 2017, 364, 253–257 CrossRef CAS.
- B. Ren, D. Li, Q. Jin, H. Cui and C. Wang, J. Mater. Chem. A, 2017, 5, 19072–19078 RSC.
- S. S. Nkabinde, P. V. Mwonga, S. Mpelane, Z. B. Ndala, T. Kolokoto, N. P. Shumbula, O. Nchoe, R. R. Maphanga, K. I. Ozoemena, K. P. Mubiayi and N. Moloto, New J. Chem., 2021, 45, 15594–15606 RSC.
- M. Pi, D. Zhang, S. Wang and S. Chen, Mater. Lett., 2018, 213, 315–318 CrossRef CAS.
- X.-D. Wang, Y.-F. Xu, H.-S. Rao, W.-J. Xu, H.-Y. Chen, W.-X. Zhang, D.-B. Kuang and C.-Y. Su, Energy Environ. Sci., 2016, 9, 1468–1475 RSC.
- A. Maiti and S. K. Srivastava, ACS Appl. Nano Mater., 2021, 4, 7675–7685 CrossRef CAS.
- M. Yang, L. Jiao, H. Dong, L. Zhou, C. Teng, D. Yan, T.-N. Ye, X. Chen, Y. Liu and H.-L. Jiang, Sci. Bull., 2021, 66, 257–264 CrossRef CAS PubMed.
- M. Zhao, H. Li, W. Li, J. Li, L. Yi, W. Hu and C. M. Li, Chem. – Eur. J., 2020, 26, 17091–17096 CrossRef CAS PubMed.
- H. Zhu, Q. Yang, D. Liu, D. Liu, W. Zhang, Z. Chu, X. Wang, S. Yan, Z. Li and Z. Zou, J. Phys. Chem. Lett., 2020, 11, 9184–9194 CrossRef CAS PubMed.
- X. Zheng, P. Cui, Y. Qian, G. Zhao, X. Zheng, X. Xu, Z. Cheng, Y. Liu, S. X. Dou and W. Sun, Angew. Chem., Int. Ed., 2020, 59, 14533–14540 CrossRef CAS PubMed.
- Q. Gong, Y. Wang, Q. Hu, J. Zhou, R. Feng, P. N. Duchesne, P. Zhang, F. Chen, N. Han, Y. Li, C. Jin, Y. Li and S. T. Lee, Nat. Commun., 2016, 7, 13216 CrossRef CAS PubMed.
- Z. Huang, S. Yuan, T. Zhang, B. Cai, B. Xu, X. Lu, L. Fan, F. Dai and D. Sun, Appl. Catal., B, 2020, 272, 118976 CrossRef CAS.
- D. Chen, H. Zhao, R. Yu, K. Yu, J. Zhu, J. Jiao, X. Mu, J. Yu, J. Wu and S. Mu, Energy Environ. Sci., 2024, 17, 1885–1893 RSC.
- D. Chen, R. Yu, D. Wu, H. Zhao, P. Wang, J. Zhu, P. Ji, Z. Pu, L. Chen, J. Yu and S. Mu, Nano Energy, 2022, 100, 107445 CrossRef CAS.
- C. Szczuka, R. A. Eichel and J. Granwehr, RSC Adv., 2023, 13, 14565 RSC.
- C. H. Lee, S. Pahari, N. Sitapure, M. A. Barteau and J. S. Kwon, ACS Catal., 2023, 13, 8336 CrossRef CAS.
- Y. Luo, Y. Guan, G. Liu, Y. Wang, J. Li and L. Ricardez-Sandoval, ACS Catal., 2024, 14, 2696–2708 CrossRef CAS.
- J. Di, C. Chen, C. Zhu, R. Long, H. Chen, X. Cao, J. Xiong, Y. Weng, L. Song, S. Li, H. Li, Y. Xiong and Z. Liu, Adv. Energy Mater., 2021, 11, 2102389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01010h |
| This journal is © The Royal Society of Chemistry 2024 |