
A wearable AuNP enhanced metal–organic gel (Au@MOG) sensor for sweat glucose detection with ultrahigh sensitivity†
Dengfeng
Zhou
 ab,
Shuangbin
Zhang
ab,
Atta Ullah
Khan
ab,
Lan
Chen
*a and
Guanglu
Ge
*a
ab,
Shuangbin
Zhang
ab,
Atta Ullah
Khan
ab,
Lan
Chen
*a and
Guanglu
Ge
*a
aCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, No. 11 ZhongguancunBeiyitiao, Beijing 100190, PR China. E-mail: chenlan@nanoctr.cn; gegl@nanoctr.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 27th November 2023
Abstract
The demand for sensitive and non-invasive sensors for monitoring glucose levels in sweat has grown considerably in recent years. This study presents the development of a wearable sensor for sweat glucose detection with ultrahigh sensitivity. The sensor was fabricated by embedding Au nanoparticles (AuNPs) and metal–organic gels (MOGs) on nickel foam (NF). A non-enzymatic electrocatalytic glucose sensor has been developed to combine the three-dimensional network of MOGs with more active sites favourable for glucose diffusion and the transfer of electrons from glucose to the electrode. These results show that the sensor has an ultrahigh sensitivity of 13.94 mA mM−1 cm−2, a linear detection range between 2 and 600 μM, and a lower detection limit as low as 1 μM (signal/noise = 3) with comparable accuracy and reliability under non-alkaline conditions to those of high-pressure ion chromatography (HPIC). Furthermore, a wearable sweat glucose sensor has been constructed by sputtering an Au conductive layer on a flexible polydimethylsiloxane (PDMS) substrate and coating it with Au@MOGs. Our work demonstrates that the combination of Au NPs and MOGs can enhance the sensitivity and activity of these materials, making them useful for electrocatalytic glucose monitoring with ultrahigh sensitivity.
Introduction
Diabetes has long been regarded as a chronic disease that threatens human health. The abnormal increase of glucose in the body needs rapid and accurate determination to diagnose and treat the condition faster.1 Therefore, the development of stable, efficient, and convenient glucose sensors has been intensively investigated, particularly non-invasive and wearable sensors.2,3 85% of the glucose sensors on the market are based on natural enzymes due to their high sensitivity and excellent selectivity, such as glucose oxidase or dehydrogenase. However, using natural enzymes also brings many problems, such as high costs, low temperature, and pH stability, significantly limiting their applications. In addition, the activity of these enzymes decreases quickly during use, causing shorter serving time or shelf life.4,5 Therefore, it is significant to develop and design non-enzymatic glucose sensors with prompt response, long serving time, and high sensitivity.6,7At present, non-enzyme catalysis mainly involves the use of precious metals (Au, Ag, and Pt),8–10 transition metals (Ni, Co, and Cu),11,12 metal oxides (NiO, Co3O4),13,14 and composites such as NiO/CuO,15etc. Although precious metal catalysts present higher electrocatalytic activity, their cost is also significantly higher. Among transition metals, nickel-based materials are widely used to manufacture electrochemical glucose sensors due to their low cost, significant electrocatalytic activity, and excellent stability.16–18 However, complete nickel sensors are easily poisoned.19 Among metal–organic frameworks (MOFs), supported Fe, Co, and Ni sensors have been developed.20–22 Lu's group has developed an array of Ni-based metal–organic framework nanosheets on nickel foam to detect glucose in NaOH. Ni-MOF/NF offers excellent performance including a wide linear range (0.04 to 2 mM) and detection limits as low as 85 nM,23 but MOFs have low catalytic activity due to their poor conductivity. Besides, MOFs are usually doped with highly conductive metal components.24,25
Metal–organic gels (MOGs) are fibrous, flaky, or granular gels that have extensive interleaved three-dimensional networks through intermolecular forces such as intermolecular hydrogen bonding, π–π stacking, and van der Waals forces with well-defined metal centers and adjustable porous structures.26 Compared with conventional MOF materials, the synthesis conditions for MOGs are milder, e.g., they can be synthesized in an aqueous phase at room temperature and ambient pressure in a short time. These make MOG materials competitive as confined catalysts with lower cost and have more broad potential applications. Wang et al.27 have prepared organometallic gels (MOGs) for the electrochemical hydrolysis of water for the first time with better catalytic performance than the polymer-based ones. However, bimetallic Fe and Ni MOGs synergize more in electrocatalytic hydrolysis.28,29 Therefore, this study aims to explore the combination of bimetallic MOGs and metal nanoparticles synergistically to enhance the electrocatalytic detection of glucose.
In this work, a metal–organic gel (Ni/Fe-MOG) catalyst has been developed by a simple one-step aqueous method using nickel foam-supported MOGs and loading with different amounts of Au nanoparticles, labeled as Au@MOG/NF. This electrochemical electrode performs better for the non-enzymatic detection of glucose under alkaline conditions. The experimental results show that the noble metal nanoparticles improve the performance of the electrochemical sensor, exhibiting a high sensitivity of 13.94 mA mM−1 cm−2 and a low detection limit of 1 μM within a detection range of 2–600 μM. In addition, the sensor exhibits higher selectivity, long-term stability, and better reproducibility. Moreover, to achieve non-invasive and real-time glucose monitoring in human sweat, the Au@MOG has been grown on a flexible PDMS film to construct a wearable glucometer, which shows better temperature tolerance and reliable results compared to those obtained by high-pressure ion chromatography (HPIC).
Experimental
Chemical reagents
H3TATAB (4,4′,4′′-s-triazine-1,3,5-triyltri-p-aminobenzoic acid) was purchased from Shanghai Bide Pharmaceutical Technology Co., Ltd. NiCl2·6H2O (99%) and FeCl3·6H2O (99%) were obtained from China National Pharmaceutical Group Corporation. DMSO and Nafion solution (5 wt%) was obtained from Sigma-Aldrich. D-Glucose, sucrose, sodium hydroxide (NaOH), and ascorbic acid (AA) were purchased from Innochem (Beijing, China). Uric acid (UA) and lactose were sourced from Macklin (Shanghai, China). Xylose was supplied by Energy Chemical (Shanghai, China). Dopamine was obtained from Adamas (Shanghai, China). Nickel foam was purchased from Tianjin Annuohe New Energy Technology Co., Ltd. The electrodes were purchased from Tianjin Aida Instrument Company. All reagents were of analytical grade and used without further purification.Characterization
The morphology of the electrodes was observed through a scanning electron microscope (SEM) (Hitachi SU8220). X-ray diffraction (XRD) analysis was performed using the Bruker D8 Advance. Thermo Fisher Scientific K-Alpha was used to analyze the electronic state of the product by X-ray photoelectron spectroscopy (XPS). The presence of Au nanoparticles in the electrode was studied by transmission electron microscopy (TEM) (G2 T20), while high-resolution transmission electron microscopy (HRTEM) and elemental mapping were performed with FEI Tecnai G2 F20. For electrochemical measurement, the prepared electrodes were measured by cyclic voltammetry (CV), chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS) using a three-electrode system – Autolab PGSTAT302N. Ag/AgCl was used as the reference electrode, and platinum sheet was used as the counter electrode in 0.1 M NaOH electrolyte. CV was performed at a sweep rate of 50 mV s−1 in a potential window of 0–0.8 V to obtain the resistance of different electrodes. The EIS test was performed in the frequency range of 100 kHz–0.01 Hz, containing 0.1 M NaOH and 10 mM [Fe (CN)6]3− and [Fe (CN)6]4−, and the amplitude of the sine wave signal was set to 5 mV AC. The electrode was scanned from 0 to 0.8 V (vs. Ag/AgCl) at 50 mV s−1 in 0.1 M NaOH electrolyte for 20 cycles before glucose measurement. And CA was performed at different potentials (0.5–0.7 V) to monitor glucose.Synthesis of the Ni/Fe-MOG
The synthesis of the MOGs was modified according to the previous literature.27,28 In a typical synthesis, the precursor with 200 mM Ni2+ and 100 mM Fe3+ was prepared by dissolving both NiCl2·6H2O and FeCl3·6H2O in deionized water. A given amount of H3TATAB was dissolved in 250 μL of DMSO solution to improve the hydrotropism and coagulability of the hydrogel. Then, by homogeneously mixing 250 μL of Fe3+/Ni2+ solution and 250 μL of H3TATAB solution in a 1 ml tube, the mixture was gelated for 30 min, followed by freeze-drying to form the Ni/Fe-MOG.Fabrication of the Au@MOG/NF electrode
Nickel foam (NF) was cut into square pieces with dimensions of 1 cm × 1 cm × 0.05 cm, then oscillated in 3 M HCl for 10 min, and washed with acetone and water under ultra-sonication for 10 min, respectively. Finally, the cleaned nickel foam squares were placed in an oven and dried for later use.The preparation of Au@MOG/NF or Pt@MOG/NF electrodes is shown in Scheme 1. Firstly, the prepared Ni/Fe-MOG was dispersed in an aqueous solution to remove the unreacted reactant, followed by the addition of chloroauric acid (HAuCl4) or chloroplatinic acid (HPtCl4) and changing the concentration of ascorbic acid (AA) of the reducing agent and hydrothermal treatment at 120 °C for 10 h. The MOG-loaded nickel foam was rinsed thrice with deionized water and dried at 60 °C in the oven. The same procedure is also applied to blank NF electrodes.
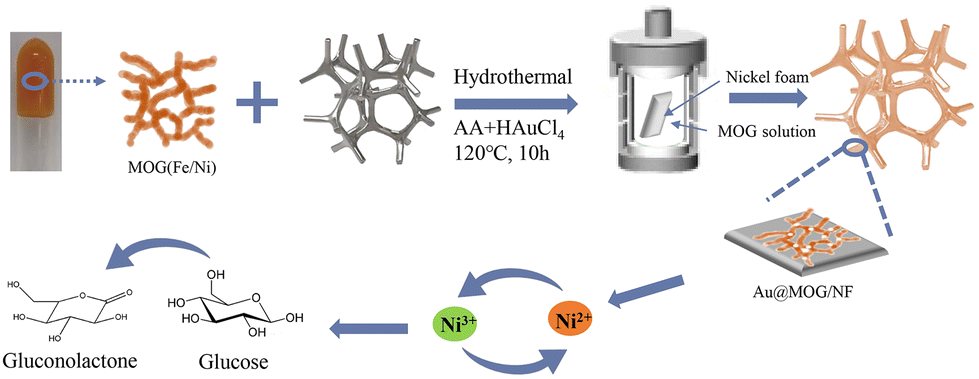 | ||
| Scheme 1 Schematic illustration of the fabrication process of the Au@MOG/NF electrode. | ||
Construction of PDMS-supported Au@MOG flexible electrodes
For preparing flexible electrodes, the selected PDMS was the substrate, where the Cr adhesion layer of 10 nm thickness was sputtered on PDMS by electron beam evaporation. Then 100 nm of Au was sputtered as the conductive layer on the Cr surface, as shown in Scheme 2. The Ag/AgCl reference electrode was made by applying 5 μl of Ag/AgCl ink droplets to the patterned PDMS substrate and drying the substrate at 60 °C for 30 min. The working electrodes were prepared by dispersing Au@MOGs in 5% Nafion solution to form a stable suspension through sonication, the prepared suspension was dropped on the different areas of the substrate as shown in Scheme 2, and dried at 60 °C for 30 min. However, the Au electrode acts as the counter electrode of the glucose sensor, and cotton fibers with excellent water absorption are used as a reservoir for the glucose solution, covering the surface of the whole sensor.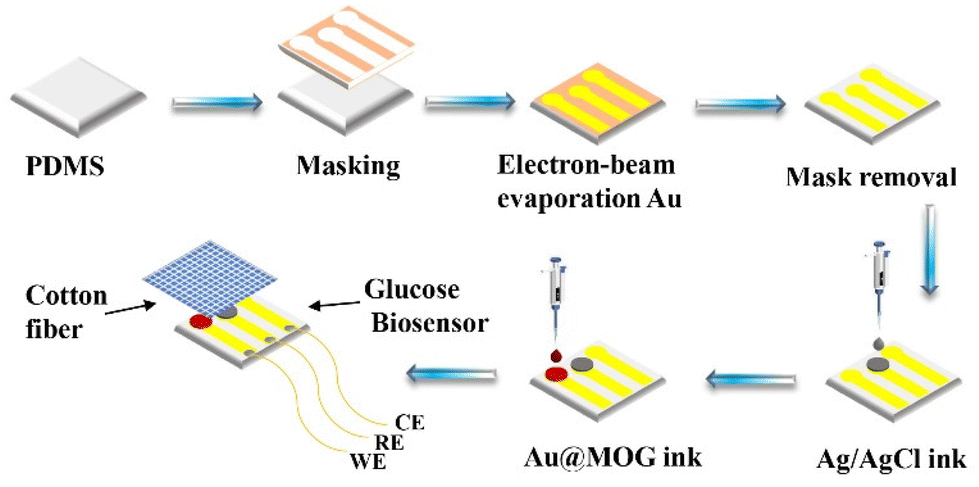 | ||
| Scheme 2 The Au@MOG/PDMS-based flexible sensor preparation process for glucose detection in sweat. | ||
Results and discussion
Fabrication of the glucose sensor
As shown in Fig. 1a, the pre-treated nickel foam exhibits a smooth morphology, and the cross-linked-MOG and Au@MOG nanofibers are fabricated on the nickel foam surface, as shown in Fig. 1b–e. Comparing Fig. 1c with Fig. 1e, it can be seen that the surface modified by AuNPs shows a denser and unique nanofiber network structure, producing a large number of voids exposing more active specific surface area, which is more conducive to promoting electron transport and the capture of glucose molecules.27 However, the woven network is shown at the microscopic level, significantly improving the sensor's conductivity, as shown in Fig. 1f. | ||
| Fig. 1 (a–e) SEM images of bare Ni, MOG/NF, and Au@MOG/NF electrodes, (f and g) SEM and TEM images of Au@MOGs, (h–k) mapping results of the elemental Ni (green), Fe (red), Au (yellow), (l) EDS spectrum. | ||
As can be seen from the TEM image in Fig. 1g, AuNPs were successfully grown in situ in Ni/Fe-MOGs. Based on the element mapping (Fig. 1h–k) analysis, AuNPs were embedded in the material, consistent with the TEM results. The EDS spectrum in Fig. 1(i–k) indicates that elements Fe, Ni, and Au are present together, and Au accounts for 3% of the total elemental content. However, Ni/Fe-MOG and Au clusters work synergistically to enhance the catalytic oxidation of glucose.30,31 As seen in Fig S1,† Ni is the majority component of the sensor with diffraction angles of less than 40° and only a wide-angle diffraction peak at 2θ = 24.8°, indicating that this was an amorphous structure of the gel. The d spacing value corresponding to 3.58 Å demonstrates that the presence of π–π stacking interactions in the MOGs promotes gel formation.
The surface valence state and bonding configuration of Au@MOG/NF have been studied by XPS, where the survey spectrum indicates the presence of elements Ni, Fe, O, N, C, and Au (Fig. S2†). In contrast, the feature spectra of Ni 2p, Fe 2p, O1 s, C1 s, and Au 4f are shown in Fig. 2. In the Ni 2p range (Fig. 2a.), the peaks at 855.9 and 873.2 eV were attributed to Ni 2p3/2 and Ni 2p1/2, respectively while the two satellite peaks at 879.5 eV and 861.9 eV indicate the presence of both Ni2+ and Ni3+.32,33 Similarly, Fig. 2b confirms the existence of both Fe(II) (710 and 715.9 eV) and Fe(III) (724.2 eV and 729.4 eV).34,35 In Fig. 2c, two peaks at 530.7 and 531.8 eV represent the presence of Ni–O/Fe–O and hydroxyl groups (O–H), respectively. The N 1s exhibits four separated peaks at 398.6, 400.1, 401.2, and 406.1 eV, attributed to the overlapping of pyridinic N, pyrrolic N, graphitic-N, and slightly oxidized N (Fig. 2d),36 which may conform to the composition of the gel. A large amount of pyrrolic N was believed to improve the adsorption of OOH* intermediates.37 The C 1s XPS spectrum (Fig. 2e) was split into four peaks at 284.5, 285.7, 288.2, and 290.6 eV, corresponding to the presence of C–C, C–N/C–N, C–O, and O–C–O functional groups.23 The signals at 84.1 and 87.7 eV are the characteristic peaks of Au 4f 7/2 and Au 4f5/2 (Fig. 2f). It is speculated that the poor signal-to-noise ratio and low intensity indicate that only a tiny number of AuNPs are present in the MOG material, accounting for less than 5% of the total elemental content according to EDS results.38
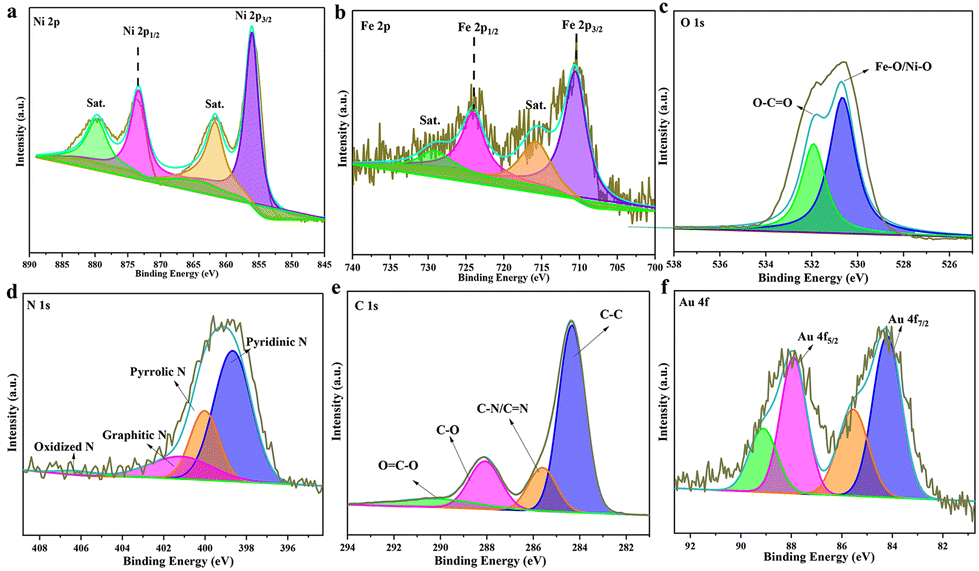 | ||
| Fig. 2 High-resolution XPS spectra of Au@MOG(Fe/Ni): (a) Ni 2p, (b) Fe 2p, (c) O1 s, (d) N1 s, (e) C1 s and (f) Au 4f. | ||
Electrocatalytic performance of the glucose sensor
The electrocatalytic performance of the Au@MOG layer on the electrode for glucose oxidization has been evaluated. According to previous literature, the MOGs doped with a small amount of Fe have higher catalytic activity, so we set the ratio of Fe3+ to Ni2+ as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in this work.27,39 As shown in Fig. S3a,† the catalytic effect for different precious metal loadings is different, where the catalytic impact of Au@MOG/NF was better than that of Pt@MOG/NF. The redox peaks in Fig. 3a represent two reactions, as shown in eqn (1) and (2):
:2 in this work.27,39 As shown in Fig. S3a,† the catalytic effect for different precious metal loadings is different, where the catalytic impact of Au@MOG/NF was better than that of Pt@MOG/NF. The redox peaks in Fig. 3a represent two reactions, as shown in eqn (1) and (2):| Ni(II) − Au@MOG + OH− → Ni(III) − Au@MOG + H2O + e− | (1) |
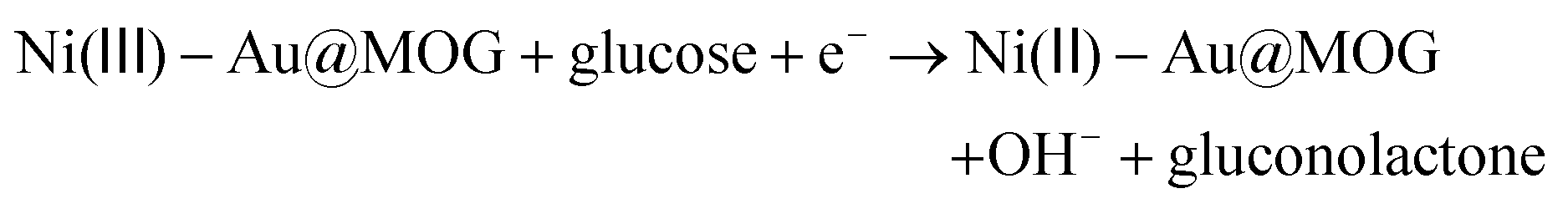 | (2) |
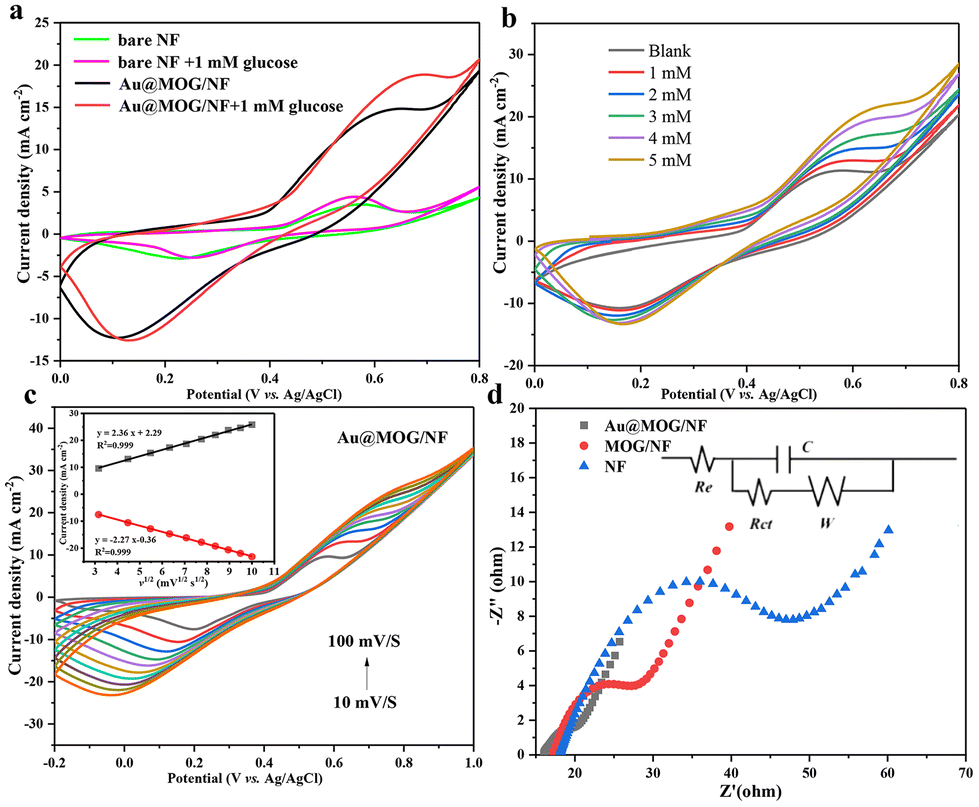 | ||
| Fig. 3 (a) CV in the absence and presence of 1 mM glucose with Au@MOG/NF and blank Ni foam at a scan rate of 50 mV s−1, (b) CV at different glucose concentrations with a scan rate of 50 mV s−1, (c) CV curves of Au@MOG/NF electrode in 0.1 M NaOH in the presence of 1 mM glucose with different scan rates and the inset shows a fitting linear curve of the peak current vs. square root of the scan rate. Black: oxidation peak current (Ipa) vs. square root of scan rate, red: reduction peak current (Ipc) vs. square root of the scan rate, (d) EIS spectra of NF, MOG/NF, and Au@MOG/NF and the equivalent circuit (inset). | ||
Ni(II)-Au@MOG is first electrochemically oxidized to Ni(III)-Au@MOG; an electron is released simultaneously, resulting in an oxidative peak current at the anode. Ni(III)-Au@MOG oxidizes glucose to gluconolactone under alkaline conditions to generate Ni(II)-Au@MOG, resulting in the generation of a peak cathodic reduction current.40 The presence of Fe in the Au@MOG material as a cocatalyst can effectively enhance the catalytic oxidation effect of Ni on glucose.30
Fig. 3b shows that the current oxidation increases with the increase of glucose concentration, indicating that Au@MOG/NF has good scalability in electrocatalytic activity. According to previous literature, the redox peak caused by the introduction of glucose suggests that the glucose oxidation process is irreversible.41 In addition, with the increase of the glucose concentration, the anodic oxidation potential slightly shifts to the right (0.1 V), indicating that the limiting of diffusion of glucose to the electrode was almost negligible.42 At the same time, efficient electron transfer occurs in the electrode, which may be attributed to the high conductivity of the electrode material.43 The CV response of Au@MOG/NF is recorded at different sweep rates of 10–100 mV s−1 in 0.1 M NaOH, as shown in Fig. 3c and Fig. S5;† as the sweep speed increases, the peak current of the anode increases and its potential shifts positively, while the cathode shows a reverse change. According to the illustration in Fig. 3c, the anode peak and cathode peak currents are linearly related to the square root (ν1/2) of the scan rate, indicating that this electrochemical redox kinetics is surface diffusion controlled.44
As shown in Fig. 3d, Nyquist plots record the EIS spectrum of the NF, MOG/NF, and Au@MOG/NF electrodes varying in 0.1 M NaOH with 10.0 mM [Fe (CN)6]3− and [Fe (CN)6]4− redox as a probe, where the equivalent circuit is shown in the inset. Among them, the Au@MOG/NF electrode had the lowest charge transfer and interface contact resistance, indicating that Au@MOG/NF has a high level of electron conductivity and excellent electrochemical performance, consistent with the electrochemical test results described above.
Amperometric response of the glucose sensor
Different applied potentials have a different impact on the current response in glucose detection, so the applied potential must be able to ensure both the formation of Ni(III)-Au@MOG and the oxidation of glucose and, at the same time, avoid a strong oxygen evolution reaction.45 Fig. S6† shows the current density for the glucose (100 μM) catalysis on the Au@MOG/NF electrode at potentials from 0.5 to 0.7 V in 0.1 M NaOH. The current density increases with the increase of the applied potential until 0.7 V, where O2 is seen; so 0.65 V was chosen as the working potential.In Fig. 4a, it can be observed that the current density increases with the increase of the glucose concentration from 1 μM to 600 μM. The calibration curve of the current density in response to the glucose concentration exhibits good linear (correlation coefficient R2 = 0.991) dependence in the range of 2–600 μM with a high sensitivity of 13.94 mA mM−1 cm−2 (slope). Based on the signal-to-noise ratio of S/N = 3, the lower detection limit (LOD) is determined as 1 μM. As shown in Fig. S7a and b,† the current densities obtained by NF and MOG/NF electrodes under the same conditions indicate that the sensitivity of NF and MOG/NF is 3.92 and 5.69 mA mM−1 cm−2, respectively, which is much lower than that of Au@MOG/NF electrodes. The porous MOG structure is attached to the three-dimensional nickel foam in a large area, which provides a large number of glucose catalytic active sites, where the combination of Au NPs and MOG material can quickly transfer electrons, resulting in high sensitivity. As seen in Fig. 4b, the Au@MOG/NF electrode only needs 3 seconds to respond to the glucose addition, confirming the rapid electron transfer through the electrode.
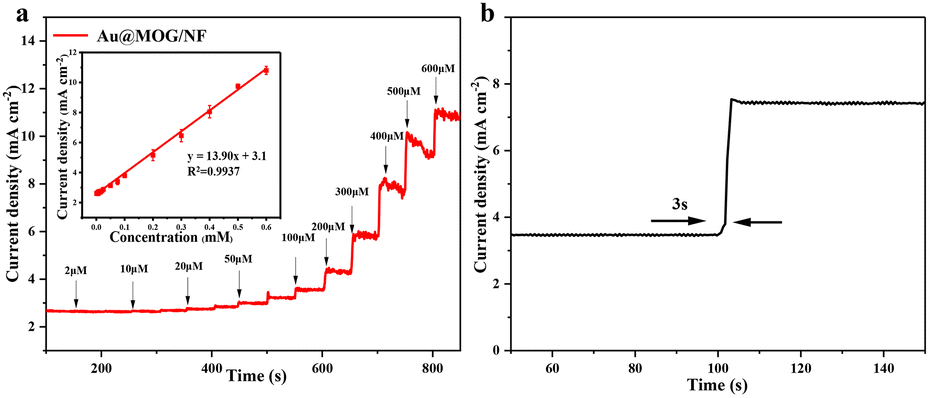 | ||
| Fig. 4 (a) Ampere response of the Au@MOG/NF electrode upon continuous injection of different concentrations of glucose with 0.1 M NaOH at 0.65 V, where in the inset is a calibration curve for current density against glucose concentration, (b) the time required for the current flattening on the Au@MOG/NF electrode after the addition of glucose. | ||
Robustness and stability of the glucose sensor
For better anti-interference, the sensor should be able to distinguish glucose from another monosaccharides in blood or sweat, which may produce current signals similar to that of glucose. However, the amount of glucose in human blood is much higher than that of other monosaccharides. To mimic the actual situation, various monosaccharides such as xylose, lactose, sucrose, fructose, uric acid, dopamine, and ascorbic acid are used to mix with glucose in 1:10 ratio. As shown in Fig. 5a, the glucose added either at the beginning or at the end of the sensing always brings a very significant and sharp jump for the current response. In contrast, that caused by other monosaccharides is relatively flat and almost negligible. The results show that the Au@MOG/NF electrode has excellent glucose selectivity and anti-interference for other monosaccharides.
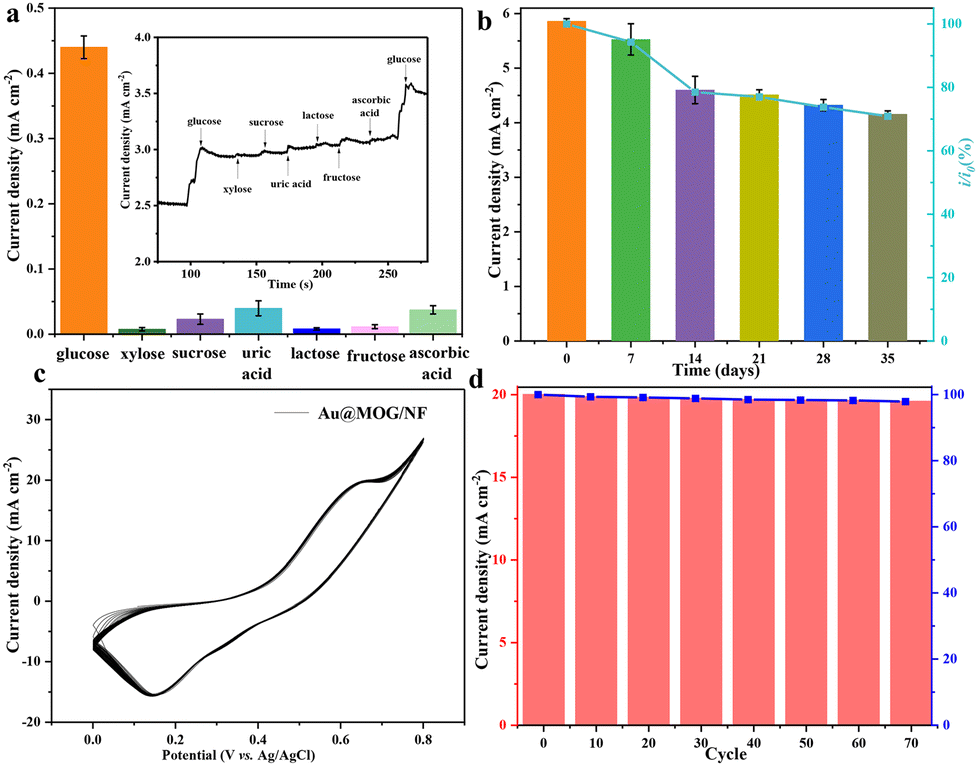 | ||
| Fig. 5 (a) Amperometric response of the Au@MOG/NF electrode to the alternating addition of 100 μM glucose and 10 μM interfering substances, (b) stability of Au@MOG/NF electrode upon storage, (c) CV response of 70 consecutive cycles on Au@MOG/NF electrode and their (d) current response for the specified cycle. | ||
In practical applications, the long-term stability and reproducibility of the glucose sensor are essential, where the Au@MOG/NF electrodes are evaluated at 7-day intervals for 35 days using 1 mM glucose in 0.1 M NaOH (Fig. 5b). The Au@MOG/NF electrode shows high long-term stability with 85% current response capacity remaining after 35 days of storage. In addition, the CV curves for the Au@MOG/NF electrode at different cycling times are shown in Fig. 5c. The oxidation peak current density decreases slightly from 100% to 97.8% after 70 cycles. These results indicate that the long-term stability and reproducibility of the Au@MOG/NF electrode are quite significant as a non-enzymatic glucose sensor, as shown in Fig. 5d. Fig. S8† shows that the current response for parallel three electrodes is essentially the same under the comparable test conditions, which proves that the repeatability of our Au@MOG/NF electrode is quite good.
Wearable monitoring of sweat glucose
The electrocatalytic activity of flexible Au@MOG/PDMS electrodes on artificial sweat solutions was studied. As seen in Fig. 6a, an apparent oxidation current occurs in a solution containing 0.1 M NaOH and 1 mM glucose. This indicates that the flexible Au@MOG/PDMS electrode has an excellent electrocatalytic effect on glucose. A potential of 0.65 V was applied to evaluate the flexible glucose sensor's detection sensitivity. The results (Fig. 6b) show that the current response of the Au@MOG/PDMS electrode increases with increasing glucose, and a well-fitted linear relationship between the current response and the glucose content in the range of 50–1100 μM of glucose is established, which fully covers the glucose range in human sweat. For practical applications, glucose selectivity and temperature tolerance should also be considered besides the sweat constituent, such as uric acid (UA), dopamine (DA), ascorbic acid (AA), and NaCl. Fig. 6c shows that the response current changes significantly upon adding glucose. At the same time, we tested the current response by directly adding the actual concentration of interfering substances without adding glucose in order to evaluate the anti-interference ability of the flexible sensor against the interfering substances in sweat. The experimental results are shown in Fig. S9,† and it can be observed that the responding currents of uric acid and ascorbic acid at a certain concentration of anti-interference substances in sweat are smaller compared to that at the same concentration of glucose in sweat; however, the corresponding current becomes higher with an increase in their concentration.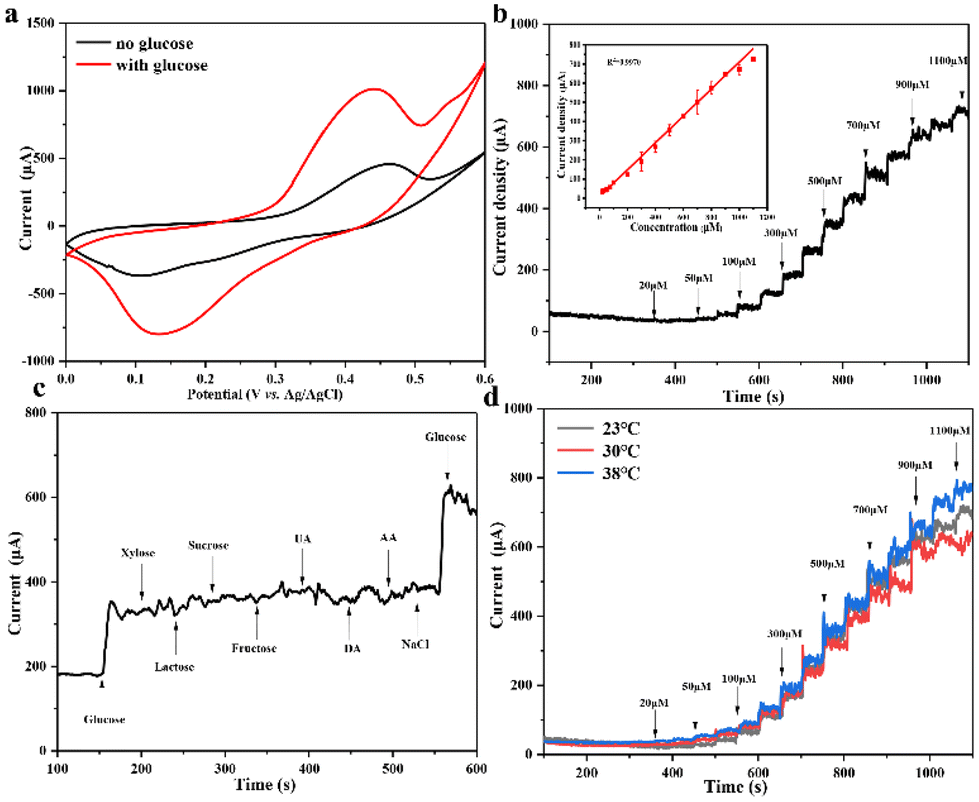 | ||
| Fig. 6 (a) CV curves of the Au@MOG/PDMS flexible sensor in the absence and presence of 1 mM glucose, (b) current response of the Au@MOG/PDMS adjustable sensor to the interval injection of different concentrations of glucose NaOH solution under 0.65 V, inset with a calibration curve of current density vs. glucose concentration, (c) amperometric response of the Au@MOG/PDMS flexible sensor to the alternating addition of 100 μM glucose and 10 μM interfering substances, (d) CA response at different temperatures in 0.1 M NaOH. | ||
In contrast, adding other interfering substances does not change the amperometric response of the sensor significantly, indicating our sensor has good selectivity for glucose. The skin temperature may vary at different times. At the same time, the current response value is similar at different temperatures, such as 23, 30, and 38 °C, respectively, as shown in Fig. 6d, confirming that the sensor has good temperature tolerance.
Most non-enzymatic glucose sensors typically require alkaline conditions to achieve high sensitivity and selectivity, but the detection of sweat glucose must be conducted under non-alkaline conditions. Applying a three-step voltage pulse strategy, including increased alkaline voltage, medium detection voltage, and positive cleaning voltage, can avoid using an alkaline solution and chloride poisoning.46,47 First of all, we need to consider that the pH near the electrode surface will have a significant impact on the wearable sensing signal, where we apply electrochemical pretreatment to increase the environmental pH surrounding the electrode through the proton reduction reaction in order to improve its catalytic performance for glucose. Fig. 7(a) shows a good negatively linear correlation between pH and the potential with pH increase, where we can estimate the pH surrounding the electrode surface by applying a certain voltage. When the voltage of −2 V is applied, pH is about 7 and close to that of human sweat (4–7), so we use PBS buffers with pH 4, 5, 6 and 7 under −2 V voltage. In Fig. 7(b), it can be seen that the pH on the surface gradually increases with time, reaching a maximum value of 10 within 20 s; so we choose 20 s as the pretreatment time.
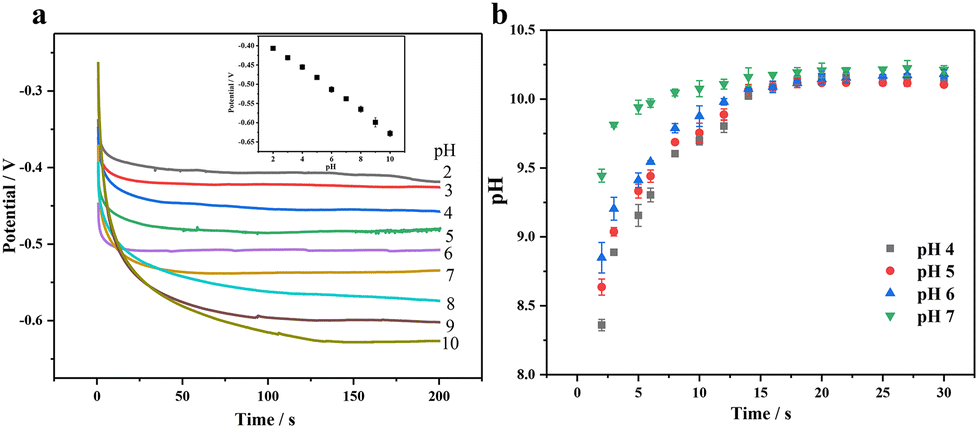 | ||
| Fig. 7 (a) Determination of the relationship between pH and potential by the constant current method and (b) the pH trend plot after pretreatment of different pH buffers. | ||
When a high voltage of −2.0 V is applied for 20 s (Fig. 8a, E1) to the Au@MOG/PDMS working electrode, a local alkaline micro-environment can be generated due to H2 evolution. The glucose content in artificial sweat can be detected well by in situ applying a medium voltage of 0.65 V for 5 s (Fig. 8a, E2). Finally, the electrode surface can be cleaned using a positive voltage of 1.0 V for 2 s (Fig. 8a, E3). As shown in Fig. 8b, the i–t curve shows that the detectable glucose concentration in artificial sweat is 2–400 μM. According to Fig. 8e, the current obtained at 5 s (E2) is linearly related to the glucose concentration. The lower detection limit is 2.0 μM determined from the standard deviation of the blank test results divided by 3 times that of the slope of the calibration curve.
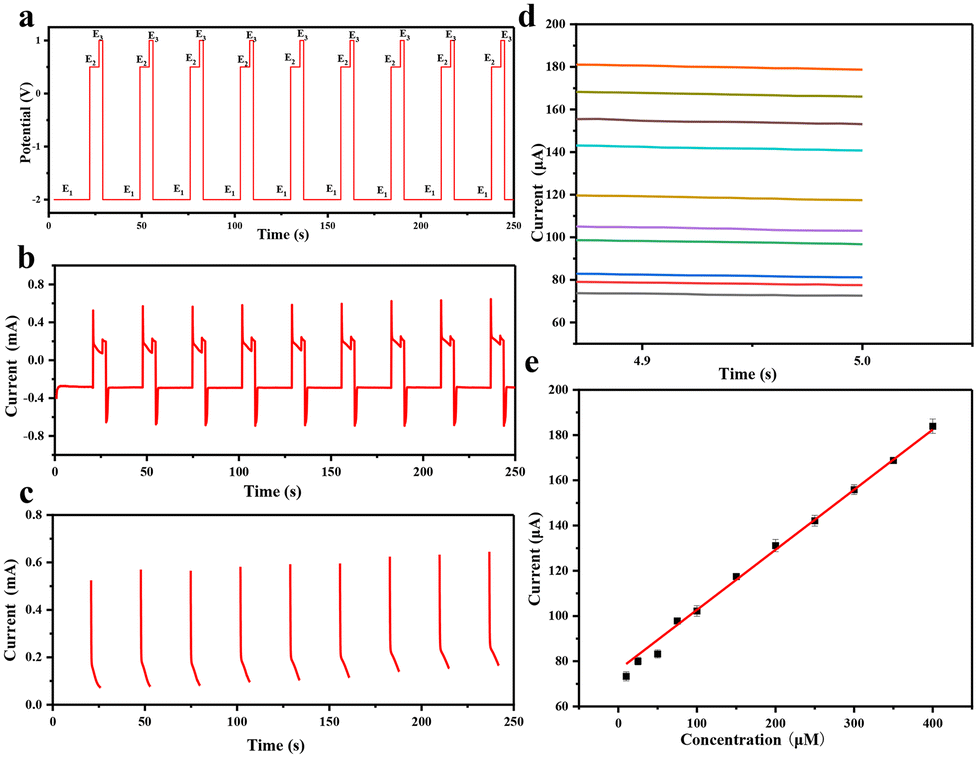 | ||
| Fig. 8 (a) potential response vs. time for the Au@MOG/PDMS flexible sensor, including (E1) pretreatment potential, glucose detection potential (E2), and cleaning potential (E3), (b) measured current over time for the glucose content in artificial sweat from (a), (c) the measured current over time during the glucose oxidation step (E2 step) from (b), (d) i–t curve of E2 in step (c), (e) calibrated current vs. glucose concentration with error bars of three repeated measurements. | ||
Finally, a volunteer exercised on a treadmill for 30 minutes before or after a meal to generate sweat for sampling. The volunteer was asked to collect sweat at 9 a.m. and 3 p.m., respectively, and take a meal at 12 noon. Sweat-absorbing cotton cloth was completely soaked with sweat and used to test CA current signals. The results of human sweat glucose before and after meals are shown in Fig. 9a. A clear difference in the current signal between the pre-meal and post-meal tests can be seen. The glucose concentration in the volunteer's sweat is 54.43 ± 2.50 μM before meals and 130.96 ± 2.14 μM after meals, fitting within the normal range. To confirm the validity of the detected results, we used a third-party technology, e.g., HPIC. The result in Fig. 9b shows that our flexible sensor's measured glucose content in human sweat is very close to that measured by HPIC, which again confirms the excellent accuracy and reliability of our Au@MOG/PDMS-based wearable sweat glucose sensor for glucose detection in sweat.
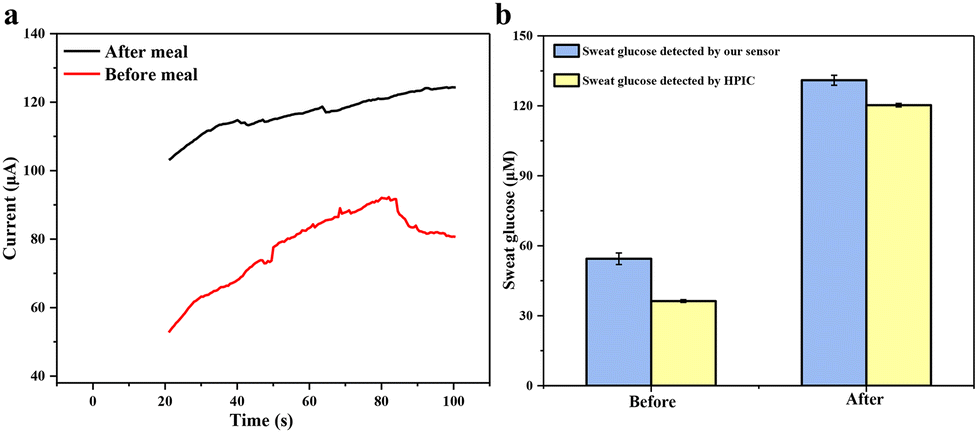 | ||
| Fig. 9 (a) Glucose fluctuation before and after meals was measured by Au@MOG/PDMS wearable electrochemical sensors, (b) comparison of the sweat glucose in volunteer's sweat determined using both the Au@MOG/PDMS wearable electrochemical sensor and HPIC technology. | ||
Conclusion
In summary, an Au@MOG has been grown on nickel foam under hydrothermal conditions to fabricate a binder-free non-enzymatic glucose sensor with ultrahigh electrocatalytic activity. The formed MOG three-dimensional network can provide many active sites for glucose catalysis combined with improved conductivity due to the AuNP modification. The resultant Au@MOG/NF sensor shows excellent electrocatalytic performance under a wide linear range of 2–600 μM with an ultrahigh sensitivity of 13.94 mA mM−1 cm−2, long standby stability of up to 5 weeks, and fast response of less than 5 seconds. Furthermore, the flexible Au@MOG/PDMS can be fabricated as a wearable monitor to monitor the glucose in sweat under non-alkaline conditions through a three-step potential strategy (−2 V, 0.65 V, and 1 V). This wearable sensor has excellent glucose selectivity and high-temperature tolerance suitable for continuous in situ glucose monitoring in sweat with accuracy and reliability comparable with that of HPIC. This sensor is useful for the screening of non-clinical glucose abnormality early and non-invasive in situ glucose monitoring.Author contributions
Dengfeng Zhou: software, validation, and writing – original draft. Atta Ullah Khan: software and methodology. Shuangbin Zhang: software, visualization, and investigation. Lan Chen: methodology, project administration, resources, and formal analysis. Guanglu Ge: supervision, funding acquisition, project administration, resources, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2021YFA1202804) and the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB36000000).References
- D. Yach, D. Stuckler and K. D. Brownell, Nat. Med., 2006, 12, 62–66 CrossRef CAS PubMed.
- S. K. Vashist, Anal. Chim. Acta, 2012, 750, 16–27 CrossRef CAS.
- J. T. La Belle, A. Adams, C.-E. Lin, E. Engelschall, B. Pratt and C. B. Cook, Chem. Commun., 2016, 52, 9197–9204 RSC.
- H. Lee, Y. J. Hong, S. Baik, T. Hyeon and D. H. Kim, Adv. Healthcare Mater., 2018, 7, 1701150 CrossRef.
- J.-S. Ye, C.-W. Chen and C.-L. Lee, Sens. Actuators, B, 2015, 208, 569–574 CrossRef CAS.
- D. Wang and Y. Chang, Nanomaterials, 2022, 12, 572 CrossRef CAS.
- W. Zhou, X. Zhou, Y. Rao, R. Lin, L. Ge, P. Yang, H. Zhang, C. Zhu, H. Ying and W. Zhuang, Colloids Surf., B, 2022, 217, 112585 CrossRef CAS.
- L. Wang, W. Zhu, W. Lu, X. Qin and X. Xu, Biosens. Bioelectron., 2018, 111, 41–46 CrossRef CAS.
- K. Tian, S. Alex, G. Siegel and A. Tiwari, Mater. Sci. Eng., C, 2015, 46, 548–552 CrossRef CAS.
- C. Li, H. Wang and Y. Yamauchi, Chem. – Eur. J., 2013, 19, 2242–2246 CrossRef CAS PubMed.
- Z. Gong, N. Hu, W. Ye, K. Zheng, C. Li, L. Ma, Q. Wei, Z. Yu, K. Zhou and N. Huang, J. Electroanal. Chem., 2019, 841, 135–141 CrossRef CAS.
- Z. Ren, H. Mao, H. Luo and Y. Liu, Carbon, 2019, 149, 609–617 CrossRef CAS.
- Y. Su, H. Guo, Z. Wang, Y. Long, W. Li and Y. Tu, Sens. Actuators, B, 2018, 255, 2510–2519 CrossRef CAS.
- Y.-L. T. Ngo, J. S. Chung and S. H. Hur, J. Alloys Compd., 2017, 712, 742–751 CrossRef CAS.
- Z. Lotfi, M. Gholivand and M. Shamsipur, Anal. Biochem., 2021, 616, 114062 CrossRef CAS PubMed.
- Y. Zhang, D. Zheng, S. Liu, S. Qin, X. Sun, Z. Wang, C. Qin, Y. Li and J. Zhou, Appl. Surf. Sci., 2021, 552, 149529 CrossRef CAS.
- S. Peng, T. Lai, Y. Kong, Y. Ran, L. Su, D. Ma, X. Xiao and Y. Wang, Nanotechnology, 2021, 32, 405502 CrossRef CAS.
- Q. Bu, J. Cai, S. V. Vasudevan, J. Ni and H. Mao, Electrochim. Acta, 2021, 398, 139319 CrossRef CAS.
- Y. Zhou, J. Chen, Y. Xu and Z. Liu, J. Mater. Sci. Technol., 2013, 29, 168–174 CrossRef CAS.
- F. Wang, X. Chen, L. Chen, J. Yang and Q. Wang, Mater. Sci. Eng., C, 2019, 96, 41–50 CrossRef CAS PubMed.
- Y. Li, M. Xie, X. Zhang, Q. Liu, D. Lin, C. Xu, F. Xie and X. Sun, Sens. Actuators, B, 2019, 278, 126–132 CrossRef CAS.
- Y. Qiao, Q. Liu, S. Lu, G. Chen, S. Gao, W. Lu and X. Sun, J. Mater. Chem. B, 2020, 8, 5411–5415 RSC.
- W. Lu and X. Wu, New J. Chem., 2018, 42, 3180–3183 RSC.
- J. Chen, Q. Xu, Y. Shu and X. Hu, Talanta, 2018, 184, 136–142 CrossRef CAS.
- P.-H. Huang, C. P. Hong, J. F. Zhu, T.-T. Chen, C.-T. Chan, Y.-C. Ko, T.-L. Lin, Z.-B. Pan, N.-K. Sun and Y.-C. Wang, Dalton Trans., 2017, 46, 6985–6993 RSC.
- Y. Qi, C.-T. He, J. Lin, S. Lin, J. Liu, J. Huang, W. Xue, G. Yu, H.-Y. Chao, Y. Tong and Z. Qiao, Nano Res., 2017, 10, 3621–3628 CrossRef CAS.
- X. Zhang, H. Zhao, C. Li, S. Li, K. Liu and L. Wang, Appl. Catal., B, 2021, 299, 120641 CrossRef CAS.
- Z. Cao, Z. Jiang, Y. Li, C. Huang and Y. Li, ChemSusChem, 2019, 12, 2480–2486 CrossRef CAS PubMed.
- T. T. Zhao, Z. W. Jiang, S. J. Zhen, C. Z. Huang and Y. F. Li, Microchim. Acta, 2019, 186, 1–8 CrossRef CAS PubMed.
- P. Kannan, T. Maiyalagan, E. Marsili, S. Ghosh, J. Niedziolka-Jönsson and M. Jönsson-Niedziolka, Nanoscale, 2016, 8, 843–855 RSC.
- C. Bao, Q. Niu, X. Cao, C. Liu, H. Wang and W. Lu, New J. Chem., 2019, 43, 11135–11140 RSC.
- Z. Fu, J. Hu, W. Hu, S. Yang and Y. Luo, Appl. Surf. Sci., 2018, 441, 1048–1056 CrossRef CAS.
- Y. Wang, S. Tao, H. Lin, S. Han, W. Zhong, Y. Xie, J. Hu and S. Yang, RSC Adv., 2020, 10, 33475–33482 RSC.
- W. Jiao, C. Chen, W. You, J. Zhang, J. Liu and R. Che, Small, 2019, 15, 1805032 CrossRef.
- P. S. Bagus, C. J. Nelin, C. Brundle, B. V. Crist, N. Lahiri and K. M. Rosso, Phys. Chem. Chem. Phys., 2022, 24, 4562–4575 RSC.
- O. Adeniyi, N. Nwahara, D. Mwanza, T. Nyokong and P. Mashazi, Sens. Actuators, B, 2021, 348, 130723 CrossRef CAS.
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 2000789 CrossRef CAS.
- Q. Xu, X. Liao, W. Hu, W. Liu and C. Wang, J. Mater. Chem. B, 2021, 9, 9606–9614 RSC.
- L. He, Z. W. Peng, Z. W. Jiang, X. Q. Tang, C. Z. Huang and Y. F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 31834–31840 CrossRef CAS.
- L. Zhang, Y. Ding, R. Li, C. Ye, G. Zhao and Y. Wang, J. Mater. Chem. B, 2017, 5, 5549–5555 RSC.
- D. Cui, L. Su, H. Li, M. Li, C. Li, S. Xu, L. Qian and B. Yang, J. Electroanal. Chem., 2019, 838, 154–162 CrossRef CAS.
- X. Xuan, C. Pérez-Ràfols, C. Chen, M. Cuartero and G. A. Crespo, ACS Sens., 2021, 6, 2763–2771 CrossRef CAS PubMed.
- H. Wei, Q. Xue, A. Li, T. Wan, Y. Huang, D. Cui, D. Pan, B. Dong, R. Wei, N. Naik and Z. Guo, Sens. Actuators, B, 2021, 337, 129687 CrossRef CAS.
- N. Gholamalizadeh, S. Mazinani, M. Abdouss, A. M. Bazargan and F. Fatemi, Prog. Org. Coat., 2022, 172, 107083 CrossRef CAS.
- M. A. Kachouei, S. Shahrokhian and M. Ezzati, Sens. Actuators, B, 2021, 344, 130254 CrossRef.
- X. Zhu, S. Yuan, Y. Ju, J. Yang, C. Zhao and H. Liu, Anal. Chem., 2019, 91, 10764–10771 CrossRef CAS.
- X. Zhu, Y. Ju, J. Chen, D. Liu and H. Liu, ACS Sens., 2018, 3, 1135–1141 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05179j |
| This journal is © The Royal Society of Chemistry 2024 |