
Blocking tau transmission by biomimetic graphene nanoparticles†
Runyao
Zhu
 a,
Kamlesh M.
Makwana
b,
Youwen
Zhang
a,
Benjamin H.
Rajewski
b,
Juan R.
Del Valle
*b and
Yichun
Wang
*a
a,
Kamlesh M.
Makwana
b,
Youwen
Zhang
a,
Benjamin H.
Rajewski
b,
Juan R.
Del Valle
*b and
Yichun
Wang
*a
aDepartment of Chemical & Biomolecular Engineering, University of Notre Dame, Indiana 46556, USA. E-mail: ywang65@nd.edu
bDepartment of Chemistry & Biochemistry, University of Notre Dame, Indiana 46556, USA
First published on 4th July 2023
Abstract
Tauopathies are a class of neurodegenerative diseases resulting in cognitive dysfunction, executive dysfunction, and motor disturbance. The primary pathological feature of tauopathies is the presence of neurofibrillary tangles in the brain composed of tau protein aggregates. Moreover, tau aggregates can spread from neuron to neuron and lead to the propagation of tau pathology. Although numerous small molecules are known to inhibit tau aggregation and block tau cell-to-cell transmission, it is still challenging to use them for therapeutic applications due to poor specificity and low blood-brain barrier (BBB) penetration. Graphene nanoparticles were previously demonstrated to penetrate the BBB and are amenable to functionalization for targeted delivery. Moreover, these nanoscale biomimetic particles can self-assemble or assemble with various biomolecules including proteins. In this paper, we show that graphene quantum dots (GQDs), as graphene nanoparticles, block the seeding activity of tau fibrils by inhibiting the fibrillization of monomeric tau and triggering the disaggregation of tau filaments. This behavior is attributed to electrostatic and π–π stacking interactions of GQDs with tau. Overall, our studies indicate that GQDs with biomimetic properties can efficiently inhibit and disassemble pathological tau aggregates, and thus block tau transmission, which supports their future developments as a potential treatment for tauopathies.
Introduction
Pathogenic aggregation of tau proteins into neurofibrillary tangles (NFTs) is a characteristic feature of tauopathies, which are a group of neurodegenerative diseases, including Alzheimer's disease (AD),1 frontotemporal dementias (FTDP-17),2 Pick's disease,3 and progressive supranuclear palsy.4 The normal function of tau is to stabilize microtubules by binding with tubulin in neurons. These microtubules provide a structural backbone for axons and dendrites and serve as ‘railways’ for the transport of proteins and organelles in axons and dendrites.5 Pathogenic fibrillization of tau reduces cytoskeletal stability, interferes with synaptic transmission and axonal transport, and causes aberrant communication between neurons.6 These insoluble tau deposits can arise due to gene mutations or aberrant post-translational modifications such as hyperphosphorylation and acetylation.2,7,8 In addition, tau aggregates can spread from neuron to neuron and induce the misfolding of normal soluble tau, leading to the propagation of tau pathology in a prion-like manner.9,10 Small molecule library screening campaigns have identified several compounds that either directly or indirectly inhibit tau aggregation.11,12 These molecules include modulators of tau post-translational modification,8 microtubule stabilizers,13 tau assembly inhibitors,13,14 and degradation promoters.15 However, there are no disease-modifying drugs that target tau aggregation. Moreover, many drug candidates suffer from limited penetration of the blood-brain barrier (BBB) and off-target effects, reducing central nervous system bioavailability and limiting their clinical utility.15,16 Furthermore, compounds targeting kinases upstream of tau may disrupt other physiological processes resulting in undesirable side effects such as an increased risk of diabetes and arrhythmia.17 Therefore, developing novel strategies to overcome current challenges is critical for the treatment of neurodegenerative tauopathies.Nanoparticles (NPs), as nanocarriers, have demonstrated the ability to deliver drugs into the brain and are capable of controlled release and targeted delivery.18,19 Graphene quantum dots (GQDs), as graphene-based NPs, are single or a-few layered graphene with lateral dimensions typically less than 20 nm.20 These nanosheets have garnered much attention in biomedicine, including bioimaging, biosensor, and cancer phototherapy, due to their unique optical, electrochemical, and physicochemical properties, high biocompatibility, and low cytotoxicity.21–24 Moreover, GQDs can be used as nanocarriers in drug delivery systems upon conjugation with target ligands and chemotherapeutic agents via either covalent or noncovalent bonds because of their surface functional groups and π-conjugated planar structure.25 GQD-based drug delivery systems26–28 were previously demonstrated to penetrate the BBB and specifically target brain tumor regions, suggesting great potential in neuroscience therapeutics. Most importantly, GQDs exhibit a wide spectrum of biomimetic properties, including polarizability, amphiphilic character, and participation in electrostatic interactions, hydrophobic interactions, hydrogen bonding, and π–π stacking.29,30 Thus, GQDs can self-assemble31 or assemble with various proteins, such as amyloid-β (Aβ) peptides,32,33 biofilm amyloids,34 and α-synuclein,28 demonstrating their potential to protect against the neuronal damage associated with AD and Parkinson's disease. Furthermore, the properties of GQDs, such as size,35 charge,36 functional groups,37 and chirality,38 can be engineered precisely, affording opportunities to modulate their assembly with proteins.39,40
Here, we report that biomimetic GQDs are able to block the seeding activity of tau fibrils in an in vitro cellular tau biosensor propagation model. To investigate the effect of GQDs on tau propagation, we engineered GQDs with different sizes and charges by post-separation technologies and functionalization of GQDs with cysteine (Cys) and ethylenediamine (EDA) and studied the interaction between tau and GQDs in vitro. By monitoring the amount of tau aggregates, the secondary structure, and the morphology of tau, we found that negatively charged GQDs with a larger size inhibited tau aggregation more effectively. In addition, the presence of GQDs induced the disaggregation of tau filaments in vitro. These findings were potentially attributed to the fact that aromatic residues on the aggregation-prone region of tau can interact with GQDs via π–π stacking interactions, as verified by the fluorescence quenching assay. The positively charged microtubule-binding domain of tau can interact with carboxyl groups on the edge of GQDs via electrostatic interactions. Furthermore, a cellular propagation assay employed to test the seeding activity of tau fibrils demonstrated that GQDs blocked the aggregation of endogenous tau induced by extracellular seeds. Overall, our findings suggest that engineered biomimetic GQDs are a promising and effective platform to prevent the cellular transmission of tau by inhibition and disassembly of tau aggregates.
Results and discussion
As-synthesized GQDs carrying carboxyl groups on the edges in an average diameter of 8 nm were obtained by the previously reported method (Fig. 1a–c, Fig. S1 and S2a, ESI†). The heights of as-synthesized GQDs are between 0.5 and 2 nm, corresponding to 1–3 graphene layers (Fig. S2b, ESI†).35 The zeta potential of as-synthesized GQDs was −19.9 ± 2.30 mV (Fig. 1d and Fig. S4, ESI†). To obtain GQDs with different charges, we reduced the negative charge by conjugating as-synthesized GQDs with cysteine (Cys) and ethylenediamine (EDA) via carboxyl groups (Fig. 1a and Fig. S1 and S3, ESI†). In the Fourier transform infrared (FTIR) spectra (Fig. 1c), due to the carboxyl groups in the structure of Cys, the peak at 1706 cm−1 assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching was more obvious in the spectrum of Cys-GQDs than those of as-synthesized GQDs and EDA-GQDs.35 The FTIR spectra of Cys-GQDs and EDA-GQDs also appeared peaks at 1250 and 1184 cm−1 attributed to C–N stretching.19 The zeta potentials of Cys-GQDs and EDA-GQDs were −1.46 ± 0.42 mV and 1.63 ± 0.55 mV (Fig. 1d and Fig. S4, ESI†), due to the pKa value of Cys and EDA, 1.71 and 10.7, respectively.
O stretching was more obvious in the spectrum of Cys-GQDs than those of as-synthesized GQDs and EDA-GQDs.35 The FTIR spectra of Cys-GQDs and EDA-GQDs also appeared peaks at 1250 and 1184 cm−1 attributed to C–N stretching.19 The zeta potentials of Cys-GQDs and EDA-GQDs were −1.46 ± 0.42 mV and 1.63 ± 0.55 mV (Fig. 1d and Fig. S4, ESI†), due to the pKa value of Cys and EDA, 1.71 and 10.7, respectively.
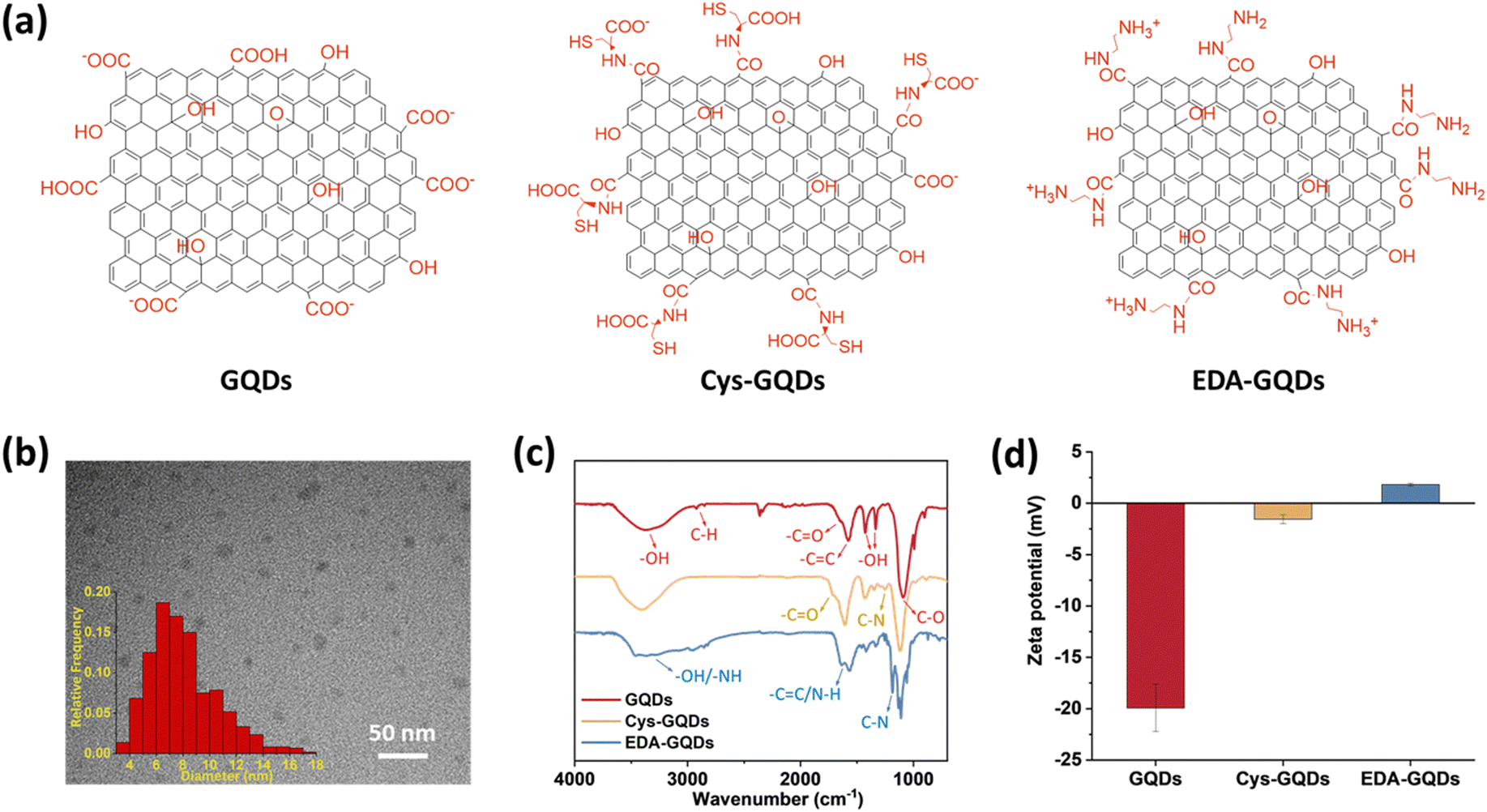 | ||
| Fig. 1 (a) Molecular structure, (b) transmission electron microscope (TEM) image and size distribution of GQDs. The average diameter is 8.1 ± 2.5 nm. (Scale bar: 50 nm.) (c) Fourier transform infrared (FTIR) spectra and (d) zeta potentials of GQDs, Cys-GQDs, and EDA-GQDs. | ||
Following the synthesis, characterization, and functionalization of GQDs, we investigated the charge effect of GQDs on the aggregation of a recombinant 0N4R isoform of tau. Tau used in these studies includes a P301L mutation (tauP301L) which is frequently observed in patients with FTDP-17 (Fig. S5, ESI†).41 This missense mutation leads to a two-fold increase in aggregation rate compared with the full-length tau.42 TauP301L contains four microtubule-binding repeat domains (R1–R4) that harbor two key aggregation-prone motifs: PHF6* (VQIINK, residues 217–222) and PHF6 (VQIVYK, residues 248–253).7 A large number of basic residues result in an overall positive charge in the repeat domains at physiological pH (Fig. 2a). Firstly, the aggregation of tauP301L was monitored by a ThT fluorescence assay (Fig. 2b). ThT is a benzothiazole dye that exhibits enhanced fluorescence upon binding to tau fibers with β-sheet secondary structure.43 In the presence of heparin sulfate, tauP301L was induced to aggregate, resulting in a sigmoidal curve from increased ThT fluorescence (Fig. 2c). Incubation of tauP301L in the presence of heparin and 6.25 μM GQDs or Cys-GQDs resulted in 99% inhibition of endpoint ThT fluorescence (Fig. 2c and d). However, EDA-GQDs with overall positive charge exhibited 88.8% inhibitory activity, less effective than the negatively charged GQDs and Cys-GQDs.
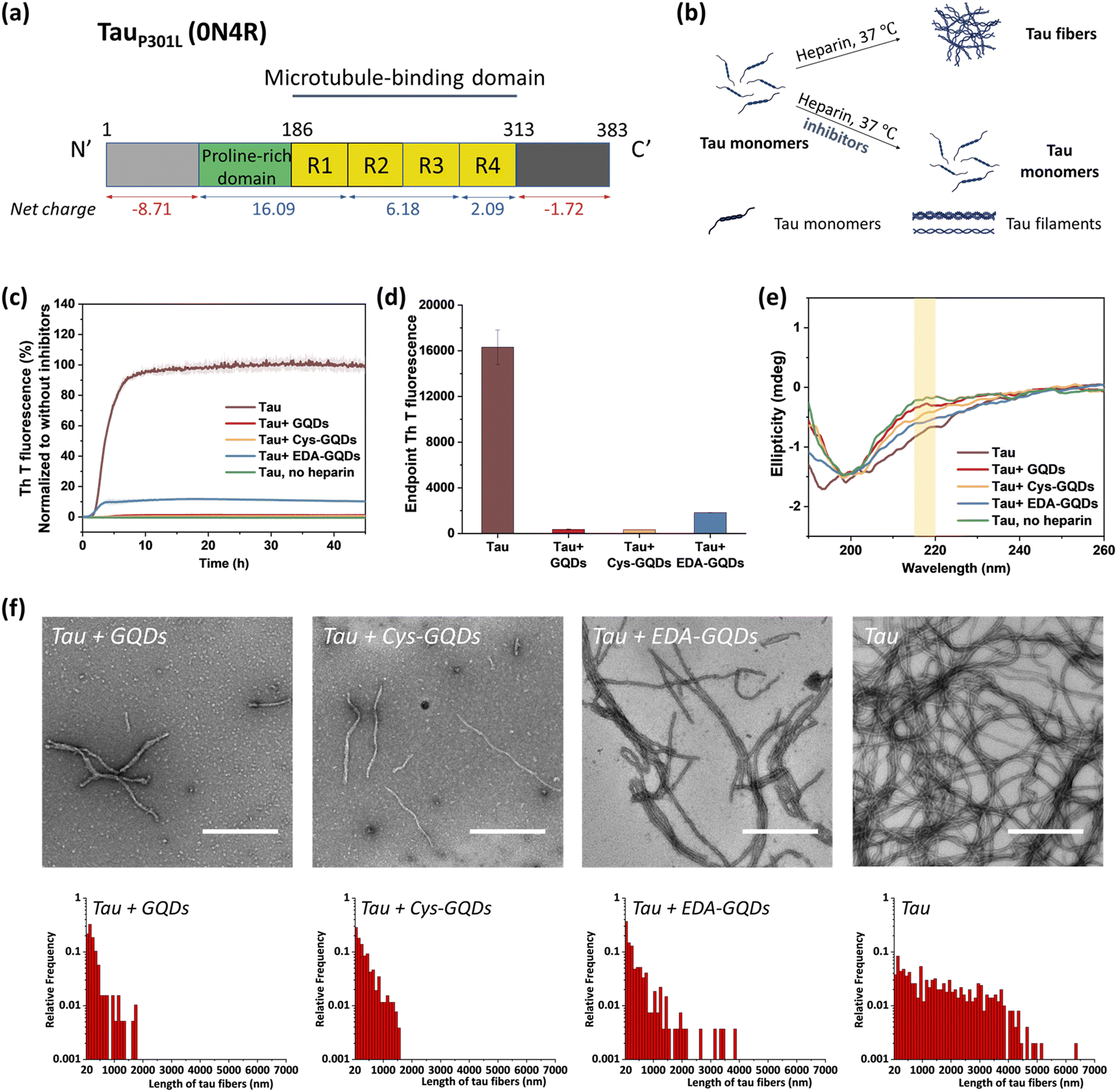 | ||
| Fig. 2 TauP301L aggregation was inhibited by GQDs, Cys-GQDs, and EDA-GQDs. (a) Aggregation-prone tauP301L sequence. (b) Schematic representation of the in vitro aggregation of recombinant tau (10 μM) in the presence and absence of inhibitors at a concentration of 6.25 μM. (c) and (d) In the presence of GQDs or functionalized GQDs, thioflavin T (ThT) fluorescence of tau aggregation was reduced significantly. The inhibitory effect of EDA-GQDs is weaker than GQDs and Cys-GQDs. (e) Circular dichroism (CD) spectra, (f) TEM images, and length distributions of tau protein incubated with/without 6.25 μM GQDs or functionalized GQDs for 4 days. (Scale bars: 500 nm) | ||
The mechanism of tau aggregation in the presence of heparin is widely described as ligand-induced nucleation-dependent polymerization (NDP).44 The lag time (tlag) and the apparent elongation rate constant (kapp) of the aggregation process were derived by fitting each kinetic data to a Gompertz growth function (Table S1, ESI†).45,46 The lag time represents the time of nucleation process (i.e., initial seed formation of tau) and the apparent elongation rate constant represents the rate of growth process (i.e., the elongation of tau fibrils). The tau aggregation without GQDs as a control showed a tlag of 1.84 ± 0.01 h and a kapp of 0.76 ± 0.008 h−1. In the presence of GQDs or Cys-GQDs, tlag was increased to nearly double that of the control sample, while kapp was reduced to 0.70 ± 0.039 and 0.38 ± 0.021 h−1, a decrease of 7.9% and 50% compared to the control sample, respectively. These results suggest that GQDs and Cys-GQDs impeded both seed formation and the fibril elongation processes. In contrast, tau incubated with EDA-GQDs led to a significant decrease in tlag, potentially because the presence of EDA-GQDs increased the local concentration of soluble monomeric tau, facilitating the formation of aggregation-prone tau seeds. Despite a decreased tlag, kapp was slightly increased, possibly because a portion of the aggregation-inducer polyanion heparin interacted with EDA-GQDs with positive charge, reducing the formation of tau fibers by 88.8% after 45 h incubation compared to the control sample.
We detected the effect of GQDs and functionalized GQDs on the secondary structure transition of tauP301L using circular dichroism (CD) spectroscopy (Fig. 2e). In the absence of heparin, soluble tau monomers showed a negative peak at ∼200 nm, indicative of a random coil conformation. After heparin-induced fibrillization, a growing CD signal at ∼218 nm, typical of β-sheet conformation, emerged.47 Upon incubation with GQDs, Cys-GQDs, or EDA-GQDs for 4 days, the CD intensity of tau at 218 nm was reduced by 61%, 39%, and 18% compared to the control sample, respectively, suggesting inhibition of transition to a β-rich tau assembly. The secondary structure of tau had less β-sheet content in the presence of GQDs or Cys-GQDs than in the presence of EDA-GQDs, which is consistent with the ThT assay results (Fig. 2c).
The morphological features of tauP301L fibrils were examined using transmission electron microscopy (TEM) after incubation for 4 days. To quantify the tau fibers in each sample, we measured their length and density using ImageJ (Fig. 2f and Table S2, ESI†). The aggregation of tau in the control sample resulted in the formation of filaments with an average length of 1623 ± 1255 μm and an average density of 4.53 ± 2.08 fibers per μm2. In the presence of GQDs or functionalized GQDs, heparin-induced tauP301L assembled into shorter fibers with lower density. When tauP301L monomers were incubated with GQDs, Cys-GQDs, or EDA-GQDs, the average length of formed fibers was decreased to 17%, 20%, and 35% of the control sample, while the average density of fibers was decreased to 30%, 34%, and 78%, compared to that of the control sample. Among samples incubated with the three GQDs, tau incubated with EDA-GQDs resulted in the formation of 3 to 4-fold more fibers per unit area than tau with GQDs or Cys-GQDs. The collective results indicate that GQDs and Cys-GQDs with negative charges have a better inhibitory effect on the fibrillization of tau than EDA-GQDs with positive charge.
To further investigate whether the degree of negative charge on GQDs can influence their inhibitory efficiency, we monitored the ThT fluorescence of tauP301L incubated with GQDs or Cys-GQDs at different concentrations. GQDs and Cys-GQDs inhibited aggregation in a dose-dependent manner (Fig. 3a). We obtained IC50 value for GQDs and Cys-GQDs by fitting the dose-response results to a sigmoidal function (Fig. 3b). The IC50 of GQDs and Cys-GQDs are 0.70 μM, and 0.89 μM.12,14 These results suggest that when the zeta potential of GQDs is between −20 mV and −1.5 mV, the amount of negative charges on GQDs does not affect the inhibitory efficiency significantly.
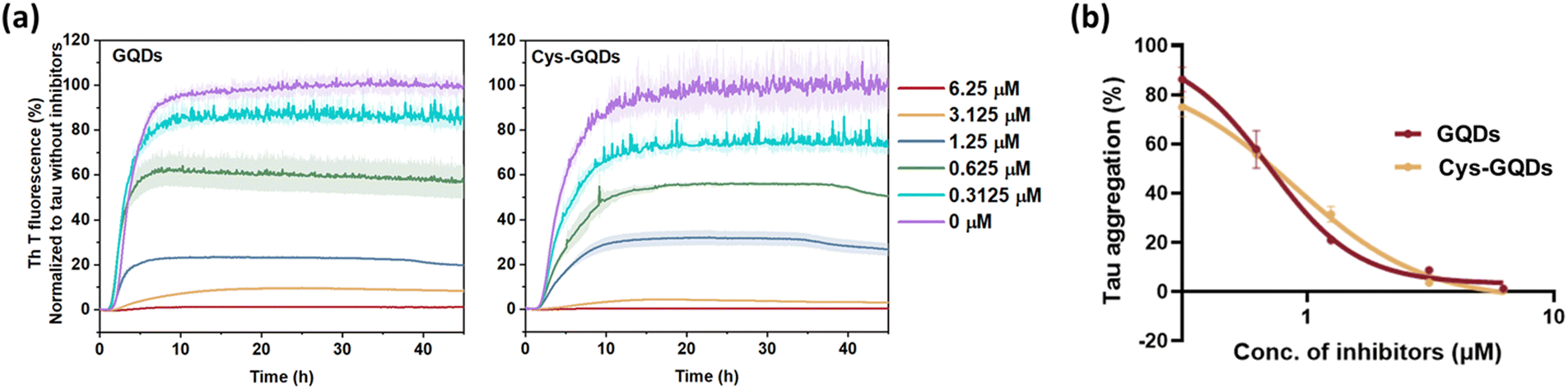 | ||
| Fig. 3 (a) ThT fluorescence of tau aggregation was decreased with the increased concentration of GQDs and Cys-GQDs. (b) Dose-response results of ThT fluorescence assay of GQDs and Cys-GQDs were fitted to a sigmoidal model. The IC50 of GQDs and Cys-GQDs is 0.70 μM and 0.89 μM, respectively, obtained from the dose-response curve. | ||
Before investigating the effect of GQDs on tau propagation in the cellular assay, we determined the cytotoxicity of GQDs, Cys-GQDs, and EDA-GQDs by employing the CCK-8 assay with SH-SY5Y human neuroblastoma cells. GQDs and Cys-GQDs exhibited no appreciable toxicity toward SH-SY5Y cells up to 5 μM, whereas 5 μM of EDA-GQDs reduced cell viability by 90.7% after 48 h incubation (Fig. S6, ESI†). It is likely that positively charged EDA-GQDs interact with the negatively charged cell membrane, resulting in higher cell uptake and disruption of membrane integrity.48 Therefore, we only considered the nontoxic GQDs and Cys-GQDs in the following cellular assay.
One of the key characteristics of tau NFTs is cellular transmission from one diseased neuron to adjacent healthy neurons, inducing the propagation of tau pathology.9,10 To examine whether GQDs and Cys-GQDs can inhibit the cellular seeding of recombinant tau, we employed HEK293 biosensor cells that stably expressed a tau-yellow fluorescent protein fusion [tau-RD(LM)-YFP] (Fig. 4a).49 After incubating preformed tauP301L fibrils with these cells for 48 h, the aggregation of endogenous tau induced by extracellular seeds can be monitored by detection of focal puncta with green fluorescence. The quantitative measurements of intracellular aggregates were provided by automated image analysis of green puncta. Monomeric tau has no capacity to seed the aggregation of endogenous tau and the cells remain diffusely fluorescent with no focal puncta. Pre-treatment of tauP301L monomers with 0.12 μM of GQDs or Cys-GQDs in the presence of heparin decreased the intracellular aggregates by 60% relative to the control treatment. As concentrations of GQDs and Cys-GQDs increased, the seeding activity of tauP301L decreased. With 0.6 μM of GQDs or Cys-GQDs, the seeding activity of tauP301L was almost completely abolished (Fig. 4b). These results demonstrate that GQDs and Cys-GQDs prevent the fibrillization of tau monomers and thus block the cellular seeding activity of recombinant tau.
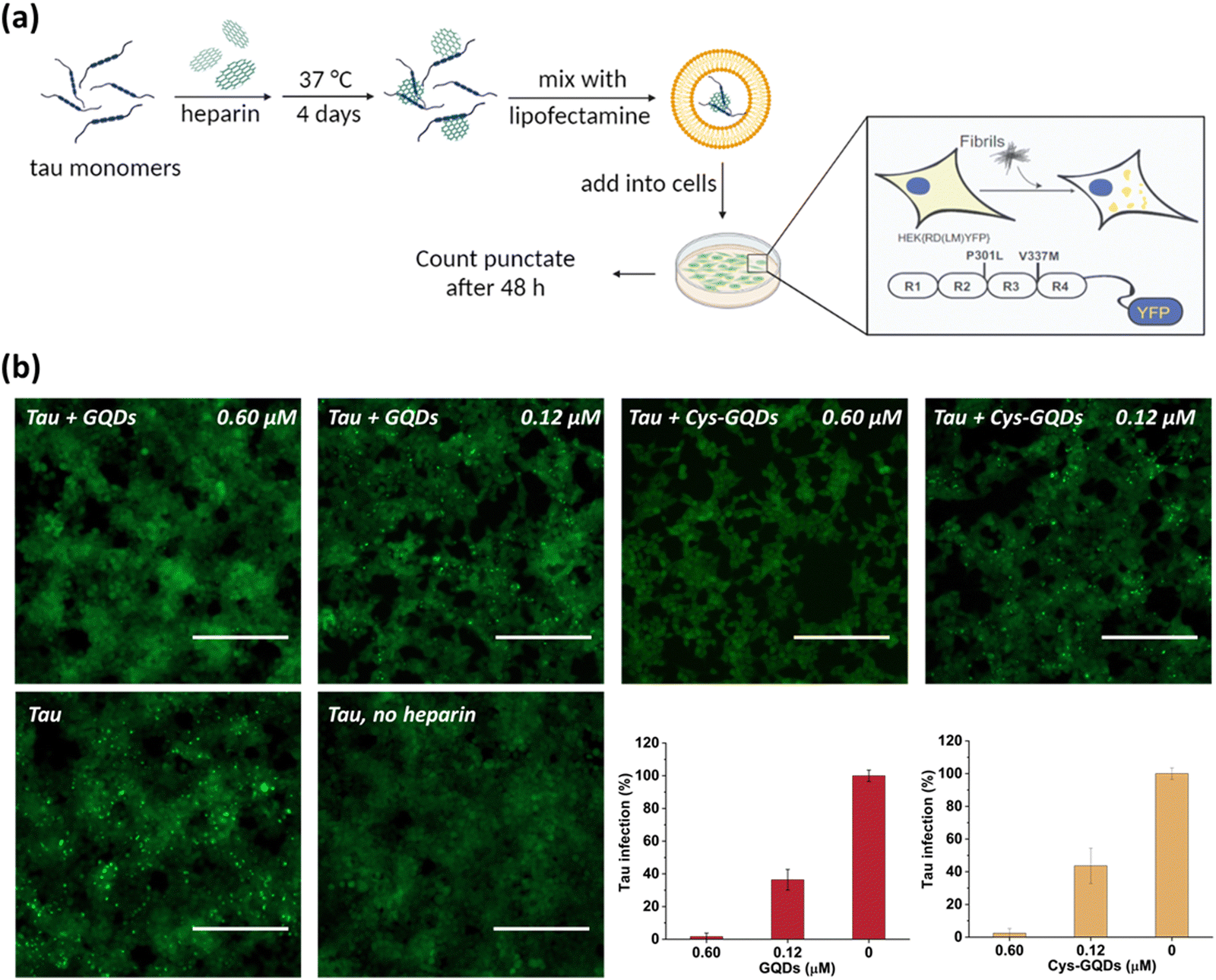 | ||
| Fig. 4 (a) Scheme of HEK293 cellular tau biosensor propagation assay. (b) GQDs and Cys-GQDs prevented the cellular seeding of tauP301L monomers. Soluble monomeric tauP301L (0.19 μM) in the presence of heparin was incubated with GQDs or Cys-GQDs for 4 days and then added to HEK293 cells stably expressing tau-RD (P301L/V337M)-YFP. Representative images of cells were taken at 20× magnification under fluorescein isothiocyanate (FITC) channel (ex: 469 nm/em: 525 nm). The green puncta with high fluorescence represented the aggregation of tau in cells induced by exogenous tau fibers. Scale bar: 200 μm. Bar graphs show the number of intracellular fluorescent puncta relative to control infection wells without inhibitors. | ||
The presence of NFTs in the brain, which are composed of tau aggerates, is the primary feature of tauopathies. Promoting the disaggregation of tau fibrils is thus considered a potential strategy for therapeutic development.15 We investigated the effect of GQDs on the disaggregation of mature tau fibrils using the ThT fluorescence assay (Fig. 5a). After incubation with GQDs, Cys-GQDs, or EDA-GQDs, the intensity of ThT fluorescence in the presence of tau fibrils decreased with time (Fig. 5b), suggesting that tau fibrils could be dissociated into tau monomers and oligomers. Based on endpoint ThT fluorescence after 30 h (Fig. 5c), GQDs, Cys-GQDs, and EDA-GQDs induced 56%, 81%, and 92% disassembly of tau fibrils. TEM images of tau fibrils treated with GQDs and engineered GQDs also revealed the disaggregation of tau filaments into fibril fragments (Fig. S7, ESI†). To quantify the tau NFTs after the disassembly by GQDs, we measured the area of tau NFTs in each sample using ImageJ (Table S3, ESI†). In the presence of GQDs, Cys-GQDs, or EDA-GQDs, tau NFTs were 33%, 89%, and 52% disassembled. This is potentially due to the negatively charged fuzzy coat of tauP301L fibrils comprised of the unfolded C-terminal and N-terminal domains (Fig. 2a) that surrounds the fibrillar tau core.50 The negative charges on the outermost layer of tau fibers could prevent GQDs from approaching, whereas Cys-GQDs and EDA-GQDs with fewer charges could pass through the fuzzy coat and reach the fibrillar tau core more easily. In addition, negatively charged Cys-GQDs are more likely to interact with the positively charged fibrillar core and disaggregate the tau fibrils, compared to EDA-GQDs with positive charge. In the ThT fluorescence assay, the presence of EDA-GQDs could affect ThT binding with tau fibers due to their positive charge, and thus ThT fluorescence of tau fibrils treated by EDA-GQDs was reduced much more than the actual fiber disassembly obtained from TEM images. Due to the toxicity of some types of soluble tau dimer, trimer, and oligomers on neurons,51 we tested the cytotoxicity of tau NFTs after disaggregation by GQDs and engineered GQDs on SH-SY5Y cells. The disaggregated fibrils did not exhibit cytotoxicity toward SH-SY5Y cells after 48 h incubation (Fig. S8, ESI†).
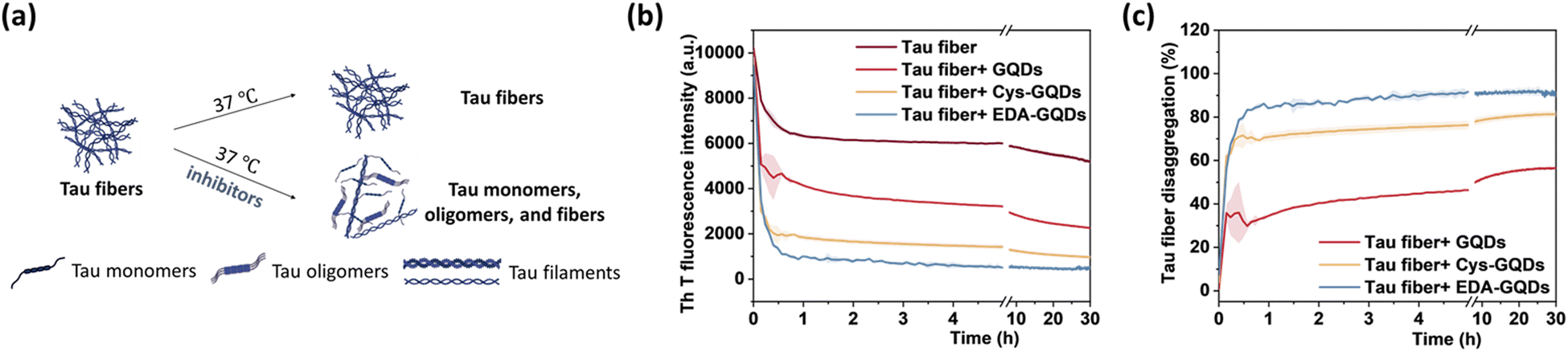 | ||
| Fig. 5 (a) Scheme of the in vitro disaggregation assay of tau fibers (5 μM) in the presence and absence of inhibitors (6.25 μM). (b) ThT fluorescence of tau fibers and (c) the percentage of disaggregation of preformed tau fibers after incubation with GQDs, Cys-GQDs, or EDA-GQDs. | ||
To further investigate the effect of GQDs and Cys-GQDs on tau propagation, we tested if they can block the cellular transmission of mature tau fibrils in the tau biosensor seeding assay. After 36 h treatment of GQDs or Cys-GQDs with preformed tau fibrils, HEK293 cells expressing tau-RD(LM)-YFP were incubated with the pre-treated tau fibrils. With the increase in concentration from 0.12 μM to 0.5 μM, GQDs reduced the aggregation of endogenous tau from 18% to 80%, while Cys-GQDs reduced the intracellular aggregates from 6% to 88% compared to the control sample without GQDs or Cys-GQDs (Fig. 6). To confirm whether GQDs or Cys-GQDs simply affect lipofectamine-mediated cellular uptake of tau fibrils rather than the seeding capacity of the fibrils themselves, we mixed tau fibrils with GQDs or Cys-GQDs without incubation and then incubated the mixture with cells for 48 h. GQDs and Cys-GQDs did not reduce the aggregation of endogenous tau without prior incubation, suggesting that GQDs and Cys-GQDs did not interfere with lipofectamine-mediated cellular uptake of tau fibrils (Fig. S9, ESI†) and the decrease of intracellular aggregates was solely attributed to fibril disassembly into seed incompetent monomers after 36 h incubation with GQDs or Cys-GQDs, effectively blocking the seeding capacity of mature tau fibrils.
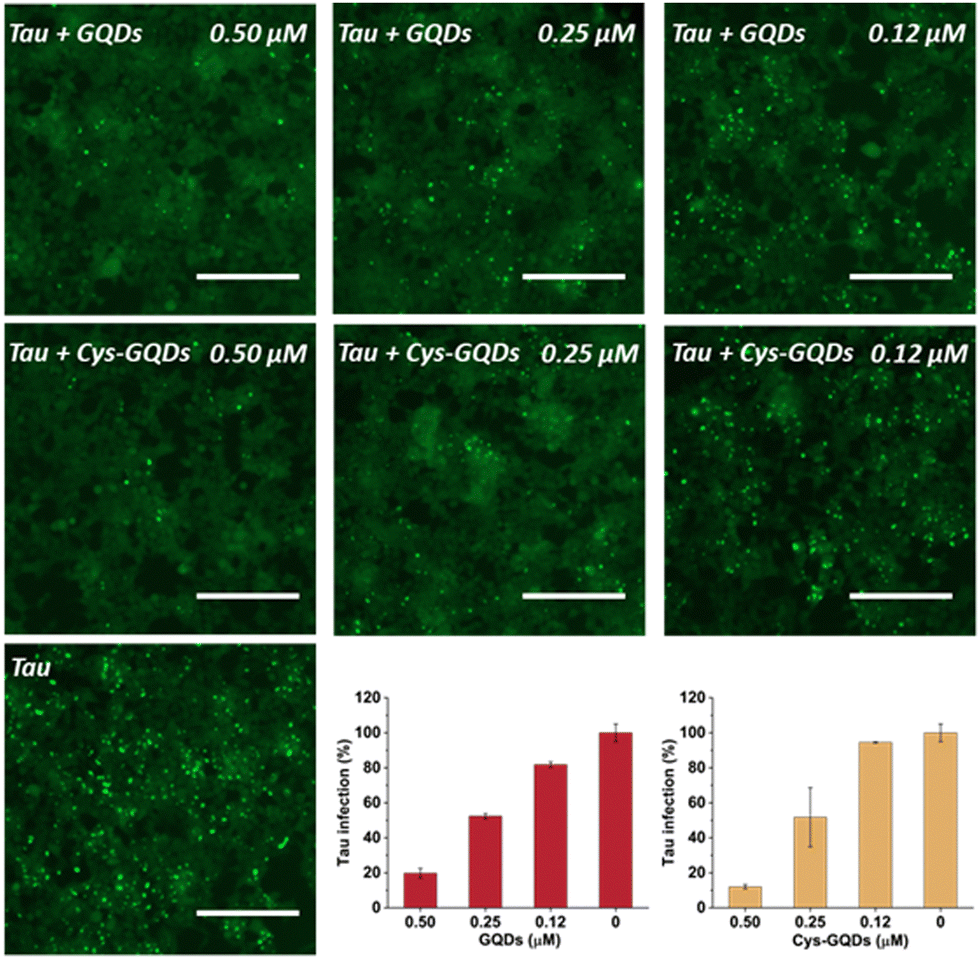 | ||
| Fig. 6 GQDs and Cys-GQDs prevented the cellular seeding of mature tau fibers. Preformed tau fibrils (0.19 μM) were treated with GQDs or Cys-GQDs for 36 h, then added to cells, and incubated for an additional 48 h. Representative images of cells were taken at 20× magnification under FITC channel (ex: 469 nm/em: 525 nm). The green puncta with high fluorescence represented the aggregation of tau in cells induced by exogenous tau fibers. Scale bar: 200 μm. Tau infection (%) in the bar graph shows the number of intracellular fluorescent puncta relative to control infection wells lacking the inhibitors. | ||
Based on our experimental results, negatively charged GQDs can inhibit the fibrillization of tau proteins more efficiently than GQDs with positive charge. This suggests that the positively charged domains of tau, especially the aggregation-prone R2 and R3 domains, prefer to engage in more electrostatic interactions with the carboxyl groups on the edge of GQDs rather than form hydrogen bonds that are critical for the formation of tau aggregates.
Moreover, based on ThT aggregation assay results (Fig. 2c), there are other factors in addition to the negative charge of GQDs that may result in the inhibition of tau aggregation. We investigated the other potential interactions between tau and GQDs using fluorescence spectroscopy. Generally, the tau exhibits intrinsic fluorescence emission near 302 nm when excited at 265 nm, resulting from tyrosine (Tyr) fluorophores in the tau structure.52,53 When 10 μM of tau was mixed with GQDs or Cys-GQDs in increasing concentrations, the fluorescence intensity of tau was significantly quenched (Fig. 7), suggesting that the π-conjugated planar structure of GQDs binds with Tyr residues in tau, probably via π–π stacking interactions.
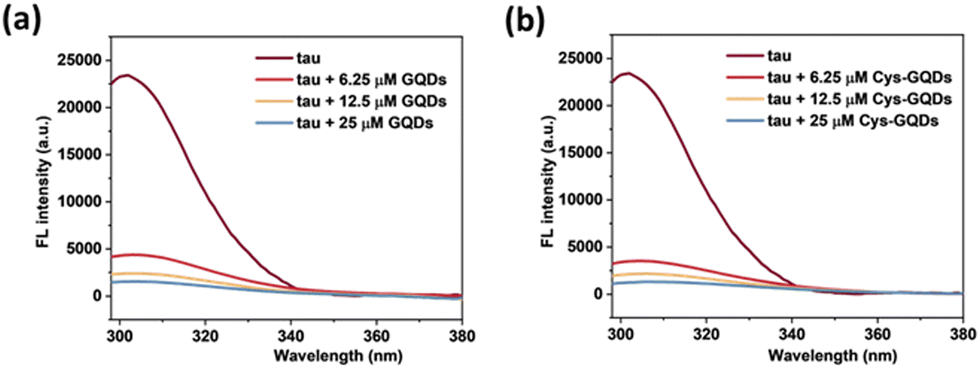 | ||
| Fig. 7 Fluorescence spectra of tau protein (10 μM) incubated with 6.25, 12.5, and 25 μM of (a) GQDs and (b) Cys-GQDs (Ex: 265 nm). The fluorescence of tau was quenched significantly in the presence of GQDs or Cys-GQDs. | ||
Besides the effect of charge, we also found that the inhibitory efficiency of GQDs increased significantly as the diameter of GQDs increased from 6.5 nm to 13.5 nm based on ThT fluorescence assay (Fig. S10, ESI†). The diameter of the full-length tau monomer is 13 ± 0.6 nm.54 As the diameter of GQDs increases from 6.5 nm to 13.5 nm, single GQDs with a larger diameter are able to interact with more tau monomers on the edge via electrostatic interaction than the ones with a smaller diameter. Also, when interacting with larger GQDs, the spacing between two tau monomers increases, reducing the local concentration of tau and enhancing inhibitory efficiency. In addition, increasing the size of GQDs promotes more aromatic residues in tau to bind to the surface of GQDs via π–π stacking, thereby increasing the binding strength between tau and GQDs.
Overall, the potential mechanism of the interaction between GQDs and tau protein can be summarized in Fig. 8. For the aggregation of tauP301L (Fig. 8a), negatively charged GQDs with carboxyl groups can interact with repeat domains of tau with positive charge via electrostatic interactions. Meanwhile, GQDs with π-conjugated planar structure can interact with amino acids containing aromatic rings in tau proteins via π–π stacking, including histidine, phenylalanine, and tyrosine. The carbon-conjugated plane of GQDs could also interact with positively charged amino acids in tau via cation–π interactions. For the disassembly of tau fibrils (Fig. 8b), GQDs with more negative charges can be prevented from penetrating the negatively charged fuzzy coat and accessing the fibrillar tau core, thereby reducing their efficacy in disassembly, compared to Cys-GQDs and EDA-GQDs with fewer charges. Negatively charged Cys-GQDs have a higher potential to interact with positively charged fibrillar cores compared to EDA-GQDs, which enhances their ability to disaggregate the tau fibrils.
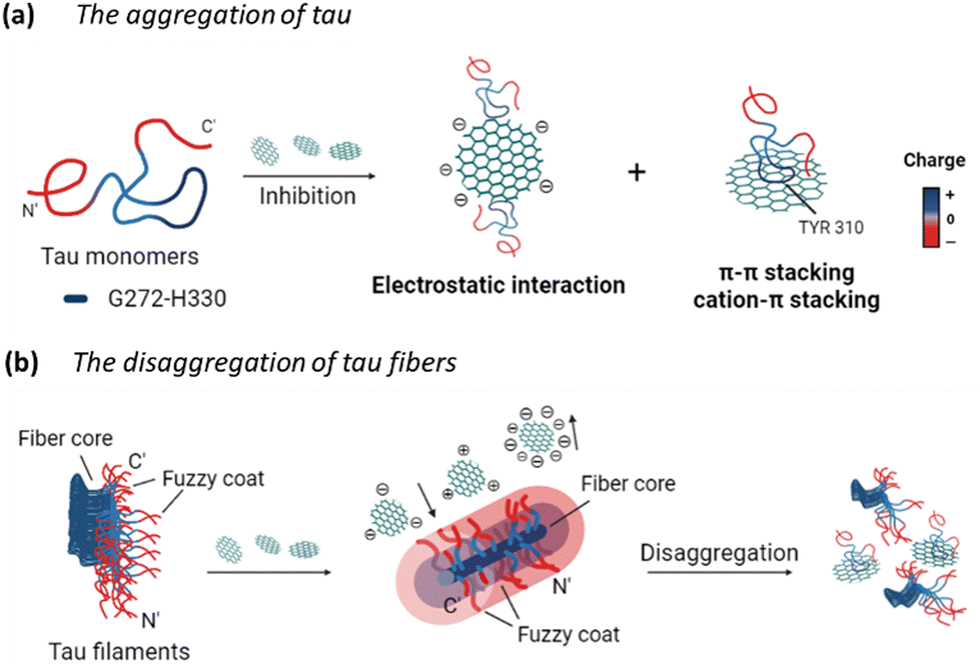 | ||
| Fig. 8 Schematics of the charge effect of GQDs on their interactions with tau proteins. GQDs can interact with tau proteins via electrostatic interactions and π–π stacking, thereby (a) preventing the tau aggregation, and (b) inducing the disassembly of tau fibrils. | ||
Experimental
Materials
Sulfuric acid (95–98%) and nitric acid (69–70%) were purchased from VWR. Carbon nanofibers, N-hydroxysulfosuccinimide sodium salt (Sulfo-NHS, >98%), EDA (>99%), sodium acetate (>99%), ThT, heparin sodium salt, and DL-dithiothreitol (DTT, 97%) were purchased from Sigma-Aldrich. The dialysis membrane tubing (MWCO: 1kD) was purchased from Spectrum. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) was purchased from ThermoFisher Scientific. D-Cysteine was purchased from AmBeed. DMEM medium, Opti-MEM medium, DMEM/F12 medium, fetal bovine serum (FBS), and penicillin/streptomycin were obtained from Gibco. Cell Counting Kit-8 was purchased from Enzo Life Sciences. Lipofectamine 2000 transfection reagent was obtained from Invitrogen.Synthesis and characterization of GQDs
GQDs were prepared using a modified Hummers’ method.35 0.45 g of carbon fiber was added into 90 mL of concentrated H2SO4 (98%) and stirred for 1.5 h. After stirring, 30 mL of concentrated HNO3 (68%) was added into the above mixture solution and sonicated for 1 h. Then, the mixture was reacted at 120 °C for 20 h. Next, the solution was neutralized by a sodium hydroxide solution. The final product was further dialyzed for 3 days in a dialysis bag (retained molecular weight: 1000 Da) for purification. The size and shape of GQDs were characterized by TEM (JEOL 2011). 3 μL droplet of the GQD solution (5 μM) was placed on the carbon-coated copper TEM grid (Purchased from Electron Microscopy Sciences) and allowed to dry in the air. The imaging was performed with a TEM instrument under an accelerating voltage of 200 kV. The size distribution of the GQDs was analyzed using ImageJ software. The chemical composition was performed by ATR-FTIR using Bruker Tensor 27 FTIR Spectrometer (Bruker Optics Inc., USA) with a diamond lens Attenuated Total Reflectance (ATR) module. 3 μL of 100 μM GQDs was dried in the air and each spectrum was measured as the accumulation of 64 scans at a spectral resolution of 2 cm−1 within the range 4000–700 cm−1. The fluorescence emission property of GQDs was measured by a plate reader (Tecan infinite 200Pro). The fluorescence of GQD solution was detected at an excitation wavelength of 365 nm and an emission wavelength from 400 nm to 700 nm. The zeta potential of GQDs (6.25 μM) was calculated as the average of three test runs using Malvern Zetasizer Nano ZS (Malvern Panalytical; Worcestershire, United Kingdom). The absorption of GQDs was tested by CD spectroscopy (Jasco J-1700 Spectrometer). The GQD solution was diluted to 2.5 μM followed by spectra scanning from 200 nm to 400 nm with 0.1 nm intervals, 5 nm bandwidth, and a scan speed of 50 nm min−1.Synthesis of Cys-GQDs and EDA-GQDs
The synthesis of Cys-GQDs and EDA-GQDs was carried out using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)/N-hydroxysuccinimide (NHS) coupling reaction.38,55 1 mL of EDC with a concentration of 100 mM was mixed with 25 mL GQDs (12.5 μM). After 30 min stirring, 1 mL of NHS (500 mM) was added into the mixture solution and stirred for 30 min. For Cys-GQDs, 1 mL of D-cysteine (100 mM) was added into the reaction and the mixture was reacted for 16 h. For EDA-GQDs, 0.78 mL of EDA was added into the solution and the mixture was reacted for 48 h. The product was purified by a dialysis bag with 1 K MWCO.TauP301L expression and purification
Human tauP301L (0N4R) with an N-terminal His6 tag was purified following the previous protocol with a slight modification.56 Briefly, transformed BL21 (DE3) cells were grown in LB + Kanamycin media at 37 °C until OD600 reached between 0.6–0.8 and were then induced with 0.5 mM IPTG overnight at 16 °C. Cells were then harvested, resuspended, and lysed by probe sonication in the lysis buffer containing 20 mM Tris, 500 mM NaCl, 10 mM imidazole, and 5 mM serine protease inhibitor PMSF, adjusted to pH 8.0. The lysate was then boiled for 20 minutes in a water bath and the debris was pelleted by centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for about 40 min at 4 °C. The supernatant obtained was then injected into a 5 mL IMAC Ni-Charged affinity column and eluted over a gradient of 10–200 mM imidazole. Eluted tau-containing fractions were further purified using GE HiPrep 16/60 Sephacryl S-200 high-resolution size exclusion chromatography into a storage buffer containing 20 mM Tris, 150 mM NaCl, and 1 mM DTT, adjusted to pH 7.6. The purity of the protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, and the concentration was estimated using bicinchoninic acid (BCA) assay.
000 g for about 40 min at 4 °C. The supernatant obtained was then injected into a 5 mL IMAC Ni-Charged affinity column and eluted over a gradient of 10–200 mM imidazole. Eluted tau-containing fractions were further purified using GE HiPrep 16/60 Sephacryl S-200 high-resolution size exclusion chromatography into a storage buffer containing 20 mM Tris, 150 mM NaCl, and 1 mM DTT, adjusted to pH 7.6. The purity of the protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, and the concentration was estimated using bicinchoninic acid (BCA) assay.
ThT aggregation assay
Recombinant tauP301L (10 μM final concentration) and GQDs, Cys-GQDs, or EDA-GQDs (final concentration: 6.25, 3.125, 1.25, 0.625, and 0.3125 μM) were mixed in an aggregation buffer (100 mM sodium acetate, 10 μM ThT, 10 μM heparin and 2 mM DTT, pH: 7.4). 200 μL of the same solution was added into two wells of a 96-well plate for each sample. The plate was then sealed with a clear sealing film and allowed to incubate at 37 °C in a Tecan infinite 200Pro plate reader. An automated method was used to carry out ThT fluorescence measurements after 30 s shaking at an excitation wavelength of 450 nm and an emission wavelength of 485 nm at an interval of every 5 min for 45 h. Every experiment included control wells that lacked tauP301L, heparin, or GQDs. The aggregation curves were fitted to a Gompertz growth function according towhere y is defined as the normalized ThT fluorescence at time t, ti is the inflection point corresponding to the time of maximum growth rate, A is the maximum normalized ThT fluorescence for a given sample, and k is the apparent elongation rate constant (kapp), in units of h−1. The lag time of the aggregation curve can be calculated by
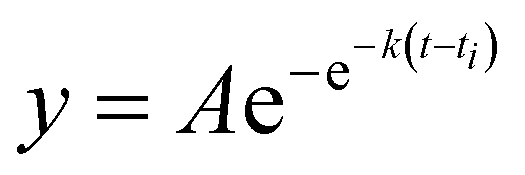
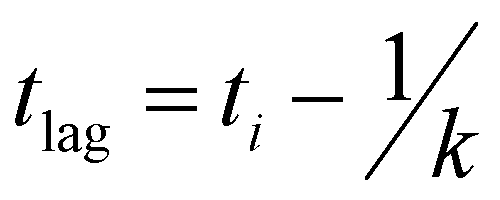 .
.
IC50 was obtained by fitting the dose-response results using a four-parameter logistic regression model, shown in the following equation.
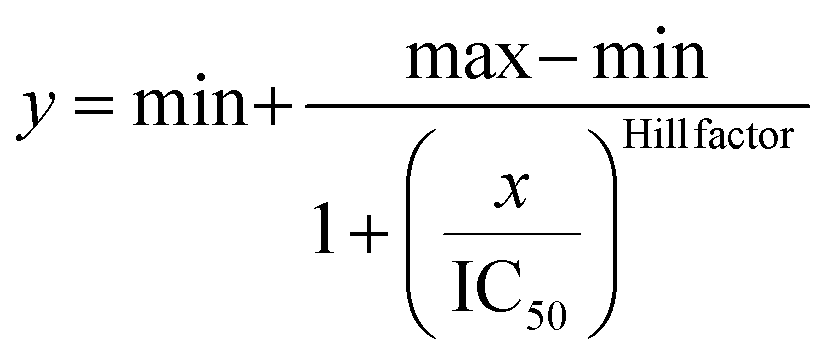
Circular dichroism spectrometry
CD spectra were measured by CD spectroscopy (Jasco J-1700 Spectrometer) at 20 °C. 10 μM of recombinant tauP301L and 6.25 μM of GQDs, Cys-GQDs, or EDA-GQDs were mixed in an aggregation buffer (100 mM sodium acetate, 10 μM ThT, 10 μM heparin and 2 mM DTT, pH: 7.4) and incubated for 4 days at 37 °C. Then, the tau mixture solution was diluted to 0.5 μM for measurement. Spectra were scanned from 190 nm to 260 nm, at 0.1 nm intervals, 5 nm bandwidth, and a scan speed of 50 nm min−1.Transmission electron microscopy
The preparation procedure of tau aggregation for TEM was the same as CD measurement above. A 3 μL droplet of the tau aggregation solution (10 μM) was placed on the carbon-coated copper TEM grid and wicked using filter paper after 45 s. Then, the grid was washed with 5 μL distilled water twice, and stained with 5 μL of 2% (w/v) uranyl acetate for 45 s. Grids were blotted and air-dried before imaging. A JEOL 2011 TEM was used for imaging at an accelerating voltage of 120 kV. Using ImageJ software, the lengths of amyloid fibrils were measured from the average of 16 TEM images per sample.ThT disaggregation assay
The tauP301L was diluted to a final concentration of 10 μM in an aggregation buffer (100 mM sodium acetate, 10 μM heparin, 2 mM DTT, and 10 μM ThT, pH: 7.4). The protein was incubated in a microcentrifuge tube for 4 days at 37 °C. After incubation, 200 μL of the same mixture solution (5 μM tau fibers and 6.25 μM GQDs, Cys-GQDs, or EDA-GQDs) was added into two wells of a 96-well plate for each sample. The plate was then sealed with a clear sealing film and allowed to incubate at 37 °C in a Tecan infinite 200 Pro plate reader. An automated method was used to carry out ThT fluorescence measurements after 30 s shaking at an excitation wavelength of 450 nm and an emission wavelength of 485 nm at an interval of every 5 min for 30 h. Every experiment included control wells that lacked tauP301L fibrils or GQDs.Cellular seeding assay
HEK293 cells stably expressing tau-RD (LM)-YFP were cultured in DMEM complete medium containing 10% FBS, 1% penicillin/streptomycin, and 1% Glutamax under 5% CO2 at 37 °C. 90 μL of cells were plated at a density of 15000 cells per well in a 96-well tissue culture plate.
For seeding by monomeric tau, 10 μM of tauP301L was incubated in an aggregation buffer containing 100 mM sodium acetate, 10 μM heparin, 2 mM DTT, and GQDs or Cys-GQDs (final concentration: 6.25 and 31.25 μM), pH: 7.4 for 4 days at 37 °C. Control vials included those in which buffer was added in place of tauP301L or heparin. Following tau incubation, 8 μL of tau aggregation solution was mixed with 32 μL of low-serum Opti-MEM medium and 2 μL of Lipofectamine 2000, and the mixture solution was incubated for an additional 20 min at room temperature.
For seeding by fibrillar tau, the tauP301L was diluted to a final concentration of 10 μM in an aggregation buffer containing 100 mM sodium acetate, 10 μM heparin, and 2 mM DTT, pH: 7.4. The protein was incubated for 4 days at 37 °C. Control vials included those in which buffer was added in place of tauP301L or heparin. Following incubation, 8 μL of tau fibrils were mixed with 28 μL of low-serum Opti-MEM medium and 4 μL of GQDs or Cys-GQDs with different concentrations. The mixture was incubated at 37 °C for 0 or 36 h, mixed with 2 μL of Lipofectamine 2000, and incubated for 20 min at room temperature.
Then, 10 μL of mixture solution was added to HEK-293 cells and incubated for 48 h at 37 °C. Every experiment included control wells that lacked tauP301L, heparin, or GQDs. After that, cells were detected by a BioTek Cytation 5 cell imager and a microplate reader. 10 × 10 pictures per well were taken at 20× magnification under a fluorescein isothiocyanate (FITC) channel (ex: 469 nm/em: 525 nm), and the punctate counting was carried out using built-in software.
Fluorescence quenching assay
10 μM of tau301L in 100 mM sodium acetate buffer was incubated with different concentrations of GQDs or Cys-GQDs (6.25, 12.5, and 25 μM) at room temperature in a 96-well plate. After 10-min incubation, the fluorescence of the tau mixture solution was detected at an excitation wavelength of 265 nm and an emission wavelength from 295 nm to 380 nm using a Tecan infinite 200 Pro plate reader. All spectra were subtracted with GQD background solutions.CCK-8 assay
SH-SY5Y human neuroblastoma cells were cultured in the DMEM/F12 complete medium containing 10% FBS and 1% penicillin/streptomycin under 5% CO2 at 37 °C. 100 μL of cells were plated at a density of 10000 cells per well in 96-well tissue culture plates and incubated overnight. Control wells contained only medium for background measurement. Then, add 10 μL of GQDs, Cys-GQDs, or EDA-GQDs with a concentration of 15 μM and 50 μM into cells, and add 10 μL of PBS as the negative control. Incubate the 96-well microplates for 24 or 48 h in a CO2 incubator at 37 °C. After that, change the medium, add 10 μL of Cell Counting Kit-8 reagent to cells and incubate the 96-well microplates for 3 h at 37 °C. The absorbance was measured at 450 nm using a Tecan infinite 200 Pro plate reader. The cell viability was calculated using the following equation.where AS and AC are the absorbances measured at 450 nm for cells added GQDs and PBS, respectively, and Ab is the absorbance measured at 450 nm for only medium.
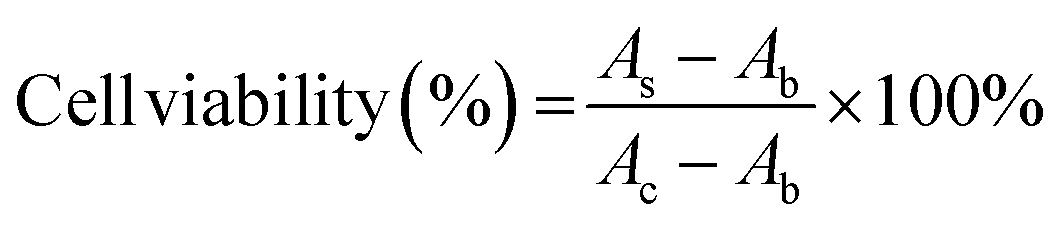
Conclusions
The aggregation of tau proteins plays a key role in the propagation of tau pathology and the observed clinical symptoms of tauopathies. However, current drug candidates for the treatment of tauopathy have shown limited success in clinical trials. Therefore, there is an urgent need for novel modulators of tau fibrillization and propagation. Here, we demonstrate that GQDs are able to prevent the seeding activity of tau fibrils by inhibiting the aggregation of monomeric tau and promoting the disassembly of tau mature fibrils. In addition, we engineered the size and charge of GQDs and investigated their interactions with tau in vitro. We found that larger GQDs with negative charges inhibited the aggregation of tau more efficiently, primarily via electrostatic and π–π stacking interactions. Our findings lay the groundwork for future investigations into the potential use of biomimetic GQDs as therapeutics for tauopathies.Author contributions
Y. W. originated and conceptualized the study. R. Z. designed and carried out the experiments. Y. Z. developed the methodology of synthesis and functionalization of GQDs. K. M. M. and B. H. R. expressed and purified tauP301L. K. M. M. developed the methodology of tau biosensor cellular seeding assay. R. Z. applied statistical and mathematical techniques to analyze data. Y. W. and J. R. D. supervised all the work. R. Z. prepared the manuscript, and all authors provided input on data interpretation, discussions, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from an American Cancer Society Institutional Research Grant (ACS IRG-17-182-04 to Y.W.) and funding to Y. W. from the National Science Foundation Industry-University Cooperative Research Center (The Center for Bioanalytic Metrology). This work was also supported by a grant from the National Institutes of Health (R01AG074570 to J. R. D.) and an Indiana Clinical and Translational Sciences Institute TL1 predoctoral fellowship (to B. R.). TEM images were carried out in part in the Integrated Imaging Facility, University of Notre Dame, using JEOL 2011 TEM. We thank Maksym Zhukovskyi for the knowledge and expertise as well as time towards this research. The authors would like to thank James Johnston, Hyunsu Jeon, and Gaeun Kim for the helpful discussion on the results and experiments. The authors are also grateful to Syrah Starnes and Isaac Angera for their assistance with experiments. Fig. 2b, 4a, 5a, and 8 were created using BioRender.com and used with permission.References
- C. Ballatore, V. M. Y. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2007, 8, 663–672 CrossRef CAS PubMed.
- R. Rademakers, M. Cruts and C. van Broeckhoven, Hum. Mutat., 2004, 24, 277–295 CrossRef CAS PubMed.
- B. Falcon, W. Zhang, A. G. Murzin, G. Murshudov, H. J. Garringer, R. Vidal, R. A. Crowther, B. Ghetti, S. H. W. Scheres and M. Goedert, Nature, 2018, 561, 137–140 CrossRef CAS PubMed.
- D. R. Williams, J. L. Holton, C. Strand, A. Pittman, R. de Silva, A. J. Lees and T. Revesz, Brain, 2007, 130, 1566–1576 CrossRef PubMed.
- P. W. Baas, A. N. Rao, A. J. Matamoros and L. Leo, Cytoskeleton, 2016, 73, 442–460 CrossRef CAS PubMed.
- T. Guo, W. Noble and D. P. Hanger, Acta Neuropathol., 2017, 133, 665–704 CrossRef CAS PubMed.
- B. Nizynski, W. Dzwolak and K. Nieznanski, Protein Sci., 2017, 26, 2126–2150 CrossRef CAS PubMed.
- M. P. Mazanetz and P. M. Fischer, Nat. Rev. Drug Discovery, 2007, 6, 464–479 CrossRef CAS PubMed.
- B. B. Holmes, J. L. Furman, T. E. Mahan, T. R. Yamasaki, H. Mirbaha, W. C. Eades, L. Belaygorod, N. J. Cairns, D. M. Holtzman and M. I. Diamond, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4376–4385 CAS.
- A. Mudher, M. Colin, S. Dujardin, M. Medina, I. Dewachter, S. M. Alavi Naini, E. M. Mandelkow, E. Mandelkow, L. Buee, M. Goedert and J. P. Brion, Acta Neuropathol. Commun., 2017, 5, 99 CrossRef PubMed.
- B. Bulic, M. Pickhardt, E. M. Mandelkow and E. Mandelkow, Neuropharmacology, 2010, 59, 276–289 CrossRef CAS PubMed.
- L. Wang, Bharti, R. Kumar, P. F. Pavlov and B. Winblad, Eur. J. Med. Chem., 2021, 209, 112915 CrossRef CAS PubMed.
- M. Pickhardt, Z. Gazova, M. von Bergen, I. Khlistunova, Y. Wang, A. Hascher, E. M. Mandelkow, J. Biernat and E. Mandelkow, J. Biol. Chem., 2005, 280, 3628–3635 CrossRef CAS PubMed.
- B. Bulic, M. Pickhardt, I. Khlistunova, J. Biernat, E. M. Mandelkow, E. Mandelkow and H. Waldmann, Angew. Chem., Int. Ed., 2007, 46, 9215–9219 CrossRef PubMed.
- K. R. Brunden, J. Q. Trojanowski and V. M. Lee, Nat. Rev. Drug Discovery, 2009, 8, 783–793 CrossRef CAS PubMed.
- K. Tatiparti, S. Sau, M. A. Rauf and A. K. Iyer, Drug Discovery Today, 2020, 25, 2110–2129 CrossRef CAS PubMed.
- I. Aprahamian, F. S. Santos, B. dos Santos, L. Talib, B. S. Diniz, M. Radanovic, W. F. Gattaz and O. V. Forlenza, J. Clin. Psychiatry, 2014, 75, e672–678 CrossRef CAS PubMed.
- D. M. Teleanu, C. Chircov, A. M. Grumezescu, A. Volceanov and R. I. Teleanu, Pharmaceutics, 2018, 10, 269 CrossRef CAS PubMed.
- W. Zhang, N. Kandel, Y. Zhou, N. Smith, B. C. L. B. Ferreira, M. Perez, M. L. Claure, K. J. Mintz, C. Wang and R. M. Leblanc, J. Colloid Interface Sci., 2022, 617, 20–31 CrossRef CAS PubMed.
- M. Bacon, S. J. Bradley and T. Nann, Part. Part. Syst. Charact., 2014, 31, 415–428 CrossRef CAS.
- M. K. Kumawat, M. Thakur, R. B. Gurung and R. Srivastava, Sci. Rep., 2017, 7, 15858 CrossRef PubMed.
- D. Iannazzo, A. Pistone, M. Salamò, S. Galvagno, R. Romeo, S. V. Giofré, C. Branca, G. Visalli and A. Di Pietro, Int. J. Pharm., 2017, 518, 185–192 CrossRef CAS PubMed.
- J. Ruan, Y. Wang, F. Li, R. Jia, G. Zhou, C. Shao, L. Zhu, M. Cui, D.-P. Yang and S. Ge, ACS Appl. Mater. Interfaces, 2018, 10, 14342–14355 CrossRef CAS PubMed.
- M. C. Biswas, M. T. Islam, P. K. Nandy and M. M. Hossain, ACS Mater. Lett., 2021, 3, 889–911 CrossRef CAS.
- S. Xiao, D. Zhou, P. Luan, B. Gu, L. Feng, S. Fan, W. Liao, W. Fang, L. Yang, E. Tao, R. Guo and J. Liu, Biomaterials, 2016, 106, 98–110 CrossRef CAS PubMed.
- G. Perini, V. Palmieri, G. Ciasca, M. De Spirito and M. Papi, Int. J. Mol. Sci., 2020, 21, 3712 CrossRef CAS PubMed.
- M. Suh and D. S. Lee, Nucl. Med. Mol. Imaging, 2018, 52, 407–419 CrossRef PubMed.
- D. Kim, J. M. Yoo, H. Hwang, J. Lee, S. H. Lee, S. P. Yun, M. J. Park, M. Lee, S. Choi, S. H. Kwon, S. Lee, S. H. Kwon, S. Kim, Y. J. Park, M. Kinoshita, Y. H. Lee, S. Shin, S. R. Paik, S. J. Lee, S. Lee, B. H. Hong and H. S. Ko, Nat. Nanotechnol., 2018, 13, 812–818 CrossRef CAS PubMed.
- Y. Yan, J. Gong, J. Chen, Z. Zeng, W. Huang, K. Pu, J. Liu and P. Chen, Adv. Mater., 2019, 31, e1808283 CrossRef PubMed.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Small, 2015, 11, 1620–1636 CrossRef CAS PubMed.
- Z. B. Qu, W. J. Feng, Y. Wang, F. Romanenko and N. A. Kotov, Angew. Chem., Int. Ed., 2020, 59, 8542–8551 CrossRef CAS PubMed.
- Y. Liu, L. P. Xu, W. Dai, H. Dong, Y. Wen and X. Zhang, Nanoscale, 2015, 7, 19060–19065 RSC.
- H. Tang, Y. Li, A. Kakinen, N. Andrikopoulos, Y. Sun, E. Kwak, T. P. Davis, F. Ding and P. C. Ke, Phys. Chem. Chem. Phys., 2021, 24, 86–97 RSC.
- Y. Wang, U. Kadiyala, Z. Qu, P. Elvati, C. Altheim, N. A. Kotov, A. Violi and J. S. VanEpps, ACS Nano, 2019, 13(4), 4278–4289 CrossRef CAS PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J. J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- B. Geng, F. Fang, P. Li, S. Xu, D. Pan, Y. Zhang and L. Shen, Chem. Eng. J., 2021, 417, 128125 CrossRef CAS.
- L. Li, G. Wu, G. Yang, J. Peng, J. Zhao and J. J. Zhu, Nanoscale, 2013, 5, 4015–4039 RSC.
- N. Suzuki, Y. Wang, P. Elvati, Z. B. Qu, K. Kim, S. Jiang, E. Baumeister, J. Lee, B. Yeom, J. H. Bahng, J. Lee, A. Violi and N. A. Kotov, ACS Nano, 2016, 10, 1744–1755 CrossRef CAS PubMed.
- J. Wang, Y. Cao, Q. Li, L. Liu and M. Dong, Chemistry, 2015, 21, 9632–9637 CrossRef CAS PubMed.
- M. Zhou, Q. Shen, J. W. Shen, L. Jin, L. Zhang, Q. Sun, Q. Hu and L. Liang, Colloids Surf., B, 2019, 174, 575–581 CrossRef CAS PubMed.
- M. Hutton, C. L. Lendon, P. Rizzu, M. Baker, S. Froelich, H. Houlden, S. Pickering-Brown, S. Chakraverty, A. Isaacs, A. Grover, J. Hackett, J. Adamson, S. Lincoln, D. Dickson, P. Davies, R. C. Petersen, M. Stevens, E. de Graaff, E. Wauters, J. van Baren, M. Hillebrand, M. Joosse, J. M. Kwon, P. Nowotny, L. K. Che, J. Norton, J. C. Morris, L. A. Reed, J. Trojanowski, H. Basun, L. Lannfelt, M. Neystat, S. Fahn, F. Dark, T. Tannenberg, P. R. Dodd, N. Hayward, J. B. J. Kwok, P. R. Schofield, A. Andreadis, J. Snowden, D. Craufurd, D. Neary, F. Owen, B. A. Oostra, J. Hardy, A. Goate, J. van Swieten, D. Mann, T. Lynch and P. Heutink, Nature, 1998, 393, 702–705 CrossRef CAS PubMed.
- D. Chen, K. W. Drombosky, Z. Hou, L. Sari, O. M. Kashmer, B. D. Ryder, V. A. Perez, D. R. Woodard, M. M. Lin, M. I. Diamond and L. A. Joachimiak, Nat. Commun., 2019, 10, 2493 CrossRef PubMed.
- C. Xue, T. Y. Lin, D. Chang and Z. Guo, R. Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed.
- G. Ramachandran and J. B. Udgaonkar, J. Biol. Chem., 2011, 286, 38948–38959 CrossRef CAS PubMed.
- C. L. Moore, M. H. Huang, S. A. Robbennolt, K. R. Voss, B. Combs, T. C. Gamblin and W. J. Goux, Biochemistry, 2011, 50, 10876–10886 CrossRef CAS PubMed.
- M. Necula and J. Kuret, Anal. Biochem., 2004, 333, 205–215 CrossRef CAS PubMed.
- M. von Bergen, S. Barghorn, J. Biernat, E.-M. Mandelkow and E. Mandelkow, Biochim. Biophys. Acta, Mol. Basis Dis., 2005, 1739, 158–166 CrossRef CAS PubMed.
- M. Weiss, J. Fan, M. Claudel, T. Sonntag, P. Didier, C. Ronzani, L. Lebeau and F. Pons, J. Nanobiotechnol., 2021, 19, 5 CrossRef CAS PubMed.
- K. M. Makwana, M. P. Sarnowski, J. Miao, Y. S. Lin and J. R. Del Valle, ACS Chem. Neurosci., 2021, 12, 3928–3938 CrossRef CAS PubMed.
- S. Wegmann, I. D. Medalsy, E. Mandelkow and D. J. Muller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E313–321 CrossRef CAS PubMed.
- C. Cowan and A. Mudher, Front. Neurol., 2013, 4, 114 Search PubMed.
- H. A. Zeinabad, A. Zarrabian, A. A. Saboury, A. M. Alizadeh and M. Falahati, Sci. Rep., 2016, 6, 26508 CrossRef CAS PubMed.
- S. Alali, G. Riazi, M. R. Ashrafi-Kooshk, S. Meknatkhah, S. Ahmadian, M. Hooshyari Ardakani and B. Hosseinkhani, Cells, 2021, 10, 3521 CrossRef CAS PubMed.
- E. Mylonas, A. Hascher, P. Bernadó, M. Blackledge, E. Mandelkow and D. I. Svergun, Biochemistry, 2008, 47, 10345–10353 CrossRef CAS PubMed.
- H. Park and S. Y. Park, ACS Appl. Mater. Interfaces, 2022, 14, 26733–26741 CrossRef CAS PubMed.
- C. Barbereau, A. Yehya, M. Silhol, N. Cubedo, J.-M. Verdier, T. Maurice and M. Rossel, Pharmacol. Res., 2020, 158, 104865 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00850a |
| This journal is © The Royal Society of Chemistry 2023 |