
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed enantioselective C–H/N–H annulation of aryl sulfonamides with allenes or alkynes: facile access to C–N axially chiral sultams†
Xiao-Ju
Si‡
,
Xiaofang
Zhao‡
,
Jianli
Wang‡
,
Xinhai
Wang
,
Yuanshuo
Zhang
,
Dandan
Yang
 *,
Mao-Ping
Song
and
Jun-Long
Niu
*
*,
Mao-Ping
Song
and
Jun-Long
Niu
*
College of Chemistry, Zhengzhou University, Zhengzhou, Henan 450001, P. R. China. E-mail: yangdandan@zzu.edu.cn; niujunlong@zzu.edu.cn
First published on 8th June 2023
Abstract
Herein we report a cobalt-catalyzed enantioselective C–H/N–H annulation of aryl sulfonamides with allenes and alkynes, using either chemical or electrochemical oxidation. By using O2 as the oxidant, the annulation with allenes proceeds efficiently with a low catalyst/ligand loading of 5 mol% and tolerates a wide range of allenes, including 2,3-butadienoate, allenylphosphonate, and phenylallene, resulting in C–N axially chiral sultams with high enantio-, regio-, and position selectivities. The annulation with alkynes also exhibits excellent enantiocontrol (up to >99% ee) with a variety of functional aryl sulfonamides, and internal and terminal alkynes. Furthermore, electrochemical oxidative C–H/N–H annulation with alkynes is achieved in a simple undivided cell, demonstrating the versatility and robustness of the cobalt/Salox system. The gram-scale synthesis and asymmetric catalysis further highlight the practical utility of this method.
Introduction
Atropisomerism, a type of chirality arising from sterically hindered rotation around a single sigma bond, has become an interesting and cutting-edge field due to its intriguing characteristics.1 In this context, the enantioselective assembly of C–N axially chiral skeletons, which possess appealing agricultural and medicinal activities2,3 (Fig. 1a), has received considerable attention in recent years.4–9 While numerous methods have been developed for the synthesis of C–N axially chiral anilides,10–13 the straightforward construction of C–N axially chiral sulfonamide continues to be underdeveloped despite its promising applications in the treatment of pain.14 Currently, the known method remains predominantly confined to the atroposelective N–H functionalization of sulfonamides by employing organic or transition-metal catalysts,15–20 pioneered by Taguchi21 and Curran,22 for Pd-catalyzed enantioselective N-allylation. Therefore, given the importance of chiral sulfonamides and their limited synthetic methods, the development of efficient and practical strategies to access enantioenriched C–N axially chiral sulfonamides is highly desirable.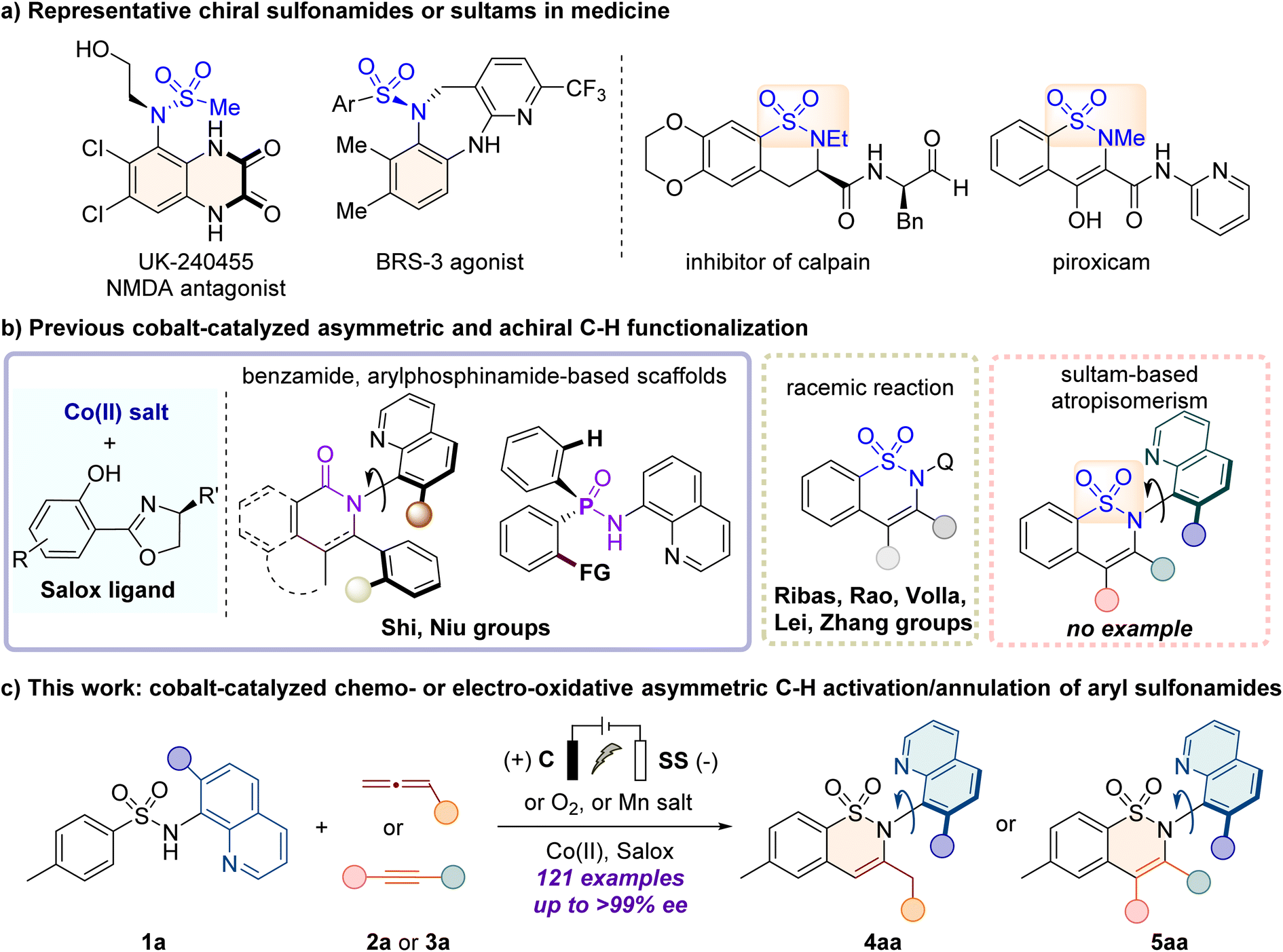 | ||
| Fig. 1 Background and project synopsis. | ||
Transition metal-catalyzed enantioselective C–H functionalization is a powerful tool for accessing valuable chiral molecules in a straightforward and atom-economic manner.23–29 Among the first-row 3d transition metals, cobalt-catalyzed enantioselective C–H activation has gained increasing attention due to its abundant reserves, low toxicity, and unique reactivity.30–36 The Ackermann,37 Shi,38–40 Cramer,41–43 Yoshino and Matsunaga44–47 groups have made significant advances by using a high-valent CpXCoIII catalyst with the carboxylic acid (CCA) ligand. The Lautens,48 Yoshikai,49,50 and Wencel-Delord51 groups developed asymmetric low-valent cobalt-catalyzed systems. Recently, growing attention has been turned to the use of readily available cobalt salt catalysts and flexibly tunable chiral ligands (such as, salicyl-oxazoline, Salox). In 2022, the Shi group52–56 and our group57–59 have independently developed cobalt/Salox-catalyzed asymmetric C–H functionalization to assemble axially chiral, C- and P-stereogenic frameworks, using carboxylic amides or arylphosphinamides as substrates (Fig. 1b). Recently, the Shi,55,56 Ackermann,60–62 and Ling63 groups have accomplished these transformations under electrochemical conditions. However, the direct activation of sulfonamide derivatives to construct enantioenriched sultam derivatives has not been reported yet.
Inspired by the pioneering achiral cobalt-catalyzed C–H activation/annulation reported by Ribas,64 Rao,65 Volla,66 Lei67 and Zhang,68 herein we aimed to develop a CoII/Salox catalyzed enantioselective C–H annulation of 8-aminoquinoline-directed69,70 aryl sulfonamides with allenes or alkynes, for the construction of C–N axially chiral sultam-based scaffolds,71,72 which are promising motifs in agrochemicals, pharmaceuticals (e.g. piroxicam) and biologically active molecules.73–75 Nevertheless, this asymmetric system faces several challenges: (1) the non-aromatic nature and the flexibility of sultam frameworks make the construction of axial chirality more challenging; (2) the regioselective annulation across two different C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds of allenes76 can lead to the feasible synthesis of either C-stereogenic or C–N axially chiral scaffolds. Controlling both the regio- and enantio-selectivity is therefore crucial. For example, the Volla and Maiti group reported that the variation of the electronic nature of allenes resulted in the change of the annulation pattern for cobalt catalysis.77 (3) Simultaneous compatibility of both allene and alkyne coupling partners in one protocol is appealing yet challenging. Herein, we report an efficient cobalt catalyzed enantioselective C–H/N–H activation/annulation of aryl sulfonamides with allenes and alkynes to access C–N axially chiral sultams (Fig. 1c). The reaction proceeded smoothly with cheap cobalt(II) salt as the catalyst, and environmental green O2 or commercial Mn salts as the oxidant. The annulation with alkynes was also achieved under constant-current electrolysis (CCE) conditions with high efficiency and enantiocontrol. Typically, a broad class of electron-poor and electron-rich allenes (e.g. 2,3-butadienoate, allenylphosphonate, and phenylallene), and internal or terminal alkynes were well tolerated to deliver C–N axially chiral sultams with high enantio-, regio- and position selectivities (121 examples, up to >99% ee).
C double bonds of allenes76 can lead to the feasible synthesis of either C-stereogenic or C–N axially chiral scaffolds. Controlling both the regio- and enantio-selectivity is therefore crucial. For example, the Volla and Maiti group reported that the variation of the electronic nature of allenes resulted in the change of the annulation pattern for cobalt catalysis.77 (3) Simultaneous compatibility of both allene and alkyne coupling partners in one protocol is appealing yet challenging. Herein, we report an efficient cobalt catalyzed enantioselective C–H/N–H activation/annulation of aryl sulfonamides with allenes and alkynes to access C–N axially chiral sultams (Fig. 1c). The reaction proceeded smoothly with cheap cobalt(II) salt as the catalyst, and environmental green O2 or commercial Mn salts as the oxidant. The annulation with alkynes was also achieved under constant-current electrolysis (CCE) conditions with high efficiency and enantiocontrol. Typically, a broad class of electron-poor and electron-rich allenes (e.g. 2,3-butadienoate, allenylphosphonate, and phenylallene), and internal or terminal alkynes were well tolerated to deliver C–N axially chiral sultams with high enantio-, regio- and position selectivities (121 examples, up to >99% ee).
Results and discussion
Optimization of the reaction conditions
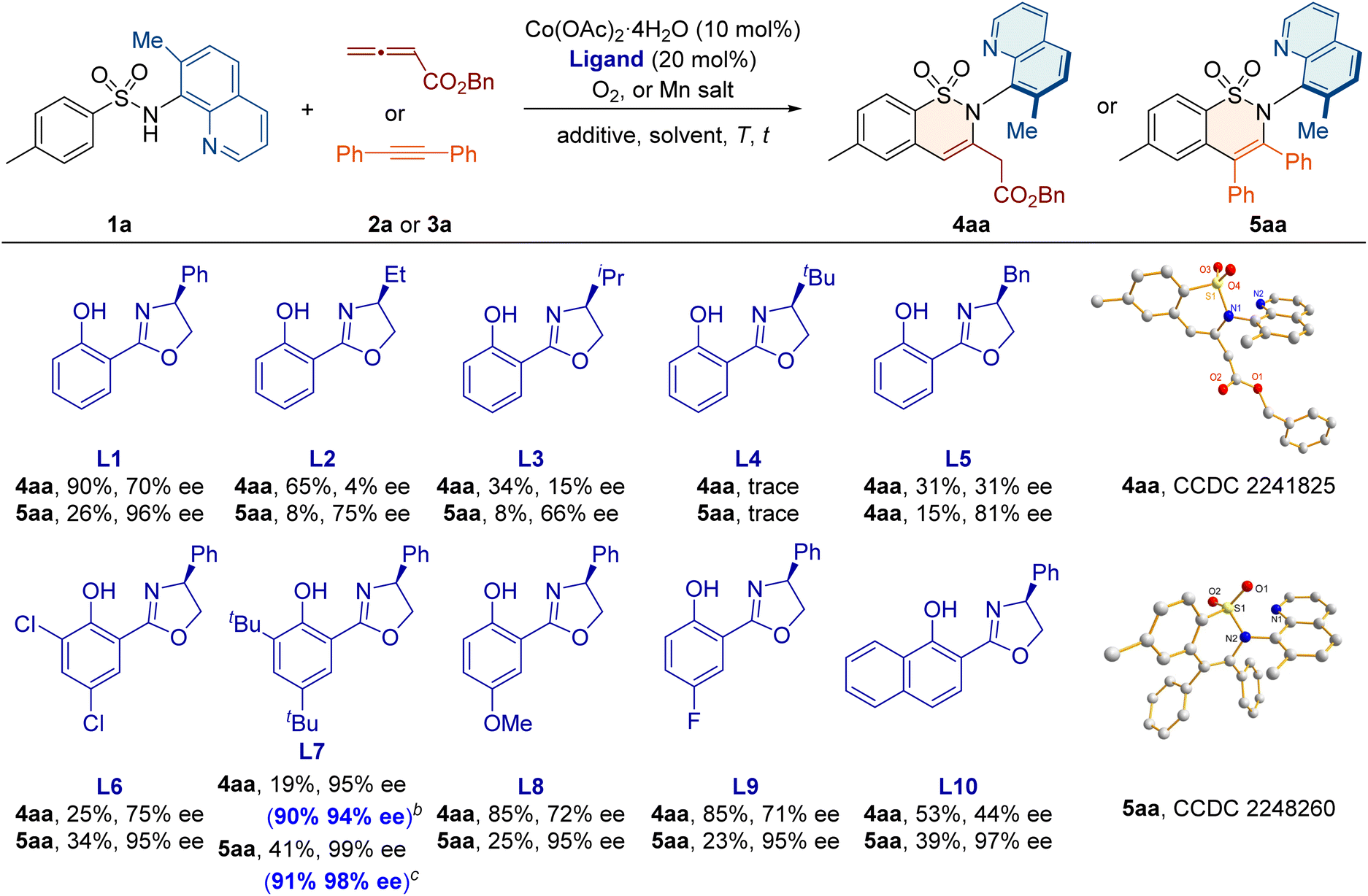
The reaction was initiated using sulfonamide 1a and ester-substituted allene 2a, with Co(OAc)2·4H2O as the catalyst and O2 as the oxidant (Table 1). When the chiral ligand salicyl-oxazoline L1 was used, the desired C–N axially chiral sultam 4aa was obtained as a single regioisomeric product with 90% yield and 70% ee. Further evaluation of the Salox ligand showed that L7, bearing bulky tert-butyl substituents on the phenol-ring, gave a promising enantioselectivity of 95% ee. Moreover, after screening solvents, additives, and catalyst/ligand loadings, the desired 4aa was obtained in 90% yield and 94% ee with a low catalyst loading of 5 mol% and O2 as the oxidant. When alkyne 3a was employed as the coupling partner, the C–N axially chiral product 5aa was formed with 91% yield and 98% ee under optimized conditions, with L7 as the chiral ligand and Mn(OAc)3·2H2O as the oxidant. The absolute configurations of 4aa and 5aa were confirmed as Ra by X-ray crystallography. Control experiments suggest that the methyl group at the C7 position of the quinoline ring is crucial for improving efficiency and meanwhile generating the axial chirality in the transformation (Scheme S1 in the ESI†).| a Reaction conditions for 4aa: 1a (0.1 mmol), 2a (0.3 mmol), Co(OAc)2·4H2O (10 mol%), L (20 mol%), dioxane (1.0 mL), O2, 100 °C, and 5 h; reaction conditions for 5aa: 1a (0.1 mmol), 3a (0.2 mmol), Co(OAc)2·4H2O (10 mol%), L (20 mol%), Mn(OAc)3·2H2O (0.1 mmol), MeOH (1 mL), air, 100 °C, and 6 h. Isolated yields. b 1a (0.2 mmol), 2a (0.3 mmol), Co(OAc)2·4H2O (5 mol%), L (5 mol%), PivOH (1.0 equiv.), (CHCl2)2 (2.0 mL), O2, 100 °C, and 5 h. c 1a (0.2 mmol), 3a (0.4 mmol), Co(OAc)2·4H2O (10 mol%), L (20 mol%), Mn(OAc)3·2H2O (1.0 equiv.), NaOPiv (1.0 equiv.), MeOH (2 mL), air, 100 °C, and 36 h. The ee values were determined by chiral HPLC analysis. |
|---|
|
Scope of the substrates
With the optimized conditions in hand, we evaluated the potential of sulfonamide substrates for annulation with allenes (Table 2). A variety of functional groups, including electron-donating (–alkyl, –Ph, –OMe, and –NHAc) or electron-withdrawing (–F, –Cl, –Br, and –CF3) groups at the para-position of the aryl ring, were well tolerated, and the resulting products 4aa–4ja were obtained with good yields and enantioselectivities. Ortho-substituted sulfonamides 1k–1m also reacted smoothly, delivering 4ka–4ma with excellent enantiopurities of 96–98% ee. Substitution on the meta-position allowed the reaction to occur exclusively on the less hindered position, giving products 4na and 4oa with 71% yields and 92% ee values. Additionally, disubstituted sulfonamides were applicable to deliver the products with good efficiency. Generally, the protocol was also compatible with naphthyl, heterocyclic, and coumarin sulfonamides, producing the desired 4ra–4ta with good yields and enantioselectivities. To further investigate the method's applicability, we evaluated sulfonamide substrates with diverse groups at the C5 or C7 position of the quinoline ring. We were pleased to find that substrates bearing alkyl (–Et and –nBu) or halogen groups (–Cl and –Br) at the C7 position of the quinoline ring reacted smoothly, giving the C–N axially chiral products 4ua–4xa with good yields (67–91%) and excellent enantioselectivities (93–97% ee). The introduction of bulky substituents, such as phenyl, 4-methoxyphenyl, styryl, and thienyl groups, also gave the desired products 4ya–4zba with comparable efficiency. Furthermore, substrates with functional groups (–Cl and –Br) at the C5 position or at both C5 and C7 positions of the quinoline ring were also applicable in this protocol, with a high level of enantioselectivities (4zca–4zfa, 93–94% ee).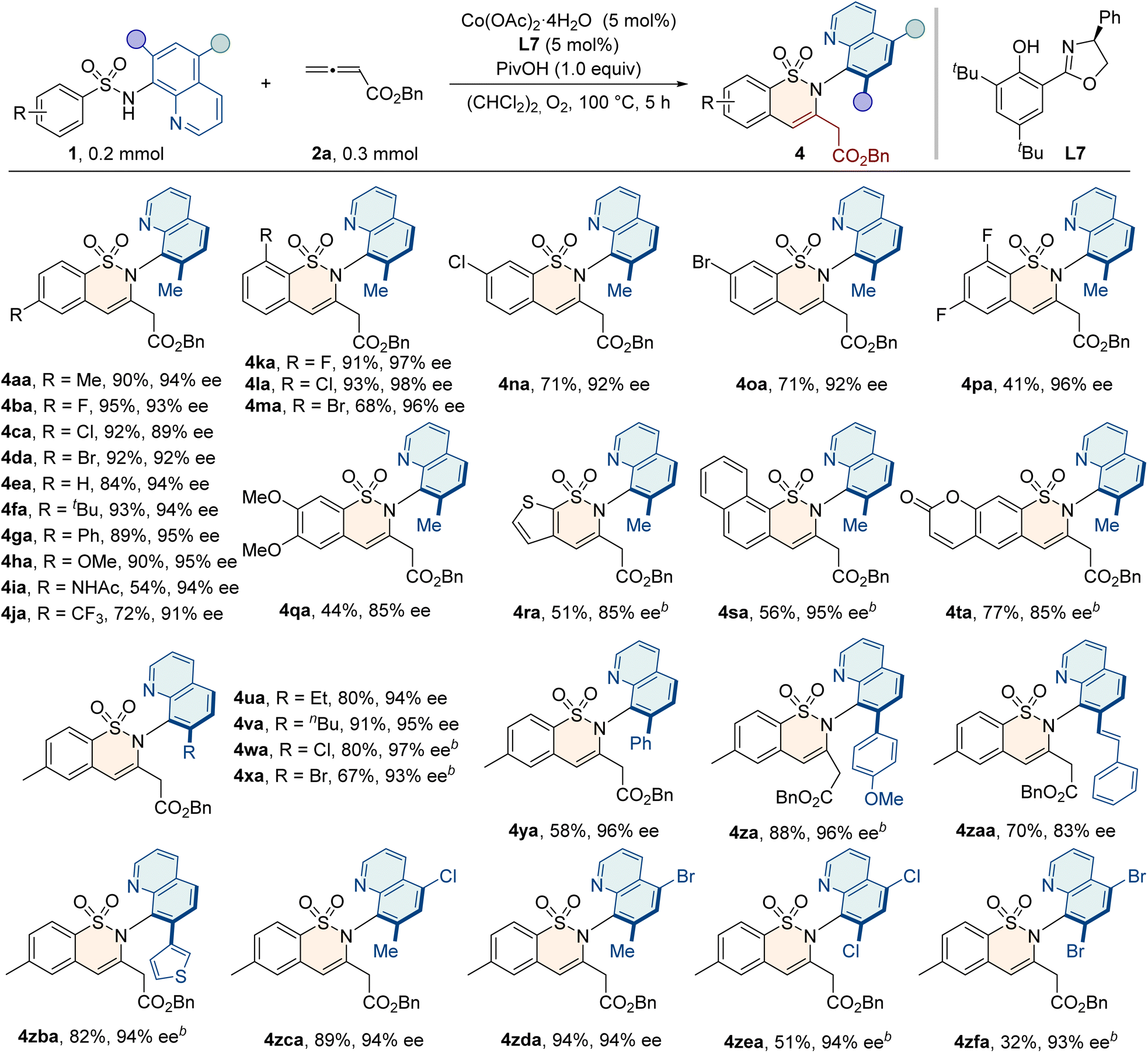
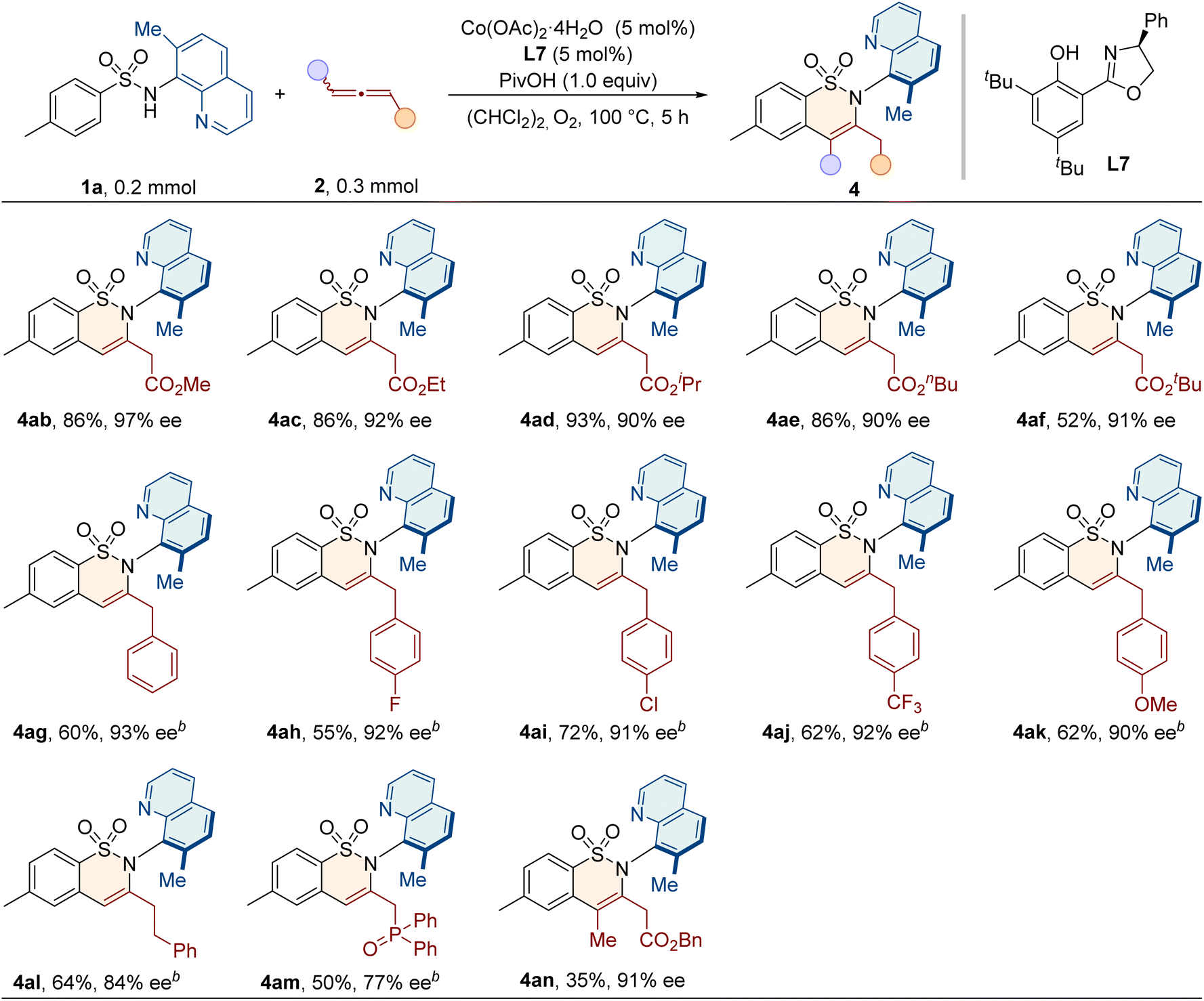
We proceeded to evaluate the scope of allenes, as shown in Table 3. When the benzyl group was replaced with other alkyl groups, such as methyl, ethyl, isopropyl, n-butyl and t-butyl, the products were obtained in good yields (52–93%) and high enantioselectivities (90–97% ee). Notably, in addition to ester-substituted allenes, electron-rich allenes were also examined in this protocol. Phenylallenes 2g and 2h–2k bearing electron-withdrawing or donating groups all reacted smoothly under slightly modified conditions to give products 4ag–4ak with high enantiopurities of 90–93% ee. Benzyl-substituted allene 2l was also compatible in this reaction. Additionally, other electron deficient allene partners, such as allenyl phosphonate 2m, underwent annulation to give 4am as the single regioisomeric product, albeit with a slightly lower ee value. For the internal disubstituted allene 2n, the methyl group and the ester group were accurately recognized, and the C–N axially chiral product 4an was formed with a high enantioselectivity of 91% ee.
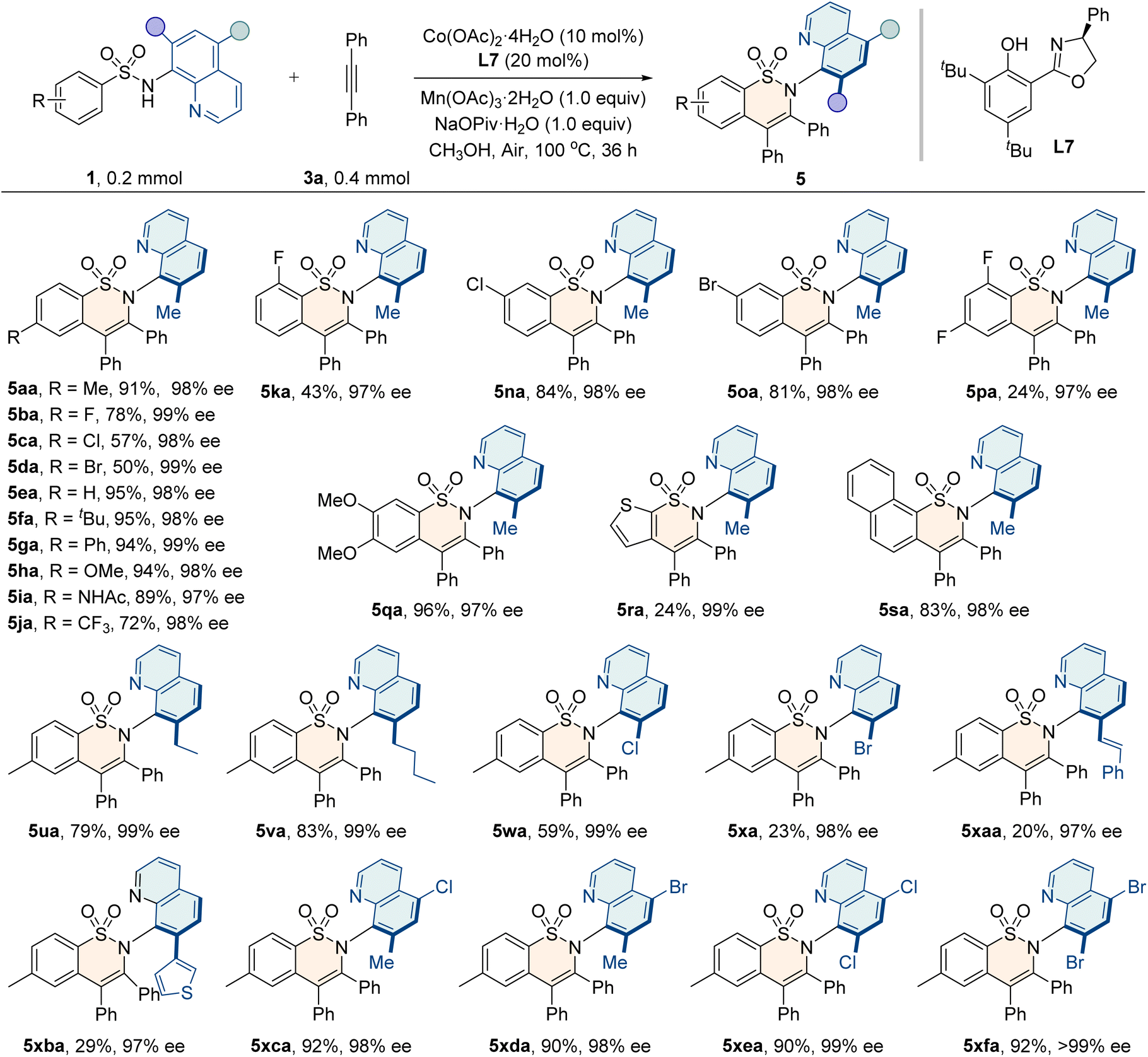
Furthermore, the scope for the annulation of sulfonamides with alkynes was examined, as shown in Table 4. Generally, sulfonamide substrates bearing electron-donating (–alkyl, –Ph, –OMe, and –NHAc) or electron-withdrawing groups (–X and –CF3) at the para-, ortho-, and meta-positions of the aryl ring reacted smoothly to give the corresponding products 5aa–5oa in moderate to good yields (43–95%) and excellent enantioselectivities (97–99% ee). The protocol also tolerated disubstituted sulfonamides, furnishing the desired products with high enantiocontrol (97% ee). Additionally, naphthyl and heterocyclic thienyl sulfonamides were compatible with this method. Moreover, sulfonamides bearing substituents at the C5 or both the C5 and C7 positions of the quinoline ring could also furnish the products 5ua–5xfa with a high level of enantioselectivities (97–>99% ee).
| a Reaction conditions: 1 (0.2 mmol), 3a (0.4 mmol), Co(OAc)2·4H2O (10 mol%), L7 (20 mol%), Mn(OAc)3·2H2O (0.2 mmol), and NaOPiv·H2O (0.2 mmol) in MeOH (2 mL) at 100 °C for 36 h. |
|---|
|
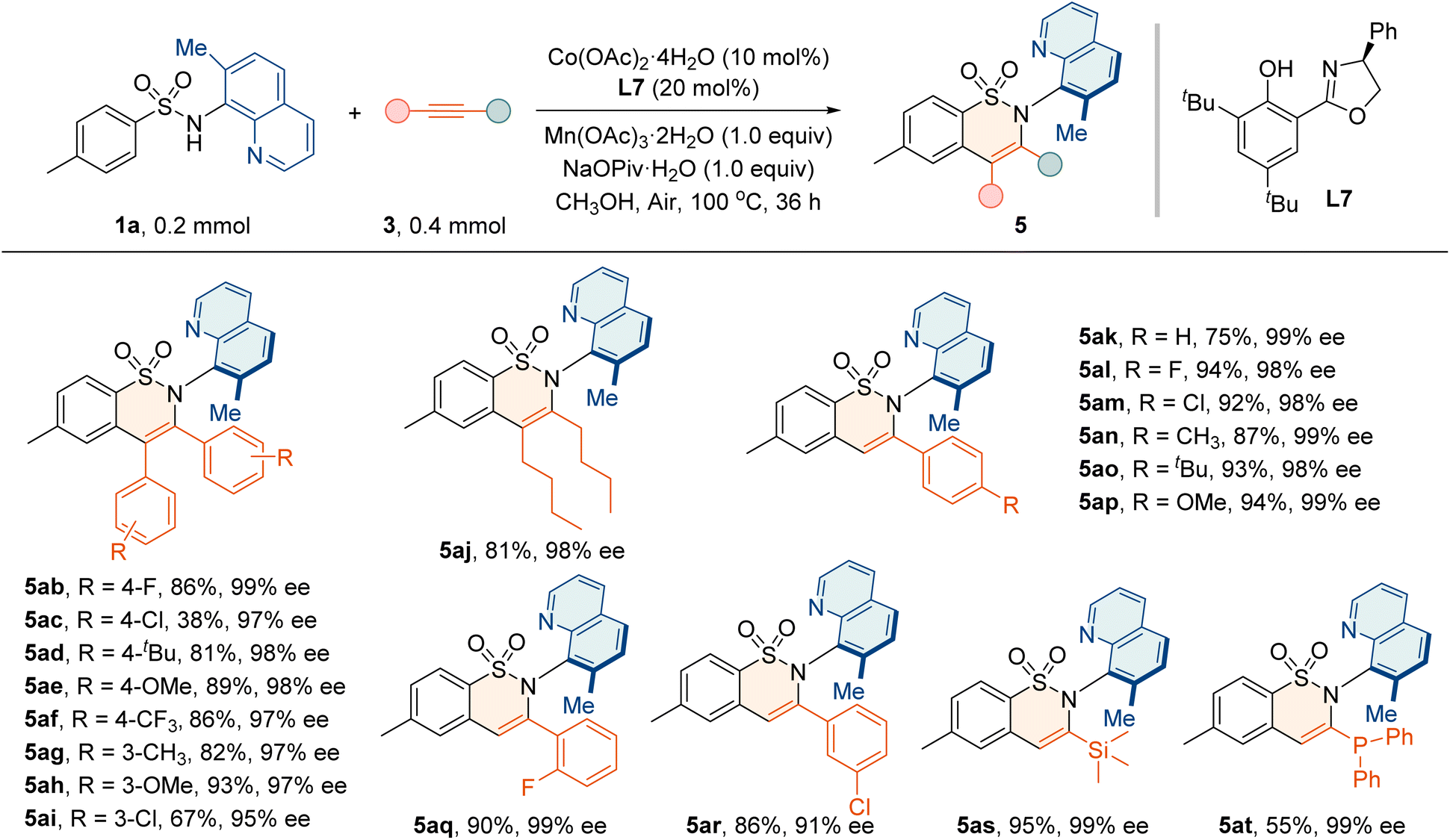
Next, the generality of alkynes 3a with various substitutions was investigated (Table 5). A wide range of diarylacetylenes bearing electron-rich or -deficient groups underwent the reaction with high enantioselectivities (5ab–5ai, 95–99% ee). The aliphatic internal alkynes also reacted smoothly to deliver 5aj in 81% yield and 98% ee. Moreover, the annulation proceeded effectively with terminal alkynes 3k–3r containing electron-withdrawing and electron-donating substituents, affording 5ak–5ar in good yields (75–94%) and high enantiopurities (91–99% ee). The trimethylsilylacetylene 3s and diphenylphosphinoacetylene 3t were also well tolerated, giving annulation products 5as and 5at with 99% ee values, which provides an opportunity for further asymmetric transformation and application.
| a Reaction conditions: 1a (0.2 mmol), 3 (0.4 mmol), Co(OAc)2·4H2O (10 mol%), L7 (20 mol%), Mn(OAc)3·2H2O (0.2 mmol), and NaOPiv·H2O (0.2 mmol) in MeOH (2 mL) at 100 °C for 36 h. |
|---|
|
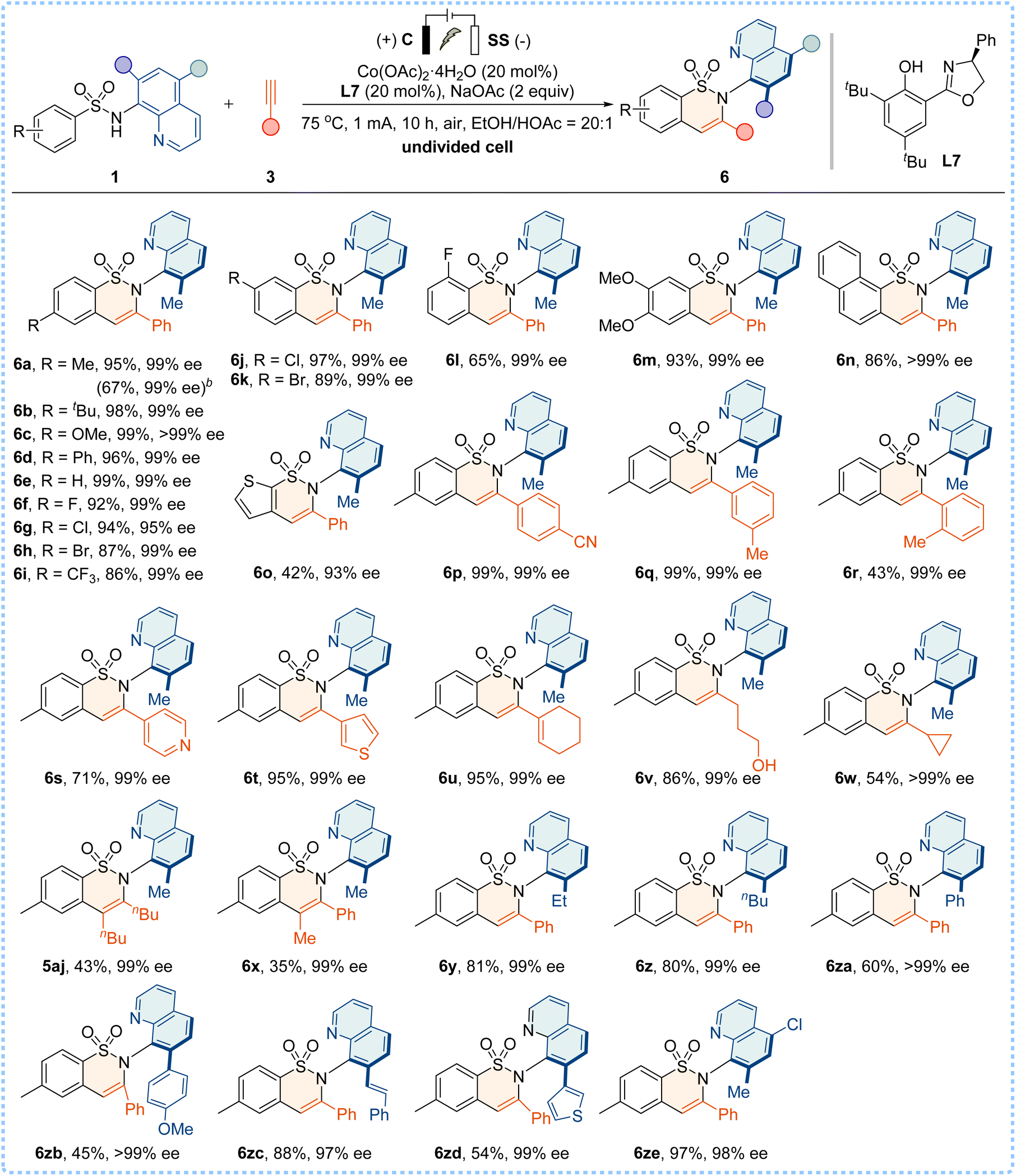
We were pleased to discover that the electrooxidative Co/Salox catalyzed enantioselective C–H annulation of 1a with alkyne 3k yielded the desired C–N axially chiral product 5ak with 95% yield and an excellent enantioselectivity of 99% ee, when conducted in an undivided cell setup equipped with a carbon cloth (C) anode and a stainless steel (SS) plate cathode under optimized conditions (for details, see the ESI†). Control experiments confirmed that the omission of electricity or the cobalt catalyst resulted in complete inactivity of the asymmetric annulation. Subsequently, the robustness of the electrochemical C–H annulation was demonstrated by using a broad range of sulfonamides and terminal alkynes (Table 6). Both electron-donating and electron-withdrawing substituents were compatible, delivering products 6a–6l with 65–99% yields and 95–>99% ee values. Disubstituted, naphthyl and thienyl sulfonamides were also suitable partners to give 6m–6o with high enantioselectivities. In addition, phenylacetylene alkynes bearing functional groups, heterocycle-substituted alkynes (such as 4-ethynylpyridine and 3-ethynylthiophene), and conjugated enyne and aliphatic alkynes all reacted smoothly to afford the desired products 6p–6w with good yields and excellent enantiocontrol. Moreover, the annulation proceeded effectively with internal alkynes (5-decyne or phenylpropyne), affording 5aj and 6x in high enantiopurities (99% ee). However, the use of diphenylacetylene is ineffective in this protocol, probably due to the steric hindrance. Furthermore, sulfonamides bearing substituents at the C7 position (such as alkyl, aryl, styryl, and heteroaromatic thienyl groups) or at the C5 position were well tolerated under this electrochemical system, and the corresponding products 6y–6ze were obtained with good efficiency. The reaction was conducted on a gram scale using 3 mmol of 1a and the product 6a was obtained with a 67% yield and 99% ee (see the ESI†).
| a Reaction conditions: carbon cloth (15 mm × 20 mm × 0.33 mm) anode, stainless steel plate (15 mm × 20 mm × 1.0 mm) cathode, constant current = 1 mA, sulfonamides 1 (0.25 mmol) and 3 (0.5 mmol), Co(OAc)2·4H2O (20 mol%), L7 (20 mol%), EtOH (10.0 mL), HOAc (0.5 mL), NaOAc (2.0 equiv.), 75 °C, 10 h, air, and isolated yields. b Gram scale reaction using 3 mmol of 1a. |
|---|
|
Study on the product stability
To investigate the stereochemical stability of the C–N axially chiral sulfonamides, we conducted racemization experiments (Fig. 2). First, we heated the compound 4aa in DMSO at 100 °C, 120 °C and 130 °C and recorded the ee values at different times ranging from 1–12 h. The ee value of 4aa decreased by 11% after 12 hours at 130 °C (Fig. 2a). The rotational energy barrier of 4aa was calculated to be 34.7 kcal mol−1, and its half-life (t1/2) was up to 4.7 × 104 years at 25 °C. The effect of the substituted groups on stereochemical stability was also investigated. Accordingly, we calculated the rotational energy barrier and the t1/2 of 4ag, 4ya, 5aa, 5aq, and 5xca in DMSO at 130 °C (Fig. 2b). The results showed that the C–N axially chiral sulfonamides feature high atropostabilities.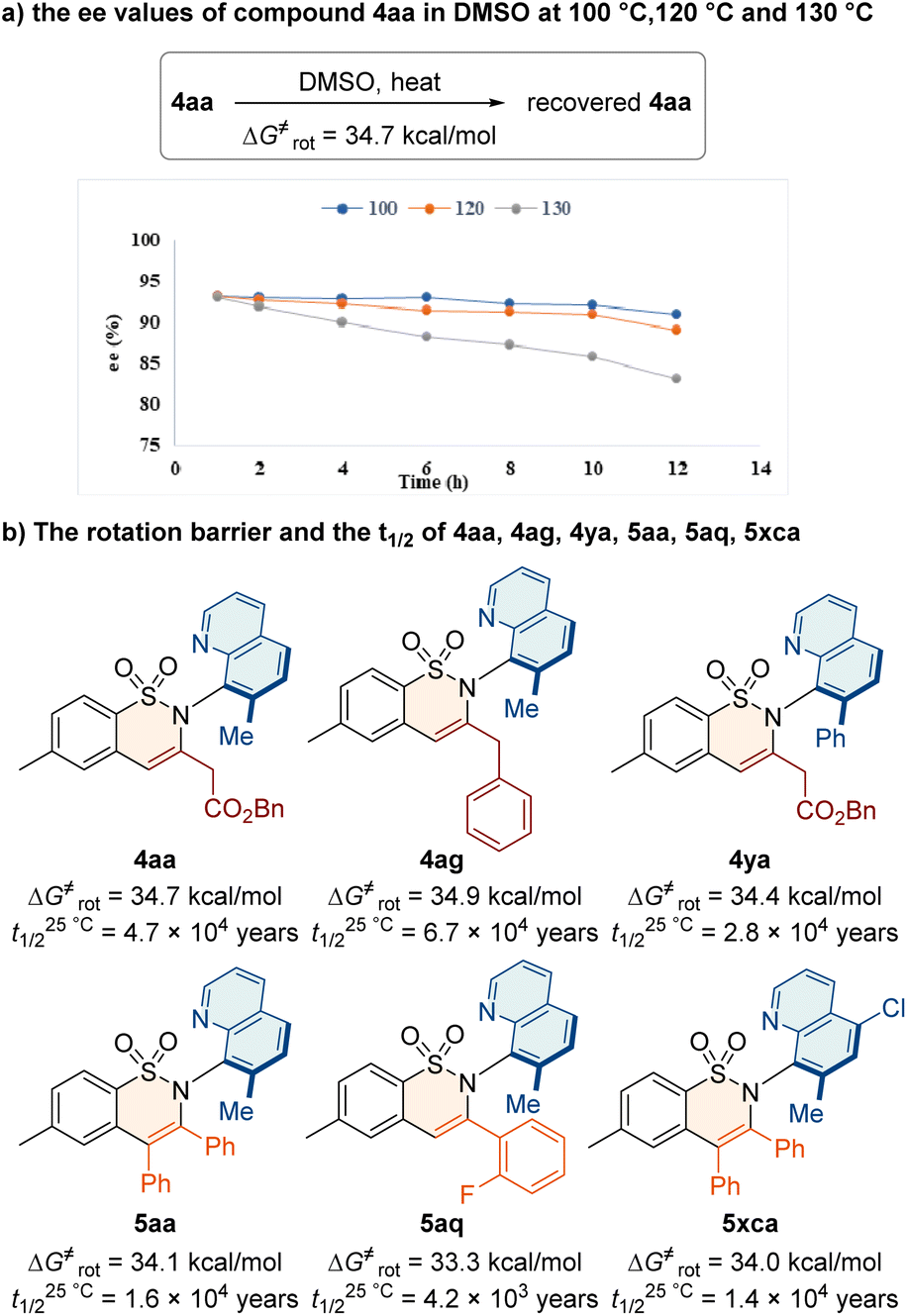 | ||
| Fig. 2 Study on stereochemical stability. | ||
Mechanistic investigation and synthetic application
To elucidate the mechanism, we conducted a series of control experiments (Fig. 3). The atroposelective synthesis of 4aa and 5aa was successfully carried out on gram scales without any loss of enantioselectivities (Fig. 3a). H/D exchange experiments under standard conditions for the synthesis of 4aa indicate that the C–H bond cleavage was irreversible (Fig. 3b, left). The competitive kinetic isotope effect (KIE) value of 1.6 and the parallel KIE value of 1.8 suggested that the C–H bond cleavage might be the rate-determining step (Fig. 3c, left).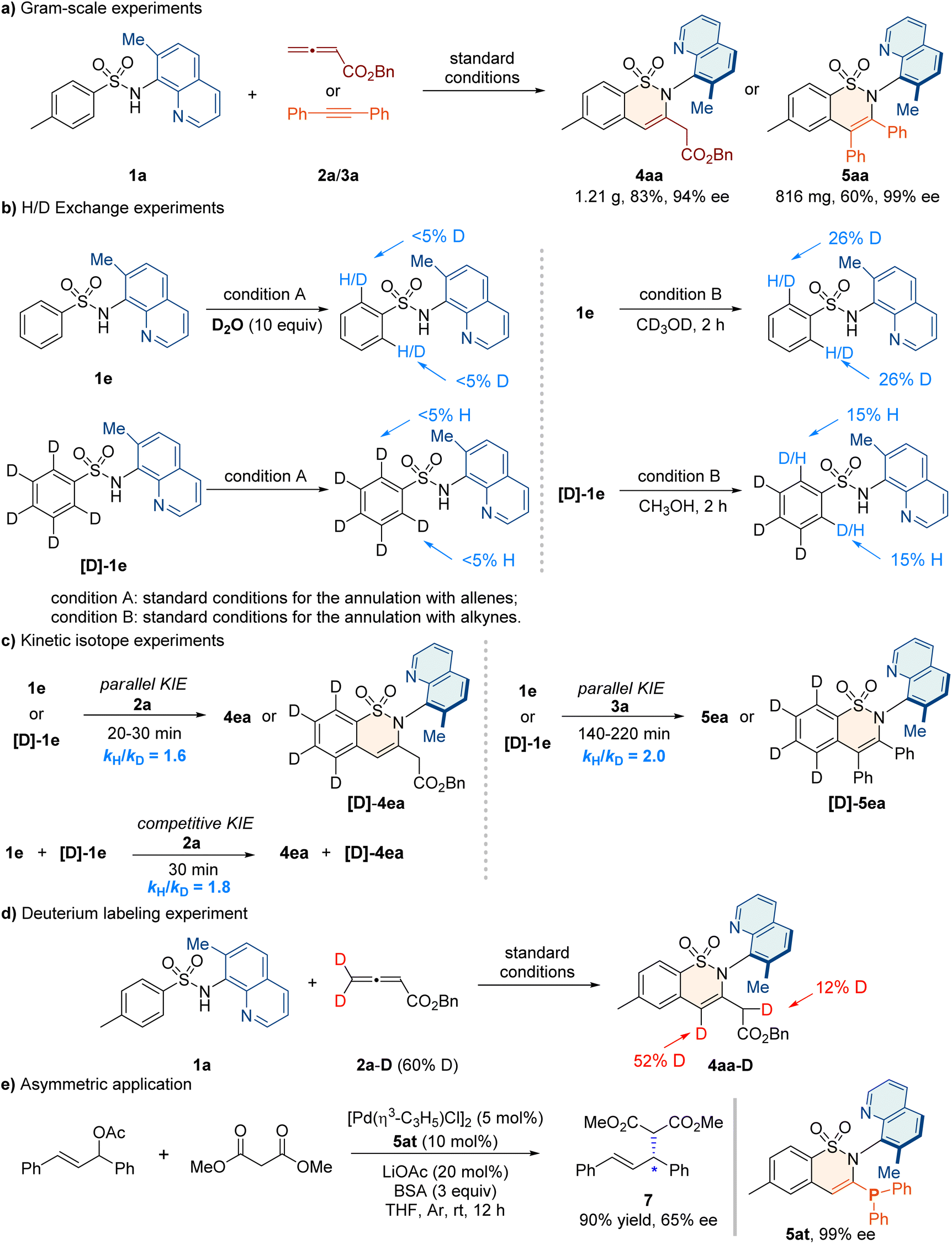 | ||
| Fig. 3 Mechanistic studies and synthetic application. | ||
Similarly, H/D exchange experiments under conditions for the synthesis of 5aa were performed and a competitive KIE value of 2.0 was observed, indicating that the C–H bond cleavage might also be involved in the rate-determining step (Fig. 3b and c, right). Furthermore, a control experiment by using deuterated allenes 2a–D was carried out, and the deuterium labeling of product 4aa was found at the α-position of the ester group (Fig. 3d). This illustrated that the formation of 4aa might involve a 1,3-hydrogen migration step. Additionally, we demonstrated the asymmetric application of the C–N axially chiral product 5at containing a monophosphine skeleton as a suitable chiral ligand for the Pd-catalyzed asymmetric Tsuji–Trost reaction, and the product 7 was obtained with a 90% yield and 65% ee (Fig. 3e).78
Based on the previous cobalt-catalyzed C–H activation of aryl sulfonamides,64–68 the electrochemical C–H/N–H annulations79 and the above control experiments, we propose a plausible mechanism for annulation with allenes or alkynes (Fig. 4). Initially, the oxidation of cobalt salt (II) produces Co(III)-species, which undergoes ligand exchange and C–H activation to form the key intermediate A. Subsequently, the allene 2a or alkyne 3 coordinates with A and inserts into the C–Co bond to from the seven-membered intermediate B or B′. The reductive elimination of B leads to the cyclic compound C, which undergoes 1,3-hydrogen migration to deliver product 4aa. The reductive elimination of B′ releases product 5 or 6. Finally, the generated Co(I) species is further oxidized to Co(III) for the new catalytic cycle.
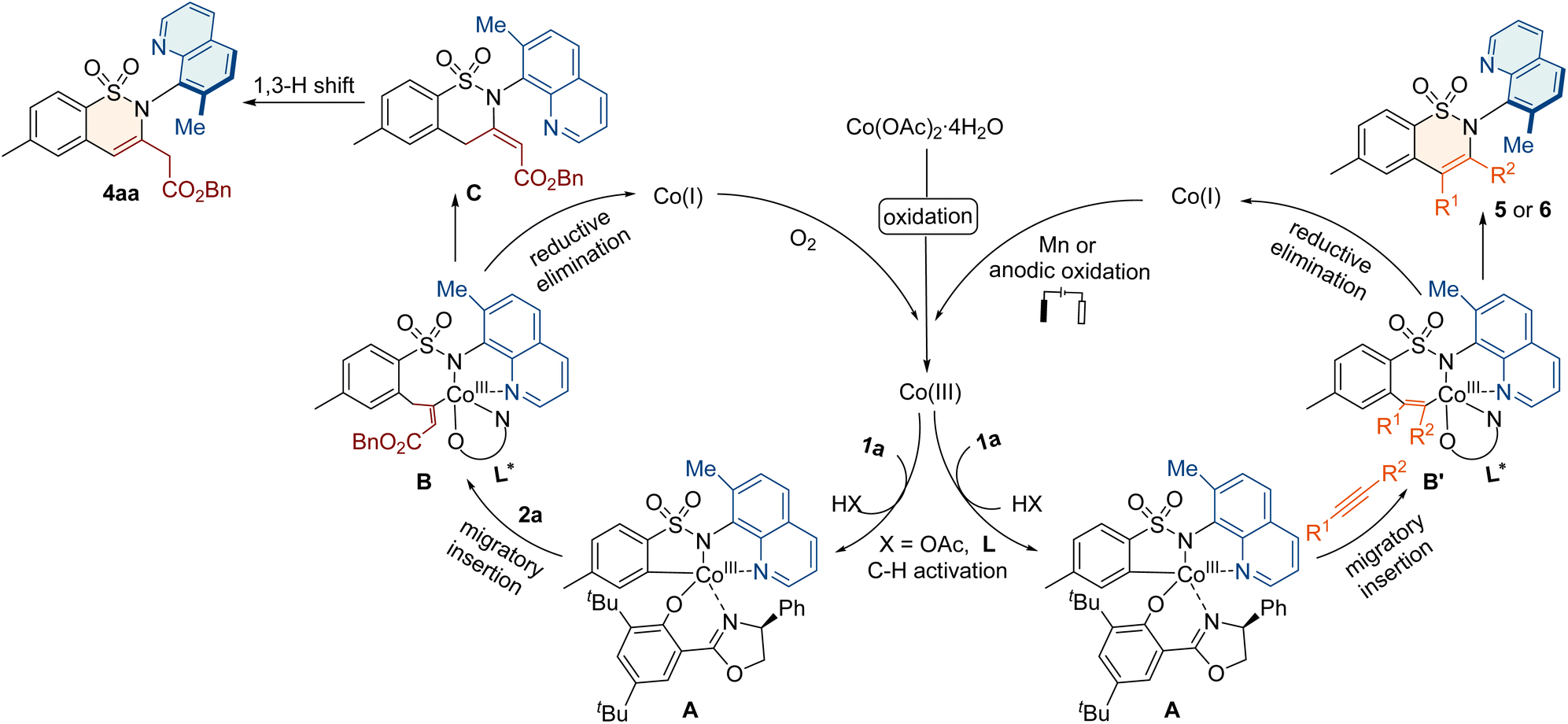 | ||
| Fig. 4 Proposed mechanism. | ||
Conclusions
To conclude, we have developed a highly efficient Co/Salox-catalyzed enantioselective C–H activation/annulation of sulfonamides with allenes or alkynes for the synthesis of C–N axially chiral sultams. In terms of the annulation with allenes, the reaction proceeds with O2 as the oxidant and a low catalyst loading of 5 mol% and tolerates broad allene substrates with high regio- and enantio-selectivities. For the annulation with alkynes, the protocol exhibited good functional group tolerance and excellent enantiocontrol. Typically, by utilizing anodic oxidation in an undivided cell to recycle the cobalt catalyst, the asymmetric C–H/N–H annulation of sulfonamides with alkynes was efficiently achieved for the construction of C–N axially chiral skeletons. These results demonstrate the versatility and robustness of cobalt/Salox asymmetric catalysis, and we are actively exploring its further applications in our laboratory.Data availability
The data supporting the findings of this work are available within the main article and the ESI.†Author contributions
X.-J. S., X. Z., and J. W. contributed equally to this work. J.-L. N. and D. Y. conceived the concept and prepared the manuscript. X.-J. S., X. Z., J. W., X. W., and Y. Z. conducted the experiments and analyzed the data. M.-P. S. provided revisions. J.-L. N. and D. Y. directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the National Natural Science Foundation of China (22271260 to J.-L. N.), and Natural Science Foundation of Henan Province (222300420291 to D. Y.).Notes and references
- G. H. Christie and J. J. Kenner, J. Chem. Soc., Trans., 1922, 121, 614–620 RSC.
- G. Bringmann, S. Tasler, H. Endress, J. Kraus, K. Messer, M. Wohlfarth and W. Lobin, J. Am. Chem. Soc., 2001, 123, 2703–2711 CrossRef CAS PubMed.
- P. W. Glunz, Bioorg. Med. Chem. Lett., 2018, 28, 53–60 CrossRef CAS PubMed.
- E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed.
- Z. Li and S. Yu, Sci. Sin.: Chim., 2020, 50, 509–525 Search PubMed.
- T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed.
- O. Kitagawa, Acc. Chem. Res., 2021, 54, 719–730 CrossRef CAS PubMed.
- G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1–39 Search PubMed.
- P. Rodríguez-Salamanca, R. Fernández, V. J. Hornillos and M. Lassaletta, Chem.–Eur. J., 2022, 28, e202104442 Search PubMed.
- O. Kitagawa, M. Takahashi, M. Yoshikawa and T. Taguchi, J. Am. Chem. Soc., 2005, 127, 3676–3677 CrossRef CAS PubMed.
- S.-L. Li, C. Yang, Q. Wu, H.-L. Zheng, X. Li and J.-P. Cheng, J. Am. Chem. Soc., 2018, 140, 12836–12843 CrossRef CAS PubMed.
- S. Lu, S. V. Ng, K. Lovato, J.-Y. Ong, S. B. Poh, X. Q. Ng, L. Kürti and Y. Zhao, Nat. Commun., 2019, 10, 3061 CrossRef PubMed.
- Z.-S. Liu, P.-P. Xie, Y. Hua, C. Wu, Y. Ma, J. Chen, H.-G. Cheng, X. Hong and Q. Zhou, Chem, 2021, 7, 1917–1932 CAS.
- C. Deur, A. K. Agrawal, H. Baum, J. Booth, S. Bove, J. Brieland, A. Bunker, C. Connolly, J. Cornicelli, J. Dumin, B. Finzel, X. Gan, S. Guppy, G. Kamilar, K. Kilgore, P. Lee, C.-M. Loi, Z. Lou, M. Morris, L. Philippe, S. Przybranowski, F. Riley, B. Samas, B. Sanchez, H. Tecle, Z. Wang, K. Welch, M. Wilson and K. Yates, Bioorg. Med. Chem. Lett., 2007, 17, 4599–4603 CrossRef CAS PubMed.
- Y. Kikuchi, C. Nakamura, M. Matsuoka, R. Asami and O. Kitagawa, J. Org. Chem., 2019, 84, 8112–8120 CrossRef CAS PubMed.
- Z. Li, H. Zhang and S. Yu, Org. Lett., 2019, 21, 4754–4758 CrossRef CAS PubMed.
- J.-Y. Ong, X. Ng, S. Lu and Y. Zhao, Org. Lett., 2020, 22, 6447–6451 CrossRef CAS PubMed.
- D. Li, S. Wang, S. Ge, S. Dong and X. Feng, Org. Lett., 2020, 22, 5331–5336 CrossRef CAS PubMed.
- Z. Gao, C.-X. Yan, J. Qian, H. Yang, P. Zhou, J. Zhang and G. Jiang, ACS Catal., 2021, 11, 6931–6938 CrossRef CAS.
- X. Xiao, Y.-Y. Lu, H.-Y. Tian, H.-J. Zhou, J.-W. Li, Y.-P. Yao, M.-L. Kea and F.-E. Chen, Org. Chem. Front., 2022, 9, 2830–2839 RSC.
- O. Kitagawa, M. Kohriyama and T. Taguchi, J. Org. Chem., 2002, 67, 8682–8684 CrossRef CAS PubMed.
- J. Terauchi and D. P. Curran, Tetrahedron: Asymmetry, 2003, 14, 587–592 CrossRef CAS.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed.
- T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed.
- M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898–12919 CrossRef CAS.
- T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS.
- T. Yoshino, S. Satake and S. Matsunaga, Chem.–Eur. J., 2020, 26, 7346–7357 CrossRef CAS PubMed.
- Q. Zhang, L.-S. Wu and B.-F. Shi, Chem, 2022, 8, 384–413 CAS.
- Y. Kommagalla and N. Chatani, Coord. Chem. Rev., 2017, 350, 117–135 CrossRef CAS.
- Ł. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef.
- A. Baccalini, S. Vergura, P. Dolui, G. Zanoni and D. Maiti, Org. Biomol. Chem., 2019, 17, 10119–10141 RSC.
- J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed.
- T. Yoshino and S. Matsunaga, ACS Catal., 2021, 11, 6455–6466 CrossRef CAS.
- Y. Zheng, C. Zheng, Q. Gu and S.-L. You, Chem Catal., 2022, 2, 2965–2985 CrossRef CAS.
- R. Mandal, B. Garai and B. Sundararaju, ACS Catal., 2022, 12, 3452–3506 CrossRef CAS.
- F. Pesciaioli, U. Dhawa, J. C. A. Oliveira, R. Yin, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 15425–15429 CrossRef CAS PubMed.
- Y.-H. Liu, P.-X. Li, Q.-J. Yao, Z.-Z. Zhang, D.-Y. Huang, M. D. Le, H. Song, L. Liu and B.-F. Shi, Org. Lett., 2019, 21, 1895–1899 CrossRef CAS PubMed.
- Y.-H. Liu, P.-P. Xie, L. Liu, J. Fan, Z.-Z. Zhang, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 19112–19120 CrossRef CAS PubMed.
- Y.-B. Zhou, T. Zhou, P.-F. Qian, J.-Y. Li and B.-F. Shi, ACS Catal., 2022, 12, 9806–9811 CrossRef CAS.
- K. Ozols, Y.-S. Jang and N. Cramer, J. Am. Chem. Soc., 2019, 141, 5675–5680 CrossRef CAS PubMed.
- K. Ozols, S. Onodera, Ł. Woźniak and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 655–659 CrossRef CAS PubMed.
- A. G. Herraiz and N. Cramer, ACS Catal., 2021, 11, 11938–11944 CrossRef CAS.
- D. Sekine, K. Ikeda, S. Fukagawa, M. Kojima, T. Yoshino and S. Matsunaga, Organometallics, 2019, 38, 3921–3926 CrossRef CAS.
- S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS PubMed.
- T. Kurihara, M. Kojima, T. Yoshino and S. Matsunaga, Asian J. Org. Chem., 2020, 9, 368–371 CrossRef CAS.
- Y. Hirata, D. Sekine, Y. Kato, L. Lin, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2022, 61, e202205341 CrossRef CAS PubMed.
- A. Whyte, A. Torelli, B. Mirabi, L. Prieto, J. F. Rodriguez and M. Lautens, J. Am. Chem. Soc., 2020, 142, 9510–9517 CrossRef CAS PubMed.
- J. Yang and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 16748–16751 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1209 CrossRef CAS PubMed.
- N. Jacob, Y. Zaid, J. C. A. Oliveira, L. Ackermann and J. Wencel-Delord, J. Am. Chem. Soc., 2022, 144, 798–806 CrossRef CAS PubMed.
- B.-J. Wang, G.-X. Xu, Z.-W. Huang, X. Wu, X. Hong, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202208912 CrossRef CAS PubMed.
- Q.-J. Yao, J.-H. Chen, H. Song, F.-R. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202202892 CAS.
- J.-H. Chen, M.-Y. Teng, F.-R. Huang, H. Song, Z.-K. Wang, H.-L. Zhuang, Y.-J. Wu, X. Wu, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202210106 CAS.
- Q.-J. Yao, F.-R. Huang, J.-H. Chen, M.-Y. Zhong and B.-F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202218533 CrossRef CAS PubMed.
- G. Zhou, J. H. Chen, Q. J. Yao, F. R. Huang, Z. K. Wang and B. F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202302964 CrossRef CAS PubMed.
- X.-J. Si, D. Yang, M.-C. Sun, D. Wei, M.-P. Song and J.-L. Niu, Nat. Synth., 2022, 1, 709–718 CrossRef.
- The professional comment on ref. 53, J. Kikuchi and N. Yoshikai, Nat. Synth., 2022, 1, 674–675 CrossRef.
- D. Yang, X. Zhang, X. Wang, X.-J. Si, J. Wang, D. Wei, M.-P. Song and J.-L. Niu, ACS Catal., 2023, 13, 4250–4260 CrossRef CAS.
- T. Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Science, 2023, 379, 1036–1042 CrossRef PubMed.
- T. v. Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, ChemRxiv, 2023, DOI: DOI:10.26434/chemrxiv-2023-dzw3d.
- Y. Lin, T. v. Münchow and L. Ackermann, ChemRxiv, 2023, DOI: DOI:10.26434/chemrxiv-2023-7g4z2.
- T. Liu, W. Zhang, C. Xu, Z. Xu, D. Song, W. Qian, G. Lu, C.-J. Zhang, W. Zhong and F. Ling, Green Chem., 2023, 25, 3606–3614 RSC.
- O. Planas, C. J. Whiteoak, A. Company and X. Ribas, Adv. Synth. Catal., 2015, 357, 4003–4012 CrossRef CAS.
- T. Lan, L. Wang and Y. Rao, Org. Lett., 2017, 19, 972–975 CrossRef CAS PubMed.
- N. Thrimurtulu, R. Nallagonda and C. M. R. Volla, Chem. Commun., 2017, 53, 1872–1875 RSC.
- Y. Cao, Y. Yuan, Y. Lin, X. Jiang, Y. Weng, T. Wang, F. Bu, L. Zeng and A. Lei, Green Chem., 2020, 22, 1548–1552 RSC.
- Y. Ran, Y. Yang and L. Zhang, Tetrahedron Lett., 2016, 57, 3322–3325 CrossRef CAS.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- M. V. Pham, B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 10610–10614 CrossRef CAS PubMed.
- H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318–7322 CrossRef CAS PubMed.
- K. Lei, X.-W. Hua, Y.-Y. Tao, Y. Liu, N. Liu, Y. Ma, Y.-H. Li, X.-H. Xu and C.-H. Kong, Bioorg. Med. Chem., 2016, 24, 92–103 CrossRef CAS PubMed.
- G. J. Wells, M. Tao and K. A. Josef, J. Med. Chem., 2001, 44, 3488–3503 CrossRef CAS PubMed.
- S. Andrea, O. Takashi, M. Antonio and T. S. Claudiu, Curr. Med. Chem., 2003, 10, 925–953 CrossRef PubMed.
- S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef PubMed.
- N. Thrimurtulu, A. Dey, D. Maiti and C. M. R. Volla, Angew. Chem., Int. Ed., 2016, 55, 12361–12365 CrossRef CAS PubMed.
- Y. An, X.-Y. Zhang, Y. N. Ding, Y. Li, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2022, 24, 7294–7299 CrossRef CAS PubMed.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, experimental section, X-ray crystallographic data, compound characterization, and NMR spectra (PDF). CCDC 2241825 (for 4aa) and 2248260 (for 5aa). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01787g |
| ‡ These authors contributed equally: Xiao-Ju Si, Xiaofang Zhao, and Jianli Wang. |
| This journal is © The Royal Society of Chemistry 2023 |