
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled introduction of functional groups at one P atom in [Cp*Fe(η5-P5)] and release of functionalised phosphines†
Stephan
Reichl
,
Felix
Riedlberger
,
Martin
Piesch
,
Gábor
Balázs
,
Michael
Seidl
and
Manfred
Scheer
 *
*
Institute of Inorganic Chemistry, University Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: manfred.scheer@ur.de
First published on 30th May 2023
Abstract
By salt metathesis reactions of the anionic complexes of the type [Cp*Fe(η4-P5R)]− (R = tBu (1a), Me (1b), –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh (1c); Cp* = 1,2,3,4,5-pentamethylcyclopentadienyl) with organic electrophiles (XRFG; X = halogen; RFG = (CH2)3Br, (CH2)4Br, Me) a variety of organo-substituted polyphosphorus ligand complexes of the type [Cp*Fe(η4-P5RRFG)] (2) are obtained. Thereby, organic substituents with different functional groups (FG), such as halogens or nitriles, are introduced. In [Cp*Fe(η4-P5RR′)] (2a: R = tBu, R′ = (CH2)3Br), the bromine substituent can be easily substituted, leading to functionalized complexes [{Cp*Fe(η4-P5tBu)}(CH2)3{Cp*Fe(η4-P5Me)}] (4) and [Cp*Fe(η4-P5RR′)] (5) (R = tBu, R′ = (CH2)3PPh2) or by abstraction of a phosphine to the asymmetric substituted phosphine tBu(Bn)P(CH2)3Bn (6). The reaction of the dianionic species [K(dme)2]2[Cp*Fe(η4-P5)] (I’) with bromo-nitriles leads to [Cp*Fe{η4-P5((CH2)3CN)2}] (7), allowing the introduction of two functional groups attached to one phosphorus atom. 7 reacts with ZnBr2 in a self-assembly reaction to form the supramolecular compound [Cp*Fe{η4-P5((CH2)3CN)2}ZnBr2]n (8).
CPh (1c); Cp* = 1,2,3,4,5-pentamethylcyclopentadienyl) with organic electrophiles (XRFG; X = halogen; RFG = (CH2)3Br, (CH2)4Br, Me) a variety of organo-substituted polyphosphorus ligand complexes of the type [Cp*Fe(η4-P5RRFG)] (2) are obtained. Thereby, organic substituents with different functional groups (FG), such as halogens or nitriles, are introduced. In [Cp*Fe(η4-P5RR′)] (2a: R = tBu, R′ = (CH2)3Br), the bromine substituent can be easily substituted, leading to functionalized complexes [{Cp*Fe(η4-P5tBu)}(CH2)3{Cp*Fe(η4-P5Me)}] (4) and [Cp*Fe(η4-P5RR′)] (5) (R = tBu, R′ = (CH2)3PPh2) or by abstraction of a phosphine to the asymmetric substituted phosphine tBu(Bn)P(CH2)3Bn (6). The reaction of the dianionic species [K(dme)2]2[Cp*Fe(η4-P5)] (I’) with bromo-nitriles leads to [Cp*Fe{η4-P5((CH2)3CN)2}] (7), allowing the introduction of two functional groups attached to one phosphorus atom. 7 reacts with ZnBr2 in a self-assembly reaction to form the supramolecular compound [Cp*Fe{η4-P5((CH2)3CN)2}ZnBr2]n (8).
Introduction
Chela originates from the ancient Greek language and means claw or pincer and is related to the word chelos – crab. Chelate ligands, similar to a crab's prey, surround the metal centre not with only one donor atom, but with two or more coordinating bonds separated from each other. The chelate effect1 can be explained by using principles of thermodynamics and favours in general the chelated complex, featuring a two, tri or multidentate ligand. Chemists took advantage of this phenomenon and developed, synthesised and tuned a variety of different chelate or pincer ligands which are very important in coordination chemistry and catalysis.2–8 Prominent classes are e.g. BINAP [2,2′-bis(diphenylphosphino)-1,1′-binaphthyl],9 DPPE (1,2-bis(diphenylphosphino)-ethane),10,11 DPPF (1,1′-bis(diphenylphosphino)ferrocene)12 or [P,N]4,13 ligands (Scheme 1). These are used e.g. for asymmetric catalysis,4,14 hydrogenation,6,15,16 and coupling reactions.17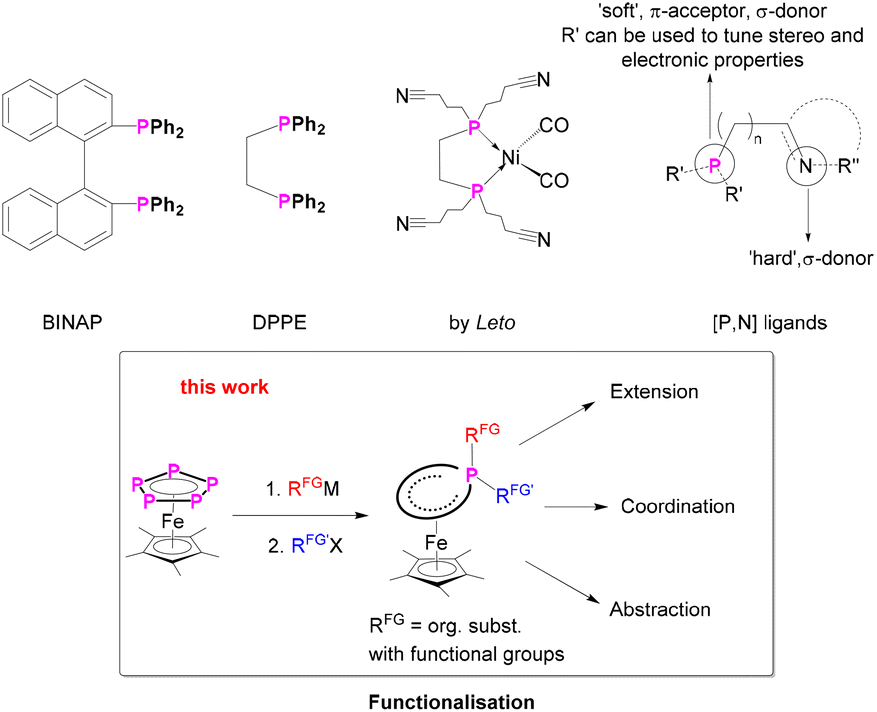 | ||
| Scheme 1 (Top) Selected bidentate phosphine ligands,9–11,25 (bottom) functionalisation of pentaphosphaferrocene: introduction of functional groups and subsequent chemical reactions. | ||
Although polydentate ligands are an important class of ligands for the stabilisation of metal complexes and catalysis, their synthesis is not trivial. Specifically, phosphorus-based ligands9,18,19 are synthesised from PCl3, obtained by chlorinating of white phosphorus. By doing this, in general, PCl3 is converted to the corresponding phosphine by stepwise salt metathesis with a suitable lithium-organyl/Grignard-reagent. The downsides of this route are the poor selectivity and the difficulties in the separation of the reaction mixture. Furthermore, this route does not commonly tolerate functional groups in the “backbone”, which, however, is crucial when it comes to adapting the desired ligand properties. In principle, multidentate ligands are synthesised according to electronic and steric requirements in the first place, then coordinated to a transition metal and, later, tested for their use in subsequent reactions and catalysis.20–22 Especially the synthesis of new and particularly unsymmetrically substituted organophosphorus compounds is a challenge.15 There are only very few poly- or especially cyclic phosphines known that carry functional groups and allow coordination and subsequent reactions.23–26 Therefore, the search for alternative synthetic routes is an active topic in chemistry.27
This prompted us to study whether it is possible to tune and functionalise a polyphosphorus ligand, already in the coordination-sphere of a transition metal, and then use it for subsequent reactions. Inter alia different coordination sites towards other Lewis-acidic metals, synthetic modification and finally the removal of the prebuilt ligand from the transition metal complex were targeted (Scheme 1).
Pentaphosphaferrocene [Cp*Fe(η5-P5)] (I) as a carrier of plenty of P atoms seemed to be an ideal candidate for such an investigation28 as this compound is readily accessible in a wide variety of different CpR ligands29 and capable of releasing functionalised phosphines by abstraction.28a,b
Herein we report the synthesis of a large variety of neutral diorganyl-substituted complexes of the type [Cp*Fe(η4-P5RR’)] (2a: R = tBu, R′ = (CH2)3Br; 2b: R = Me, R′ = (CH2)3Br; 2c: R = –CCPh, R′ = (CH2)3CN; 2d: R = Me, R′ = (CH2)3CN; 2e: R = Me, R′ = (CH2)4Br) featuring functional groups, with ethynyl, nitrile and/or bromine substituents. The obtained complexes readily react with nucleophiles as e.g. KPPh2 or KBn (Bn = benzyl), resulting in the selective formation of complexes in which the Br substituent is replaced by a terminal PPh2 or Bn unit, which was exemplified for 2a. Furthermore, complex 7 ([Cp*Fe{η4-P5((CH2)3CN)2}]), containing two nitrile units, proved to be a versatile ligand towards ZnBr2 leading to the unprecedented coordination polymer 8 ([Cp*Fe{η4-P5((CH2)3CN)2}ZnBr2]n) in the solid state.
Results and discussion
In a first step, the scope of anionic starting materials [Cp*Fe(η4-P5R)]− (1a: R = tBu; 1b: R = Me) was extended by introducing an ethynyl group by synthesising the complex [K(18c6)(thf)2][Cp*Fe(η4-P5-CCPh)] (1c) (Scheme 2). When ethynylbenzene is deprotonated with benzyl potassium and added to a solution of [Cp*Fe(η5-P5)] (I) in THF, a colour change from green to brown is observed. The 31P{1H} NMR spectrum of the reaction solution shows only one set of signals corresponding to an ABB′XX′ spin system of the anion of 1c with resonances centred at 37.3, 27.9 and 58.4 ppm (Fig. 1 and S16†). Crystals of 1c are obtained from a concentrated THF solution layered with n-hexane. Single crystal X-ray structure analysis revealed the molecular structure of the resulting product 1c in the solid-state (Fig. 1 and S34†). The main structural feature of 1c is an η4-coordinated cyclo-P5 ligand in an envelope conformation with a phenylethynyl substituent attached to the out-of-the-plane phosphorus atom. All P–P bonds are in the expected range and reveal double bond character with P–P bond lengths of 2.127(4)–2.187(3) Å,30,31 whereas the newly formed P–C bond is with 1.782(7) Å in the range of a single bond.30
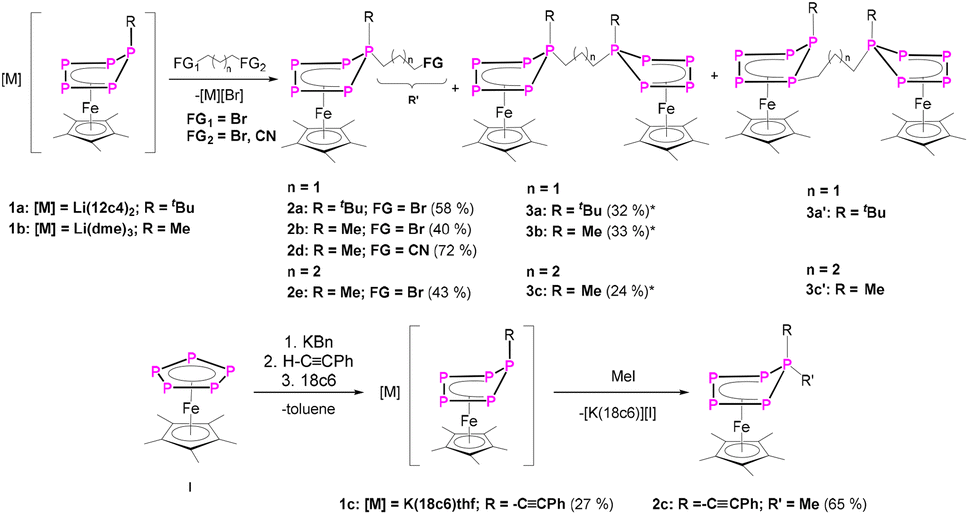 | ||
| Scheme 2 Reaction of 1a,b with 1,3-dibromopropane/1,4-dibromobutane/4-bromobutyronitrile (upper part) and I with 1. KBn, 2. ethynylbenzene and consecutive reaction with MeI (lower part), respectively. Yields are given in parentheses; *complexes 3 and 3’ co-crystallise, so the yields are summed up. | ||
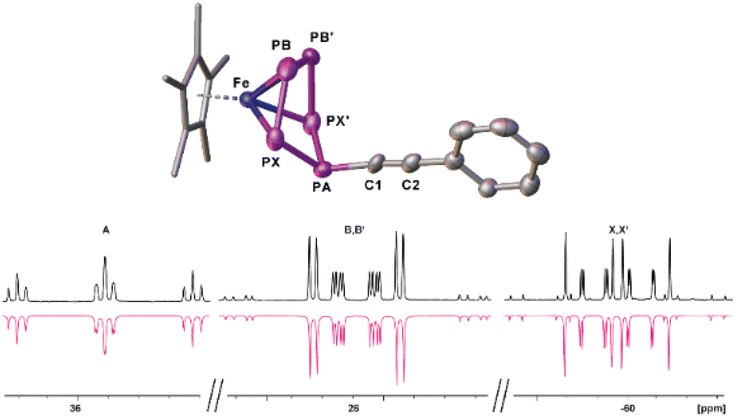 | ||
| Fig. 1 (Top) Solid-state molecular structure of 1c with thermal ellipsoids at 50% probability level. Cations and hydrogen atoms are omitted for clarity, the Cp* ligand is drawn in a wire frame model. (Bottom) Experimental (upwards) and simulated (downwards) 31P{1H} NMR (161.98 MHz, THF-d8) spectrum of the anion of 1c. | ||
Similarly to 1c, the anionic complexes [Cp*Fe(η4-P5R)]− (R = tBu (1a) and R = Me (1b)) can be synthesised.27 Due to the nucleophilic character of the out-of-plane phosphorus atom in these complexes, which possess a lone pair, 1a–c were reacted with carbon-centred electrophiles, which, however, contain a functional group.
The reaction of 1a,b with 1,3-dibromopropane results in the formation of a mixture of 2a,b and 3a,b (Scheme 2) which can be separated by column chromatography under an inert atmosphere. 2a,b can be isolated as brown needles in moderate crystalline yields of 53% (2a) and 50% (2b), respectively, while 3a,b afford yields of 32% (3a) and 33% (3b). Single crystal X-ray structure analyses show the expected structure of [Cp*Fe(η4-P5RR′)] (2a: R = tBu, R′ = (CH2)3Br; 2b: R = Me, R′ = (CH2)3Br) containing a 1-bromopropane substituent attached to the out-of-plane phosphorus atom (Scheme 2, Fig. 2). This simple method can be extended to introducing a nitrile group (2d) by using 4-bromobutyronitrile, or to increasing the alkyl chain length by using 1,4-dibromobutane (2e).
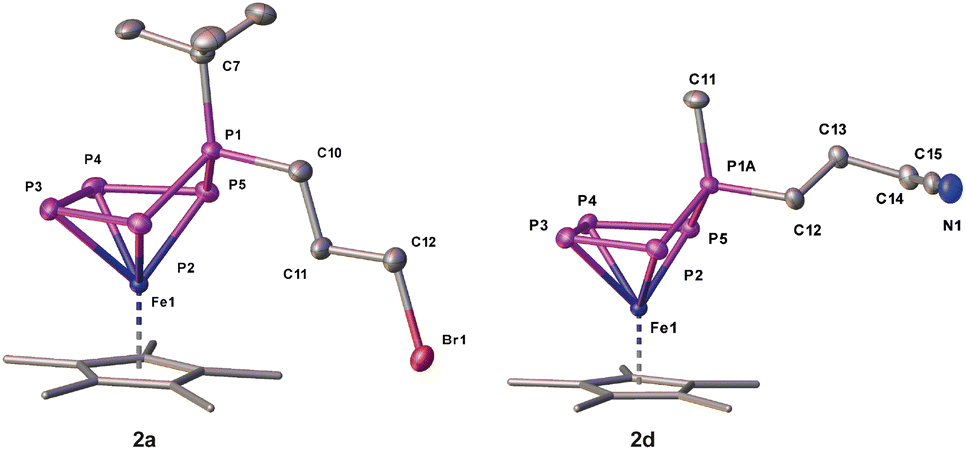 | ||
| Fig. 2 Molecular structures of 2a and 2d with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. The Cp* ligands are drawn in a wire frame model. | ||
In all of these products 2a–e (Fig. 2), the phosphorus-carbon distances are close to a single bond,30 whereas the P–P bond lengths lie within the range of a single and double bond (Tables S20–29†).30,31
The second fraction of the chromatographic workup was identified as 3a and b, revealing two [Cp*Fe(η4-P5)R] (3a: R = tBu; 3b: R = Me) moieties linked by a propane-1,3-diyl-group. In the major product 3a, the n-propyl group is attached to the out-of-plane phosphorus atom P1 (Fig. S42†). In addition to 3a, a minor product 3a’ co-crystallises in a ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :92. In 3a’, the electrophile is attached to the P atom next to the out-of-plain P atom leading to a 1,2-substitution pattern, instead of the 1,1-substitution (for X-ray structure of 3a’ see Fig. S43†). However, the separation of 3a and 3a’via column chromatography or other methods failed. Therefore, it was investigated by 31P NMR spectroscopy whether the reaction temperature, the order of addition of the starting materials or the chain length (1,4-dibromobutane instead of 1,3-dibromopropane) have an influence on the reaction progress. Unfortunately, it was not possible to completely hamper the formation of 3a’ as was possible for 3b. But a minimum amount of 3a’ is formed when slowly adding 1,3-dibromopropane to a −80 °C pre-cooled solution of 1a (Fig. S55†). Within the same reaction conditions, the amount of 2a decreases significantly (Fig. S55†). DFT calculations (B3LYP/def2-TZVP level of theory) predict 3a to be with 29 kJ mol−1 more stable than 3a’ (Table S46†). Since very little amount of 3a' is formed when changing the starting material to [Li(dme)3][Cp*Fe(η4-P5Me)] (1b) (Fig. S56†), the formation of 3a’ is probably also due to the steric bulk of the tert-butyl group in contrast to that of the significantly smaller methyl group in 3b.
:92. In 3a’, the electrophile is attached to the P atom next to the out-of-plain P atom leading to a 1,2-substitution pattern, instead of the 1,1-substitution (for X-ray structure of 3a’ see Fig. S43†). However, the separation of 3a and 3a’via column chromatography or other methods failed. Therefore, it was investigated by 31P NMR spectroscopy whether the reaction temperature, the order of addition of the starting materials or the chain length (1,4-dibromobutane instead of 1,3-dibromopropane) have an influence on the reaction progress. Unfortunately, it was not possible to completely hamper the formation of 3a’ as was possible for 3b. But a minimum amount of 3a’ is formed when slowly adding 1,3-dibromopropane to a −80 °C pre-cooled solution of 1a (Fig. S55†). Within the same reaction conditions, the amount of 2a decreases significantly (Fig. S55†). DFT calculations (B3LYP/def2-TZVP level of theory) predict 3a to be with 29 kJ mol−1 more stable than 3a’ (Table S46†). Since very little amount of 3a' is formed when changing the starting material to [Li(dme)3][Cp*Fe(η4-P5Me)] (1b) (Fig. S56†), the formation of 3a’ is probably also due to the steric bulk of the tert-butyl group in contrast to that of the significantly smaller methyl group in 3b.
However, the formation of 2a/b can be completely suppressed in favour of 3a/b by using a ratio of 2:1 in the reaction between 1a/b and 1,3-dibromopropane at −80 °C. Increasing the carbon chain length, i.e. by using 1,4-dibromobutane, does have a relevant influence on the ratio of the 1,1- and 1,2-substitution products, according to 31P NMR spectroscopic investigations (Fig. S57†), and favours the 1,2-substituted product 3c’, although 3c remains the major isomer. The reaction of compound 1b with 1,4-dibromobutane still leads to the formation of 2e (Scheme 2) and, as described above, to the bridged compounds 3c/c’ (Fig. S58†). The 1,2-substitution product 3c’ is formed in minor amounts only, but a minor amount of 2e' can be observed in the crystal structure of 2e (ratio 5:95, Fig. S41†). When the reaction is conducted at room temperature, 2e can be isolated as a spectroscopically pure compound (Fig. S6 and S21†). Note, also 2b’ was detected as the 1,2-substituted minor product in a ratio of 2b:2b’ = 90:10 (cf. Fig. S37†).
In order to extend the scope of functional groups, we used 4-bromobutyronitrile to attach an extra terminal nitrile group (Scheme 2). 31P NMR spectroscopic investigations of the reaction mixtures of 1a–c with 4-bromobutyronitrile show selective conversions. However, single crystals of 2d (Scheme 2) could only be obtained by using 1b. Compound 2d can be isolated as dark brown blocks in 72% yield. 2d co-crystalises with traces of I (5%), which can be washed with hexane to yield the spectroscopically pure compound 2d (Fig. 2). The rather easy introduction of a CN functional group is very remarkable and shows the high potential of the presented method of functionalisation.
To prove the accessibility of the bromine functionality in 2a for consecutive reactions, complex 2a was reacted with the nucleophile 1b. Indeed, this reaction leads to the formation of the asymmetric organo-substituted polypnictogen ligand complex 4 as dark green blocks in 80% yield (Scheme 3). The solid-state molecular structure is depicted in Fig. 4 and exhibits two organo-substituted [Cp*Fe(η4-P5R)] (R = tBu, Me) moieties which are linked via an n-propyl group. The formation of different isomers by migration of the organic groups was not observed.
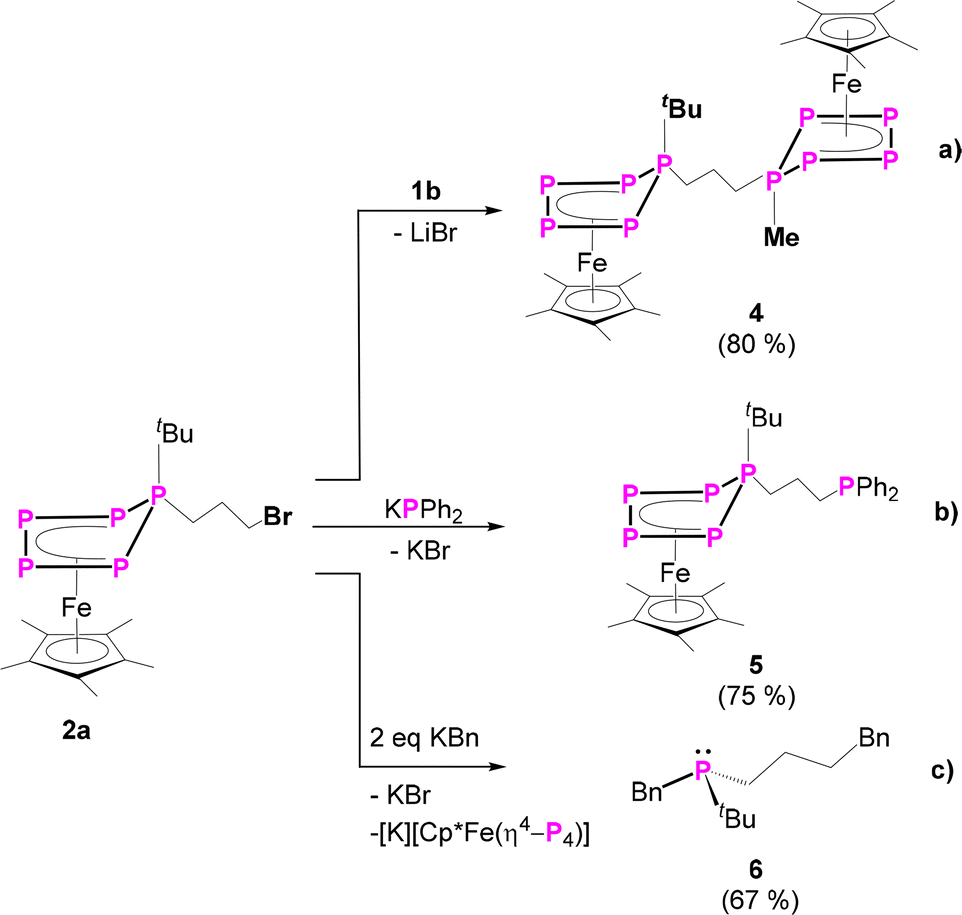 | ||
| Scheme 3 Consecutive reactions of 2a: (a) reaction with 1b to form 4; (b) functionalisation with KPPh2 to form 5; (c) first reaction with one equivalent of KBn, then abstraction of the functionalised phosphine 6 by a second equivalent of KBn. Yields are given in parentheses. | ||
The 31P{1H} NMR spectrum of 4 reveals two different AMM′XX′ spin systems, corresponding to the two inequivalent P5 units (Fig. 3). Based on the presence of different types of P–H coupling in the 31P NMR spectrum, an unambiguous assignment of the spin systems to the differently substituted P5 units could be made (Fig. S25 and S26†). Both spin systems show multiplets centred at −120.3, 41.0 and 164.3 ppm as well as at −129.9, 29.8 and 128.9 ppm, respectively.
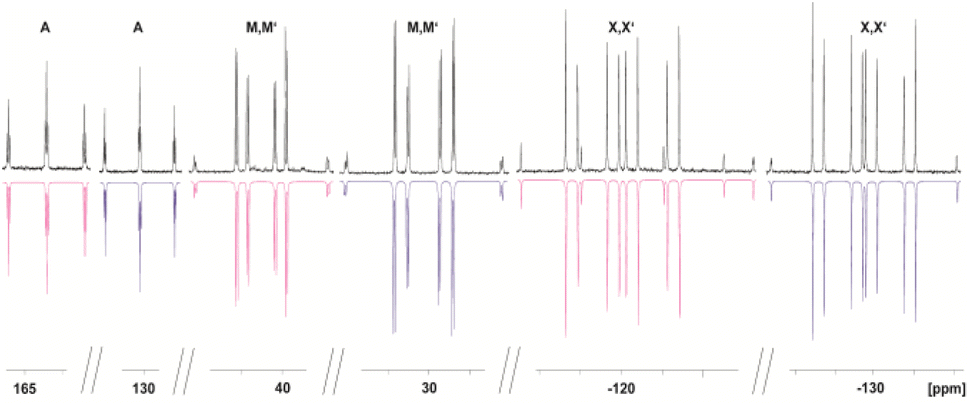 | ||
| Fig. 3 Experimental (upwards) in CD2Cl2 at 293 K and simulated (downwards) 31P{1H} NMR spectrum of 4. | ||
Since compound 4 shows two different η4-P5RR’ (R = Me, tBu; R’ = n-propyl) ligands, its cyclovoltammogram (CV) was measured to investigate the redox chemistry and check whether the two iron atoms are electrochemically inequivalent. Indeed, the CV (Fig. S63,† in o-DFB, referring to [Cp2Fe]/[Cp2Fe]+) shows three oxidation processes, one of them reversible (Erev = −0.21 V) and two irreversible (E1irev = 0.90 V; E2irev = 1.01 V), as well as two irreversible reductions (E1irev = −2.70 V; E2irev = −2.57 V), revealing the inequivalence of both Cp*Fe moieties.
Since the complexes 2 can be rationalised as formally being bromo-alkanes, their reactivity towards charged nucleophiles was investigated. Thus, compound 2a was reacted with potassium diphenylphosphanide (KPPh2) leading to the formation of complex 5 (Scheme 3, Fig. 4). The 31P{1H} NMR spectrum of the reaction solution shows an AMM’XX’ spin system and an additional singlet located at δ = −17.9 ppm, which evidences the formation of the desired product 5, featuring a terminal phosphine unit. Crystallisation from n-pentane at r.t. under reduced pressure leads to pure 5 in 75% yield. The molecular structure proves the identity of 5 (Fig. 4). Note that the nature of the phosphanide can be varied. The use of LiPCy2 (Cy = cyclo-hexyl) does also lead to the desired product, according to 31P NMR spectroscopic (Fig. S59†) and mass spectrometric investigations.
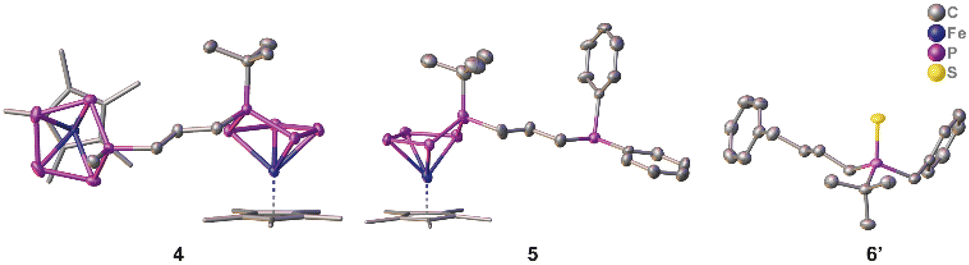 | ||
| Fig. 4 Molecular structures of 4, 5 and 6’ in the solid state with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. The Cp* ligands are drawn in a wire frame model. | ||
Interestingly, the 31P NMR spectrum of the reaction mixture does not show any signs of a phosphine abstraction or the formation of [Cp*Fe(η4-P4)]− as reported for the reaction of [Cp*FeP5R2)] (R = Me, tBu) with KBn.27 Instead, KPPh2 reacts selectively, under salt metathesis, with the bromine in 2a. Formally, the functional group can be stated to act as a protecting group and to shield the P5 ligand from a possible nucleophilic attack. Inspired by this observation and the reported phosphine abstraction,27 compound 2a was reacted with two equivalents (in portions) of benzyl potassium (KBn). According to 31P{1H} NMR spectroscopic investigations (Fig. S60†), the first equivalent of KBn reacts with 2avia salt metathesis, i.e. the substitution of the bromine group, and the second equivalent leads to the elimination of the asymmetrically substituted phosphine tBuPBn((CH2)3Bn) 6 (Scheme 3 and Fig. S60†) and the formation of [Cp*Fe(η4-P4)]−. The identity of the phosphine 6 (Scheme 3) was proven by NMR spectroscopy (δ(31P{1H}) = 4.5 ppm, which is in the typical range of phosphines)32 and, after oxidation with sulphur to the corresponding phosphine sulphide 6’, also by single-crystal X-ray diffraction analysis (Fig. 4). Note that 6 and 6’ are obtained as racemates.
This result shows that is not only possible to selectively functionalise one phosphorus atom in [Cp*Fe(η4-P5)] (I) but also to introduce different substituents containing functional groups, which can be further converted to other functionalities and finally cleave off the corresponding phosphine or bis-phosphine. In principle, this route represents an easy and convenient way to synthesise any desired functionalised phosphine.
To increase the number of functional groups attached to the P5-platform, namely such that do not tolerate strong nucleophiles, the dianionic species [K(dme)2]2[Cp*Fe(η4-P5)] (I’) was reacted with two equivalents of 4-bromobutyronitrile (Scheme 4). This reaction results quantitatively in the formation of the desired product [Cp*Fe{η4-P5((CH2)3CN)2}] (7) in 67% isolated yield. The solid-state molecular structure reveals an η4-P5 moiety, bearing two n-propyl substituents with terminal nitrile groups (Fig. S51†).
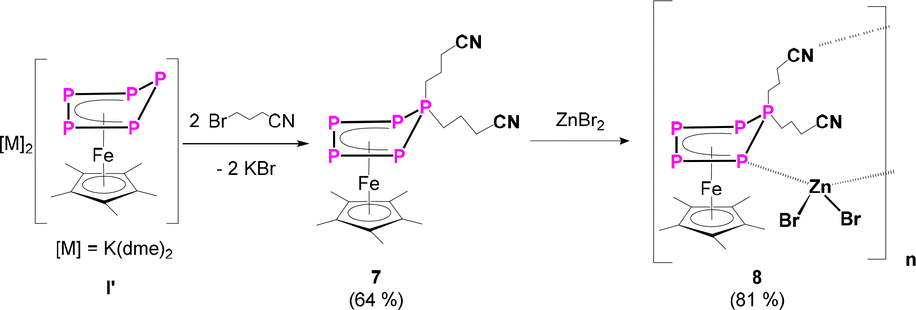 | ||
| Scheme 4 Reaction of [K(dme)2]2[Cp*Fe(η4-P5)] (I’) with 4-bromobutyronitrile leading to complex 7 and consecutive reaction of 7 with zinc(II)bromide. Yields are given in parentheses. | ||
The presence of the nitrile functionalities in 7 renders it a suitable starting material for the synthesis of coordination compounds containing polyphosphorus units, with potentially uncommon structures. Based on our expertise in the supramolecular chemistry of polypnictogen ligand complexes,29 coordinating groups, such as nitriles, are feasible ligands in self-assembly reactions with transition metal halides.29 Therefore, in a preliminary study, 7 was reacted with zinc(II)bromide in THF (Scheme 4). After three days, green/brownish plate-shaped crystals of 8 were obtained in 81% yield, exhibiting a linear 1D structure in the solid state (Fig. 5). Within the strain, monomers of [Cp*Fe{η4-P5((CH2)3CN)2}] are connected by zinc(II)bromide. That way, the endo-(with respect to the envelope P5 unit) nitrile group coordinates to the zinc atom (dN-Zn = 2.053(6) Å) (Fig. 5, Table S44†). The latter is coordinated in a tetrahedral coordination environment on the opposite side towards one phosphorus atom (dP-Zn = 2.430(2) Å) (Fig. 5, Table S44†) next to the P atom that is out of the plane which bears the nitrile linking unit. The zinc-phosphorus/nitrogen distances and angles are in line with similar complexes.33,34 The sum of the covalent radii corresponds approximately to the bond distances in the solid state, indicating a bonding interaction between phosphorus-zinc and nitrogen-zinc, respectively.33 To the best of our knowledge, compound 8 represents the first complex featuring a ZnBr2 unit, which is coordinated by a phosphorus atom and a nitrile group. Surprisingly, the exo nitrile group does not coordinate (Fig. 5). This opens the way for further coordination reactions, by using e.g. a different stoichiometry or reactions with e.g. copper halides. As this would go beyond the scope of this report, it will be topic of future work. Nevertheless, the reaction of 7 with zinc(II)bromide shows the versatility of the system and the possibility for consecutive reactions in which other than [Cp*Fe(η5-P5)] (I) are used, which does not reveal any reactivity towards zinc(II)bromide.
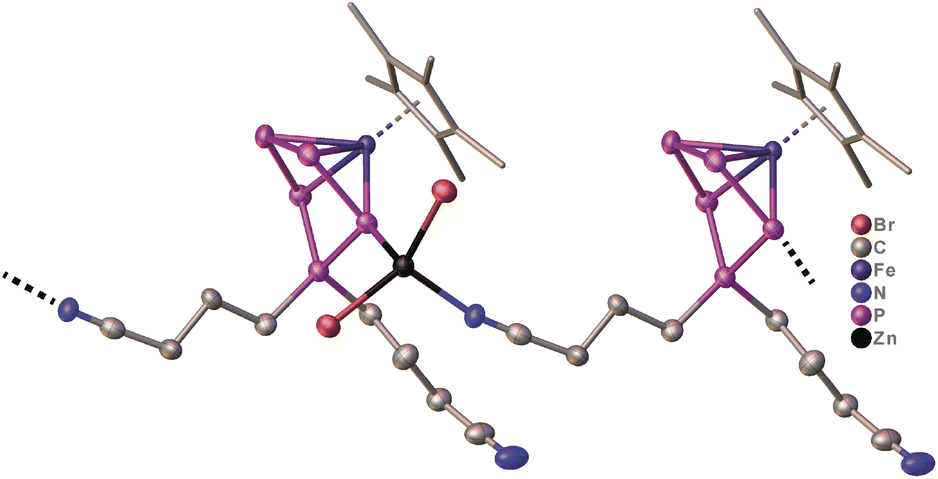 | ||
| Fig. 5 Solid-State molecular structure of 8 with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. The Cp* ligands are drawn in a wire frame model. | ||
The composition of 8 was also proven by elemental analysis and NMR spectroscopy, with the latter showing an AMM′XX′ spin system in THF-d8 at room temperature (Fig. S31†). The 31P and 1H NMR spectra are similar to those of complex 7 but slightly shifted (Fig. S61 and S62†), indicating no phosphorus-zinc-interaction, but a nitrogen-zinc-coordination in solution which is in accordance with the HSAB principle.35 Notably, after crystallisation, complex 8 is completely insoluble in non-coordinating solvents such as CH2Cl2, but highly soluble in coordinating ones as for instance THF and DME which indicates a partial depolymerization and a coordination of the solvent towards the zinc atom. This is in agreement with ESI-MS (in DME) which exclusively shows a molecular ion peak at m/z = 482.03, corresponding to complex 7.
Conclusions
In summary, it was shown that pentaphosphaferrocene (I) represents a very versatile platform for the successful functionalisation of the cyclo-P5 ligand. In alternating reactions with nucleophiles and electrophiles, a variety of functional groups can be easily introduced to one P atom at this polyphosphorus ligand complex, which had not been achieved before. Thus, a range of different compounds were obtained containing various functional groups (2a–e) including e.g. bromides and nitriles. This new modular system can even be further modified synthetically by using a Br-substituted derivative such as 2a to introduce afterwards additional functional groups like phosphines and others (4 and 5) but remain coordinated in the coordination sphere of Cp*Fe. Moreover, in a consecutive nucleophilic attack, the functionalised phosphine can be finally abstracted from the Cp*Fe unit to give access to an unprecedented, functionalised phosphine (6) demonstrating the versatility of this method. In addition, it was possible to synthesise polyphosphorus ligand complexes that bear two functional groups at one P atom (7), which can be used for consecutive reactions in supramolecular chemistry as was exemplified in the yield of a first unique ZnBr2 1D-coordination polymer 8. Current investigations focus on the introduction of different functional groups as well as their modifications and final release from the complex.Data availability
All experimental procedures, spectroscopic data, information on the theoretical calculations and crystallographic data can be found in the ESI.†Author contributions
The conceptualization (together with GB and MS), experimental work assisted by FR and MP and writing of the manuscript of this work were achieved by SR. M. Seidl accomplished the solution and refinement of X-ray structural data. MP and GB performed the DFT calculations. The entire work was supervised, guided, and revised by MS, who also acquired funding for the project. The final manuscript was reviewed and edited by SR, GB and MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/38-3. S.R. is grateful to the Studienstiftung des Deutschen Volkes and M. P. to the Fonds der Chemischen Industrie for PhD fellowships.References
- G. Schwarzenbach, Helv. Chim. Acta, 1952, 35, 2344–2359 CrossRef CAS.
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- S. Lühr, J. Holz and A. Börner, ChemCatChem, 2011, 3, 1708–1730 CrossRef.
- M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819–833 RSC.
- C. Redshaw and Y. Tang, Chem. Soc. Rev., 2012, 41, 4484 RSC.
- H. Wang, J. Wen and X. Zhang, Chem. Rev., 2021, 121, 7530–7567 CrossRef CAS PubMed.
- H. Han and S. A. Johnson, Organometallics, 2006, 25, 5594–5602 CrossRef CAS.
- H. Han, M. Elsmaili and S. A. Johnson, Inorg. Chem., 2006, 45, 7435–7445 CrossRef CAS PubMed.
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- W. Hewertson and H. R. Watson, J. Chem. Soc., 1962, 1490 RSC.
- J. Dogan, J. B. Schulte, G. F. Swiegers and S. B. Wild, J. Org. Chem., 2000, 65, 951–957 CrossRef CAS PubMed.
- G. Marr and T. Hunt, J. Chem. Soc. C Org., 1969, 1070 RSC.
- D. Peng, X. Yan, C. Yu, S. Zhang and X. Li, Polym. Chem., 2016, 7, 2601–2634 RSC.
- Privileged Chiral Ligands and Catalysts, ed. Q. Zhou, Wiley, 2011 Search PubMed.
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS PubMed.
- H. Brunner, C. Zettler and M. Zabel, Monatsh. Chem., 2003, 134, 1253–1269 CrossRef CAS.
- A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- H. Gali, S. R. Karra, V. S. Reddy and K. V Katti, Angew. Chem., Int. Ed., 1999, 38, 2020–2023 CrossRef CAS PubMed.
- M. M. Rauhut, I. Hechenbleikner, H. A. Currier, F. C. Schaefer and V. P. Wystrach, J. Am. Chem. Soc., 1959, 81, 1103–1107 CrossRef CAS.
- H. Li, B. Zheng and K.-W. Huang, Coord. Chem. Rev., 2015, 293–294, 116–138 CrossRef CAS.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
- M. Sietzen, S. Batke, L. Merz, H. Wadepohl and J. Ballmann, Organometallics, 2015, 34, 1118–1128 CrossRef CAS.
- W. A. Henderson, M. Epstein and F. S. Seichter, J. Am. Chem. Soc., 1963, 85, 2462–2466 CrossRef CAS.
- L. S. Meriwether, M. F. Leto, E. C. Colthup and G. W. Kennerly, J. Org. Chem., 1962, 27, 3930–3941 CrossRef CAS.
- L. S. Meriwether and J. R. Leto, J. Am. Chem. Soc., 1961, 83, 3192–3196 CrossRef CAS.
- A. K. Adhikari, C. G. P. Ziegler, K. Schwedtmann, C. Taube, J. J. Weigand and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 18584–18590 CrossRef CAS PubMed.
- For recent P4 conversion to phosphines by main group compounds cf. e.g.: (a) D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed; (b) M. Donath, K. Schwedtmann, T. Schneider, F. Hennersdorf, A. Bauza, A. Frontera and J. J. Weigand, Nat. Chem., 2022, 14, 384–391 CrossRef CAS PubMed; (c) F. Chen, M. Bai, Y. Zhang, W. Liu, X. Huangta, Y. Liu, G. Tang and Y. Zhao, Angew. Chem., Int. Ed. Engl., 2022, 61, e202210334 CAS; (d) D. J. Scott, Angew. Chem., Int. Ed. Engl., 2022, 61, e202205019 CrossRef CAS PubMed; (e) S. K. Ghosh, C. C. Cummins and J. A. Gladysz, Org. Chem. Front., 2018, 5, 3421–3429 RSC.
- (a) S. Reichl, E. Mädl, F. Riedlberger, M. Piesch, G. Balázs, M. Seidl and M. Scheer, Nat. Commun., 2021, 12, 5774 CrossRef CAS PubMed; (b) S. Reichl, G. Balázs and M. Scheer, Chem. Sci., 2023, 14, 3834–3838 RSC; (c) S. B. Dinauer, M. Piesch, R. Szlosek, M. Seidl, G. Balázs and M. Scheer, Chem. Eur J., 2023, 29, e202300459 CrossRef CAS PubMed; (d) Ch. Riesinger, G. Bálazs, M. Seidl and M. Scheer, Chem. Sci., 2021, 12, 13037–13044 RSC; (e) Ch. Riesinger, G. Balázs, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2020, 59, 23879–23884 CrossRef CAS; (f) D. J. Jones, E. M. O'Leary and T. P. O'Sullivan, Adv. Synth. Catal., 2020, 362, 2801–2846 CrossRef CAS.
- E. Peresypkina, A. Virovets and M. Scheer, Coord. Chem. Rev., 2021, 446, 213995 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- O. Kühl, Phosphorus-31 NMR Spectroscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009 Search PubMed.
- U. Siemeling, T. Klemann, C. Bruhn, J. Schulz and P. Tpnika, Dalton Trans., 2011, 40, 4722–4740 RSC.
- T. Tsukuda, C. Nishigata, K. Arai and T. Tsubomura, Polyhedron, 2009, 28, 7–12 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2247948–2247956 and 2041982–2041986. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01488f |
| This journal is © The Royal Society of Chemistry 2023 |