
Donor–acceptor complex formation by social self-sorting of polycyclic aromatic hydrocarbons and perylene bisimides†‡
Simon
Soldner
a,
Olga
Anhalt
a,
Menyhárt B.
Sárosi
 ab,
Matthias
Stolte
ab and
Frank
Würthner
*ab
ab,
Matthias
Stolte
ab and
Frank
Würthner
*ab
aCenter for Nanosystems Chemistry (CNC), Universität Würzburg, Theodor-Boveri-Weg, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
bInstitut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 31st August 2023
Abstract
Self-assembly versus complexation with polycyclic aromatic hydrocarbon (PAH) guest molecules is studied for a series of perylene bisimides (PBIs). Bulky imide substituents at the PBI guide their self-assembly into dimer aggregates with null-type exciton coupling. Host–guest titration experiments with perylene and triphenylene PAHs afford 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 complexes whose properties are studied by single crystal X-ray analysis and UV/Vis and fluorescence spectroscopy.
:1 and 1:2 complexes whose properties are studied by single crystal X-ray analysis and UV/Vis and fluorescence spectroscopy.
Electron-donor–acceptor (EDA) complexes between electron-rich and electron-poor π-scaffolds are most desirable dye aggregates as they afford light-induced charge carrier generation as required for many applications.1–3 However, whilst the formation of EDA complexes is common for smaller π-systems such as tetracyanoethene or nitrobenzenes in combination with electron-rich polycyclic aromatic hydrocarbons (PAHs, e.g. tetramethyl-p-phenylenediamine, 1,3,5-triaminobenzenes, anthracene, carbazole) in thin films and solid state, the association constants between the two counterparts in solution remain rather modest, typically around 10 M−1.4 Unfortunately, such low binding affinities cause severe limitations for photophysical studies that typically have to be carried out in dilute solution. For the enhancement of the binding affinities for such EDA complexes, enlargement of the π-scaffolds appears as an obvious approach because by this means dispersion interactions between planar π-scaffolds should provide the desired binding strength.5 Recent progress in this field, albeit with binding affinities remaining still modest, was accomplished in particular for naphthalene bisimide based electron acceptors, opening up research avenues in the fields of foldamer science6,7 and anion–π-catalysis.8
Among the common π-systems, perylene bisimides (PBIs) appear as a perfect choice with an expanded π-scaffold of 24 sp2-hybridized carbon and two additional nitrogen atoms. PBIs are also versatile dyes for photophysical studies due to intense absorption bands in the visible range, excellent (photo-)stability, outstanding fluorescence and reversible reduction at modest potential.9 However, these dyes are prone to self-aggregation which is most apparent in their main industrial application as high grade colour pigments.10 The strong cohesive forces between PBIs are also manifested in a multitude of dye aggregates, liquid-crystalline mesophases and supramolecular polymers.11 As a consequence of the strong intermolecular interactions between PBIs, primarily by dispersion and electrostatic forces,12 early attempts in our laboratory for preparing EDA complexes between the electron-poor π-scaffold of PBIs and electron-rich PAHs failed because of narcissistic self-sorting, i.e. the formation of pristine PBI aggregates that did not incorporate the electron-rich PAHs.13 Only more recently, such complexes could be observed in solution with PBI cyclophane supramolecular hosts14 as well as in the solid state as cocrystals.15–18
Here, following our recent research on larger nanographene multi-imides,19,20 in this communication we introduce PBIs bearing bulky imide groups as supramolecular hosts for PAHs. Even for this design, our research reveals the preference of PBI for self-aggregation into dimer aggregates via PBI-PBI π-contacts along with weak C–H⋯π-interactions that are considerably stronger than those between electron-rich PAHs and PBI. Accordingly, our results showcase that EDA interactions are weak compared to the stronger non-covalent forces between electron-poor PBIs.
For our research we chose PBI receptors 3a–c (Fig. 1a) whose synthesis was accomplished from perylene tetraester 1 and aromatic amines 2a–c (Scheme S3, ESI‡),21 which vary in the para-position of the terphenyl wedges. 3a21 has a bulky tert-butyl group while 3b and 3c have flexible n-hexyl or branched alkyl chains, respectively. The bulky imide substituents not only endow the PBIs with excellent solubility even in low polarity solvents like methylcyclohexane (MCH) but also establish recognition sites for guest molecules like smaller PAHs. All three PBIs were characterized by high-resolution mass spectrometry (HRMS) as well as 1H- and 13C-NMR, UV/Vis and fluorescence spectroscopy (for details see ESI‡).
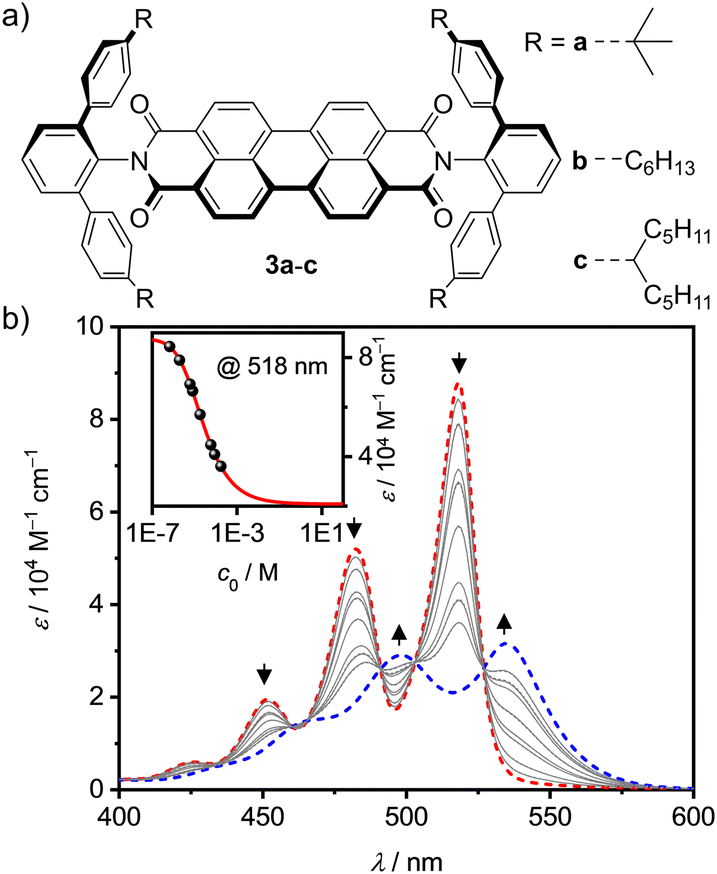 | ||
| Fig. 1 (a) Chemical structures of the investigated PBIs 3a–c. (b) Concentration-dependent UV/Vis absorption spectra (grey solid lines) for PBI 3b in methylcyclohexane (c0 = 1.75 × 10−4 − 1.50 × 10−7 M) at 298 K. The dashed lines represent the calculated monomer (red) and dimer (blue) spectra according to a global fit analysis by the monomer–dimer model. The arrows indicate the spectral changes with increasing concentration. The inset depicts the monomer–dimer fit (red line) at λmax = 518 nm (black symbols). | ||
Next, we explored the effect of the bulky imide substituents on the self-assembly of these dyes in solvents of different polarity. To appreciate the effect of these substituents it is helpful to compare the studies to those previously performed for PBI dyes bearing sterically non-demanding imide substituents that enable the formation of extended dye aggregates by isodesmic self-assembly with aggregation constants (K) from about 200–600 M−1 in chloroform (CHCl3) up to 100000 M−1 in MCH for PBIs functionalized with simple alkyl or trialkyl- and trialkoxyphenyl substituents.22 Other PBIs tailored for dimerization have K-values in a similar range of 910 M−1 in CDCl3/MCH (1/5),23 2800 M−1 in n-heptane24 and 14100 M−1 in CHCl3.25 As shown by concentration-dependent UV/Vis studies, 3c shows no aggregate growth which is explained by the steric hindrance imparted by the flexible branched alkyl chains (Fig. S3, ESI‡). However, 3b self-assembles into discrete dimers (Fig. 1b) with an appreciably high dimerization constant of 43000 M−1 in MCH.26 The aggregation behavior was further investigated in different solvents, namely CHCl3 and tetrachloroethane (TCE, Fig. S4–S7, ESI‡). In the more polar chlorinated solvents aggregation is less pronounced, i.e. K = 140 M−1 in CHCl3. In comparison, 3a shows an about sixfold stronger dimerization (K = 870 M−1) than 3b in CHCl3 (Table S2, ESI‡).
This dimerization is explained by the structural features as disclosed by single crystal X-ray analyses of 3a and 3b (Fig. 2 and Fig. S8, S9, CCDC 2284853 and 2284854, ESI‡). Here two PBIs are closely stacked at 3.4–3.6 Å with a significant π–π-contact and an almost orthogonal rotational displacement of their respective N,N′-axes (3a). The spectral changes upon dimerization (Fig. 1b) are more pronounced than expected for such cross-stacked structures composed of two almost orthogonally arranged dyes where the long-range Coulomb coupling (JCoul) of their molecular transition dipoles (μeg = 8.1 D) should be small.27 However as emphasized in recent work by Spano and co-workers, low-lying charge transfer (CT) states can play a major role in PBI aggregates and should not be ignored as the resultant short-range coupling (JCT) can even outperform JCoul.28 Therefore, for analyzing the PBI–PBI interactions we started with geometry optimizations of each dimer that afforded an angle between the two PBI N,N′-axes of 86° (3a) and 90° (3b) (Fig. S11, ESI‡). A negligible negative overall coupling of only −12 cm−1 is calculated for the perfect orthogonal case of 3b that is, however, impressively decreased to −273 cm−1 for dimers of 3a upon a minor change of the rotational offset to 86° (Table S4, ESI‡). Here, the computed J-type short-range coupling JCT is considerably stronger than the H-type long-range Coulomb coupling JCoul. This aggregate can accordingly be classified as a hJ dimer according to Hestand and Spano,28 and explains the observed bathochromic shift of 610 cm−1 for the UV/Vis absorption spectra of the dimers compared to their monomers (Fig. 1b). For the dimers of 3b in MCH solution as well as the dimers of 3a in single crystals we also observe a bathochromically shifted unstructured excimer-type emission band as well as a significant increase of the fluorescence lifetime from 4.0 ns (monomer) up to 19.9 ns (dimer, Fig. S30–S32, ESI‡).
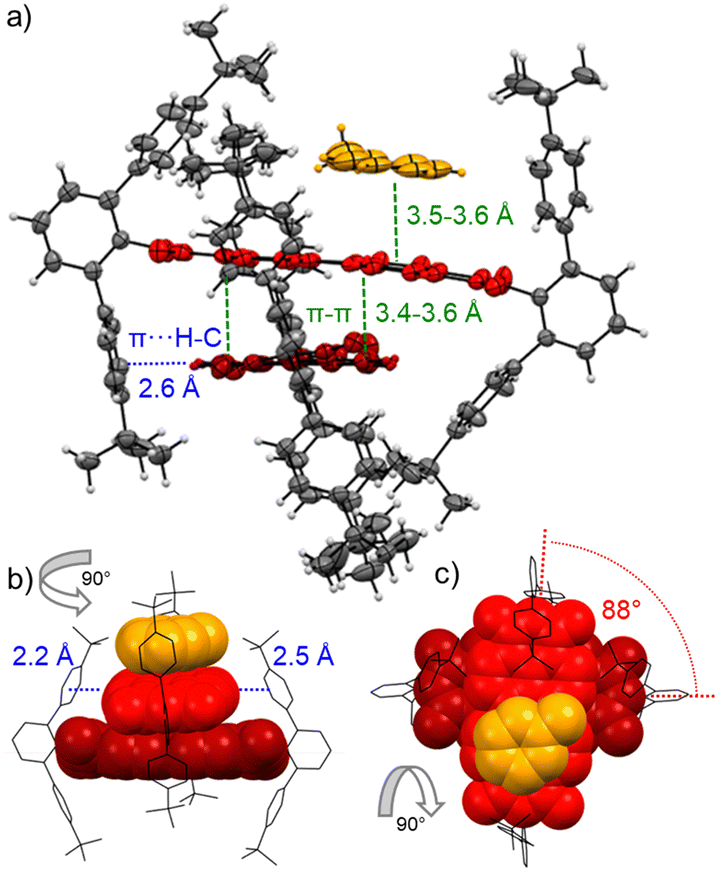 | ||
| Fig. 2 Packing of PBI 3a (red) in dimer π-stacks including a π-stacked toluene molecule (orange) in the single crystals grown from toluene and methanol in side (a and b) as well as top (c) view onto the PBIs π-system. C–H⋯π (blue) and π–π (green) interactions as well as the rotational displacement (red) between the PBI's N,N′-axes are indicated. Ellipsoids are set to 50% probability in (a) and the PBIs are highlighted in red. Further solvent molecules and molecular disorder are omitted for clarity. | ||
The major interest of our study was, however, the utilization of 3a–c as supramolecular receptors for the complexation of PAHs like perylene (P) and triphenylene (T). Here, the most obvious choice was P, whose π-surface should be more similar to the host and accordingly provide more tightly bound complexes. The binding studies were performed in the three solvents CHCl3, TCE and MCH, which not only affect the PBIs self-assembly into dimers (competing equilibrium) but also the solubility of the guest molecules. Accordingly, a low host concentration was chosen for all titrations to ensure only minor presence of dimeric aggregate species (<2%).
All three PBIs 3a–c show distinct changes in their respective UV/Vis absorption spectra with the addition of guest PAHs – however to different extend, pending their respective binding affinity due to the solvent polarity as well as the guest's solubility. A representative complexation study of 3a with P in TCE is shown in Fig. 3 (for all other titrations, see ESI‡). The addition of guest leads to a bathochromic shift of the absorption maximum by 280 cm−1 to 539 nm, accompanied by an overall significant decrease of the intensity and a small increase of absorbance in the spectral range 580–700 nm. Similar results are obtained for the other hosts 3b,c (Fig. S14–S19, ESI‡). Thus, some of the oscillator strength for the PBI S0–S1 transition is transferred into a low-intensity CT transition between the electron-rich P guest and the electron-poor PBI. In contrast, the titrations with T, whilst also showing a bathochromic shift, do not reveal such CT signatures (Fig. S21, S25 and S26, ESI‡). Likewise, whilst the emission for the P complex is entirely quenched, a new bathochromically shifted exciplex-type emission band is observed at 600 nm for the host–guest complex with T (Fig. S27, ESI‡).
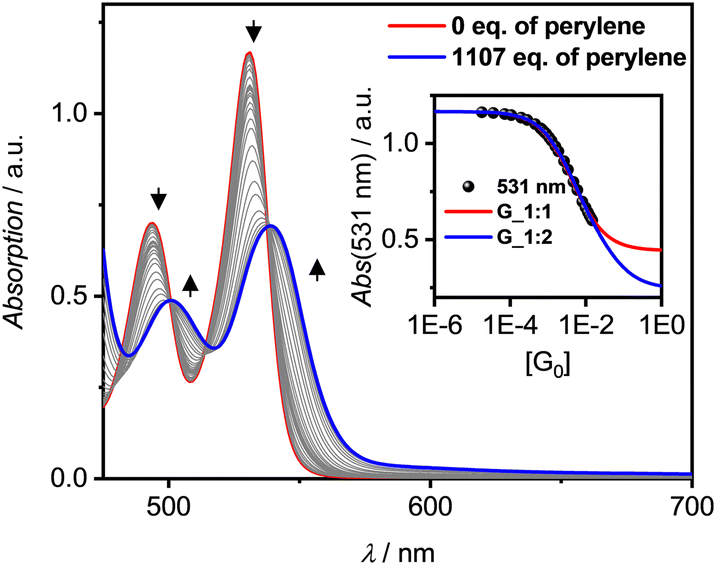 | ||
| Fig. 3 UV/Vis absorption spectra (solid lines) for a solution of PBI 3a as host (c0 = 1.34 × 10−5 M, red line) and changes upon addition of P as guest (grey to blue lines, 1107 eq.) in TCE at 298 K. Resulting plot of the absorption at λmax = 531 nm (black symbol) with nonlinear curve fits by the 1:1 (red line) and 1:2 (blue line) global (500–550 nm) models. Arrows depict spectral changes with increasing equivalents (eq.) of the P guest. | ||
Due to the weak binding affinity between the supramolecular receptors and the PAHs the in-depth analyses of the UV/Vis titration studies for the prevalence of 1:1 or 1:2 complexes are limited by the solubility of the guest molecules and the resultant maximal achievable degree of complexation (αC). Only for the highly soluble PBIs 3b and 3c binding constants (K1) of up to 2262 M−1 for 3b (1:1) in MCH with P at 298 K were determined. Unfortunately, here the poor solubility of P as well as the PBI's competing dimerization do not allow for a reliable analysis, whether also 1:2 complexes are formed. Similarly, NMR titration studies with the non-aggregating PBI 3c could not discriminate between 1:1 and 1:2 complex formation, even at low temperature of 223 K in deuterated MCH (Fig. S28 and S29, ESI‡) like it was possible for PAH complexes of larger C64 nanographene tetraimide receptors.19
Still, in the more polar solvents CHCl3 and TCE the higher excess of guests enabled a more accurate evaluation due the higher achievable αC (> 60%; Fig. S12 and S23, ESI‡) whether also 1:2 complexes are formed. Studies with T in TCE show that the isosbestic points disappear after a certain amount of guest addition (Fig. 3 and Fig. S21, S22, ESI‡), which is indicative that 1:2 complexes are present in solution. Additionally, the global 1:2 fit determined with bindfit29,30 as well as SIVVU31 show much higher accuracy and more homogenous residual than the respective 1:1 analysis (Fig. S13/24, ESI‡). As for the dimerization, PBI 3a shows the higher affinities for PAHs at 298 K compared to 3b with values of K1 = 254 M−1 and K2 = 23 M−1 for P and K1 = 81 M−1 and K2 = 13 M−1 for T in TCE (for the resp. complexes of 3a,c see Table S5, ESI‡). The observed anti-cooperativity might be explained by the imide substituents that, like a hinge, can adjust to the firstly complexed guest molecule, similarly as seen in the crystal structure of the PBI dimer shown in Fig. 2. However, opening of the hinge is possible to accommodate a second perylene guest molecule as seen in the single crystal structure of the 1:2 complex in Fig. 4a and b.
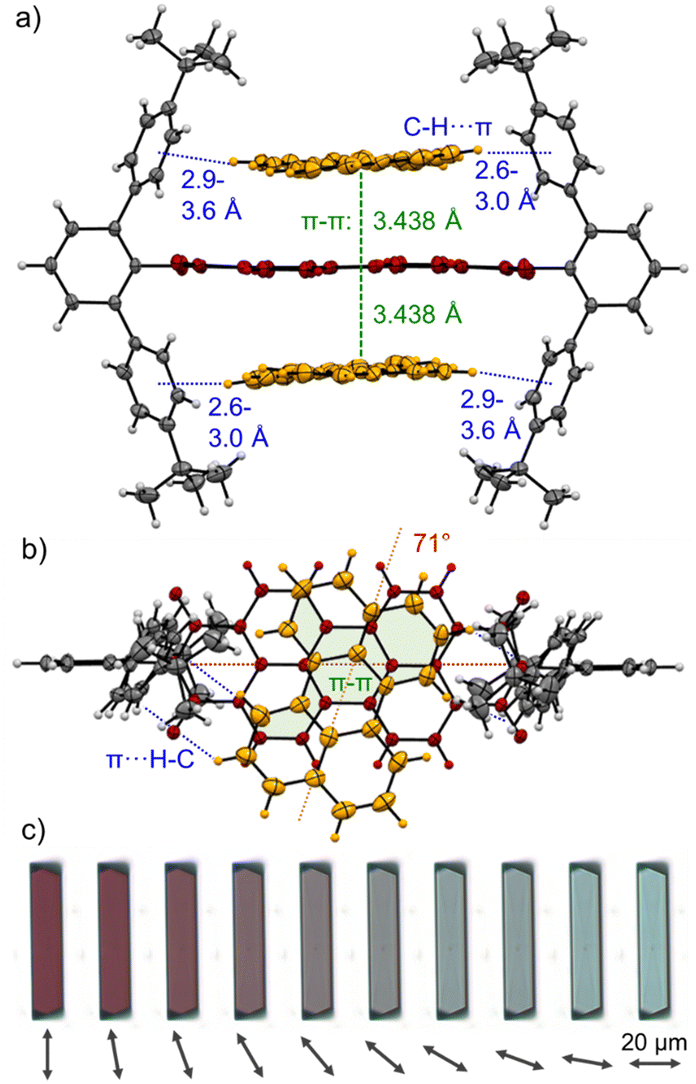 | ||
| Fig. 4 Single crystals structure of the 1:2 complex of PBI 3a (red) with two perylene guest molecules (orange) as observed by X-ray diffraction in side (a) as well as top (b) view onto the PBIs π-system. C–H⋯π (blue) and π–π (green) interactions as well as the rotational displacements (red) between the PBI N,N′-axis and the guest molecules are indicated. Ellipsoids are set to 50% probability and solvent molecules as well as molecular disorder are omitted for clarity. (c) Polarization-dependent bright-field optical microscope images of a [P·3a·P] cocrystal on a quartz substrate while rotating the polarization axis of the transmitted light in steps of 10° as indicated by the arrow. | ||
Cocrystals suitable for single crystal analysis could indeed be grown for PBI 3a in 1:2 mixing ratio with both P and T (Fig. 4a, b and Fig. S10, CCDC 2284855 and 2284856, ESI‡) by slow diffusion of n-hexane (P) and methanol (T) into a 10−3 M solution in toluene. In the [P·3a·P] cocrystal each guest molecule closely stacks coplanar on top of the PBI host with a π–π distance of 3.44 Å with significant π-overlap and a rotational displacement of 71°. Additional CH⋯π interactions between the guest and host's imide residues with distances of 2.6 to 3.6 Å further stabilize the complex. In the [T·3a·T] cocrystal the situation is similar, but each guest molecule does not have the same π-distance to the PBIs π-surface (3.2–3.6 Å) and the relative orientation varies (74°, 89°). Still, short CH⋯π interactions (2.5–2.7 Å) support also here the complexation of the PAH guests (Fig. S10, ESI‡).
Interestingly, [P·3a·P] grown on quartz substrates change their colour depending on the relative orientation towards to substrate ([101],[10–1]) as well as the polarization axis of the incident light between red, deep violet and turquoise (Fig. 4c and Fig. S33–S37, ESI‡). This is due to different absorption contributions of local excitations of the PBI host, P guests as well as a prominent CT-transition, which is perpendicularly oriented to the PBIs S0–S1 transition. Indeed, the calculated first excited state S1 of the cocrystal is of CT character (Fig. S38, S39, ESI‡). This transition is clearly observable in the UV/Vis spectrum in the range 600–750 nm (Fig. S38, ESI‡). While the pure host shows no absorption in this range, after addition of an excess of P an increase can be detected (Fig. 3). The higher excited states are localized either on the PBI host or the P guests, in order of increasing energy. The observed experimental trend is also reproduced by theoretical calculations (Fig. S40, ESI‡): compared to the computed PBI localized excitation (LE) state of 3a monomer, the corresponding absorptions of 3a dimer and [P·3a·P] are red shifted by 455 and 1121 cm−1, respectively, which is in accordance with the experimental findings. Similar calculations for the [T·3a·T] complex revealed no low-lying CT state between host and guest but only a LE state on the PBI unit. The stabilizing forces of [P·3a·P] and [T·3a·T] were characterized with energy decomposition analyses based on the absolutely localized molecular orbital approach (ALMO-EDA). All contributions are of similar magnitude in both complexes (Fig. S41, ESI‡). The major stabilizing interactions are provided by electrostatics and dispersion forces, while the CT contribution only plays a minor part. The lack of a significant CT contribution explains why intermolecular interactions between two electron-poor PBIs are stronger than those between an electron-poor PBI and an electron-rich P or T, thereby favouring narcissistic over social self-sorting.
In summary three sterically shielded PBIs 3a–c were synthesized and studied as supramolecular building blocks. Concentration-dependent UV/Vis absorption studies revealed dimerization constants up to 870 M−1 in chlorinated solvents and 43000 M−1 in MCH, as well as null-type exciton coupling which was explained by the almost orthogonal arrangement of the PBIs in single-crystal structures. 3a–c were next utilized for the binding of perylene and triphenylene which was comparably weak with binding constants up to K1 = 254 M−1 and K2 = 23 M−1 for 3a with two perylene guests in the chlorinated solvent TCE. Single crystals provided structural evidence for the 1:2 complexes [P·3a·P] and [T·3a·T] and revealed interesting polarization-dependent absorption properties for [P·3a·P] that warrant further studies for such CT-cocrystals.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- G. Briegleb, Elektronen-Donator-Acceptor-Komplexe, Springer, 1st edn, 1961 Search PubMed.
- R. Foster, J. Phys. Chem., 1980, 84, 2135–2141 CrossRef CAS.
- D. S. Weiss and M. Abkowitz, Chem. Rev., 2010, 110, 479–526 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed.
- B. A. Ikkanda and B. L. Iverson, Chem. Commun., 2016, 52, 7752–7759 RSC.
- A. Sikder, S. Chakraborty, P. Rajdev, P. Dey and S. Ghosh, Acc. Chem. Res., 2021, 54, 2670–2682 CrossRef CAS PubMed.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- R. Dubey and F. Würthner, Nat. Chem., 2023, 15, 884 CrossRef CAS PubMed.
- M. U. Schmidt and K. Hunger, Industrial organic color pigments, Wiley, 4th edn, 2004 Search PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- R. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed.
- M. L. Williams, I. Schlesinger, R. M. Jacobberger and M. R. Wasielewski, J. Am. Chem. Soc., 2022, 144, 18607–18618 CrossRef CAS PubMed.
- Y. Su, Y. Li, J. Liu, R. Xing and Y. Han, Nanoscale, 2015, 7, 1944–1955 RSC.
- I. Schlesinger, N. E. Powers-Riggs, J. L. Logsdon, Y. Qi, S. A. Miller, R. Tempelaar, R. M. Young and M. R. Wasielewski, Chem. Sci., 2020, 11, 9532–9541 RSC.
- P. Yu, Y. Li, H. Zhao, L. Zhu, Y. Wang, W. Xu, Y. Zhen, X. Wang, H. Dong, D. Zhu and W. Hu, Small, 2021, 17, 2006574 CrossRef CAS PubMed.
- M. Mahl, M. A. Niyas, K. Shoyama and F. Würthner, Nat. Chem., 2022, 14, 457–462 CrossRef CAS PubMed.
- M. A. Niyas, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2023, 62, e202302032 CrossRef CAS PubMed.
- M. Mahl, K. Shoyama, A. M. Krause, D. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 13401–13405 CrossRef CAS PubMed.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 RSC.
- C. Shao, M. Grüne, M. Stolte and F. Würthner, Chem. – Eur. J., 2012, 18, 13665–13677 CrossRef CAS PubMed.
- M. M. Salfont-Sempere, P. Osswald, K. Radacki and F. Würthner, Chem. – Eur. J., 2010, 16, 7380–7384 CrossRef PubMed.
- J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Chem. Sci., 2016, 7, 1729–1737 RSC.
- F. Fennel, S. Wolter, Z. Xie, P. A. Plötz, O. Kühn, F. Würthner and S. Lochbrunner, J. Am. Chem. Soc., 2013, 135, 18722–18725 CrossRef CAS PubMed.
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS PubMed.
- N. J. Hestand and F. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- bindfit (Supramolecular, 2020), http://supramolecular.org/, accessed July 27th, 2023.
- Vander Griend DA, SIVVU, https://sivvu.org/, accessed July 27th, 2023.
Footnotes |
| † All UV/Vis absorption spectra of the titration studies are available in the Zenodo repository https://doi.org/10.5281/zenodo.8196841. |
| ‡ Electronic supplementary information (ESI) available. Experimental and synthetic details, UV/Vis and fluorescence spectra, aggregation and complexation studies, crystallographic data, microscopy and theoretical calculations. CCDC 2284853–2284856. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03704e |
| This journal is © The Royal Society of Chemistry 2023 |