
Photocatalytic activity enhancement of Cu2O cubes functionalized with 2-ethynyl-6-methoxynaphthalene through band structure modulation†
Anindya Sundar
Patra
a,
Jui-Cheng
Kao
b,
Shang-Ju
Chan
a,
Po-Jung
Chou
a,
Jyh-Pin
Chou
 c,
Yu-Chieh
Lo
*b and
Michael H.
Huang
*a
c,
Yu-Chieh
Lo
*b and
Michael H.
Huang
*a
aDepartment of Chemistry and Frontier Research Center on Fundamental and Applied Sciences of Matters, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: hyhuang@mx.nthu.edu.tw
bDepartment of Materials Science and Engineering, National Yang Ming Chiao Tung University, Hsinchu 300093, Taiwan. E-mail: cylo@nycu.edu.tw
cDepartment of Physics, National Changhua University of Education, Changhua 50007, Taiwan
First published on 17th February 2022
Abstract
Since the exciting discovery that binding of 4-ethynylaniline on inert Cu2O cubes makes the functionalized cubes highly photocatalytically active toward methyl orange (MO) degradation, binding of 2-ethynyl-6-methoxynaphthalene (2E-6MN) molecules was found to produce a similar effect to activate Cu2O cubes. A moderate activity enhancement was observed for the functionalized Cu2O rhombic dodecahedra, while the activity of octahedra only improves slightly. Thus, the degree of photocatalytic activity enhancement is highly facet-dependent. Electron, hole, and radical scavenger tests confirm the photocatalysis results. Electrochemical impedance measurements also show a reduced charge transfer resistance after 2E-6MN modification for cubes and rhombic dodecahedra. Moreover, density functional theory (DFT) calculations have revealed the emergence of narrow bands within the band gaps of Cu2O{100} and {110} surfaces, but not on the {111} surface. Charge density distribution analysis is also insightful to understand the pronounced photocatalytic activity of the modified Cu2O cubes. Surface functionalization with conjugated molecules represents a new strategy to activate or enhance photoinduced charge transfer of semiconductor materials.
Introduction
Large facet-dependent photocatalytic activities have been observed in many semiconductor materials including Cu2O, Ag2O, SrTiO3, and Ag3PO4.1–10 For example, while {110}-bound Cu2O rhombic dodecahedra are photocatalytically more active than {111}-bound octahedra, {100}-terminated Cu2O cubes are inert even after surface Au deposition or the formation of Au–Cu2O core–shell cubes resulting from a lack of radical production upon light irradiation, attributed to a large surface band bending preventing charge carriers from exiting the crystals.11–13 Previous density functional theory (DFT) calculations have revealed the presence of a thin surface layer of less than 1.5 nm with dissimilar band structures for different crystal faces of Cu2O.14 Thus, as charge carriers exit Cu2O crystals, they are met with facet-specific barrier heights to either facilitate or inhibit charge transport. Furthermore, DFT calculations on varying numbers of Cu2O, Si, Ge, and GaAs surface planes have also indicated subtle deviations in bond length, bond geometry, and frontier orbital energy distribution between the highly conductive and less conductive faces, suggesting these orbital and bond level perturbations contribute to the band structure differences.15–18 Slight bond length variations may be observable from the XRD patterns of polyhedral Cu2O crystals.19 In addition to the inactivity of Au particle-decorated Cu2O cubes, binary semiconductor heterostructures formed between Cu2O cubes and ZnO, ZnS, CdS, and Ag3PO4 nanostructures remain photocatalytically inert despite their favorable band energy alignments, and sometimes suppressed activities are observed for the decorated Cu2O octahedra and rhombic dodecahedra.20–23 These results suggest mismatched or unfavorable interfacial band bending situations may be present depending on the contacting planes at the interface. Unexpectedly, Ag2S-deposited Cu2O cubes were found to be highly photocatalytically active possibly because the interfacial band bending is favorable, illustrating the unpredictable nature of photocatalytic activity in semiconductor heterostructures with well-defined planes for at least one material of the heterojunction.24Beyond the typical metal, graphene, and semiconductor deposition, as well as metal–semiconductor core–shell formation, to create semiconductor heterostructures for enhanced photocatalytic performance,25–31 surface molecular functionalization may be a new approach for increased photocatalytic activity.32 Recent surface functionalization of Cu2O crystals with conjugated 4-ethynylaniline (4-EA) showed an unexpected excellent photocatalytic activity for Cu2O cubes, while rhombic dodecahedra gave a notably improved activity and octahedra presented a moderate activity enhancement.33 DFT calculations have revealed a 4-EA-derived band within the band gap of Cu2O. The appearance of this band with energies close to the valence band of the {100}-terminated Cu2O, and the observed electron density distribution over the anchored molecule, are believed to make the inert cubes highly active. It is therefore highly interesting to explore the use of other conjugated molecules to promote photoexcited charge transfer.
In this study, 2-ethynyl-6-methoxynaphthalene (2E-6MN)-functionalized Cu2O cubes, octahedra, and rhombic dodecahedra were used for methyl orange photodegradation. Similar to 4-EA with an electron-donating –NH2 group, 2E-6MN contains an electron-donating –OCH3 substituent on the naphthalene ring. The photocatalytically inert Cu2O cubes became more active than that of octahedra after molecular modification. Moderate activity enhancement was generally observed for Cu2O rhombic dodecahedra, but surprisingly octahedra showed only slight activity change after 2E-6MN functionalization. Electron, hole, and radical scavenger tests confirmed the observed photocatalytic behaviors. DFT calculations have yielded very different band structure situations for the three surfaces of Cu2O to explain their photocatalytic behaviors. Interestingly, the occurrence of some activity suppression of 2E-6MN-modified Cu2O rhombic dodecahedra in one experiment was analyzed by DFT calculations, showing inadvertent hydrogen atom coverage could lead to such photocatalytic effect. This insight suggests ionic species and protons in the solution may unknowingly be involved in the photocatalytic process.
Results and discussion
In this work, cuprous oxide cubes, octahedra, and rhombic dodecahedra were synthesized following our reported procedures with slight modifications by preparing an aqueous mixture of sodium dodecyl sulfate (SDS) and CuCl2, followed by the addition of NaOH and then NH2OH·HCl as the reductant (see the ESI† for the synthesis conditions).34,35 Scanning electron microscopy (SEM) images of the synthesized Cu2O crystals are provided in Fig. S1 (ESI†), showing high size and shape uniformity. Fig. S2 (ESI†) gives the size distribution histograms of these samples. Cu2O cubes have an average edge length of 233 nm, while the average opposite corner length of octahedra is 273 nm. For rhombic dodecahedra, they have an average opposite face length of 263 nm. X-Ray diffraction (XRD) patterns of the synthesized crystals indicate formation of pure Cu2O (Fig. 1). Interestingly, while Cu2O cubes and octahedra have nearly identical diffraction peak positions, the rhombic dodecahedral crystals exhibit notable peak shifts. For example, the Cu2O(200) peak appears at 42.62° 2θ, but the (200) peak is at 42.40° 2θ for rhombic dodecahedra. Such XRD peak shifts have been observed before for Cu2O crystals.19 This may be experimental evidence of the existence of the thin surface layer with slight bond length and bond direction deviations. These average particle sizes were used to calculate the amounts of 2E-6MN molecules needed to functionalize 10 mg of each Cu2O sample with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1:10, and 1:100 molar ratios of surface copper atoms to 2E-6MN (see Fig. S3 and Table S1, ESI†). These large amounts of 2E-6MN were used to ensure dense molecular coverage on the Cu2O crystals, and photocatalysis occurs via electron transfer through the conjugated molecules. A total particle surface area of 0.03 m2 was chosen for fair photocatalytic activity comparison of different Cu2O polyhedra, and the required weights of pristine and modified Cu2O crystals were determined (Table S2, ESI†). After the molecular functionalization, no changes in the XRD patterns were observed (Fig. 1). Fig. 2 shows the preservation of particle shapes after anchoring 2E-6MN molecules.
:5, 1:10, and 1:100 molar ratios of surface copper atoms to 2E-6MN (see Fig. S3 and Table S1, ESI†). These large amounts of 2E-6MN were used to ensure dense molecular coverage on the Cu2O crystals, and photocatalysis occurs via electron transfer through the conjugated molecules. A total particle surface area of 0.03 m2 was chosen for fair photocatalytic activity comparison of different Cu2O polyhedra, and the required weights of pristine and modified Cu2O crystals were determined (Table S2, ESI†). After the molecular functionalization, no changes in the XRD patterns were observed (Fig. 1). Fig. 2 shows the preservation of particle shapes after anchoring 2E-6MN molecules.
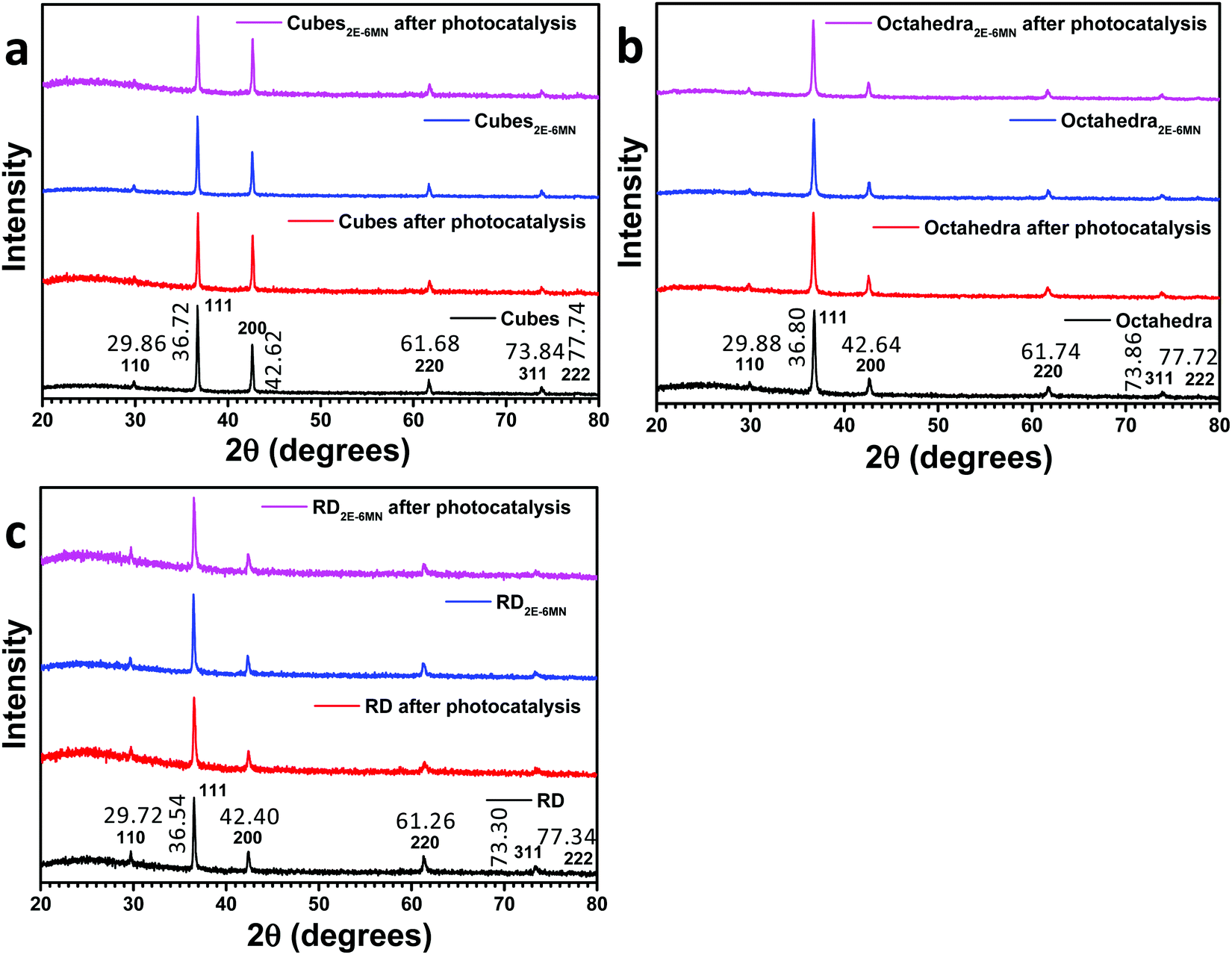 | ||
| Fig. 1 XRD patterns of Cu2O (a) cubes, (b) octahedra, and (c) rhombic dodecahedra before and after 2E-6MN functionalization and the photocatalysis experiment. | ||
 | ||
| Fig. 2 SEM images of Cu2O (a) cubes, (b) octahedra, and (c) rhombic dodecahedra functionalized with 1:100 molar ratios of surface Cu atoms to 2E-6MN molecules. | ||
To confirm molecular functionalization on the Cu2O crystals, Fourier-transform infrared (FT-IR) spectra were taken. Fig. 3 displays the FT-IR spectra of 2E-6MN and the 1:100 functionalized Cu2O samples. Fig S4 (ESI†) presents FR-IR spectrum of pristine Cu2O cubes with only a strong Cu(I)–O vibration peak at 622 cm−1. FT-IR spectra of Cu2O cubes, octahedra and rhombic dodecahedra have been reported.36 2E-6MN shows a peak at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3257 cm−1, corresponding to the stretching vibrations of the acetylenic hydrogen (C
= 3257 cm−1, corresponding to the stretching vibrations of the acetylenic hydrogen (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H).33 The peaks around 3000 cm−1 should arise from the C–H bond stretching modes of naphthalene and the methoxy group.37 Moreover, the peaks due to the C
C–H).33 The peaks around 3000 cm−1 should arise from the C–H bond stretching modes of naphthalene and the methoxy group.37 Moreover, the peaks due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the aromatic ring show up at = 1625 and 1598 cm−1. After surface modification, the acetylenic hydrogen peak disappears for all the samples, confirming successful formation of C–Cu bonds on the Cu2O crystals.19,33,38 Moreover, the C–H bond stretching peaks around 3000 cm−1 also become invisible after the molecular functionalization. The CC stretching vibration peaks and other characteristic peaks of the molecule with frequencies below 1650 cm−1 also become very weak after anchoring to the Cu2O crystals. Such spectral feature has also been observed for 4-EA binding to Cu2O.33 It is suggested that the surface band structure tuning of Cu2O after molecular functionalization causes such weakening or loss of infrared absorption peaks.
C stretching vibrations of the aromatic ring show up at = 1625 and 1598 cm−1. After surface modification, the acetylenic hydrogen peak disappears for all the samples, confirming successful formation of C–Cu bonds on the Cu2O crystals.19,33,38 Moreover, the C–H bond stretching peaks around 3000 cm−1 also become invisible after the molecular functionalization. The CC stretching vibration peaks and other characteristic peaks of the molecule with frequencies below 1650 cm−1 also become very weak after anchoring to the Cu2O crystals. Such spectral feature has also been observed for 4-EA binding to Cu2O.33 It is suggested that the surface band structure tuning of Cu2O after molecular functionalization causes such weakening or loss of infrared absorption peaks.
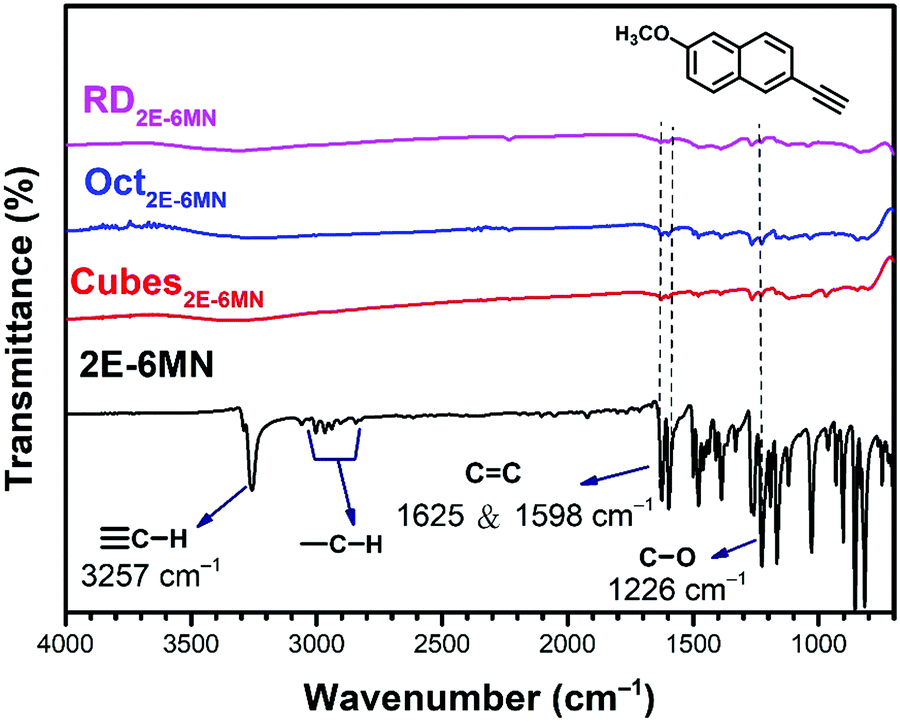 | ||
| Fig. 3 FT-IR spectra of 2E-6MN molecules and the 2E-6MN-functionalized Cu2O cubes, octahedra, and rhombic dodecahedra. The molecular structure is also shown. | ||
To further characterize the samples after 2E-6MN modification, diffuse reflectance spectra of the pristine and functionalized Cu2O crystals were taken and are shown in Fig. S5 (ESI†). Tauc plots from the diffuse reflectance spectra give their band gaps. The synthesized Cu2O cubes, octahedra, and rhombic dodecahedra have band gaps of ∼1.92, 1.83, and 1.99 eV, respectively. They also have distinctively different solution colors. After 2E-6MN functionalization, only the cube sample shows a slight band gap shift to 1.88 eV. This is expected, since molecular modification should have little effect on the band gaps of large semiconductor particles. Band gap variation is observable in these samples because band gap value has both size and facet components.39,40
UV–vis absorption spectra of methyl orange as a function of photoirradiation time in the presence of pristine and 2E-6MN-modified Cu2O cubes, octahedra, and rhombic dodecahedra are available in Fig. S6–S8 (ESI†). Fig. 4 summarizes the photocatalysis results. Cu2O cubes remained photocatalytically inert, but remarkably they exhibited a good photocatalytic activity after surface modification with 2E-6MN molecules. Increase in the molecular loading led to a considerable activity enhancement, finishing the photodegradation reaction in 120 min for cubes with a Cu:2E-6MN molar ratio of 1:5 to only 90 min for a molar ratio of 1:100. This example shows again that proper surface molecular modification can be a good strategy to boost photocatalytic activity. For rhombic dodecahedra, only moderate activity improvement was generally recorded. The reaction completion shortens from 40 min to 30 min for the 1:5 sample and 25 min for the 1:10 and 1:100 samples. Thus, there is no further enhancement beyond 1:10 usage of 2E-6MN molecules. Surprisingly, in one experiment, some activity suppression was observed for the functionalized rhombic dodecahedra, and increasing the 2E-6MN loading actually produced a greater activity suppression (Fig. S9, ESI†). This result has been analyzed by DFT calculations and will be discussed later. Unexpectedly, practically no or only slight activity enhancement was measured for Cu2O octahedra after 2E-6MN modification. Such outcome illustrates that molecular functionalization does not always produce similar effects to all the samples; the resultant photocatalytic behaviors can still be highly facet-dependent.
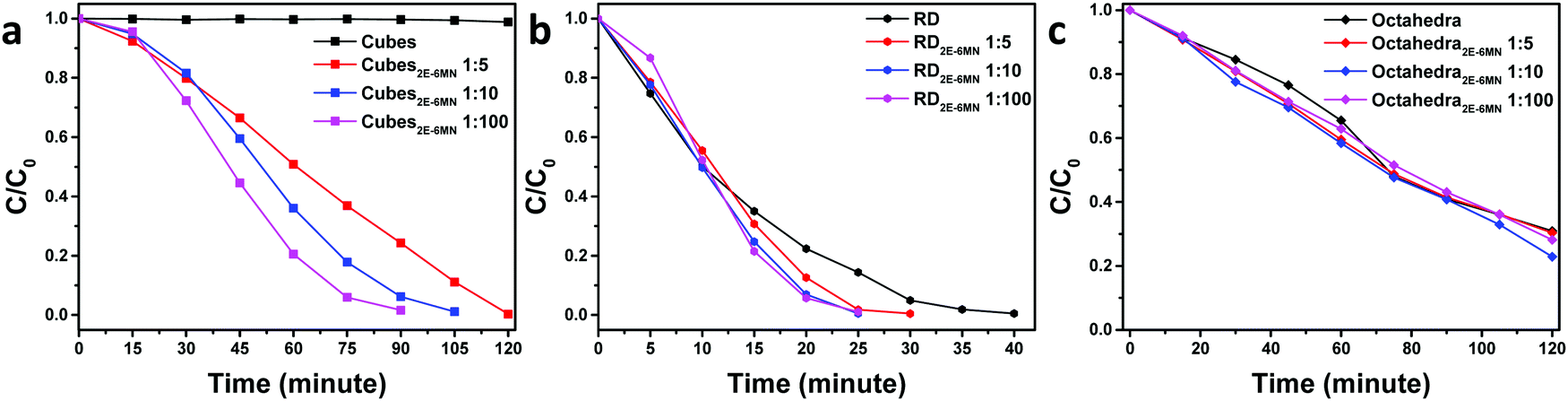 | ||
| Fig. 4 Extents of methyl orange photodegradation with respect to the irradiation time for pristine and 2E-6MN-functionalized Cu2O (a) cubes, (b) rhombic dodecahedra, and (c) octahedra. | ||
To confirm the sudden appearance of photocatalytic activity in the 2E-6MN-modified Cu2O cubes, electron, hole, and radical scavenger tests were performed on the 1:100 sample. In photocatalysis, the photogenerated electrons and holes should migrate to the crystal surfaces and react with water and/or dissolved oxygen to produce radical species, which then attack the dispersed organic molecules. Fig. 5 shows the electron and hole scavenger results with CrO3 acting as an electron scavenger, and sodium oxalate (Na2C2O4) was added to captures holes. Additional scavenger results using 1 and 5 μmol of Na2C2O4 and 1 μmol of CrO3 are provided in Fig. S10, ESI.† In the presence of 3 μmol of CrO3, the good photocatalytic activity of the functionalized Cu2O cubes became completely quenched. Adding 3 μmol of Na2C2O4, photocatalytic activity was reduced by roughly half. Use of 5 μmol of Na2C2O4 can nearly eliminate photocatalytic activity. These results show both electrons and holes are used to degrade methyl orange. The 2E-6MN modification on the surface should facilitate electron transfer through the conjugated molecular structure.
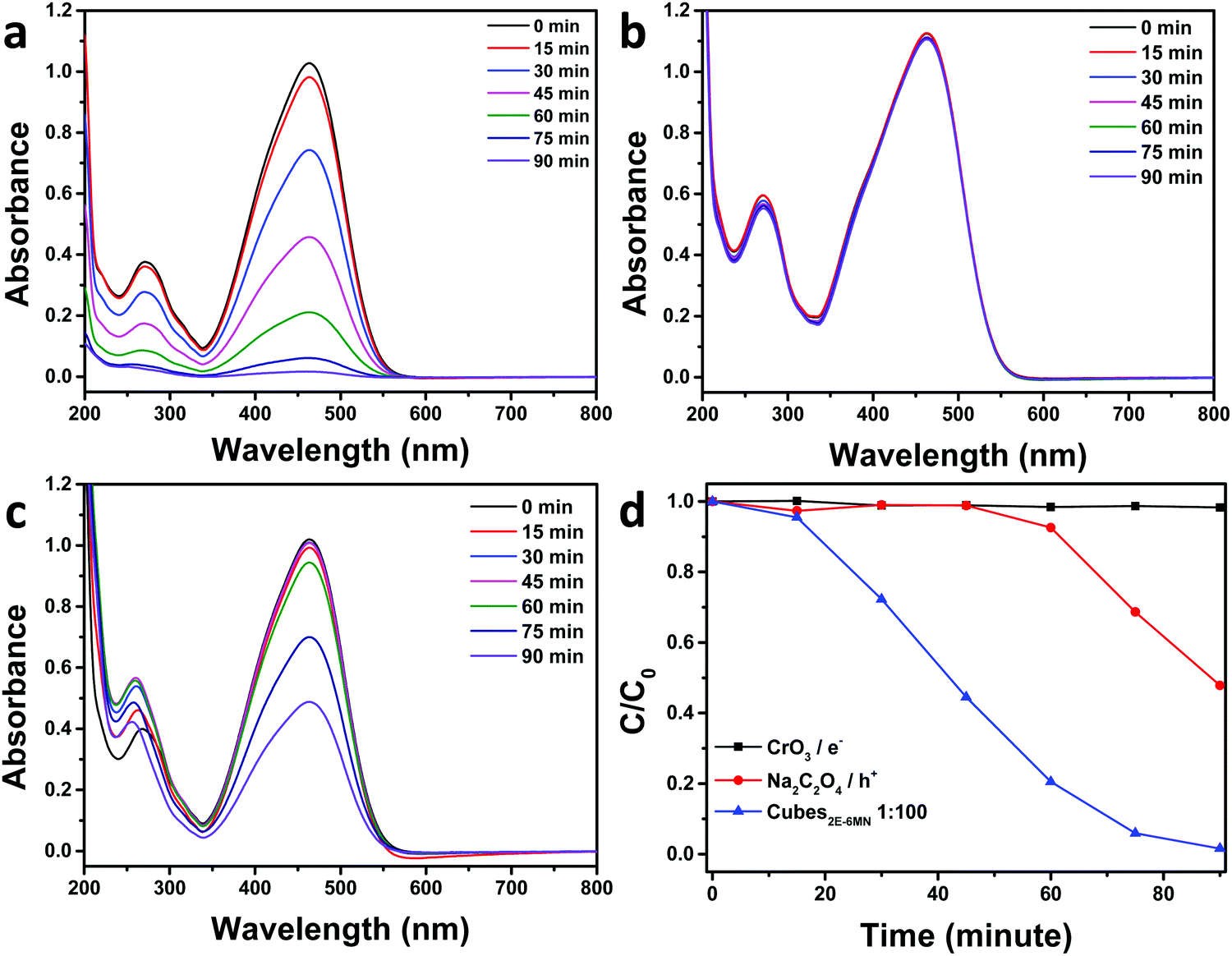 | ||
| Fig. 5 UV–vis absorption spectra of methyl orange as a function of irradiation time using (a) 1:100 2E-6MN-modified Cu2O cubes as the photocatalyst and in the presence of (b) 3 μmol of CrO3 and (c) 3 μmol of Na2C2O4. (d) Summary of the electron and hole scavenger tests. | ||
Electron paramagnetic resonance (EPR) measurements can further confirm the observed photocatalysis results. Fig. 6 presents the recorded EPR spectra using DMPO to capture radical species produced during the photocatalysis process. The spectra with a quartet of peaks indicate formation of largely DMPO–OH, so hydroxyl radicals (˙OH) are the major radical species produced under light illumination. Consistent with the photocatalytic inertness of Cu2O cubes, there were essentially no EPR signals for the pristine Cu2O cubes. After 2E-6MN functionalization, strong quartet peaks were recorded, confirming the presence of good photocatalytic activity in the modified cubes. Stronger EPR signals were observed for the modified rhombic dodecahedra, and both pristine and modified octahedra displayed similar EPR peak intensities. The EPR measurements match nicely to the photocatalysis results.
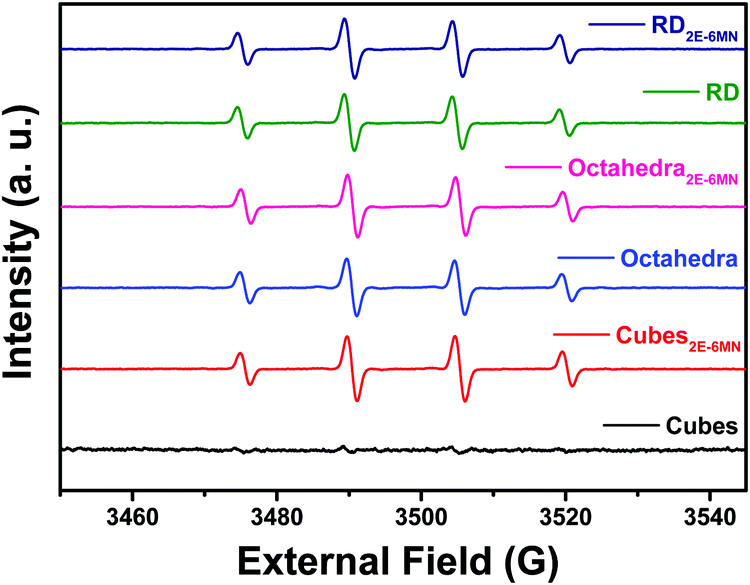 | ||
| Fig. 6 EPR spectra of photoirradiated pristine and 2E-6MN-functionalized Cu2O polyhedra. | ||
Electrochemical impedance spectroscopic (EIS) measurements were also performed to check charge transfer resistance before and after 2E-6MN functionalization. Fig. 7 is the collected Nyquist impedance data. Both the modified Cu2O cubes and rhombic dodecahedra present smaller semicircles, indicating decreased charge transfer resistance after {100} and {110} surface modification. The impedance plot for octahedra appears similar after 2E-6MN functionalization. These EIS data agree reasonably well with the photocatalysis results. Table S3 (ESI†) provides the determined charge transfer resistance (Rct) values. The dramatic change in the photocatalytic activity of Cu2O cubes after surface decoration with 2E-6MN can be rationalized by drawing the modified band diagrams for different Cu2O surfaces (Fig. S11, ESI†). A downward surface band bending is drawn for rhombic dodecahedra and octahedra to represent they are photocatalytically active. For cubes, there should be a large upward band bending to inhibit conduction band electrons from exiting the Cu2O{100} surface. The photogenerated electrons and holes then recombine inside the crystal. After functionalized with 2E-6MN, the surface is constituted of the original thin surface layer giving the various facet-dependent effects plus the dense molecular layer. Because the surface is now different, the band structure should be different, and the surface band bending can change. This is the concept of how surface molecular modification can be useful to tune the photocatalytic or charge transfer properties of semiconductor crystals. A downward band bending is presented for the {100} faces of cubes. For rhombic dodecahedra and octahedra, they also have varying degrees of downward band bending to reflect their enhanced and unchanged photocatalytic properties.
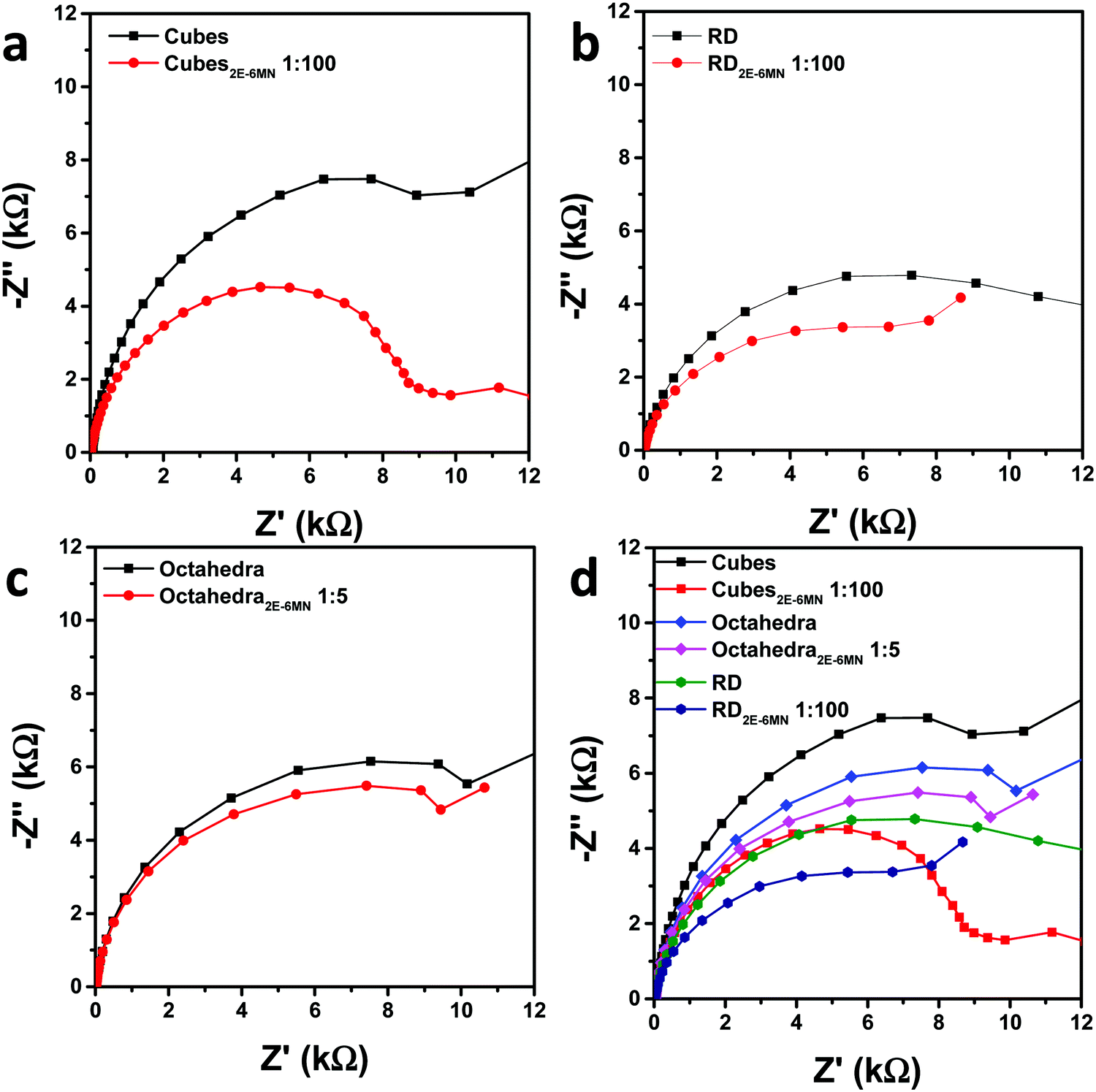 | ||
| Fig. 7 Nyquist impedance plots for pristine and 2E-6MN-functionalized (a) cubes, (b) rhombic dodecahedra, and (c) octahedra. (d) Summary of the impedance plots. | ||
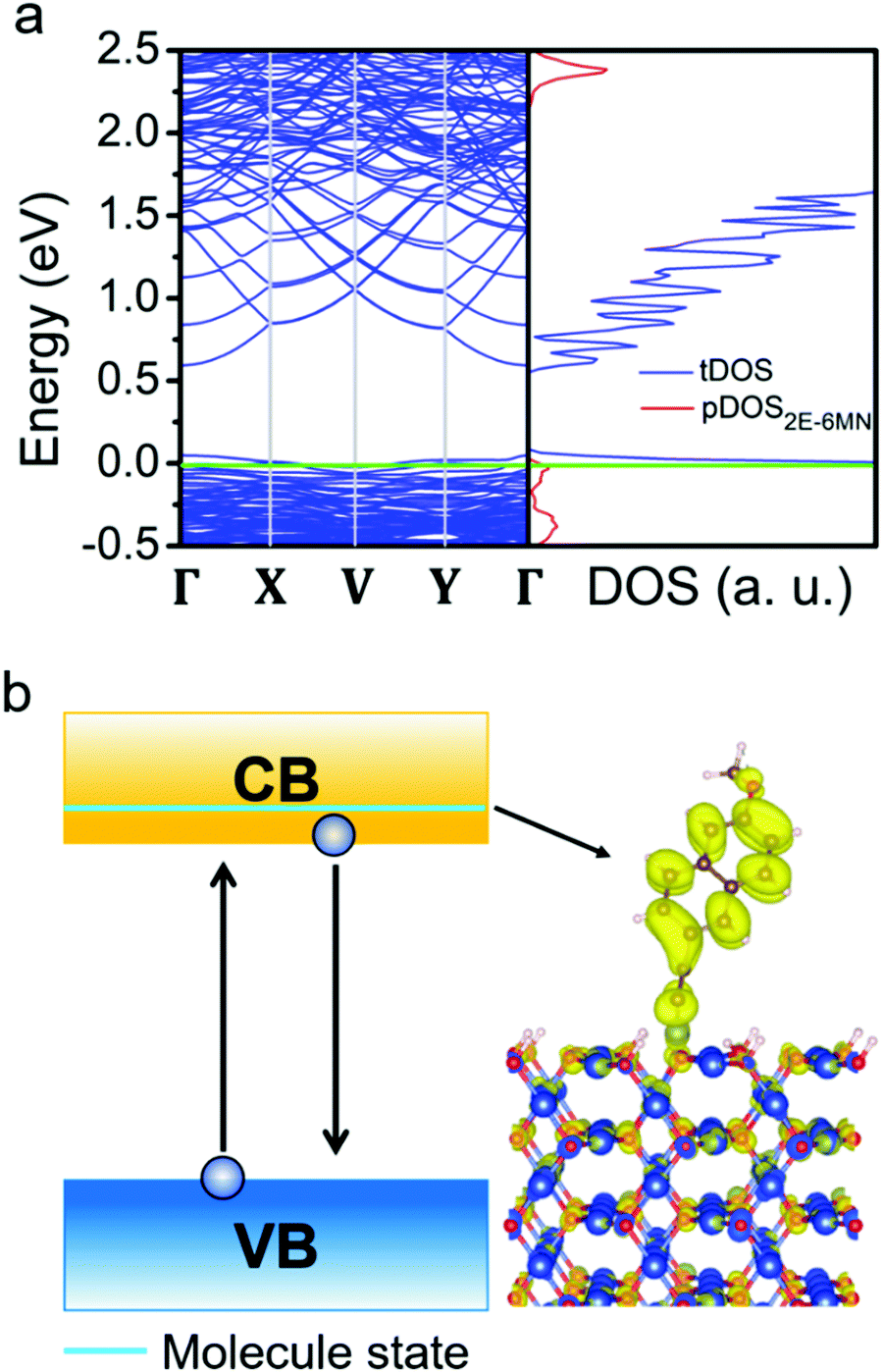
To substantiate the presumption that band structures of Cu2O surfaces are tuned upon 2E-6MN functionalization, DFT calculations were performed. Fig. 8 provides the obtained band structures and the corresponding density of states (DOS) of the Cu2O{100}, {110}, and {111} surfaces before and after surface modification with 2E-6MN molecules. The Cu2O band structures and DOS plots before surface modification are consistent with the previous works (Fig. 8a–c).33,41–44 After the molecular functionalization, significant changes in the band structures, particularly for the {100} and {110} surfaces, can be easily identified. Fig. 8d shows three new bands with values above 0.25 eV appear after molecular modification on the {100} surface. The partial density of states (pDOS) of the 2E-6MN molecule, shown as the red curves in the total DOS (tDOS) plot, confirms the molecular contribution in these three bands. This is a clear evidence that Cu2O band structure can be greatly modulated through surface molecular grafting. Likewise, a new band and its corresponding pDOS peak also appear above 0.25 eV in the modified {110} surface (Fig. 8e). For the modified {111} surface, there is no new band emerged above the valence band maximum (VBM) (Fig. 8f). Note that there are also 2E-6MN-contributed pDOS distributions within the valence bands for the three modified surfaces, especially an explicit peak in the case of {110} surface.
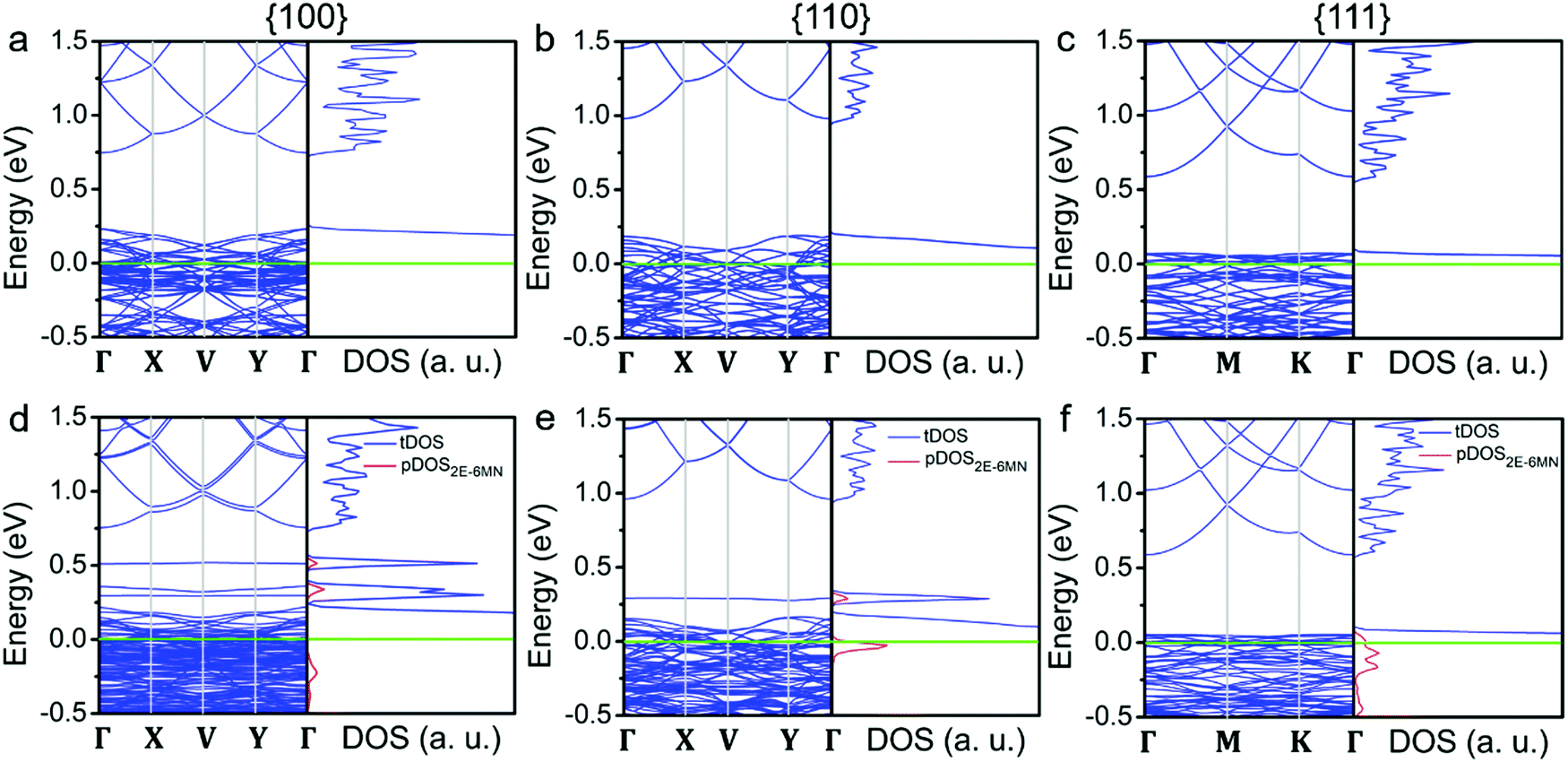 | ||
| Fig. 8 (a–c) Band structures and density of states for the three different Cu2O crystallographic surfaces and (d–f) 2E-6MN-modified surfaces. Fermi level (green line) is set at zero in all band structures and density of states. The red peaks are the partial density of states (pDOS) from 2E-6MN molecules. | ||
To determine the significance of the new bands formed in the band structures of the modified {100} and {110} surfaces, the partial charge density distribution in real space is presented in Fig. 9, using the band-decomposed charge density analysis. Fig. 9a shows the three different distributions of partial charge density in the {100} case, denoted as adsorbed band −1, −2, and −3, which should correspond to the 2E-6MN-derived bands seen in the band structure of the modified {100} surface. The charge density distribution of the adsorbed band −1 is mostly on the Cu2O surface, while the charge density distribution of the adsorbed band −3 is localized at the junction between the surface and the 2E-6MN molecule. As for the adsorbed band −2, the charge density distributes over both the molecule and partial Cu2O surface. In Fig. 9b, the partial charge distribution of the new band in the band gap of the modified {110} surface, similar to the case of the adsorbed band −3 of the {100} surface, is localized at the junction between the Cu2O surface and the molecule. An interesting finding is that an energy state corresponding to the explicit pDOS peak within the valence band in Fig. 8e produces a uniform charge density distribution covering the entire 2E-6MN molecule. Here it is denoted as the 2E-6MN binding state. For the modified {111} surface, calculation of the partial charge density for the pDOS within the valence band shows that the charge density distribution is mostly on and below the Cu2O surface and barely on the molecule (see Fig. 9c).
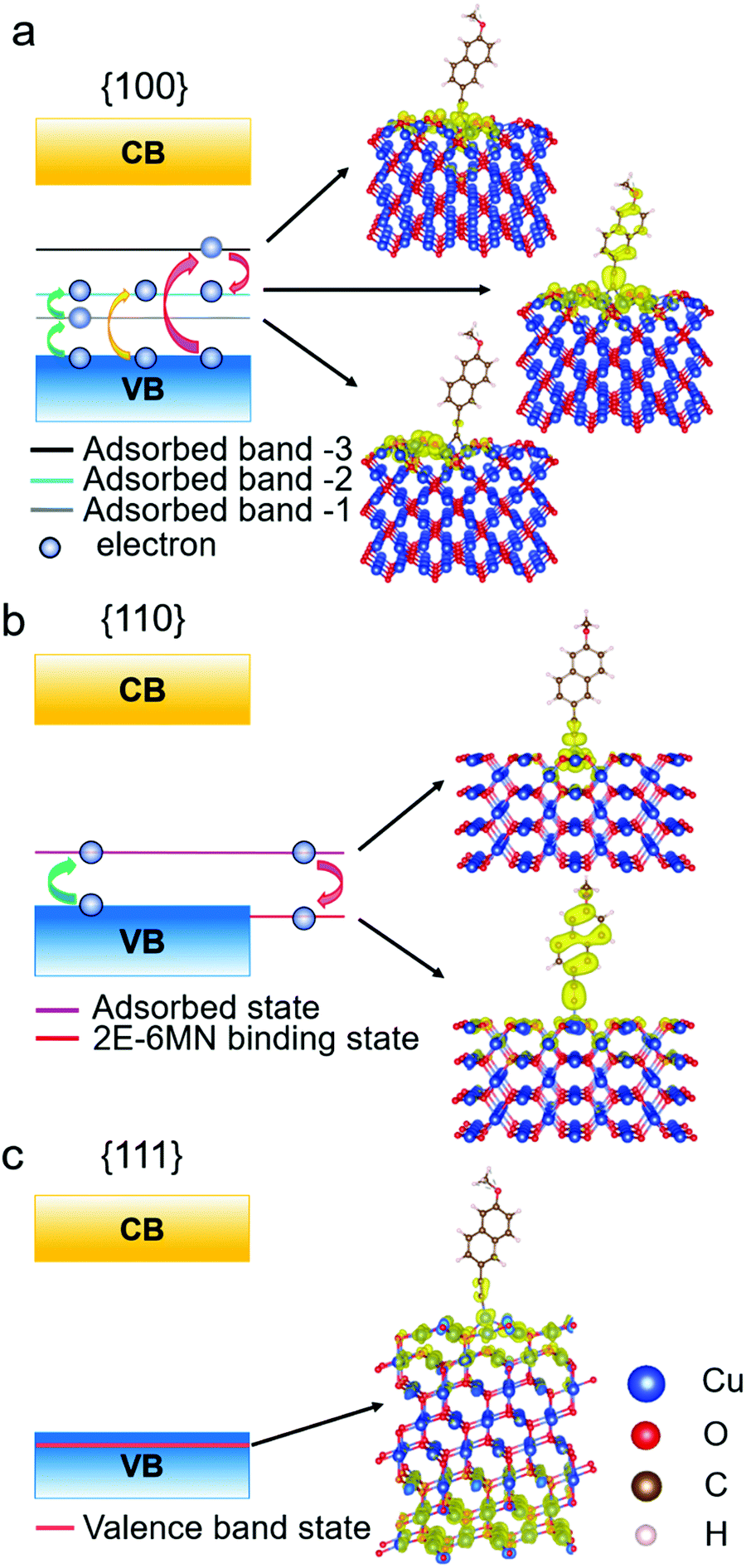 | ||
| Fig. 9 Schematic illustration of the band diagrams and partial charge density distributions in real space for functionalized Cu2O (a) {100}, (b) {110}, and (c) {111} surfaces. | ||
These DFT results are quite useful to explain the large variation in the photodegradation efficiency among the different Cu2O samples before and after modifying with 2E-6MN molecules, that there are two conditions controlling the injection of electrons after the surface modification. First, at least one adsorbed-molecule-induced band (adsorbed states) must be within the band gap. Second, their corresponding charge density needs to be distributed over the molecule. Similar phenomenon has been observed before.33 Hence, for the 2E-6MN-modified {100} surface, excited electrons are inclined to inject into the adsorbed band −2, in which the partial charge density distribution is on the molecule. Then the electrons will be able to leave the surface through the conjugated system of 2E-6MN because the escaping probability of electrons becomes high. If the 2E-6MN coverage on the surface increases, more adsorbed bands form within the band gap to facilitate charge transfer. In the {110} case, the localized charge density distribution of the adsorbed state could give rise to an electron trapping effect (see Fig. 9b). Once an electron is injected into this adsorbed state, it may be trapped at the junction between the molecule and the crystal surface. Even if the electron can disengage from or bypass the junction to arrive at the 2E-6MN molecule, it can drop back to the 2E-6MN binding state in the valence band, so overall photocatalytic activity cannot be much enhanced. The increase of surface molecular coverage will enhance this trapping effect, so it is not helpful to the photodegradation efficiency of Cu2O rhombic dodecahedra.
To investigate why moderate photocatalytic activity suppression after 2E-6MN functionalization can sometimes occur as seen in Fig. S9 (ESI†), DFT calculations show that some unintended species in the solution such as hydrogen protons can greatly alter the band structure of the surface modification, leading to the disappearance of the 2E-6MN band in the band gap (see Fig. 10). This situation can give the observed decreases in photocatalytic efficiency in the modified Cu2O rhombic dodecahedral samples. Of course, surface proton coverage may not necessarily be the cause for a lowered photocatalytic efficiency, as an acidic condition should be avoided for the structural integrity of Cu2O crystals.45 However, this interesting finding suggests that some solution species may unknowingly affect photocatalytic activity, and this could be a reason why slight photocatalytic activities have been reported for Cu2O nanocubes.46,47 For the {111} case, no adsorbed states are formed within the band gap after surface modification (Fig. 9c). Consequently, the 2E-6MN molecules cannot tune the band structure and the corresponding DOS of the Cu2O{111} surface, so the molecular functionalization does not really affect the photocatalytic properties of the modified octahedra.
 | ||
| Fig. 10 (a) Band structure and the corresponding density of states of the hydrogen atom-terminated and 2E-6MN-modified Cu2O{110} surface that is half covered with hydrogen atoms. Fermi level (green line) is set at zero in the band structure and density of states. The red peaks are the partial density of states (pDOS) of 2E-6MN molecules. (b) Schematic illustration of the band diagram and partial charge density distribution in real space of the hydrogen-terminated and 2E-6MN-modified Cu2O{110} surface. The molecular states are now promoted to the conduction band after the hydrogen atom coverage. Hence, the 2E-6MN molecules may not affect the injection of photoexcited electrons. | ||
Conclusions
The surfaces of Cu2O cubes, octahedra, and rhombic dodecahedra were modified with 2E-6MN molecules to explore the effect of this molecular functionalization on their photocatalytic properties. The inactive Cu2O cubes showed a good photocatalytic activity after 2E-6MN functionalization, demonstrating surface modification with a conjugated molecule is an effective approach to boost the photocatalytic activity of Cu2O cubes and perhaps other semiconductor materials. For Cu2O rhombic dodecahedra, only moderate activity enhancement was observed. Unexpectedly, practically no enhancement was recorded for the modified octahedra. These results reveal that the photocatalytic outcome of molecular functionalization can differ greatly depending on the anchored crystal surfaces, and the effects also vary for different molecules. Electron, hole, and radical scavenger tests were performed to confirm the photocatalytic results. EIS measurements also support the photocatalysis data. DFT calculations show notable changes to the band structures after 2E-6MN functionalization with the emergence of new bands within the band gap of Cu2O to facilitate charge transfer for the {100} and {110} surfaces. No new bands appear for the {111} surface after adding 2E-6MN, which is consistent with the experimental observation. It is anticipated that molecular functionalization should open new possibilities to photocatalytic activity enhancement beyond the traditional strategies of metal particle deposition, surface coverage with graphene sheets, and the formation of semiconductor heterojunctions.Experimental
Molecular functionalization of Cu2O crystals
Synthesis procedures of Cu2O crystals and the calculated amounts of 2E-6MN molecules needed are provided (see Tables S1 and S2, ESI†). First, 10 mg of Cu2O particles were dispersed in 2.0 mL of 99% ethanol, followed by the addition of calculated amounts of 2E-6MN in 3 mL of ethanol having surface Cu atoms to 2E-6MN molar ratios of 1:5, 1:10, and 1:100. After stirring for 4 h at room temperature, the particles were washed twice with ethanol. After centrifugation at 7000 rpm for 5 min, particles were dried by purging with nitrogen before storage. For dye photodegradation assessment, the same total surface area of 0.03 m2 for pristine and 2E-6MN-functionalized Cu2O particles was chosen. Here 6.99, 4.72, and 7.89 mg of Cu2O cubes, octahedra, and rhombic dodecahedra were used, respectively. To a cubic quartz reactor, 45 mL of aqueous MO solution with a dye concentration of 15 ppm was added. A 500 W Xenon lamp with a Y-43 filter blocking UV light as the illumination source was placed 30 cm away from the quartz reactor. A light intensity of 300 mW cm−2, measured by a power meter, illuminated the reactor. After certain time intervals, 1 mL of the solution from the reactor was taken out and centrifuged at 7500 rpm for 3 minutes. The supernatant was placed in a UV–vis spectrophotometer for spectral measurements.
Electron paramagnetic resonance analysis
DMPO (5,5-dimethyl-1-pyrolin-N-oxide, 98%, Matrix Scientific) was used to detect radical generation in the solution. Prior to an experiment, commercial DMPO was highly purified by activated charcoal and quickly checked to make sure there are practically no EPR signals. A 1.0 M DMPO solution was prepared by dissolving 1.0 g of DMPO in 8.8 mL of ice-cold Milli-Q water that has been purged with nitrogen gas overnight to remove oxygen in Milli-Q water. Aliquots of the DMPO solutions were frozen at −20 °C until use. Then 2 mL of 1.0 M DMPO solution was filtered three times with activated charcoal. After filtering completely, 0.1, 0.5, and 0.2 mL pristine and functionalized Cu2O cubes, octahedra, and rhombic dodecahedra with a concentration of 1.0 mg mL−1 were dispersed into quartz cells containing respectively 0.8, 0.4, and 0.7 mL of deionized water. Next, 0.1 mL of the purified 1.0 M DMPO solution was added to give a total solution volume of 1 mL. The solutions were irradiated by a xenon lamp with stirring for 2 min, and sent the samples for EPR analysis immediately.Electrochemical measurements
EIS data were collected using a CHI 6273E electrochemical analyzer. The electrochemical cell consists of a Pt wire as the counter electrode, photocatalyst (nearly 1 mg) coated on an indium tin oxide glass (ITO) as the working electrode, Ag/AgCl as the reference electrode, and 0.5 M K2SO4 as the electrolytic solution. Cu2O particles were coated on the ITO glass covering an area of 1 × 1 cm2. A frequency range from 50 mHz to 100 KHz and an amplitude of 5 mV was used to carry out the electrochemical impedance spectroscopic measurements.Computational details
The first-principles calculations within the density functional theory with the projector augmented wave (PAW) method48,49 were performed using the Vienna ab initio simulation package (VASP).50,51 The Perdew–Burke–Ernzerhof (PBE) functional under the generalized-gradient approximation (GGA) was used to describe the exchange and correlation interaction in the structural optimization calculations, band structure calculations, density of states (DOS) calculations, and the band-decomposed charge density analysis. The kinetic energy cut-off for the plane-wave basis set was 400 eV and the Brillouin zone was sampling with 8 × 8 × 8 Monkhorst–Pack k-point grids for the Cu2O primitive cell. For the 2E-6MN molecule, a 520 eV cut-off energy for the plane-wave and 1 × 1 × 1 k-point grid was used. The geometric structures of the Cu2O crystal and the 2E-6MN molecule were relaxed using the conjugated-gradient method. A value of 1.0 × 10−5 eV was used in the convergence accuracy of the electronic relaxation for both the Cu2O crystal and 2E-6MN molecule. Gaussian smearing was used with a smearing width of 0.01 eV. The equilibrium lattice constant of the Cu2O primitive cell was 4.31 Å by the structural optimization under the PBE functional.Subsequently, {100}, {110}, and {111} surfaces of Cu2O crystals modified with 2E-6MN molecules were created in the supercells based on the optimized primitive cell. These supercells had a vacuum layer larger than 10 Å separating the periodic boundaries in the z-direction. For those surfaces modifying with 2E-6MN molecules, the spacing between molecules is more than 8 Å to control the molecular coverage on the crystal surfaces. Those Cu2O surface models were subjected to energy minimization calculations again with the k-point mesh of 3 × 3 × 1, 3 × 2 × 1, and 3 × 3 × 1 for the {100}, {110}, and {111} surfaces, respectively. After obtaining the optimized models of the functionalized surfaces, we conducted the electronic structure calculations including band structure and DOS. In the band structure and the corresponding DOS calculations, the Fermi level was used as a reference energy in order to compare surface models with and without 2E-6MN functionalization. The band-decomposed charge density analysis was performed after the band structure calculations.
Instrumentation
A JEOL JSM-7000F scanning electron microscope was used to observe the crystals. A Shimadzu XRD-6000 diffractometer with Cu Kα radiation was used to record the XRD patterns of the samples. FT-IR spectra of the samples were recorded on a PerkinElmer Spectrum Two FT-IR spectrometer. A xenon lamp (X500 from Blue Sky Technologies) was used as the light source in the photodegradation experiments. UV–Vis diffuse reflectance spectra and absorption spectra were recorded by a JASCO V-670 spectrophotometer. EPR spectra were obtained on a Bruker ELEXSYSE 580 CW/Pulse spectrometer.Author contributions
Anindya Sundar Patra: investigation, writing – original draft. Jui-Cheng Kao: formal analysis, methodology, visualization. Shang-Ju Chan: investigation. Po-Jung Chou: investigation. Jyh-Pin Chou: formal analysis. Yu-Chieh Lo: supervision, writing – original draft. Michael H. Huang: conceptualization, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Ministry of Science and Technology, Taiwan (MOST 107-2113-M-007-013-MY3, 109-2811-M-007-516, 109-2634-F-007-023, 110-2634-F-009-027, and 109-2221-E-009-058). Y.-C. Lo also thanks the Center for Semiconductor Technology Research from The Featured Areas Research Center Program within the framework of Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. Prof. Chih-Shan Tan of National Yang Ming Chiao Tung University provided the EIS facility.References
- M. H. Huang, G. Naresh and H.-S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4–15 CrossRef CAS PubMed
.
- S. Rej, M. Bisetto, A. Naldoni and P. Fornasiero, J. Mater. Chem. A, 2021, 9, 5915–5951 RSC
- W. Chen, Q. Liu, S. Tian and X. Zhao, Appl. Surf. Sci., 2019, 470, 807–816 CrossRef CAS
- W. Tu, W. Guo, J. Hu, H. He, H. Li, Z. Li, W. Luo, Y. Zhou and Z. Zou, Mater. Today, 2020, 33, 75–86 CrossRef CAS
- Y.-J. Chen, Y.-W. Chiang and M. H. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 19672–19679 CrossRef CAS PubMed
- P.-L. Hsieh, G. Naresh, Y.-S. Huang, C.-W. Tsao, Y.-J. Hsu, L.-J. Chen and M. H. Huang, J. Phys. Chem. C, 2019, 123, 13664–13671 CrossRef CAS
- M.-S. Hsieh, H.-J. Su, P.-L. Hsieh, Y.-W. Chiang and M. H. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 39086–39093 CrossRef CAS PubMed
- H. Wang, J. Yang, X. Li, H. Zhang, J. Li and L. Guo, Small, 2012, 8, 2802–2806 CrossRef CAS PubMed
- Y. P. Xie, G. Liu, L. Yin and H.-M. Cheng, J. Mater. Chem., 2012, 22, 6746–6751 RSC
- D. Majumder, I. Chakraborty, K. Mandal and S. Roy, ACS Omega, 2019, 4, 4243–4251 CrossRef CAS PubMed
- G.-Z. Yuan, C.-F. Hsia, Z.-W. Lin, C. Chiang, Y.-W. Chiang and M. H. Huang, Chem. – Eur. J., 2016, 22, 12548–12556 CrossRef CAS PubMed
- C.-Y. Chu and M. H. Huang, J. Mater. Chem. A, 2017, 5, 15116–15123 RSC
- W.-C. Wang, L.-M. Lyu and M. H. Huang, Chem. Mater., 2011, 23, 2677–2684 CrossRef CAS
- C.-S. Tan, S.-C. Hsu, W.-H. Ke, L.-J. Chen and M. H. Huang, Nano Lett., 2015, 15, 2155–2160 CrossRef CAS PubMed
- C.-S. Tan and M. H. Huang, Inorg. Chem. Front., 2021, 8, 4200–4208 RSC
- C.-S. Tan and M. H. Huang, Chem. – Eur. J., 2017, 23, 11866–11871 CrossRef CAS PubMed
- C.-S. Tan and M. H. Huang, Chem. – Asian J., 2018, 13, 1972–1976 CrossRef CAS PubMed
- C.-S. Tan and M. H. Huang, Chem. – Asian J., 2019, 14, 2316–2321 CrossRef CAS PubMed
- M. Madasu and M. H. Huang, Catal. Sci. Technol., 2020, 10, 6948–6952 RSC
- M. H. Huang and M. Madasu, Nano Today, 2019, 28, 100768 CrossRef CAS
- J.-Y. Huang, P.-L. Hsieh, G. Naresh, H.-Y. Tsai and M. H. Huang, J. Phys. Chem. C, 2018, 122, 12944–12950 CrossRef CAS
- G. Naresh, P.-L. Hsieh, V. Meena, S.-K. Lee, Y.-H. Chiu, M. Madasu, A.-T. Lee, H.-Y. Tsai, T.-H. Lai, Y.-J. Hsu, Y.-C. Lo and M. H. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 3582–3589 CrossRef CAS PubMed
- G. Naresh, A.-T. Lee, V. Meena, M. Satyanarayana and M. H. Huang, J. Phys. Chem. C, 2019, 123, 2314–2320 CrossRef CAS
- T.-Y. Liang, S.-J. Chan, A. S. Patra, P.-L. Hsieh, Y.-A. Chen, H.-H. Ma and M. H. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 11515–11523 CrossRef CAS PubMed
- Y.-C. Pu, H.-Y. Chou, W.-S. Kuo, K.-H. Wei and Y.-J. Hsu, Appl. Catal., B, 2017, 204, 21–32 CrossRef CAS
- X. Li, D. Wei, L. Ye and Z. Li, Inorg. Chem. Commun., 2019, 104, 171–177 CrossRef CAS
- Y.-S. Chang, P.-Y. Hsieh, T.-F. M. Chang, C.-Y. Chen, M. Sone and Y.-J. Hsu, J. Mater. Chem. A, 2020, 8, 13971–13979 RSC
- Y.-H. Zhang, M.-M. Liu, J.-L. Chen, S.-M. Fang and P.-P. Zhou, Dalton Trans., 2021, 50, 4091–4111 RSC
- Y.-H. Chiu, T.-F. M. Chang, C.-Y. Chen, M. Sone and Y.-J. Hsu, Catalysts, 2019, 9, 430 CrossRef CAS
- M.-Y. Kuo, C.-F. Hsiao, Y.-H. Chiu, T.-H. Lai, M.-J. Fang, J.-Y. Wu, J.-W. Chen, C.-L. Lin, K.-H. Wei, H.-C. Lin and Y.-J. Hsu, Appl. Catal., B, 2019, 242, 499–506 CrossRef CAS
- Y.-H. Chiu, S. A. Lindley, C.-W. Tsao, M.-Y. Kuo, J. K. Cooper, Y.-J. Hsu and J. Z. Zhang, J. Phys. Chem. C, 2020, 124, 11333–11339 CrossRef CAS
- P. Wang, M. Xi, L. Qin, S.-Z. Kang, Y. Fang and X. Li, ACS Appl. Nano Mater., 2019, 2, 7409–7420 CrossRef CAS
- T.-N. Chen, J.-C. Kao, X.-Y. Zhong, S.-J. Chan, A. S. Patra, Y.-C. Lo and M. H. Huang, ACS Cent. Sci., 2020, 6, 984–994 CrossRef CAS PubMed
- J.-Y. Ho and M. H. Huang, J. Phys. Chem. C, 2009, 113, 14159–14164 CrossRef CAS
- W.-C. Huang, L.-M. Lyu, Y.-C. Yang and M. H. Huang, J. Am. Chem. Soc., 2012, 134, 1261–1267 CrossRef CAS PubMed
- K. Chanda, S. Rej and M. H. Huang, Nanoscale, 2013, 5, 12494–12501 RSC
- C. Huang, A. Li, L.-J. Li and Z.-S. Chao, RSC Adv., 2017, 7, 24950–24962 RSC
- M. Madasu, C.-F. Hsia and M. H. Huang, Nanoscale, 2017, 9, 6970–6974 RSC
- M. H. Huang, Small, 2019, 15, 1804726 CrossRef PubMed
- M. H. Huang, J. Chin. Chem. Soc., 2021, 68, 45–50 CrossRef CAS
- A. Soon, M. Todorova, B. Delley and C. Stampfl, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125420 CrossRef
- L. Li, R. Zhang, J. Vinson, E. L. Shirley, J. P. Greeley, J. R. Gues and M. K. Y. Chan, Chem. Mater., 2018, 30, 1912–1923 CrossRef CAS PubMed
- M. Soldemo, J. H. Stenlid, Z. Besharat, M. Ghadami Yazdi, A. Önsten, C. Leygraf, M. Göthelid, T. Brinck and J. Weissenrieder, J. Phys. Chem. C, 2016, 120, 4373–4381 CrossRef CAS
- R. Zhang, L. Li, L. Frazer, K. B. Chang, K. R. Poeppelmeier, M. K. Y. Chan and J. R. Guest, Phys. Chem. Chem. Phys., 2018, 20, 27456–27463 RSC
- C.-H. Kuo and M. H. Huang, J. Am. Chem. Soc., 2008, 130, 12815–12820 CrossRef CAS PubMed
- C.-H. Kuo, Y.-C. Yang, S. Gwo and M. H. Huang, J. Am. Chem. Soc., 2011, 133, 1052–1057 CrossRef CAS PubMed
- Y. Liang, L. Shang, T. Bian, C. Zhou, D. Zhang, H. Yu, H. Xu, Z. Shi, T. Zhang, L.-Z. Wu and C.-H. Tung, CrystEngComm, 2012, 14, 4431–4436 RSC
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1999, 6, 15–50 CrossRef
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05278k |
| This journal is © The Royal Society of Chemistry 2022 |