
Biological sample-compatible Au nanoparticle-containing fluorescent molecularly imprinted polymer microspheres by combining RAFT polymerization and Au–thiol chemistry†
Xiaohui
Shi
,
Wanlan
Zhang
and
Huiqi
Zhang
 *
*
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Functional Polymer Materials (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), and College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: zhanghuiqi@nankai.edu.cn
First published on 1st March 2022
Abstract
The development of biological sample-compatible fluorescent molecularly imprinted polymers (MIPs) with more functions and/or improved performance is of great importance for various bioanalytical and biomedical applications, but remains challenging. Herein, we report on a versatile strategy for preparing well-defined hydrophilic gold (Au) nanoparticle (AuNP)-containing fluorescent MIP microspheres capable of directly optosensing folic acid (FA) in undiluted urine samples and study of the effect of the incorporated AuNPs on their optosensing sensitivity. Such advanced functional fluorescent MIP particles were readily prepared by combining RAFT polymerization (including RAFT precipitation polymerization and surface-initiated RAFT polymerization) and Au–thiol chemistry [including successive attachment of AuNPs and a thiol-terminated poly(2-hydroxyethyl methacrylate) onto MIP particles after aminolysis of their surface dithioester into thiol groups]. They showed “turn-on” fluorescence and high optosensing selectivity and sensitivity toward FA in the undiluted urine sample (detection limit = 0.088 μM). They also exhibited outstanding photostability and reusability and could directly quantify FA in another undiluted urine sample with good recoveries (96.3–101.6%) and high accuracy (RSD = 0.6–3.1%), even in the presence of several interferents. Unfortunately, the incorporated AuNPs did not improve the optosensing sensitivity of AuNP-containing fluorescent MIPs. Nevertheless, introducing AuNPs onto the surfaces of fluorescent MIPs not only provides an effective new way for grafting with hydrophilic polymer brushes, but it also can endow them with certain new functions [e.g., surface-enhanced Raman scattering (SERS)], thus making them highly promising as both efficient optosensors and potential SERS sensors for rapid FA detection in applications such as clinical diagnostics and food analyses.
Introduction
Molecularly imprinted polymers (MIPs) are synthetic receptors with tailor-made molecular recognition sites.1–12 They have many prominent characteristics such as high molecular recognition ability, excellent physiochemical stability, easy preparation, and low cost, which make them highly promising in a wide range of applications such as separation and purification,2,3,5–7 enzyme-like catalysis,4 chemosensing,1,7,8 and various biomedical areas.9–12 Among them, MIP-based fluorescent chemosensors have garnered great interest in the bioanalytical and biomedical fields because of the combined advantages of MIPs and fluorescence analyses (i.e., high sensitivity, simple instruments, and easy implementation).13–19 They are typically fabricated by labelling MIPs with various fluorescent species, where the MIPs function as the recognition elements to selectively recognize the target analytes and the fluorescent species quantitatively transform such recognition processes into the fluorescent signals.So far, a great number of MIP-based fluorescent chemosensors have been developed for different purposes.13–19 However, fluorescent MIPs capable of directly and selectively optosensing small organic analytes in complex biological samples are still rare, which largely limits their broad applications in various bioanalytical and biomedical applications. Over the past few years, our group has developed some complex biological sample-compatible fluorescent MIPs via the controlled grafting of hydrophilic polymer brushes onto fluorescent MIP particles.20–22 For example, we have reported the efficient synthesis of either organic fluorescent dye or inorganic quantum dot-labelled hydrophilic MIP nanoparticles by the combined use of controlled/“living” radical polymerization and molecular imprinting techniques, which could directly and selectively detect small organic analytes [e.g., antibiotic tetracycline (Tc)20,22 and food additive folic acid (FA)21] (through fluorescence quenching) in the complex biological samples (i.e., undiluted pure milk and serum samples). Despite the significant progress made in this field, the development of new strategies for preparing complex biological sample-compatible fluorescent MIPs is still highly desirable.
Recent years have also witnessed tremendous efforts being devoted to developing fluorescent MIPs with more functions and/or improved performance. Many useful approaches have been utilized for such purposes. For instance, our group developed some complex biological sample-compatible ratiometric “turn-on” type fluorescent MIPs by their incorporation of dual fluorescent species (both organic and inorganic ones), which could directly and selectively optosense the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) in undiluted pure milk samples.23,24 The “turn-on” fluorescence of these optosensors can avoid false-positive responses owing to the less interfering effects and achieve higher sensitivity due to their lower optical background and higher signal-to-noise ratio (compared with fluorescence quenching). Moreover, the ratiometric measurements can not only eliminate external interferences because of their self-referencing ability (by using the ratio of two fluorescence intensities), but they also show higher sensitivity and reliability than single fluorescent MIPs for visual detection. Haupt and coworkers developed an efficient strategy for enhancing the optosensing sensitivity of a fluorescent MIP by its incorporation of AuNPs.25 They prepared a MIP-based fluorescent fiber optosensor, which had a micrometer-sized 2,4-D-MIP tip generated in situ at one end through the rapid photopolymerization of a molecular imprinting formulation with a “turn-on” type fluorescent monomer and AuNPs inside. This MIP sensor showed fluorescence enhancement upon exposure to 2,4-D. Importantly, the incorporated AuNPs largely improved the sensitivity of the fluorescent MIP, as revealed by a 10-fold decrease in its detection limit. Nevertheless, the general applicability of this strategy has not been demonstrated.
Herein, we report for the first time a versatile strategy for preparing complex biological sample-compatible AuNP-containing fluorescent MIP microspheres by combining reversible addition–fragmentation chain-transfer (RAFT) polymerization and Au–thiol chemistry (Scheme 1) and study of the effect of the incorporated AuNPs on their optosensing sensitivity. FA was chosen here as the model target analyte due to its great clinical importance to human health.26 Our strategy involves first the synthesis of uniform “living” polymer microspheres with surface-bound dithioester groups via RAFT precipitation polymerization (RAFTPP, which combines the advantages of traditional precipitation polymerization and “living” radical polymerization and allows the one-pot synthesis of uniform spherical polymer particles with surface-bound “living” chain-transfer groups),27 the successive grafting of a nitrobenzoxadiazole (NBD)-labelled fluorescent FA-imprinted polymer (FA-MIP) layer and a lightly crosslinked thin polymer layer (as the separation layer for the fluorescent MIP layer and AuNPs to avoid the significant reduction in its fluorescence enhancement effect) via two-step surface-initiated RAFT (SI-RAFT) polymerization, aminolysis of their surface dithioester units into thiol groups,28 and finally successive attachment of AuNPs and a well-defined thiol-terminated poly(2-hydroxyethyl methacrylate) (PHEMA-SH) via Au–thiol chemistry.29 The resulting hydrophilic AuNP-containing fluorescent MIP particles showed “turn-on” fluorescence and high optosensing selectivity and sensitivity toward FA in the undiluted urine (with a detection limit of 0.088 μM). Moreover, they exhibited outstanding photostability and reusability and could directly quantify FA in another undiluted urine sample with good recoveries (96.3–101.6%) and high accuracy (RSD = 0.6–3.1%), even in the presence of several interferents. Unfortunately, the incorporated AuNPs did not improve (or even reduce in the case of directly attaching AuNPs onto the fluorescent MIP layer) the optosensing sensitivity of the AuNP-containing fluorescent MIP particles. Nevertheless, the introduction of AuNPs onto the surfaces of fluorescent MIP particles not only affords an effective new way for their grafting of hydrophilic polymer brushes, but it can also impart them with certain new functions (e.g., SERS capability), thus making them highly promising as both efficient optosensors and potential SERS sensors30,31 for rapid FA detection in a wide range of bioanalytical and biomedical applications.
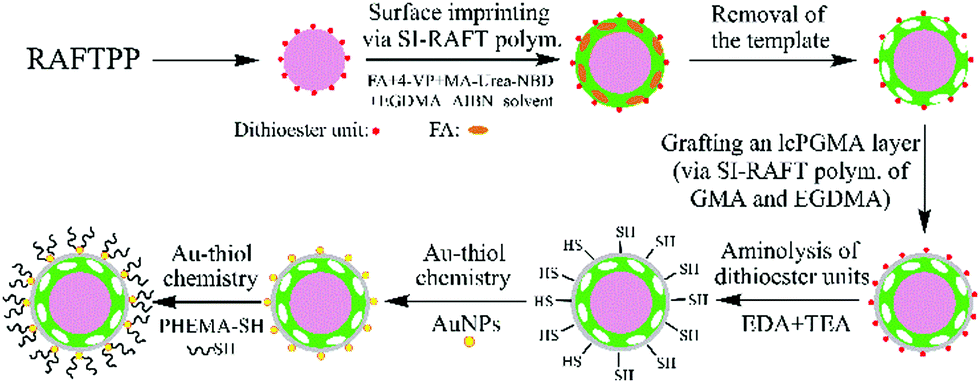 | ||
| Scheme 1 Schematic illustration of the preparation of complex biological sample-compatible AuNP-containing fluorescent MIP microspheres by combining RAFT polymerization and Au–thiol chemistry. | ||
Results and discussion
Synthesis and characterization of well-defined hydrophilic AuNP-containing fluorescent MIP/CP microspheres
The main aim of this work is to develop a versatile strategy for preparing well-defined hydrophilic AuNP-containing fluorescent MIP microspheres capable of directly and selectively optosensing small organic analytes in complex biological samples and check whether the incorporated AuNPs can improve their optosensing sensitivity. To demonstrate the proof-of-principle, the core–shell-structured fluorescent FA-MIP microspheres (polym@NBD-MIP, entry 2 in Table 1) were first prepared via controlled surface imprinting (by using SI-RAFT polymerization) on the uniform “living” poly(4-VP-co-EGDMA) particles (prepared via RAFTPP, entry 1 in Table 1), where FA, 4-vinylpyridine (4-VP), 2-(3-(4-nitrobenzo[c][1,2,5]oxadiazo-7-yl)ureido)ethyl methacrylate (MA-Urea-NBD), ethylene glycol dimethacrylate (EGDMA), and acetonitrile (ACN)/N,N-dimethylformamide (DMF) (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) were used as the template, functional monomer, fluorescent comonomer, crosslinker, and solvent, respectively (Scheme 1 and Scheme S1, ESI†). Both 4-VP and MA-Urea-NBD can form hydrogen-bonded complexes with FA via pyridine–carboxylic acid and urea–carboxylic acid interactions. Particularly, the interaction of MA-Urea-NBD with FA can result in fluorescence enhancement.23,24 A lightly crosslinked thin polymer layer (briefly lcPGMA) was then grafted onto polym@NBD-MIP particles via SI-RAFT copolymerization of glycidyl methacrylate (GMA) and EGDMA (molar ratio = 2:1, following our previous report32) to afford the core-double shell-structured fluorescent MIP particles (polym@NBD-MIP@lcPGMA, entry 4 in Table 1). Afterwards, polym@NBD-MIP@lcPGMA particles with surface-bound AuNPs (i.e., polym@NBD-MIP@lcPGMA@AuNPs, entry 8 in Table 1) (briefly the ungrafted AuNP-containing fluorescent MIP particles) were prepared by first the aminolysis of their surface dithioester units into thiol groups28 and subsequent immobilization of AuNPs via Au–thiol chemistry.29 Finally, the AuNP-containing fluorescent MIP particles with PHEMA brushes (polym@NBD-MIP@lcPGMA@AuNPs@PHEMA, entry 10 in Table 1) (briefly the hydrophilic or grafted AuNP-containing fluorescent MIP particles) were obtained by grafting a well-defined PHEMA-SH (Mn,NMR = 5840 g mol−1, see its synthesis in the ESI,† Fig. S1, ESI†) onto polym@NBD-MIP@lcPGMA@AuNPs via Au–thiol chemistry. The corresponding controls (entries 5, 7, 9 and 11, Table 1) of the above MIP particles were also prepared under the same conditions except changing polym@NBD-MIP to polym@NBD-CP (entry 3 in Table 1; it was prepared under the same conditions as polym@NBD-MIP but omitting FA during the surface imprinting process). All the polymer particles showed a certain weight increase after each surface modification step, indicating their successful grafting of the MIP/CP layer, lcPGMA layer, AuNPs, and polymer brushes, respectively. Note that an lcPGMA layer (with a thickness of 28 nm) was introduced between the MIP layer and AuNPs because the direct attachment of AuNPs onto the fluorescent MIP layer surfaces proved to significantly reduce its fluorescence enhancement effect upon exposure to FA (thus largely lowering its optosensing sensitivity) (not shown) owing to the fluorescence resonance energy transfer (FRET) from the donor (NBD) to the acceptor (AuNPs) located within a distance <10 nm (Fig. S3, ESI†).33 Moreover, the low crosslinking degree of the lcPGMA layer also allows FA to pass through it freely and reach the imprinted sites in the MIP layer.
:1 v/v) were used as the template, functional monomer, fluorescent comonomer, crosslinker, and solvent, respectively (Scheme 1 and Scheme S1, ESI†). Both 4-VP and MA-Urea-NBD can form hydrogen-bonded complexes with FA via pyridine–carboxylic acid and urea–carboxylic acid interactions. Particularly, the interaction of MA-Urea-NBD with FA can result in fluorescence enhancement.23,24 A lightly crosslinked thin polymer layer (briefly lcPGMA) was then grafted onto polym@NBD-MIP particles via SI-RAFT copolymerization of glycidyl methacrylate (GMA) and EGDMA (molar ratio = 2:1, following our previous report32) to afford the core-double shell-structured fluorescent MIP particles (polym@NBD-MIP@lcPGMA, entry 4 in Table 1). Afterwards, polym@NBD-MIP@lcPGMA particles with surface-bound AuNPs (i.e., polym@NBD-MIP@lcPGMA@AuNPs, entry 8 in Table 1) (briefly the ungrafted AuNP-containing fluorescent MIP particles) were prepared by first the aminolysis of their surface dithioester units into thiol groups28 and subsequent immobilization of AuNPs via Au–thiol chemistry.29 Finally, the AuNP-containing fluorescent MIP particles with PHEMA brushes (polym@NBD-MIP@lcPGMA@AuNPs@PHEMA, entry 10 in Table 1) (briefly the hydrophilic or grafted AuNP-containing fluorescent MIP particles) were obtained by grafting a well-defined PHEMA-SH (Mn,NMR = 5840 g mol−1, see its synthesis in the ESI,† Fig. S1, ESI†) onto polym@NBD-MIP@lcPGMA@AuNPs via Au–thiol chemistry. The corresponding controls (entries 5, 7, 9 and 11, Table 1) of the above MIP particles were also prepared under the same conditions except changing polym@NBD-MIP to polym@NBD-CP (entry 3 in Table 1; it was prepared under the same conditions as polym@NBD-MIP but omitting FA during the surface imprinting process). All the polymer particles showed a certain weight increase after each surface modification step, indicating their successful grafting of the MIP/CP layer, lcPGMA layer, AuNPs, and polymer brushes, respectively. Note that an lcPGMA layer (with a thickness of 28 nm) was introduced between the MIP layer and AuNPs because the direct attachment of AuNPs onto the fluorescent MIP layer surfaces proved to significantly reduce its fluorescence enhancement effect upon exposure to FA (thus largely lowering its optosensing sensitivity) (not shown) owing to the fluorescence resonance energy transfer (FRET) from the donor (NBD) to the acceptor (AuNPs) located within a distance <10 nm (Fig. S3, ESI†).33 Moreover, the low crosslinking degree of the lcPGMA layer also allows FA to pass through it freely and reach the imprinted sites in the MIP layer.
| Entry | Sample | ΔWa (%) | D n,SEM (μm) | U |
|---|---|---|---|---|
| a Increased weight percentage of the polymer particles after each surface modification step in comparison with the starting polymer particles. b D n,SEM and U denote the number-average diameter and size distribution index of the particles (derived from the SEM characterization results), respectively. | ||||
| 1 | Poly(4-VP-co-EGDMA) | — | 2.254 | 1.025 |
| 2 | Polym@NBD-MIP | 25.7 | 2.430 | 1.020 |
| 3 | Polym@NBD-CP | 29.9 | 2.457 | 1.029 |
| 4 | Polym@NBD-MIP@lcPGMA | 7.4 | 2.486 | 1.024 |
| 5 | Polym@NBD-CP@lcPGMA | 6.8 | 2.509 | 1.027 |
| 6 | Polym@NBD-MIP@lcPGMA-SH | 0 | 2.486 | 1.025 |
| 7 | Polym@NBD-CP@lcPGMA-SH | 0 | 2.507 | 1.021 |
| 8 | Polym@NBD-MIP@lcPGMA@AuNPs | 6.3 | 2.535 | 1.022 |
| 9 | Polym@NBD-CP@lcPGMA@AuNPs | 6.7 | 2.560 | 1.017 |
| 10 | Polym@NBD-MIP@lcPGMA@AuNPs@PHEMA | 4.2 | 2.571 | 1.026 |
| 11 | Polym@NBD-CP@lcPGMA@AuNPs@PHEMA | 4.6 | 2.596 | 1.023 |
SEM characterization revealed that all the above-obtained polymer particles were uniform spherical particles (Fig. 1a–k). Their diameters ranged from 2.254 to 2.596 μm and their size distribution indices were ≤1.029 (Table 1). Particularly, their diameters (Dn,SEM) increased after each surface modification step (Table 1), again verifying their successful surface modification. In addition, TEM characterization also confirmed the presence of AuNPs (Fig. 1l) on polym@NBD-MIP@lcPGMA@AuNPs surfaces (Fig. 1m and n).
 | ||
| Fig. 1 (a–k) SEM images of poly(4-VP-co-EGDMA) (a), polym@NBD-MIP (b), polym@NBD-CP (c), polym@NBD-MIP@lcPGMA (d), polym@NBD-CP@lcPGMA (e), polym@NBD-MIP@lcPGMA-SH (f), polym@NBD-CP@lcPGMA-SH (g), polym@NBD-MIP@lcPGMA@AuNPs (h), polym@NBD-CP@lcPGMA@AuNPs (i), polym@NBD-MIP@lcPGMA@AuNPs@PHEMA (j), and polym@NBD-CP@lcPGMA@AuNPs@PHEMA (k) (the scale bar is 5 μm). (l-n) TEM images of AuNPs (l) and polym@NBD-MIP@lcPGMA@AuNPs (m) and the amplified TEM image of polym@NBD-MIP@lcPGMA@AuNPs (n). | ||
Fig. 2 presents the FT-IR spectra of some representative polymer particles (entries 1, 2, 4, 6, 8 and 10, Table 1). It can be seen that besides the absorption peaks of poly(4-VP) around 1597/1558 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration) and 1453 cm−1 (CC stretching vibration),34 those of poly(EGDMA) around 1724 cm−1 (CO stretching vibration) and 1247/1137 cm−1 (C–O stretching vibration),32 and that of the dithioester group around 1047 cm−1 in ‘‘living’’ poly(4-VP-co-EGDMA) and polym@NBD-MIP,32 a weak peak around 905 cm−1 (corresponding to the epoxy ring) was observed in the spectrum of polym@NBD-MIP@lcPGMA,32 suggesting its successful grafting of the lightly crosslinked poly(GMA-co-EGDMA) layer. In addition, the absorption peak around 1047 cm−1 in the spectrum of polym@NBD-MIP@lcPGMA-SH decreased in comparison with that of polym@NBD-MIP@cPGMA, indicating the occurrence of aminolysis of the surface dithioester groups on polym@NBD-MIP@lcPGMA particles. Furthermore, one new rather weak and broad peak around 3492 cm−1 (corresponding to O-H stretching vibration)32 appeared in the spectrum of polym@NBD-MIP@lcPGMA@AuNPs@PHEMA, revealing its successful grafting of PHEMA brushes. It is noteworthy that a negligible change was observed for the spectra of poly(4-VP-co-EGDMA) and polym@NBD-MIP as well as those of polym@NBD-MIP@lcPGMA-SH and polym@NBD-MIP@lcPGMA-SH@AuNP, mainly because of the low contents of NBD and AuNPs in polym@NBD-MIP and polym@NBD-MIP@lcPGMA-SH@AuNP, respectively. Nevertheless, the fluorescent spectrum of polym@NBD-MIP (see the last section) and the TEM images of polym@NBD-MIP@lcPGMA-SH@AuNPs (Fig. 1m and n) clearly demonstrated their successful syntheses.
N stretching vibration) and 1453 cm−1 (CC stretching vibration),34 those of poly(EGDMA) around 1724 cm−1 (CO stretching vibration) and 1247/1137 cm−1 (C–O stretching vibration),32 and that of the dithioester group around 1047 cm−1 in ‘‘living’’ poly(4-VP-co-EGDMA) and polym@NBD-MIP,32 a weak peak around 905 cm−1 (corresponding to the epoxy ring) was observed in the spectrum of polym@NBD-MIP@lcPGMA,32 suggesting its successful grafting of the lightly crosslinked poly(GMA-co-EGDMA) layer. In addition, the absorption peak around 1047 cm−1 in the spectrum of polym@NBD-MIP@lcPGMA-SH decreased in comparison with that of polym@NBD-MIP@cPGMA, indicating the occurrence of aminolysis of the surface dithioester groups on polym@NBD-MIP@lcPGMA particles. Furthermore, one new rather weak and broad peak around 3492 cm−1 (corresponding to O-H stretching vibration)32 appeared in the spectrum of polym@NBD-MIP@lcPGMA@AuNPs@PHEMA, revealing its successful grafting of PHEMA brushes. It is noteworthy that a negligible change was observed for the spectra of poly(4-VP-co-EGDMA) and polym@NBD-MIP as well as those of polym@NBD-MIP@lcPGMA-SH and polym@NBD-MIP@lcPGMA-SH@AuNP, mainly because of the low contents of NBD and AuNPs in polym@NBD-MIP and polym@NBD-MIP@lcPGMA-SH@AuNP, respectively. Nevertheless, the fluorescent spectrum of polym@NBD-MIP (see the last section) and the TEM images of polym@NBD-MIP@lcPGMA-SH@AuNPs (Fig. 1m and n) clearly demonstrated their successful syntheses.
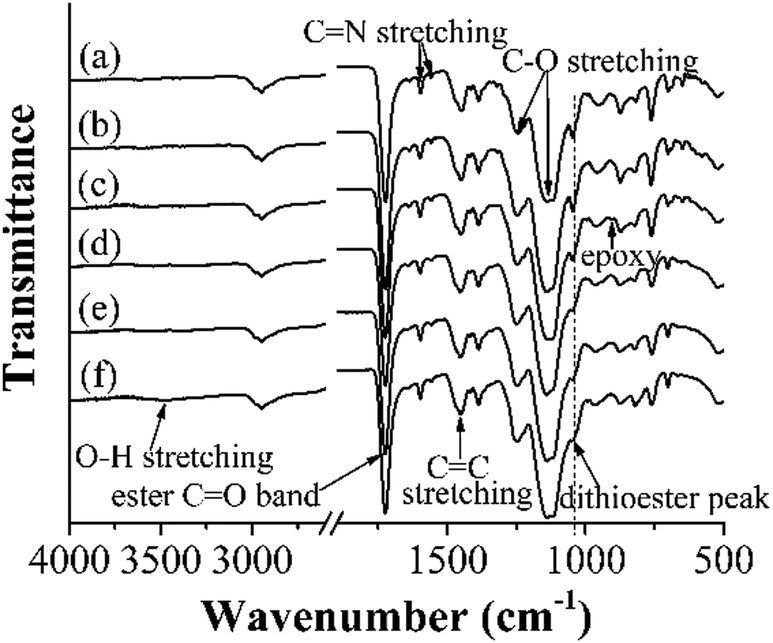 | ||
| Fig. 2 FT-IR spectra of poly(4-VP-co-EGDMA) (a), polym@NBD-MIP (b), polym@NBD-MIP@lcPGMA (c), polym@NBD-MIP@lcPGMA-SH (d), polym@NBD-MIP@lcPGMA@AuNPs (e), and polym@NBD-MIP@lcPGMA@AuNPs@PHEMA (f). | ||
The successful preparation of polym@NBD-MIP@lcPGMA@AuNPs@PHEMA and its control polymer was further demonstrated by their largely enhanced aqueous dispersion stability in comparison with all the polymer particles without PHEMA brushes (Fig. 3). In addition, the aqueous dispersions of the MIP/CP particles with attached AuNPs [i.e., the ungrafted and grafted AuNP-containing fluorescent MIPs/CPs (entries 8–11, Table 1)] showed rather different colors compared with those of the particles without attached AuNPs, which again verified their successful incorporation of AuNPs.
 | ||
| Fig. 3 Dispersion photographs of poly(4-VP-co-EGDMA) (1), polym@NBD-MIP (2), polym@NBD-CP (3), polym@NBD-MIP@lcPGMA (4), polym@NBD-CP@lcPGMA (5), polym@NBD-MIP@lcPGMA-SH (6), polym@NBD-CP@lcPGMA-SH (7), polym@NBD-MIP@lcPGMA@AuNPs (8), polym@NBD-CP@cPGMA@AuNPs (9), polym@NBD-MIP@lcPGMA@AuNPs@PHEMA (10), and polym@NBD-CP@lcPGMA@AuNPs@PHEMA (11) in pure water at 25 °C after setting for 0 h (a) and 3 h (b), respectively. | ||
Equilibrium/competitive binding properties of the MIPs/CPs
With a series of fluorescent MIPs/CPs (entries 2–11, Table 1) in hand, we first studied their equilibrium binding properties in the organic solvent (ACN/DMF = 4:1 v/v). All the MIPs (entries 2, 4, 6, 8 and 10, Table 1) showed higher FA binding capacities than their corresponding CPs (entries 3, 5, 7, 9 and 11, Table 1) (Fig. S4a–e, ESI†), revealing their presence of imprinted binding sites. It is also noteworthy that although the binding capacities of these MIPs/CPs are somewhat different because of their different surface properties, their specific bindings (i.e., the binding differences between MIPs and their CPs) are almost the same. These results indicated that the surface-modification of MIPs had negligible influence on the imprinted binding sites.
The equilibrium binding properties of the ungrafted and grafted AuNP-containing fluorescent MIPs/CPs (entries 8–11, Table 1) were then investigated in the undiluted urine sample I. As expected, the specific bindings of the ungrafted MIP (entry 8, Table 1) disappeared (Fig. S5a, ESI†), mainly owing to its high surface hydrophobicity.35,36 In sharp contrast, the specific bindings of the grafted MIP (entry 10, Table 1) in urine sample I are almost the same as those in the organic solvent (Fig. S5b, ESI†), revealing its high compatibility with the complex biological sample.
The competitive binding properties of the ungrafted and grafted AuNP-containing fluorescent MIPs/CPs toward FA and its structural analogues methotrexate hydrate (MTX) and trimethoprim (TMP) were further evaluated in ACN/DMF (4:1 v/v) and the undiluted urine sample I, respectively (Fig. S6, ESI†). A parameter named “imprinting-induced promotion of binding” (IPB) [IPB (%) = ((BMIP − BCP)/BCP) × 100, where BMIP and BCP are the equilibrium analyte bindings of MIP and CP, respectively] was chosen here to quantify the selectivity of the studied MIPs because it can normalize MIPs’ nonspecific bindings toward different analytes.37 A MIP with a larger IPB value shows higher analyte selectivity. The IPB values of the studied MIPs were derived and listed in Table S2 (ESI†). The grafted MIP showed distinct FA selectivity in both the organic and urine samples, whereas the ungrafted MIP exhibited no FA selectivity in pure urine in spite of its good selectivity toward FA in the organic medium. The above results strongly demonstrated the versatility of our strategy for developing AuNP-containing fluorescent MIPs fully compatible with complex biological samples.
Optosensing properties of the grafted AuNP-containing fluorescent MIP/CP in the undiluted urine samples
The photostability and reusability of the grafted AuNP-containing fluorescent MIP/CP (entries 10 and 11, Table 1) were first studied because of their great importance for practical uses. It can be seen from Fig. 4a and b that the fluorescence intensities of the aqueous solutions of the grafted AuNP-containing fluorescent MIP/CP hardly changed both after 10 days under an air atmosphere at room temperature and after 10 regeneration cycles in the undiluted urine, indicating their excellent photostability and reusability.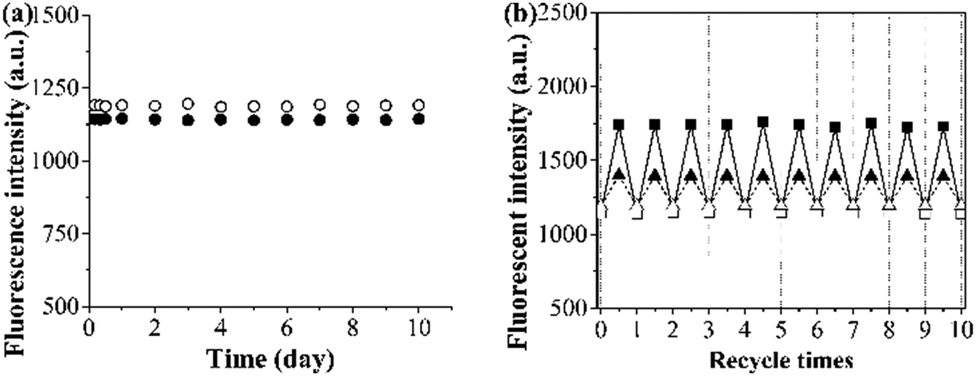 | ||
| Fig. 4 (a) Time dependence of the fluorescence intensities of the grafted AuNP-containing fluorescent MIP (filled circle)/CP (open circle) in pure water (0.25 mg mL−1) in air at 25 °C. (b) The dependence of the fluorescence intensities of the grafted AuNP-containing fluorescent MIP (square)/CP (triangle) over regeneration cycles upon adsorption (filled symbols)/desorption (open symbols) of FA (15 μM) in the undiluted urine sample I. | ||
The binding kinetics of the grafted AuNP-containing fluorescent MIP/CP toward FA in the undiluted urine samples was then studied by monitoring the time-dependence of the fluorescence intensities. The fluorescence intensities of both the grafted AuNP-containing fluorescent MIP and CP increased with time upon their exposure to a FA solution in urine and eventually levelled off after about 35 min (Fig. 5a, b and Fig. S7, ESI†), indicating their rapid binding kinetics. In addition, the fluorescence enhancement effect was much higher for the MIP than CP, again verifying the presence of imprinted binding sites in the MIP and its good complex biological sample-compatibility.
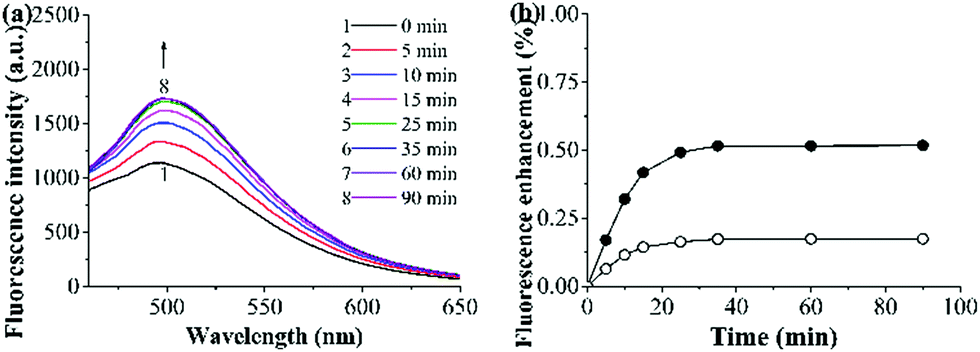 | ||
| Fig. 5 (a) Fluorescence spectra of the grafted AuNP-containing fluorescent MIP (0.25 mg mL−1) after its incubation with a FA solution (15 μM) in the undiluted urine sample I at 25 °C for different times. (b) Binding kinetics of the grafted AuNP-containing fluorescent MIP (filled symbol)/CP (open symbol) (0.25 mg mL−1) in a FA solution (15 μM) in the undiluted urine sample I at 25 °C [derived from (a) and Fig. S7 (ESI†); fluorescence enhancement (%) = [(Ft − F0)/F0] × 100, where Ft and F0 are MIP/CP's fluorescence intensity (at 500 nm) at a time of t and 0, respectively]. | ||
The fluorescence titration experiments were then carried out to evaluate the optosensing parameters of the grafted AuNP-containing fluorescent MIP. Fig. 6a and b presents the fluorescence spectra of the mixed solutions of the grafted AuNP-containing fluorescent MIP/CP and FA with varying concentrations in the undiluted urine sample I. The fluorescence intensities of both the MIP and CP systems increased with an increase in the FA concentrations, but the enhancement effect was much higher for the MIP than CP, again confirming the successful molecular imprinting process. Linear calibration curves were established for the grafted AuNP-containing fluorescent MIP and CP with the FA concentration C = 0–15 μM based on the Stern–Volmer equation [i.e., (F − F0)/F0 = KSVC, where F and F0 are the fluorescence intensities with and without FA, respectively, KSV is the Stern–Volmer enhancement constant, and C is the FA concentration] (Fig. 6c). The detection limit [or limit of detection (LOD) = 3SB/KSV, where SB is the standard deviation of the blank measurements (for 20 times)] of the studied MIP was calculated to be 0.088 μM in the undiluted urine.38 In addition, the imprinting factor (IF) of the MIP in the studied urine system was derived to be 3.2 by using the equation Ksv(MIP)/Ksv(CP), which further demonstrated the high complex biological sample-compatibility of our grafted AuNP-containing fluorescent MIP.
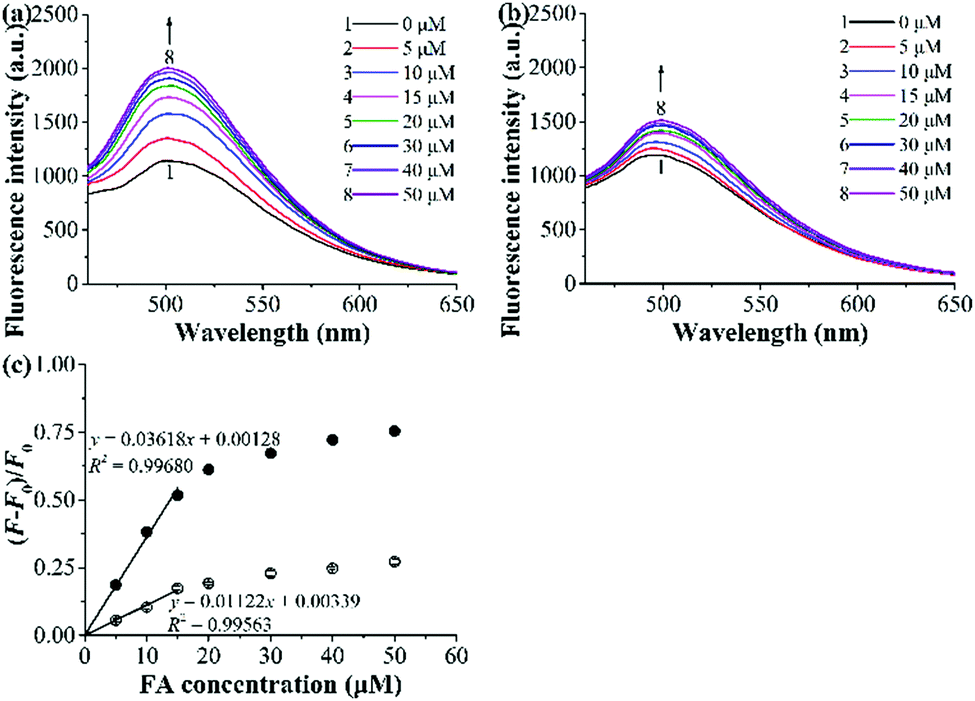 | ||
| Fig. 6 (a and b) Fluorescence spectra of the grafted AuNP-containing fluorescent MIP (a)/CP (b) (0.25 mg mL−1) upon their exposure to different concentrations of spiked FA in the undiluted urine sample I at 25 °C for 2 h. (c) Dependence of fluorescence enhancement of the grafted AuNP-containing fluorescent MIP (filled symbol) and CP (open symbol) on FA concentrations in the undiluted urine sample I (derived from (a) and (b), respectively). | ||
The optosensing selectivity of the grafted AuNP-containing fluorescent MIP and CP was then studied by measuring their fluorescence responses toward FA and its structurally related compounds MTX and TMP as well as some other compounds such as ascorbic acid (VC), Tc, glucose, L-cysteine, L-glutamic acid, and bovine serum album (BSA) (Fig. 7a–d). The grafted AuNP-containing fluorescent MIP showed much larger fluorescence enhancement toward FA than all the other compounds (Fig. 7a and b). In addition, its FA optosensing results remained almost unchanged when 10-fold of interferents (i.e., MTX, TMP, VC, Tc, glucose, L-cysteine, L-glutamic acid, or BSA) were added into FA solutions (Fig. 7c and d). These results demonstrate that the grafted AuNP-containing fluorescent MIP has high FA optosensing selectivity (even in the presence of a large excess of various interferents) and is a promising optosensor for direct FA detection in complex biological matrixes.
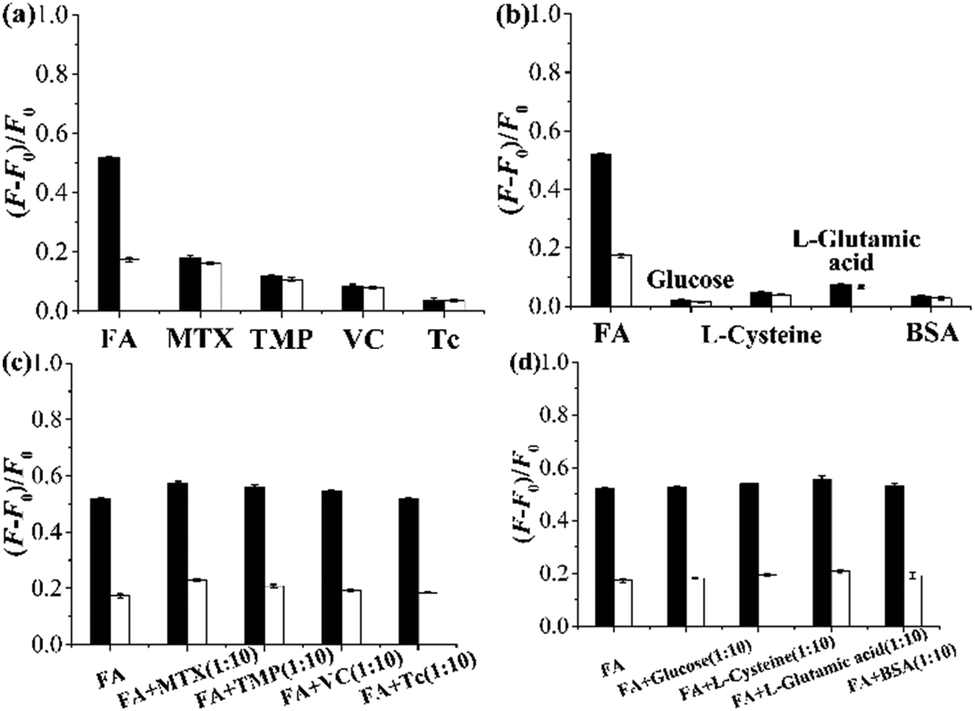 | ||
| Fig. 7 (a and b) Fluorescence enhancement of the grafted AuNP-containing fluorescent MIP (filled column)/CP (open column) (0.25 mg mL−1) after their incubation with a solution of FA, MTX, TMP, VC, or Tc (CFA,MTX,TMP,VC,orTc = 15 μM) (a) and after their incubation with a solution of FA, glucose, L-cysteine, L-glutamic acid, or BSA (CFA,glucose,L-cysteine,L-glutamic acid,orBSA = 15 μM) (b) in the undiluted urine sample I at 25 °C for 2 h. (c and d) Fluorescence enhancement of the grafted AuNP-containing fluorescent MIP (filled column)/CP (open column) (0.25 mg mL−1) after their incubation with a solution of FA in the presence of 10-fold of MTX, TMP, VC, or Tc (c) and in the presence of 10-fold of glucose, L-cysteine, L-glutamic acid, or BSA (d) in the undiluted urine sample I at 25 °C for 2 h. | ||
The application of the grafted AuNP-containing fluorescent MIP in directly detecting FA in another undiluted urine sample II was also investigated. Very good recoveries from 96.3% to 101.6% and high accuracy with RSD ranging from 0.6% to 3.1% were achieved for the urine samples spiked with different amounts of FA (entries 1–3, Table 2) or mixtures of FA and some other compounds including TMP, VC, and Tc (the concentrations of each compound are the same as FA in the solutions) (entries 4–6, Table 2), which clearly indicates that the grafted AuNP-containing fluorescent MIP is highly useful for directly, selectively, and accurately optosensing FA in real, undiluted biological samples, even in the presence of several interferents.
| Entry | Sample | Analyte | Analyte concentration (μM) | Recovery (%) (n = 3) | |
|---|---|---|---|---|---|
| Spiked | Found (FA) | ||||
| 1 | Urine II | FA | 0.5 | 0.49 ± 0.013 | 97.5 ± 2.6 |
| 2 | Urine II | FA | 5 | 4.92 ± 0.05 | 98.3 ± 1.0 |
| 3 | Urine II | FA | 10 | 10.11 ± 0.06 | 101.1 ± 0.6 |
| 4 | Urine II | FA + TMP + VC + Tc | 0.5(FA) + 0.5(TMP) + 0.5(VC) + 0.5(Tc) | 0.48 ± 0.014 | 96.3 ± 3.1 |
| 5 | Urine II | FA + TMP + VC + Tc | 5(FA) + 5(TMP) + 5(VC) + 5(Tc) | 4.90 ± 0.07 | 98.0 ± 1.3 |
| 6 | Urine II | FA + TMP + VC + Tc | 10(FA) + 10(TMP) + 10(VC) + 10(Tc) | 10.16 ± 0.10 | 101.6 ± 1.0 |
In this context, it is noteworthy that we have previously developed a series of approaches to prepare complex biological sample-compatible fluorescent MIP particles with surface-grafted hydrophilic polymer brushes, which mainly include the one-pot approach (i.e., one-pot synthesis of such MIP particles via hydrophilic macro-RAFT agent-mediated RAFTPP)20,21 and two-step approaches (including both the RAFT coupling chemistry24 and methods involving the grafting of hydrophilic polymer brushes onto “living” fluorescent MIP particles via surface-initiated controlled/“living” radical polymerizations (SI-CRPs)22,23). Similar to these previously reported approaches, our newly developed strategy can also afford well-defined complex biological sample-compatible fluorescent MIP particles. Particularly, this new strategy can further endow the complex biological sample-compatible fluorescent MIP particles with certain new function [e.g., surface-enhanced Raman scattering (SERS)], which thus largely expands the scope of their potential applications.
Effect of the incorporated AuNPs on the optosensing sensitivity of the fluorescent MIPs/CPs
To check whether the incorporation of AuNPs into our fluorescent MIPs can largely improve their optosensing sensitivity (as reported by Haupt and coworkers25), we evaluated the optosensing properties of polym@NBD-MIP/CP and polym@NBD-MIP/CP@lcPGMA@AuNPs in the organic solvent (ACN/DMF = 4:1 v/v) by performing their fluorescence titration experiments (Fig. 8a, c and Fig. S8a, b, ESI†). Polym@NBD-MIP and polym@NBD-MIP@lcPGMA@AuNPs proved to have almost the same LODs toward FA, as revealed by the rather similar slopes of their linear plots of fluorescence enhancement versus FA concentrations (Fig. 8b and d). This result indicates that the incorporation of AuNPs onto the above NBD-labelled fluorescent MIP particles has negligible influence on their optosensing sensitivity. This result, together with our observation that the direct attachment of AuNPs onto the fluorescent MIP layer surfaces significantly reduces its fluorescence enhancement effect upon exposure to FA and thus largely lowers its optosensing sensitivity (as mentioned in the synthesis and characterization section), clearly demonstrates that the incorporated AuNPs cannot improve (or even reduce) the optosensing sensitivity of our AuNP-containing fluorescent MIPs, which is in sharp contrast with the previous finding by Haupt and coworkers in another fluorescent MIP system.25 The different physical and chemical structures as well as the target analytes between our AuNP-containing fluorescent MIPs and theirs might be responsible for these different results. Further investigation is ongoing to design and study more systems to obtain a general conclusion.
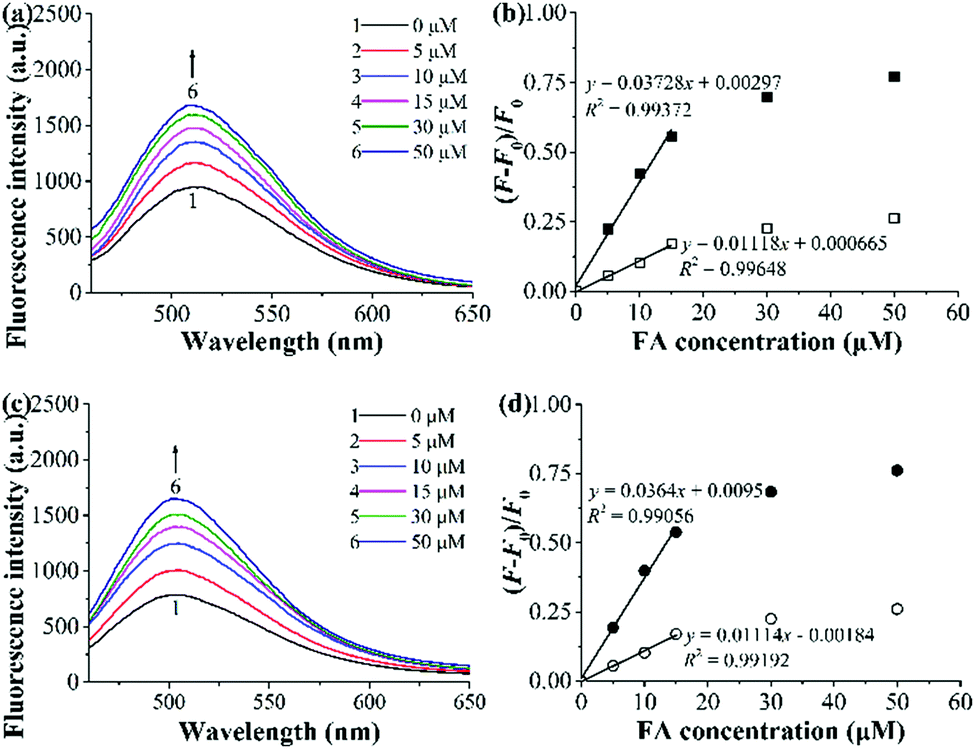 | ||
| Fig. 8 (a and c) Fluorescence spectra of polym@NBD-MIP (a) and polym@NBD-MIP@lcPGMA@AuNPs (c) (0.25 mg mL−1) upon their exposure to different concentrations of FA in ACN/DMF (4:1 v/v) at 25 °C for 2 h. (b and d) Dependence of the fluorescence enhancement of polym@NBD-MIP/CP (b) and polym@NBD-MIP/CP@lcPGMA@AuNPs (d) [MIP (filled symbols)/CP (open symbols)] on FA concentrations in ACN/DMF (4:1 v/v) at 25 °C (derived from (a), (c) and Fig. S8a, b, ESI†). | ||
Conclusions
We have demonstrated for the first time a versatile strategy for developing well-defined complex biological sample-compatible AuNP-containing fluorescent MIP microspheres and the negligible (or negative) effect of the incorporated AuNPs on their optosensing sensitivity. Such advanced functional fluorescent MIP particles were readily prepared by the combined use of RAFT polymerization and Au–thiol chemistry. The introduction of a lightly crosslinked thin polymer layer between the fluorescent MIP layer and AuNPs played a decisive role in avoiding the large reduction in the fluorescence enhancement effect of the MIP layer upon exposure to the target analyte FA (it also allowed FA to pass through it freely and reach the imprinted sites in the MIP layer because of its low crosslinking degree). They proved to function as a highly efficient “turn-on” type fluorescent chemosensor toward FA. They exhibited high FA optosensing selectivity and sensitivity in the undiluted urine samples (with a LOD of 0.088 μM) as well as outstanding photostability and reusability. Moreover, their direct use for FA quantification in another undiluted urine sample with good recoveries (96.3–101.6%) and high accuracy (RSD = 0.6–3.1%) (even in the presence of several interferents) was also demonstrated. Although the incorporated AuNPs did not improve the optosensing sensitivity of our AuNP-containing fluorescent MIP microspheres, their introduction of AuNPs not only provides an effective new way for grafting hydrophilic polymer brushes, but it also imparts them with certain new functions (e.g., SERS capability). Considering the high performance controllability of these hydrophilic AuNP-containing fluorescent MIP microspheres owing to their easy structure-tailoring via controlled RAFT polymerization and efficient Au–thiol chemistry, we believe that such advanced functional fluorescent MIP particles hold much promise as both efficient optosensors and potential SERS sensors for rapidly and accurately detecting FA in many biomedical and bioanalytical applications such as clinical diagnostics and food analyses. The development of more advanced fluorescent MIP sensors for FA detection with improved optosensing selectivity (by preparing homogeneous binding sites39) and sensitivity (by creating binding sites with higher affinity39 or introducing mesoporous structures into the fluorescent MIP layers18) is currently ongoing in our laboratories.Experimental
Materials
The chemicals used in this work and the detailed synthetic procedures for the “living” poly(4-VP-co-EGDMA) microspheres, an aqueous solution of AuNPs, and PHEMA-SH are presented in the ESI.†Synthesis of polym@NBD-MIP/CP microspheres (entries 2 and 3 in Table 1)
Polym@NBD-MIP microspheres were prepared via the controlled surface molecular imprinting by using a SI-RAFT polymerization method with the “living” poly(4-VP-co-EGDMA) particles (entry 1, Table 1) as the immobilized RAFT agent: a mixed solution of FA (0.31 mmol), 4-VP (2.45 mmol), MA-Urea-NBD (0.12 mmol), and ACN/DMF (4:1 v/v) (350 mL) was magnetically stirred at room temperature for 30 min to allow the self-assembly of the functional monomers and template. Afterwards, the “living” poly(4-VP-co-EGDMA) particles (350 mg), EGDMA (7.41 mmol), and AIBN (0.36 mmol) were added into the reaction system successively. The mixture was dispersed with ultrasonication for 5 min, degassed with argon bubbling for 30 min in an ice-water bath, sealed, and magnetically stirred (300 rpm) first at 50 °C for 2 h and then at 60 °C for 22 h. The polymer particles were obtained through centrifuging the reaction mixture. They were then washed with methanol until no FA was detectable in the washing solution. After being dried at 40 °C under a vacuum to a constant weight, the desired polym@NBD-MIP particles (entry 2, Table 1) were obtained with a weight increase of 25.7% compared with the starting “living” poly(4-VP-co-EGDMA).
The preparation and purification of polym@NBD-CP microspheres were carried out similarly except for omitting FA in the SI-RAFT polymerization system. A weight increase of 29.9% was observed for polym@NBD-CP (entry 3, Table 1) compared with the starting “living” poly(4-VP-co-EGDMA).
Synthesis of polym@NBD-MIP/CP@lcPGMA microspheres (entries 4 and 5 in Table 1)
Polym@NBD-MIP@lcPGMA and polym@NBD-CP@lcPGMA microspheres were prepared via the SI-RAFT copolymerization of GMA and EGDMA by using the above-obtained “living” polym@NBD-MIP/CP as the immobilized RAFT agent following our previous report:32 to a one-neck round-bottom flask (50 mL) with a magnetic stir bar inside were added THF (22 mL), GMA (2.20 mmol), EGDMA (1.10 mmol), AIBN (13.64 μmol), and polym@NBD-MIP/CP (220 mg) successively. After being dispersed with ultrasonication for 5 min, the reaction mixture was purged with argon for 20 min in an ice-water bath, sealed, and magnetically stirred (300 rpm) at 60 °C for 1 h. The polymer particles were collected by centrifugation, washed with methanol thrice, and then dried at 40 °C under vacuum to a constant weight. Polym@NBD-MIP@lcPGMA and polym@NBD-CP@lcPGMA showed a weight increase of 7.4% and 6.8% relative to the starting polym@NBD-MIP and polym@NBD-CP, respectively (entries 4 and 5 in Table 1).Synthesis of polym@NBD-MIP/CP@lcPGMA-SH microspheres (entries 6 and 7 in Table 1)
Polym@NBD-MIP/CP@lcPGMA-SH microspheres were prepared via the aminolysis of the above-obtained polym@NBD-MIP/CP@lcPGMA particles following a literature method:28 to a one-neck round-bottom flask (10 mL) with a magnetic stir bar inside were added DMF (2.90 mL), ethanediamine (EDA) (1.08 mmol), triethylamine (TEA) (1.09 mmol) and polym@NBD-MIP/CP@lcPGMA (175 mg) successively. After being dispersed with ultrasonication for 5 min, the reaction mixture was purged with argon for 10 min in an ice-water bath, sealed, and magnetically stirred (300 rpm) at 50 °C for 15 h. An aqueous solution of hydrochloride (0.1 M) was added into the resulting solutions to adjust their pH = 5. Afterwards, the polymer particles were collected by centrifugation, washed with distilled water until the washing solutions became neutral, and then dried at 40 °C under vacuum to a constant weight. Negligible weight changes were observed for the resulting polym@NBD-MIP@lcPGMA-SH and polym@NBD-CP@lcPGMA-SH in comparison with their corresponding precursors (entries 6 and 7, Table 1).Synthesis of polym@NBD-MIP/CP@lcPGMA@AuNPs microspheres (entries 8 and 9 in Table 1)
To a one-neck round-bottom flask (250 mL) with a magnetic stir bar inside was added the aqueous solution of AuNPs (156 mL) and polym@NBD-MIP/CP@lcPGMA-SH (130 mg). After being dispersed with ultrasonication for 5 min, the reaction mixture was purged with argon for 10 min in an ice-water bath, sealed, and magnetically stirred (300 rpm) at 25 °C for 1 h. The polymer particles were collected by centrifugation (all AuNPs were attached onto the MIP/CP particles with surface-bound thiol groups, as revealed by the absence of AuNPs in the supernatants), washed with methanol thrice, and then dried at 40 °C under vacuum to a constant weight. Polym@NBD-MIP@lcPGMA@AuNPs and polym@NBD-CP@lcPGMA@AuNPs showed a weight increase of 6.3% and 6.7% relative to the starting polym@NBD-MIP@cPGMA-SH and polym@NBD-CP@lcPGMA-SH, respectively (entries 8 and 9, Table 1).Synthesis of polym@NBD-MIP/CP@lcPGMA@AuNPs@PHEMA microspheres (entries 10 and 11 in Table 1)
To a one-neck round-bottom flask (25 mL) with a magnetic stir bar inside was added methanol (6 mL), pure water (3 mL), PHEMA-SH (0.048 mmol, 288 mg), and polym@NBD-MIP/CP@lcPGMA@AuNPs (80 mg) successively. After being dispersed with ultrasonication for 5 min, the reaction mixture was purged with argon for 20 min in an ice-water bath, sealed, and magnetically stirred (300 rpm) at 25 °C for 12 h. The polymer particles were then collected by centrifugation, washed with methanol thrice, and then dried at 40 °C under vacuum to a constant weight. Polym@NBD-MIP@lcPGMA@AuNPs@PHEMA and polym@NBD-CP@lcPGMA@AuNPs@PHEMA showed a weight increase of 4.2% and 4.6% relative to the starting polym@NBD-MIP@lcPGMA@AuNPs and polym@NBD-CP@lcPGMA@AuNPs, respectively (entries 10 and 11, Table 1).Characterization
The samples were characterized with proton nuclear magnetic resonance (1H NMR), scanning electron microscope (SEM), transmission electron microscope (TEM), Fourier-transform infrared (FT-IR) spectrometer, UV-vis scanning spectrophotometer, and spectrofluorometer. The water dispersion stability of the polymer particles was also studied.The equilibrium or competitive binding properties of the MIPs/CPs were evaluated by incubating them with FA or a mixture of FA and its analogues MTX and TMP (Scheme S1, ESI†) in different media at 25 °C for 8 h, and the amounts of the analyte(s) bound to the MIPs/CPs were determined by measuring those remaining in the solutions with high performance liquid chromatography (HPLC).
The optosensing properties of the MIPs/CPs were studied by measuring the fluorescence of their solutions or their mixed solutions with analyte(s) in different media using the spectrofluorometer.
The details of the above instruments, characterization, and measurements are included in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (21574070 and 22071121) and the Project supported by the NCC Fund (NCC2020PY12).Notes and references
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- F. Lanza and B. Sellergren, Chromatographia, 2001, 53, 599–611 CrossRef CAS.
- H. Zhang, L. Ye and K. Mosbach, J. Mol. Recognit., 2006, 19, 248–259 CrossRef CAS PubMed.
- G. Wulff and J. Liu, Acc. Chem. Res., 2012, 45, 239–247 CrossRef CAS PubMed.
- X. Shen, C. Xu and L. Ye, Ind. Eng. Chem. Res., 2013, 52, 13890–13899 CrossRef CAS.
- A. Martín-Esteban, TrAC, Trends Anal. Chem., 2013, 45, 169–181 CrossRef.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- T. Takeuchi and H. Sunayama, Chem. Commun., 2018, 54, 6243–6251 RSC.
- H. Zhang, Adv. Mater., 2020, 32, 1806328 CrossRef CAS PubMed.
- K. Haupt, P. X. Medina Rangel and B. Tse Sum Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS PubMed.
- S. Xu, L. Wang and Z. Liu, Angew. Chem., Int. Ed., 2020, 60, 3858–3869 CrossRef PubMed.
- B. Tse Sum Bui, T. Auroy and K. Haupt, Angew. Chem., Int. Ed., 2022, 61, e20210649 CrossRef PubMed.
- T. Takeuchi, T. Mukawa and H. Shinmori, Chem. Rec., 2005, 5, 263–275 CrossRef CAS PubMed.
- F. Canfarotta, M. J. Whitcombe and S. A. Piletsky, Biotechnol. Adv., 2013, 31, 1585–1599 CrossRef CAS PubMed.
- J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 207, 144–157 CrossRef CAS.
- W. Wan, S. Wagner and K. Rurack, Anal. Bioanal. Chem., 2016, 408, 1753–1771 CrossRef CAS PubMed.
- M. Dabrowski, P. Lach, M. Cieplak and W. Kutner, Biosens. Bioelectron., 2018, 102, 17–26 CrossRef CAS PubMed.
- Q. Yang, J. Li, X. Wang, H. Peng, H. Xiong and L. Chen, Biosens. Bioelectron., 2018, 112, 54–71 CrossRef CAS PubMed.
- H. Zhang, in Methods in Molecular Biology 2359: Molecularly Imprinted Polymers-Methods and Protocols, ed. A. Martín-Esteban, Humana Press (Springer Nature), New York, 2021, ch. 8, pp. 97–108 Search PubMed.
- H. Niu, Y. Yang and H. Zhang, Biosens. Bioelectron., 2015, 74, 440–446 CrossRef CAS PubMed.
- Y. Yang, Z. Wang, H. Niu and H. Zhang, Biosens. Bioelectron., 2016, 86, 580–587 CrossRef CAS PubMed.
- Y. Yang, H. Niu and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 15741–15749 CrossRef CAS PubMed.
- S. Xu, Y. Zou and H. Zhang, Talanta, 2020, 211, 120711 CrossRef CAS PubMed.
- Y. Hou, Y. Zou, Y. Zhou and H. Zhang, Langmuir, 2020, 36, 12403–12413 CrossRef CAS PubMed.
- X.-A. Ton, B. Tse Sum Bui, M. Resmini, P. Bonomi, I. Dika, O. Soppera and K. Haupt, Angew. Chem., Int. Ed., 2013, 52, 8317–8321 CrossRef CAS PubMed.
- R. M. Pitkin, Am. J. Clin. Nutr., 2007, 85(suppl.), 285S–288S CrossRef CAS PubMed.
- H. Zhang, Eur. Polym. J., 2013, 49, 579–600 CrossRef CAS.
- T. Zhou, L. Jørgensen, M. A. Mattebjerg, I. S. Chronakis and L. Ye, RSC Adv., 2014, 4, 30292–30299 RSC.
- L. F. Ribeiro, J. C. Masini and F. Svec, TrAC, Trends Anal. Chem., 2019, 118, 606–624 CrossRef CAS.
- M. Bompart, Y. De Wilde and K. Haupt, Adv. Mater., 2010, 22, 2343–2348 CrossRef CAS PubMed.
- T. Kamra, S. Chaudhary, C. Xu, L. Montelius, J. Schnadt and L. Ye, J. Colloid Interface Sci., 2016, 461, 1–8 CrossRef CAS PubMed.
- Y. Ma, J. Gao, C. Zheng and H. Zhang, J. Mater. Chem. B, 2019, 7, 2474–2483 RSC.
- S. Y. Lim, J. H. Kim, J. S. Lee, J. Ahn, M.-G. Kim and C. B. Park, J. Mater. Chem., 2011, 21, 17623–17626 RSC.
- M. Zhao, X. Chen, H. T. Zhang, H. Yan and H. Zhang, Biomacromolecules, 2014, 15, 1663–1675 CrossRef CAS PubMed.
- G. Pan, Y. Zhang, Y. Ma, C. Li and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11731–11734 CrossRef CAS PubMed.
- Y. Ma, G. Pan, Y. Zhang, X. Guo and H. Zhang, Angew. Chem., Int. Ed., 2013, 52, 1511–1514 CrossRef CAS PubMed.
- T. Hishiya, M. Shibata, M. Kakazu, H. Asanuma and M. Komiyama, Macromolecules, 1999, 32, 2265–2269 CrossRef CAS.
- D. C. Apodaca, R. B. Pernites, R. R. Ponnapati, F. R. Del Mundo and R. C. Advincula, ACS Appl. Mater. Interfaces, 2011, 3, 191–203 CrossRef CAS PubMed.
- H. Zhang, M. Zhao and Y. Yang, in Biomedical Nanomaterials, ed. Y. Zhao and Y. Shen, Wiley-VCH Verlag GmbH & Co. KGaA., Weinheim, 1st edn, 2016, ch. 11, pp. 283–326 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2tb00179a |
| This journal is © The Royal Society of Chemistry 2022 |