
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen reduction reaction catalyzed by carbon composites with ruthenium-doped iron oxide nanoparticles†
Qiming
Liu
a,
Hong Bo
Zhou
ab,
Forrest
Nichols
a,
Han-Lin
Kuo
c,
Rene
Mercado
a,
Bingzhang
Lu
a,
Weiya
Zhu
a,
Yashu
Liu
ad,
Jennifer Q.
Lu
c,
Frank
Bridges
e and
Shaowei
Chen
 *a
*a
aDepartment of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: shaowei@ucsc.edu
bSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China
cSchool of Engineering, University of California, 5200 North Lake Road, Merced, California 95343, USA
dSchool of Environmental and Chemical Engineering, Jiangsu University of Science and Technology, Zhenjiang, Jiangsu 212013, China
eDepartment of Physics, University of California, 1156 High Street, Santa Cruz, California 95064, USA
First published on 12th April 2022
Abstract
Carbon nanocomposites based on transition-metal oxides have been attracting extensive attention as cost-effective catalysts towards the oxygen reduction reaction (ORR). However, the activity remains subpar as compared to state-of-the-art platinum catalysts. One way to enhance the ORR performance is to dope a second metal into the nanocomposite to manipulate the electronic structure and hence the interactions with key reaction intermediates. Herein, dual metal (Ru and Fe) and nitrogen codoped carbon (RuFe-NC) nanocomposites were synthesized by controlled pyrolysis of a Fe–Ru–Fe trinuclear complex along with zeolitic imidazolate framework-8. The obtained porous nanocomposites consisted of Ru-doped Fe2O3 nanoparticles embedded within a carbon scaffold, and exhibited an ORR activity in alkaline media rivaling that of commercial Pt/C, which was also markedly better than those of the monometallic counterparts and nanocomposites prepared with a simple mixture of the individual monometallic compound precursors. Structural characterization suggests that the use of the trinuclear complex facilitated the atomic dispersion of ruthenium within the iron oxide nanoparticles and charge transfer between the metal centers led to a high ORR activity. Results from this study suggest that rational design of heteronuclear complexes may be a unique strategy in the structural engineering of carbon-metal nanocomposites for high-performance electrocatalysis.
Introduction
Fuel cell technologies have been attracting extensive interest as an integral part of the sustainable economy.1 In fuel cell operation, small molecule fuels are oxidized at the anode and oxygen is reduced at the cathode, where both reactions necessitate appropriate catalysts so as to boost the electron-transfer kinetics and produce a sufficiently high current density for practical applications.2–5 Of these, the oxygen reduction reaction (ORR) at the cathode has been recognized as a major bottleneck that limits the fuel cell performance, largely because of the complex reaction pathways and sluggish electron-transfer kinetics. Towards this end, platinum-based nanoparticles have been used extensively as the catalysts of choice for the ORR.4–7 Yet, the high cost and low natural abundance of platinum have significantly hampered the wide-spread application of fuel cell technologies. Thus, in recent studies, a range of strategies have been developed to reduce the metal loadings (and costs) and concurrently retain/enhance the catalytic activity.8–13 One effective route is to exploit low-cost 3d transition metals (i.e., Fe, Co, Ni, and Cu) and their derivatives (i.e., oxides, carbides, and nitrides) as the catalytic active centers.14–17Among these various candidates, transition metal oxides (TMOs) have been extensively investigated with promising advances.18–21 Nevertheless, it should be noted that the intrinsic activity of TMOs has remained mostly uncompetitive to that of the commercial Pt/C benchmark. For instance, theoretical studies based on first principles calculations have shown that the FeO5, CoO5, and NiO5 centers are too weak for the adsorption of O* species, while CrO5 and MnO5 are too strong rendering the desorption of O* difficult, owing to their different electronic structures.22 Thus, none of these is ideal for ORR electrocatalysis. To increase their activity towards ORR, a variety of strategies have been utilized to modify TMOs. For example, Wu et al.23 grew Fe3O4 nanoparticles on N-doped graphene aerogel and found that the substrate significantly increased the onset potential and cathodic current density, in contrast to those with (undoped) carbon black. Ma et al.24 also observed that with N-doped carbon black, ultrafine FeO1.4 nanoparticles exhibited an ORR activity competitive to that of Pt/C. Fan et al.25 showed that the carbon substrate could benefit FeOx with rapid mass transfer and catalyst stability.
In addition to structural engineering of the substrate, compositional manipulation of the oxide materials is another viable route with dual- or even tri-metal centers, where the metal-to-metal charge transfer can be exploited for enhanced electrical conductivity and improved ORR activity. For instance, Gao et al.26 atomically dispersed Pt on α-Fe2O3 and observed a dramatically enhanced ORR activity with a half-wave potential (E1/2) of +1.05 V vs. reversible hydrogen electrode (RHE), owning to the strong electronic coupling of the Pt–Fe atomic pairs. Such a synergistic effect facilitated the adsorption of O2 and dissociation of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. Wei et al.27 found that Ru dopants could facilitate O2 adsorption on Co3O4 and significantly improve the ORR activity with E1/2 increased from +0.32 to +0.77 V. In fact, Ru has been widely utilized as dopants of TMOs in various oxygen-involved reactions, due to its optimal affinity to oxygen intermediate species.8,28 Theoretical calculations have shown that Ru can serve as the active sites on SnO2 for the adsorption of oxygen species owning to the delocalization of electrons.29 Dong et al.30 also showed that Ru could regulate the charge transfer among the Cr, Co, and oxygen species. That is, the introduction of Ru into TMOs may tune their electronic structures and improve the ORR activity due to enhanced interactions with O*.
O bonds. Wei et al.27 found that Ru dopants could facilitate O2 adsorption on Co3O4 and significantly improve the ORR activity with E1/2 increased from +0.32 to +0.77 V. In fact, Ru has been widely utilized as dopants of TMOs in various oxygen-involved reactions, due to its optimal affinity to oxygen intermediate species.8,28 Theoretical calculations have shown that Ru can serve as the active sites on SnO2 for the adsorption of oxygen species owning to the delocalization of electrons.29 Dong et al.30 also showed that Ru could regulate the charge transfer among the Cr, Co, and oxygen species. That is, the introduction of Ru into TMOs may tune their electronic structures and improve the ORR activity due to enhanced interactions with O*.
Herein, nanocomposites based on Ru-doped Fe2O3 nanoparticles supported on porous N-doped carbon were prepared by controlled pyrolysis of a Fe–Ru–Fe trinuclear complex ([(DMAP)4RuII{(μ-NC)FeIII(CN)5}2](PPh4)4, Ru(LFe)2, with DMAP = 4-(dimethylamino)pyridine and PPh4 = tetraphenylphosphonium)31 as the metal sources and zeolitic imidazolate framework-8 (ZIF-8) as the carbon and nitrogen precursor. The resulting RuFe-NC nanocomposites exhibited a hierarchical porous structure, with abundant nitrogen dopants in the carbon scaffold and Ru homogeneously doped into the Fe2O3 nanoparticles. By contrast, for the control sample prepared with a physical mixture of ZIF-8 and individual Fe and Ru metal salts at equivalent feeds, partial phase segregation was observed between Fe and Ru. Notably, in alkaline media, RuFe-NC displayed an ORR activity rivalling that of commercial Pt/C and was the best among the metal,nitrogen-codoped carbon composites. Results from this study highlight the significance of pre-designed metal precursors in spatial control of dual metal oxide nanoparticles in carbon nanocomposites for high-performance electrocatalysis.
Experimental section
Chemicals
Dichlorotetrakis(dimethylsulfoxide)ruthenium(II) (cis-Ru(DMSO)4Cl2), tetraphenylphosphonium ferricyanide ((PPh4)3[Fe(CN)6]), Ru(LFe)2, and ZIF-8 were prepared according to methods described in the literature.31,32 All other chemicals and reagents were commercially available and used as received without further purification. Water was supplied with a Barnstead Nanopure Water system (18 MΩ cm).Sample preparation
To prepare the RuFe-NC composites, 0.1 g (0.4 mmol) of ZIF-8 was mixed with 0.01 g (0.004 mmol) of Ru(LFe)2 synthesized above in 10 mL of hexane under sonication for 10 min and then under magnetic stirring for 2 h at room temperature. The solid was then collected by centrifugation at 6000 rpm for 5 min and dried under vacuum at 50 °C for 12 h. The resulting ZIF-8@Ru(LFe)2 precursor was then loaded onto a ceramic boat, which was transferred to a tube furnace and heated at 900 °C for 2 h under a nitrogen atmosphere, producing RuFe-NC.A series of control samples were prepared in the same fashion: (i) NC by direct pyrolysis of ZIF-8, (ii) Ru-NC by a mixture of 0.1 g (0.4 mmol) of ZIF-8, 0.002 g (0.004 mmol) of cis-Ru(DMSO)4Cl2, and 0.003 g of DMAP, (iii) Fe-NC by 0.1 g (0.4 mmol) of ZIF-8 and 0.01 g (PPh4)3[Fe(CN)6], and (iv) RuFe-NCmix by 0.1 g (0.4 mmol) of ZIF-8, 0.002 g (0.004 mmol) of cis-Ru(DMSO)4Cl2, 0.003 g of DMAP and 0.01 g of (PPh4)3[Fe(CN)6].
Characterizations
Transmission electron microscopic (TEM) measurements were carried out on a JEOL JEM-2100F electron microscope. Scanning electron microscopy (SEM) and energy dispersive spectroscopic (EDS) mapping studies were carried out with an Apreo SEM microscope. X-ray diffraction (XRD) patterns were acquired with a Bruker D8 Advance diffractometer with Cu Kα radiation (λ = 0.15418 nm). X-ray photoelectron spectra (XPS) were obtained with a PHI-5400 XPS instrument with an Al Kα source operated at 350 W and 10−9 Torr. X-ray absorption spectroscopy (XAS) measurements were carried out at 10 K on beamline 4–1 at the Stanford Synchrotron Radiation Light source using an Oxford liquid helium cryostat. Nitrogen sorption isotherms were acquired with a Micromeritics ASAP 2020 porosimetry system at 77.3 K. UV-Vis absorption spectra were collected on a PerkinElmer Lambda 35 UV-vis spectrometer. Infrared spectroscopic measurements were conducted with a PerkinElmer Spectrum One FTIR spectrometer. Inductively coupled plasma-optical emission spectrometry (ICP-OES) measurements were conducted with an iCap 7400 analyzer.Electrochemistry
The electrochemical tests were carried out on a CHI 710 electrochemical workstation in a standard three-electrode configuration. A graphite rod was used as the counter electrode, a Ag/AgCl in 0.1 M KCl as the reference electrode, and a polished rotating (gold) ring-(glassy carbon) disk electrode (RRDE, from Pine Instrument Co.) as the working electrode. The Ag/AgCl electrode was calibrated against an RHE and all potentials in the present study were referenced to this RHE. During the ORR tests, the ring potential was set at +1.5 V vs. RHE. To prepare the catalyst inks, 2 mg of the samples obtained above was added into 1 mL of an isopropanol/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture and 10 μL of a 100 wt% Nafion solution. The suspension was sonicated to form a homogeneous ink. 20 μL of the ink was then dropcast onto the glassy carbon disk electrode (surface area 0.246 cm2), dried at room temperature, and coated with 5 μL of a 20 wt% Nafion solution, corresponding to a catalyst loading of 0.162 mg cm−2. A same loading of commercial 20% Pt/C was used in the test.
:1 v/v) mixture and 10 μL of a 100 wt% Nafion solution. The suspension was sonicated to form a homogeneous ink. 20 μL of the ink was then dropcast onto the glassy carbon disk electrode (surface area 0.246 cm2), dried at room temperature, and coated with 5 μL of a 20 wt% Nafion solution, corresponding to a catalyst loading of 0.162 mg cm−2. A same loading of commercial 20% Pt/C was used in the test.
Results and discussion
Synthesis and morphological characterization
The synthetic procedure of the RuFe-NC composite consists of three major steps, as shown in Scheme 1. The first step is the synthesis of the Ru(LFe)2 trinuclear complex by simple refluxing of cis-Ru(DMSO)4Cl2, DMAP, and (PPh4)3[Fe(CN)6] (Fig. S1–S3, ESI†),31 where one Ru atom is bridged by two cyanide (CN) ligands to two Fe centers. Second, rhombic dodecahedral ZIF-8 crystals were synthesized by mixing 2-methylimidazole and zinc nitrate salts,33,34 onto which was adsorbed the Ru(LFe)2 complex in hexane. Third, controlled pyrolysis of the ZIF-8@Ru(LFe)2 mixture at 900 °C for 2 h in a nitrogen atmosphere led to the production of porous carbons doped with Ru and Fe (RuFe-NC). Control samples of RuFe-NCmix, Ru-NC, Fe-NC, and NC were synthesized in the same fashion.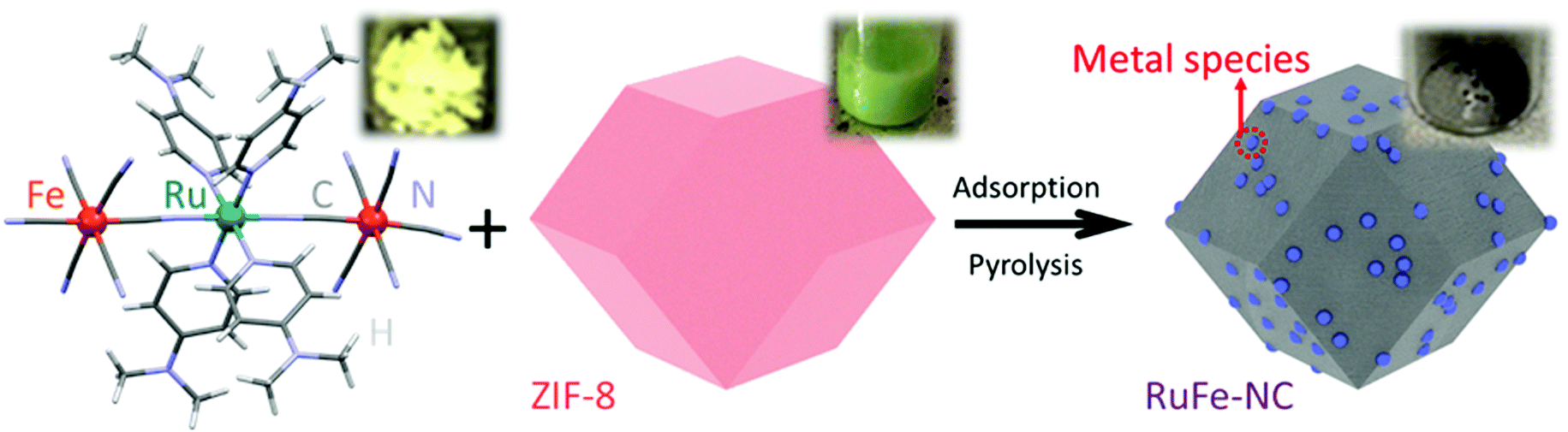 | ||
| Scheme 1 Molecular structure of the Ru(LFe)2 complex and the synthetic procedure of the RuFe-NC composite. Insets show the photographs of the products at different reaction stages. | ||
The porous structure of RuFe-NC was confirmed by Brunauer–Emmett–Teller (BET) measurements. As shown in Fig. 1a, the RuFe-NC, RuFe-NCmix, Ru-NC, Fe-NC, and NC samples all exhibited an H4-type hysteresis loop in the N2 sorption isotherms, suggesting the formation of a mesoporous structure within the carbon matrices, and RuFe-NC shows an abrupt increase of the adsorbed N2 quantity at relatively low pressures, indicative of a large number of micropores. Notably, the specific surface area varied markedly among the sample series, 507.6 m2 g−1 for RuFe-NC, 540.2 m2 g−1 for RuFe-NCmix, 452.3 m2 g−1 for Ru-NC, 442.7 m2 g−1 for Fe-NC, and only 163.2 m2 g−1 for NC, suggesting that the metal precursors actually behaved as effective porogens due to the high thermal volatility.35 In addition, by the nonlocal density functional theory (NLDFT), the pore size distributions were then derived and depicted in Fig. 1b. Both RuFe-NC and NC can be seen to entail mesopores in the range of 1 to 2 nm, while RuFe-NCmix has a large portion of micropores at 0.8 nm. As for both Ru-NC and Fe-NC, the micropore fraction was relatively small, in comparison to the dual-metal counterparts. The RuFe-NC sample also showed a larger fraction of mesopores and lower fraction of micropores than NC, as confirmed from the cumulative pore volume in Fig. S4 (ESI†). The enhanced surface area and porosity of the metal-containing nanocomposites is anticipated to facilitate mass transfer and accessibility of the catalytic active sites,35 as demonstrated below in electrochemical measurements.
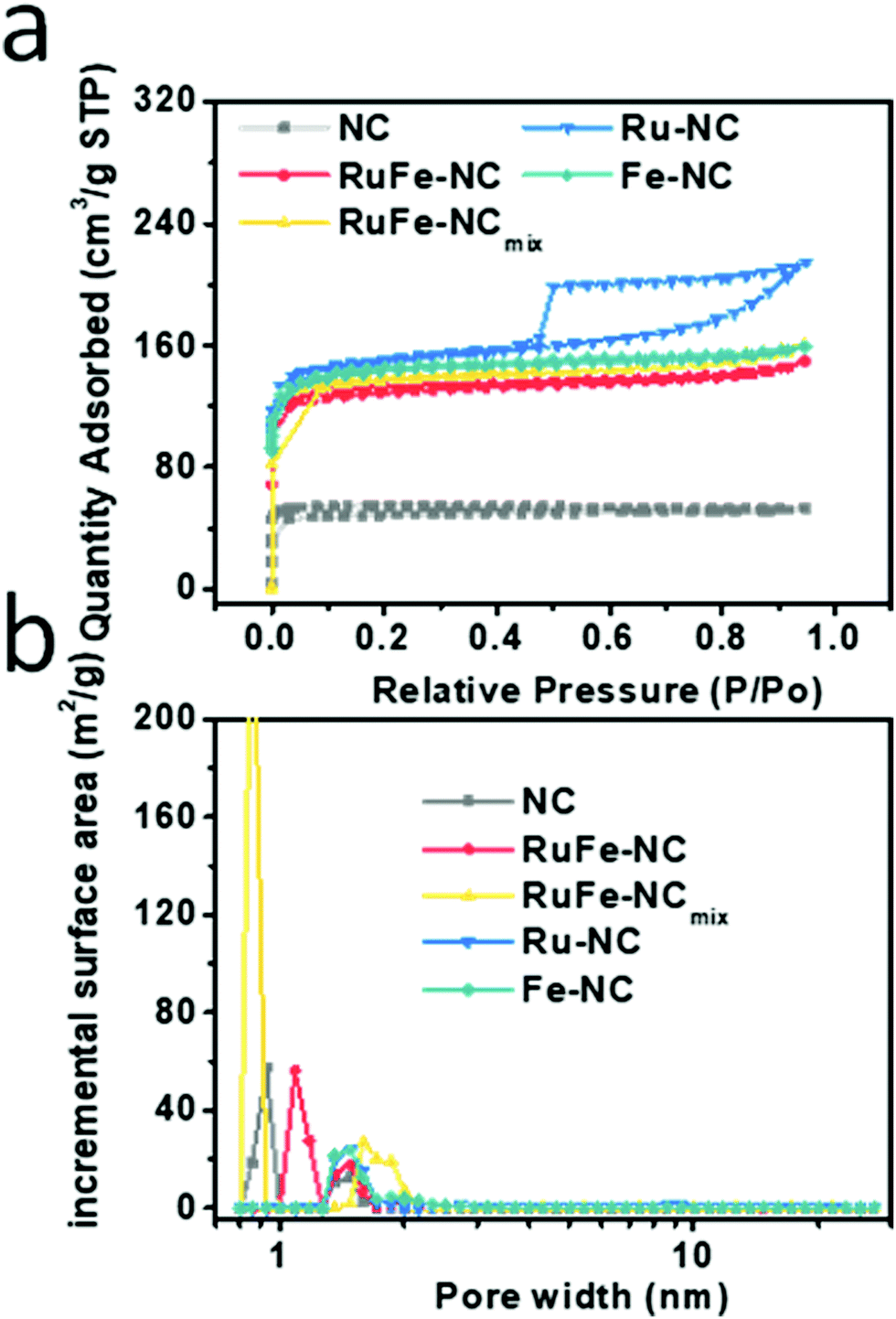 | ||
| Fig. 1 (a) Nitrogen sorption isotherms of RuFe-NC, RuFe-NCmix, Ru-NC, Fe-NC, and NC, and (b) the corresponding pore size distribution profiles. | ||
Further structural insights were obtained from TEM measurements. One can see from Fig. 2a and b that the RuFe-NC sample partially retained the dodecahedral shape of the ZIF-8 precursor, exhibiting a lateral length of several hundred nm with an apparent porous structure (as highlighted by the pink circles in Fig. 2a). At higher magnifications (Fig. 2c and Fig. S5, ESI†), one can find nanoparticles of ca. 2 nm in diameter that were encapsulated with a graphitized carbon shell. These nanoparticles exhibited well-defined lattice fringes, with a d spacing of ca. 0.220 and 0.235 nm that may be ascribed to the (113) and (400) facets of Fe2O3 (PDF#32-0469, red dashed circles),36 respectively. It should be noted that no apparent RuO2 or Ru nanoparticles can be found, and both d spacings are slightly larger than those of standard hematite Fe2O3, likely due to the doping of Ru into the iron oxide lattices (Fig. S5–S7, ESI†). Meanwhile, graphitized carbon can be seen to show a hemispherical shape with a d spacing of 0.338 nm (blue circle), corresponding to the (002) facet of graphitized carbon (JCPDS No. 01-0646).37,38 Furthermore, in elemental mapping analysis (Fig. 2d and Fig. S8, ESI†), it can be seen that Ru was mostly confined within the dark-contrast nanoparticles, consistent with the notion that Ru was doped into Fe2O3 nanoparticles. In addition, the RuFe-NC sample was found to consist of ca. 0.9 at% of Fe and 0.3 at% of Ru (Fig. S9, ESI†).
 | ||
| Fig. 2 (a–c) Representative TEM images of RuFe-NC at varied magnifications. The pink circles highlight the microporous structure of the carbon matrix. (d) Representative TEM image of RuFe-NC and the corresponding EDS elemental maps of Ru, Fe, and O. | ||
A similar structural morphology was observed with the Ru-NC (Fig. S10, ESI†), Fe-NC (Fig. S11, ESI†), and RuFe-NCmix samples (Fig. S12, ESI†). Notably, the RuFe-NCmix composite can be seen to consist of nanoparticles of ca. 4 nm in diameter embedded within the carbon sheets (Fig. S13, ESI†). Elemental mapping analysis of the selected area (Fig. S13, ESI†) showed a homogeneous distribution of C and N, while the elements of Fe and O were concentrated in the bright regions in the dark-field image, suggesting the formation of FeOx nanoparticles. Yet, the distribution pattern of ruthenium was different from that of Fe, suggesting partial segregation between these two elements. Furthermore, EDS measurements (Fig. S14, ESI†) showed that the RuFe-NCmix sample consisted of ca. 0.6 at% of Fe and 0.3 at% of Ru, consistent with the initial feed ratio. A similar sheet-like structure was observed withe both Ru-NC (Fig. S8, ESI†) and Fe-NC (Fig. S9, ESI†), which also featured metal oxide particles with a diameter of around 5 nm on the carbon sheets. Note that the nanoparticles were all markedly larger in these control samples than those in RuFe-NC, likely because of the bonding constraint in the Ru(LFe)2 complex precursor (Scheme 1) and the geometric confinement by the micropores (Fig. 1b) in the pyrolytic preparation of RuFe-NC.
X-ray spectroscopy analysis
XRD measurements were then carried out to examine the graphitization of the samples prepared by high-temperature pyrolysis. From Fig. 3, all samples can be seen to exhibit two broad diffraction peaks at 2θ ≈ 23° and 43°, which can be assigned to the (002) and (100) facets of graphite (JCPDS No. 01-0646), in good agreement with results from TEM measurements (Fig. 2c). No Additional diffraction peaks of metal or metal oxides can be discerned from the XRD profiles, most likely due to the low contents as detected in EDS measurements (Fig. S9 and S14, ESI†) and the small size of the nanoparticles (Fig. 2 and Fig. S5, ESI†).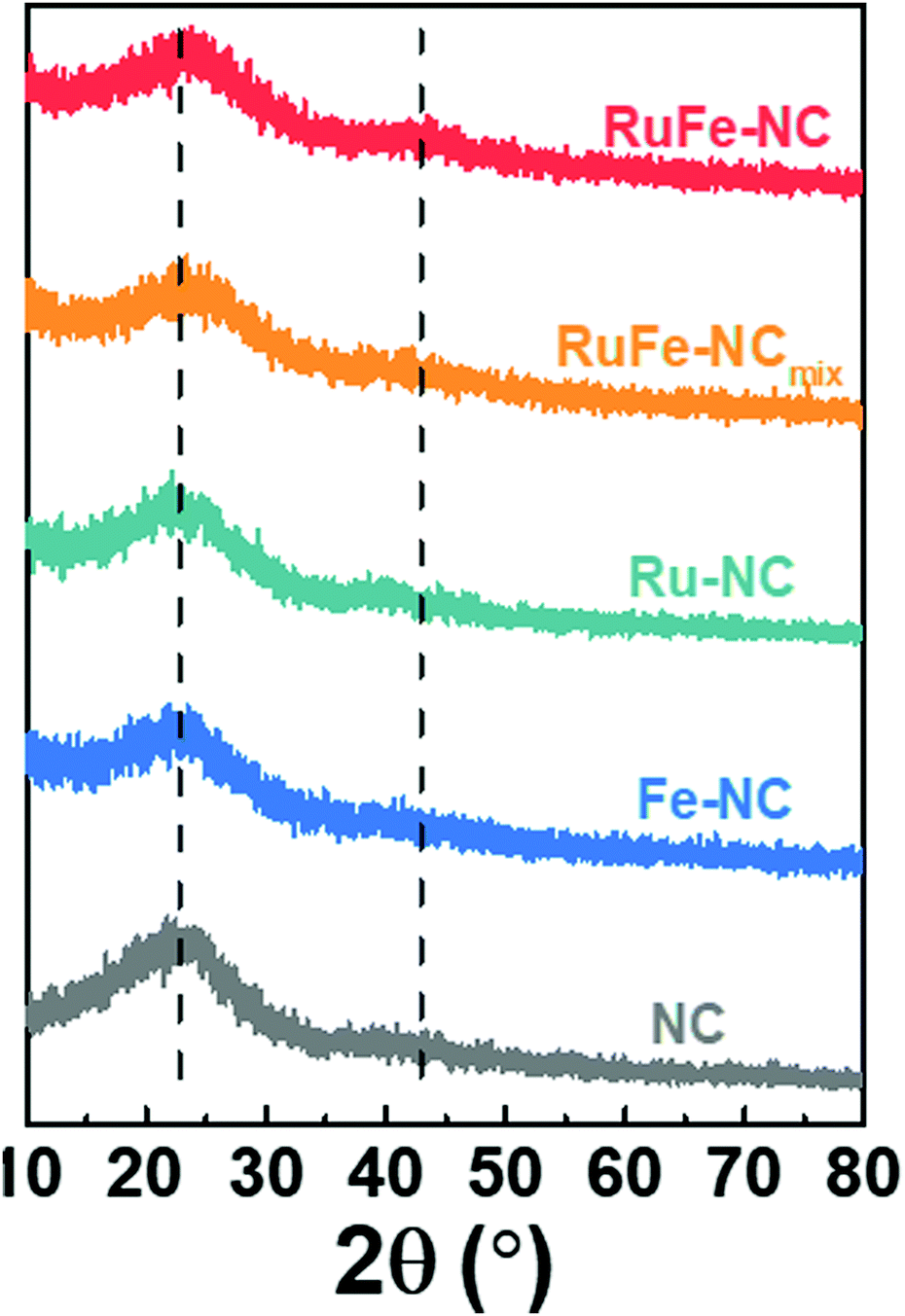 | ||
| Fig. 3 XRD patterns of RuFe-NC, RuFe-NCmix, Ru-NC, Fe-NC, and NC. The dashed lines indicate the expected 2θ position of the (002) and (100) diffractions of graphite (JCPDS No. 01-0646). Units on the y-axis have arbitrary units. | ||
XPS measurements were further performed to determine the elemental composition and valence states of the samples. From the survey spectra of RuFe-NC and RuFe-NCmix in Fig. S15 (ESI†), the C 1s, N 1s, and O 1s electrons can be readily identified at ca. 285 eV, 398 eV, and 531 eV, respectively. Fig. 4a depicts the high-resolution Fe 2p spectra of the Ru(LFe)2 complex, RuFe-NC, RuFe-NCmix, and Fe-NC, where the Fe 2p3/2 peaks of RuFe-NC (711.51 eV), RuFe-NCmix (711.26 eV) and Fe-NC (711.31 eV) can be found to blue-shift by ca. 2 eV in comparison to that of the Ru(LFe)2 complex (709.38 eV), suggesting an increase of the Fe valence state from Fe(II) in the complex to Fe(III) in the pyrolytic products.39,40
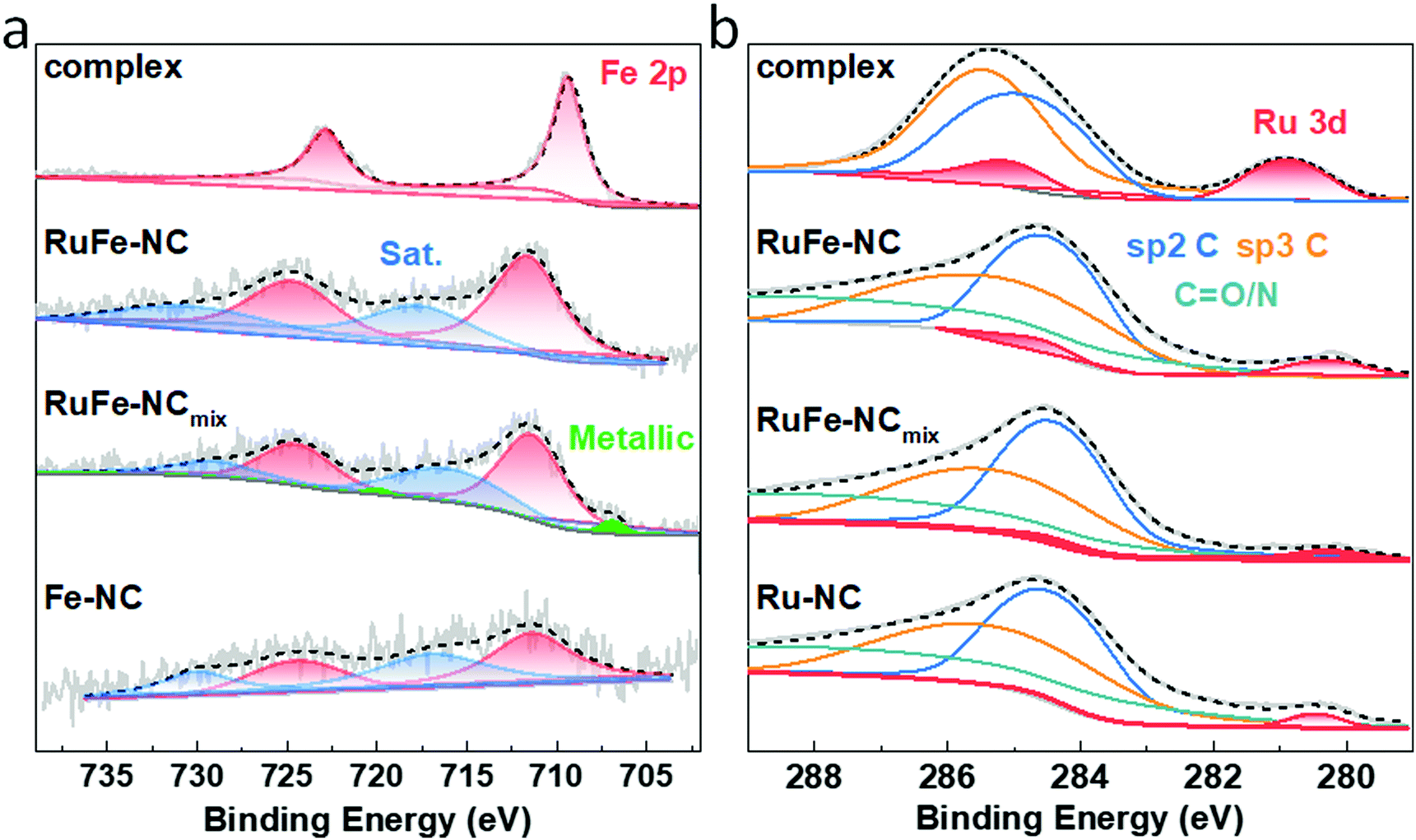 | ||
| Fig. 4 (a) High-resolution XPS spectra of the Fe 2p electrons of the RuFe complex, RuFe-NC, RuFe-NCmix, and Fe-NC. (b) High-resolution XPS spectra of the C 1s and Ru 3d electrons of RuFe complex, RuFe-NC, RuFe-NCmix, and Ru-NC. Note, the y-axis is logarithmic. | ||
As for the Ru 3d spectra in Fig. 4b, RuFe-NC, RuFe-NCmix and Ru-NC showed a small shoulder near 280.30 eV, which is ca. 0.6 eV lower than that of the Ru(LFe)2 complex (280.90 eV), suggesting partial reduction (electron enrichment) of ruthenium(II) in the complex precursor after pyrolysis (i.e., the Ru valence state in RuFe-NC was between 0 and +2).37,41 Notably, the Ru 3d binding energy of RuFe-NC and RuFe-NCmix was ca. 0.15 eV lower than that of Ru-NC (280.45 eV). This observation, in conjunction with the variation of the Fe(III) 2p3/2 binding energy of RuFe-NC (711.51 eV) > RuFe-NCmix (711.26 eV) ≈ Fe-NC (711.31 eV), suggests charge transfer from Fe to Ru in the Fe2O3 nanoparticles of RuFe-NC, which diminished in RuFe-NCmix due to segregated distributions of Fe and Ru (Fig. 2 and Fig. S11, ESI†). In fact, metallic Fe was even found in RuFe-NCmix (706.89 eV, 0.068 at%, Fig. 4a).
In addition, based on the integrated peak areas, the elemental compositions of the samples were also evaluated. The Fe content was estimated to be ca. 1.97 wt% (0.45 at%) for RuFe-NC and 2.35 wt% (0.59 at%) for RuFe-NCmix, close to the results from ICP-OES (1.69 wt% and 2.64 wt%, respectively) and EDS measurements (vide ante, Fig. S9 and S14, ESI†). The corresponding Ru content was ca. 0.50 wt% for RuFe-NC, somewhat higher than that from EDS analysis (Fig. S9, ESI†), and 0.34 wt% for RuFe-NCmix that was consistent with the EDS results (Fig. S14, ESI†). This suggests Ru enrichment on the surface of RuFe-NC as compared to that of RuFe-NCmix.
From the C 1s spectra in Fig. 4b, one can see that the samples all consists of a large amount (284.63 eV, 38.9 at%) of sp2-hybridized C, along with sp3 C (285.69 eV) and CO/N (288.80 eV), suggesting successful graphitization of the ZIF-8 precursors.34 The N 1s spectrum of RuFe-NC is shown in Fig. S16 (ESI†), which can be deconvoluted into four peaks at 398.3 eV for pyridinic N, 399.7 eV for pyrrolic N, 400.8 eV for graphitic N, and 403.4 eV for oxidized N, with an atomic content of 2.0, 1.7, 2.0, and 1.2 at%, respectively (6.9 at% in total). With such abundant pyridinic N and pyrrolic N moieties, it is possible that part of the Fe species was coordinated to those N moieties. In fact, from the O 1s spectra in Fig. S17 (ESI†), RuFe-NC can be seen to entail a small metal-O shoulder, which accounted for 0.20 at% of the sample, corresponding to 0.13 at% Fe in Fe2O3, which suggests that the rest of about 0.32 at% of Fe was in the form of FeNx moieties. Such a metal-lattice O component was markedly more pronounced with RuFe-NCmix, which was estimated to be 0.92 at%, ca. 1.75 times that of Fe in FeOx (0.53 at%), very close to the atomic ratio of 1.5 in Fe2O3. This suggests that the Fe species in RuFe-NCmix was mostly in the form of Fe2O3 (Fig. S13, ESI†), with a minor component of metallic Fe and no FeNx moieties.42 These results are also listed in Table S1–S6 (ESI†).
Further structural insights were obtained in XAS measurements. From the Fe K edge spectra in Fig. 5a, one can see that RuFe-NC exhibited a similar pre-edge profile and post-edge oscillations to Fe2O3, suggesting an analogous chemical environment of the Fe centers. In fact, both RuFe-NC and (hematite) Fe2O3 can be seen to display a small pre-edge peak at 7113 eV (magenta arrow) arising from the 1s to 3d forbidden electric dipole transition, consistent with an octahedral coordination shell in the samples,43,44 in sharp contrast to Fe foil which featured an intense shoulder in the pre-edge region. The Ru K edge spectra are depicted in Fig. 5b. One can see that the main edge energy of RuFe-NC was lower than that of RuO2, but higher than that of Ru foil, suggesting electron enrichment of Ru in RuFe-NC in comparison to RuO2. These observations are consistent with results from the XPS measurements which suggested Fe to Ru charge transfer in the composites.
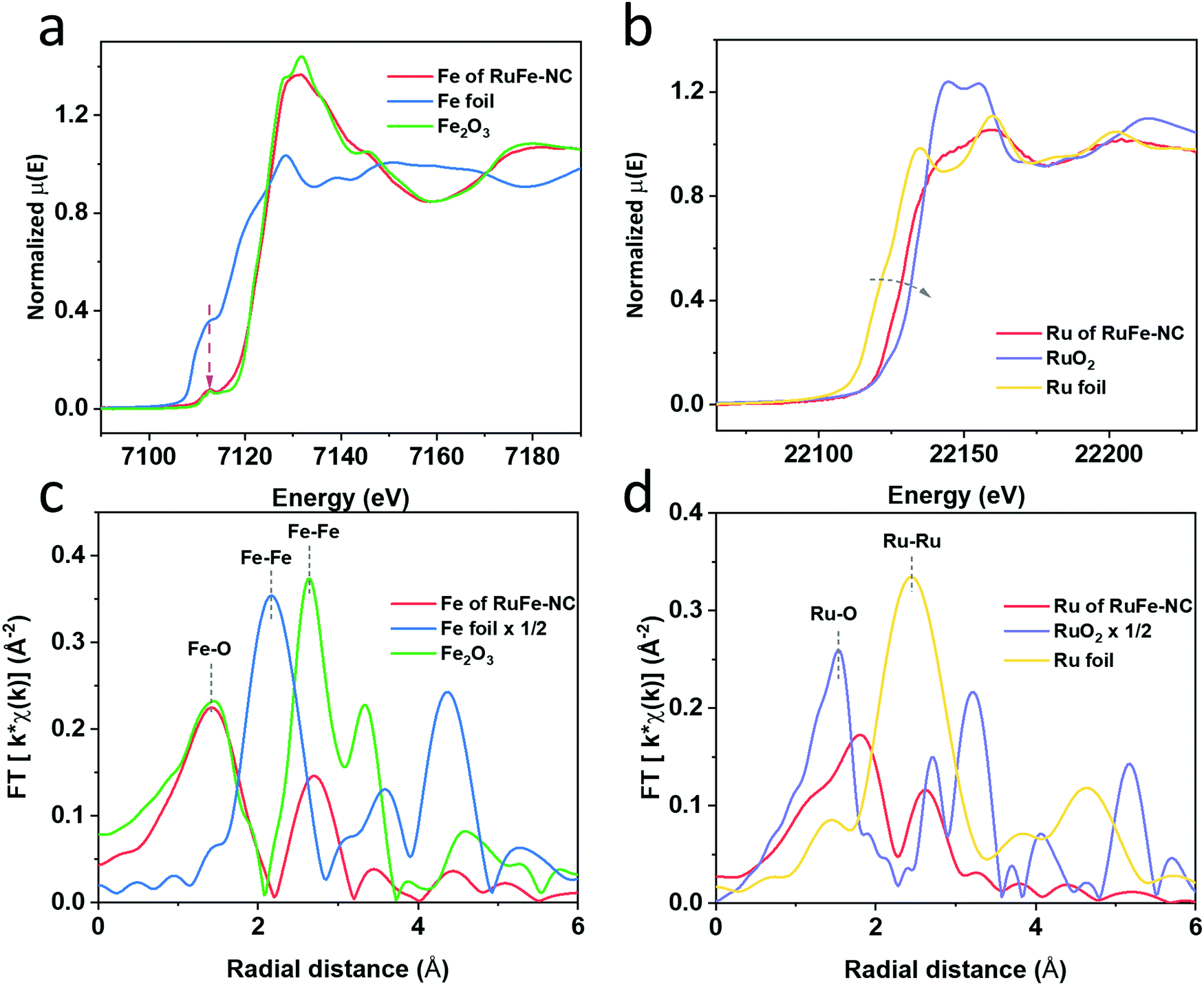 | ||
| Fig. 5 X-ray absorption near-edge spectra (XANES): (a) Fe K edge of RuFe-NC, Fe foil, and Fe2O3 (hematite); (b) Ru K edge of RuFe-NC, Ru foil, and RuO2. Corresponding extended X-ray absorption fine structure (EXAFS) spectra of (c) Fe of RuFe-NC, Fe foil, and Fe2O3; (d) Ru of RuFe-NC, Ru foil, and RuO2. | ||
The corresponding R space EXAFS spectra are shown in Fig. 5c and d. One can see that the peak patterns, again, are very similar between RuFe-NC and Fe2O3. In the Fe R space spectrum of RuFe-NC (Fig. 5c), two main peaks can be identified at ca. 1.42 and 2.70 Å, which can be assigned to the Fe–O/N bonds and second-shell Fe–Fe/Ru, respectively, in good agreement with the formation of (Ru-doped) Fe2O3 nanoparticles and FeNx moieties, as suggested in XPS measurements (Fig. 4). From the fitting results (Fig. S18a and Table S7, ESI†), one can see that that the Fe–O/N bonds in RuFe-NC possessed a coordination number of 4.8 with a bond length of 2.00 Å, while the Fe–Fe/Ru linkage featured a coordination number of 2.0 with a bond length of 2.95 Å. These fitting results are consistent with the small size and poor crystallinity of the Ru-doped Fe2O3 nanoparticles, and the formation of FeNx moieties as speculated in TEM and XPS measurements (vide ante). In the corresponding Ru EXAFS data (Fig. 5d), Ru of RuFe-NC displayed an apparently different pattern from those of Ru foil and RuO2. Three major species can be resolved in RuFe-NC, i.e., Ru–O (shoulder, ca. 1.3 Å), Ru–P (1.81 Å), and Ru–Ru (2.61 Å). These lengths are approximately 0.3 Å shorter than the realized bond length due to the well-known phase shift in EXAFS. Ru-doped α-Fe2O3 was used to calculate the FEFF functions of the Ru–O and Ru–Fe standard paths for EXAFS fitting. Data of the RuFe-NC sample was fitted by using two distinct structures, Ru-doped α-Fe2O3 and tetragonal RuP3, where the former accounted for ca. 80% in the sample and 20% for the latter.45 As shown in Fig. S18b and Table S7 (ESI†), one can see that the Ru–O bond possessed a coordination number of 2.5 with a bond length of 2.04 Å, which is larger than the typical Ru–O bond length of RuO2 (1.98 Å)46,47 and closer to that of M–O bond of hematite. The Ru–Fe path featured a coordination number of 1.16 and a bond length of 2.99 Å, which is smaller than the Ru–Ru bond length in RuO2 (3.3 Å) but closer to the one in hematite (2.97 Å), suggesting the successful incorporation of Ru into α-Fe2O3. Note that the low coordination number of Ru–O (2.51) is strong evidence for significant disorder in α-Fe2O3, likely due to the small oxide particle size and presence of amorphous iron oxide within the sample. Also, the fact that the peak at ca. 3.2 Å observed with RuO2 was absent in RuFe-NC suggests atomic dispersion of Ru into the Fe2O3 nanoparticles, as proposed in the above TEM and XPS measurements.
Electrocatalytic activity
The ORR activity of the obtained samples was then examined by electrochemical measurements. All samples displayed a similar cyclic voltammetric (CV) profile in N2-saturated 0.1 M KOH that is free of redox features, most likely due to the low content of the metal species (Fig. S19, ESI†). From the linear sweep voltammograms (LSV) acquired in rotating ring-disk electrode (RRDE) measurements in O2-saturated 0.1 M KOH (Fig. 6a), all composite samples with metal dopants can be seen to exhibit an apparently better ORR performance than the metal-free NC, and the dual-metal samples, RuFe-NC and RuFe-NCmix, display a further enhanced performance in comparison with the monometal counterparts of Ru-NC and Fe-NC. Remarkably, RuFe-NC stood out as the best ORR catalyst among the series. For instance, Ru-NC exhibited an onset potential (Eonset) of +0.90 V, ca. 60 mV more positive than that of NC, but the half-wave potential was slightly inferior (E1/2 = +0.62 V vs. +0.64 V). Fe-NC was significantly more active towards ORR, with an Eonset of +0.94 V and E1/2 of +0.81 V, which were similar to those of RuFe-NCmix (though the latter actually showed a much higher diffusion current density of ca. 4 mA cm−2 than other monometal-doped samples). This suggests that iron oxides played a dominant role in these samples in ORR electrocatalysis.23,25 Notably, an even better ORR activity was observed with RuFe-NC, where the performance (Eonset = +0.99 V, E1/2 = +0.87 V) was actually highly comparable to commercial Pt/C (Eonset = +1.00 V, E1/2 = +0.87 V), suggesting the significance of Ru doping in Fe2O3 nanoparticles in enhancing the ORR activity of RuFe-NC likely due to the Fe–Ru charge transfer. It should be noted that increasing or decreasing the Ru(LFe)2 loading in the sample preparation actually led to a diminished ORR performance, suggesting that RuFe-NC represented the optimal composite (Fig. S20, ESI†).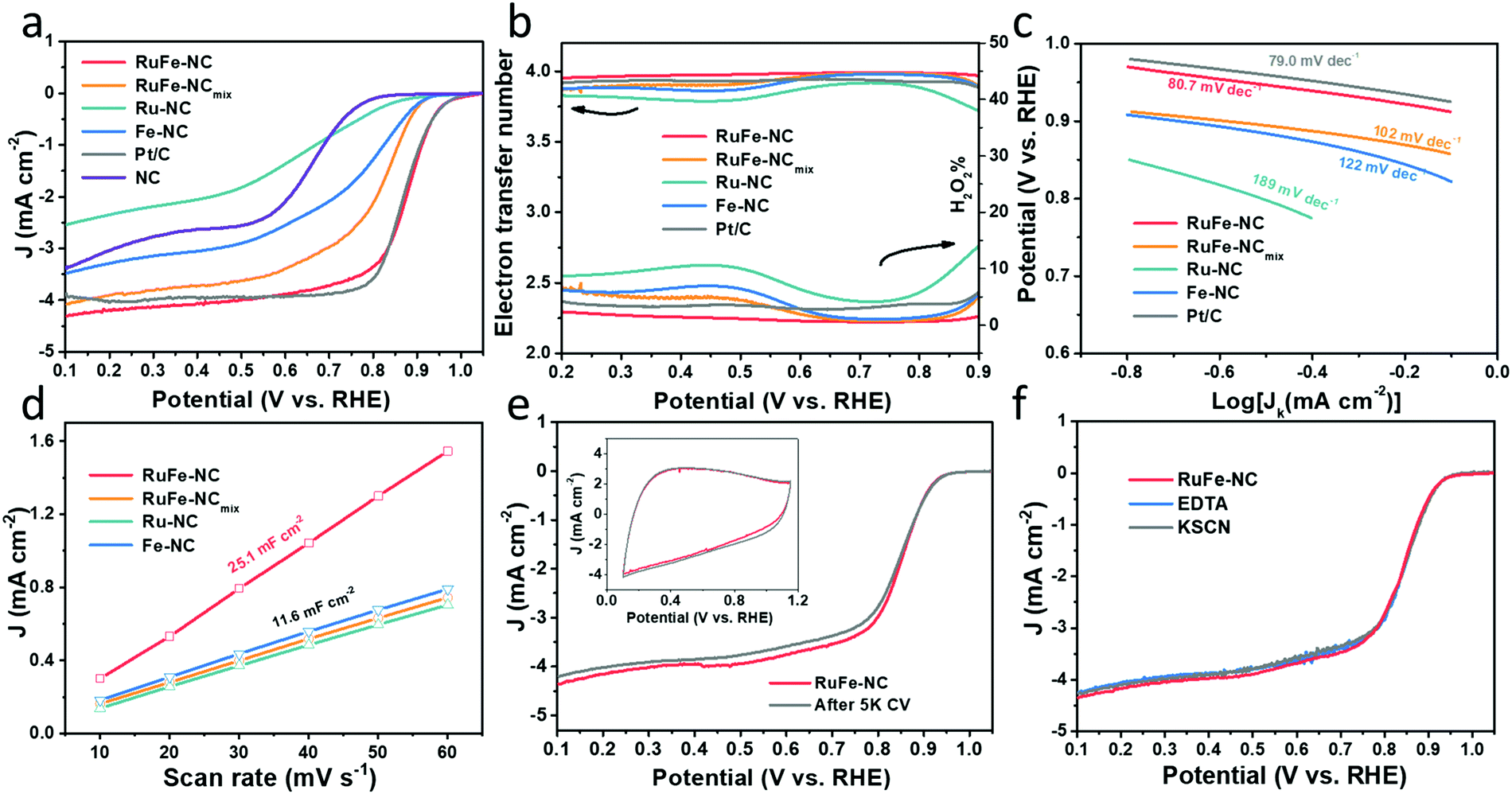 | ||
| Fig. 6 ORR performance of the RuFe-NC and control samples in oxygen-saturated 0.1 M KOH. (a) Linear sweep voltammograms (LSVs) of RuFe-NC and control samples of RuFe-NCmix, Ru-NC, Fe-NC, Pt/C, and NC at the rotation rate of 1600 rpm. (b) The corresponding electron-transfer number (n, left y-axis), the yield of H2O2 (%, right y-axis), and (c) Tafel plots with the slopes shown in mV dec−1. (d) Double layer capacitances (Cdl) of selected samples. (e) Stability test of RuFe-NC for 5000 cycles in N2-saturated 0.1 M KOH. Inset is the corresponding cyclic voltammograms at the scan rate of 50 mV s−1 before and after the stability test. (f) Poisoning test of RuFe-NC with EDTA and KSCN treatments. | ||
To gain further insights into the ORR electrocatalysis, the number of electron transfer (n) and H2O2% yield were evaluated by 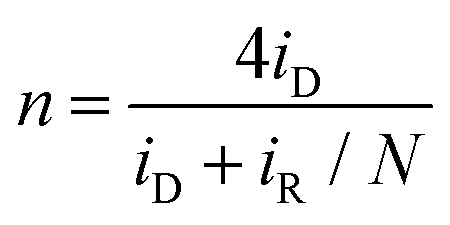 and
and 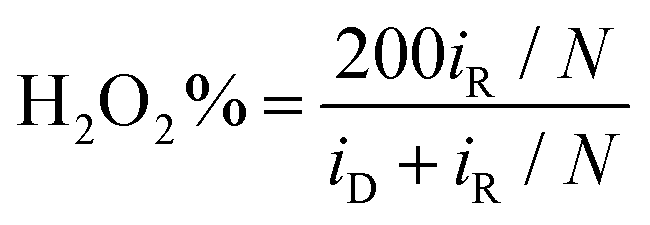 , respectively, where iR and iD are the ring current and disk current, and N is the collection efficiency of the ring electrode (0.40). One can see from Fig. 6b that at +0.8 V the n value is ca. 3.98 with an ultralow H2O2% of 0.65% for RuFe-NC, suggestive of the four-electron pathway of ORR. The performance of RuFe-NCmix was a close second, with n = 3.98 and a slightly higher H2O2% of 0.95%. Note that these are even better than that of commercial Pt/C (n = 3.92, H2O2% = 3.82%). In contrast, a substantially lower performance was observed with Fe-NC (n = 3.97 and H2O2% = 1.40%) and Ru-NC (n = 3.88, H2O2% = 5.81%). In the Tafel plots (Fig. 6c), RuFe-NC can be seen to display a Tafel slope of 80.7 mV dec−1, which is close to that of Pt/C (79.0 mV dec−1), indicating highly efficient electron-transfer kinetics where the first electron reduction of oxygen was likely the rate-determining step.34,40,48 Notably, the Tafel slope was greater for RuFe-NCmix at 102 mV dec−1, and markedly higher for Ru-NC (189 mV dec−1) and Fe-NC (122 mV dec−1), indicating a diminishing ability to cleave the O–O bonds during the ORR process.49
, respectively, where iR and iD are the ring current and disk current, and N is the collection efficiency of the ring electrode (0.40). One can see from Fig. 6b that at +0.8 V the n value is ca. 3.98 with an ultralow H2O2% of 0.65% for RuFe-NC, suggestive of the four-electron pathway of ORR. The performance of RuFe-NCmix was a close second, with n = 3.98 and a slightly higher H2O2% of 0.95%. Note that these are even better than that of commercial Pt/C (n = 3.92, H2O2% = 3.82%). In contrast, a substantially lower performance was observed with Fe-NC (n = 3.97 and H2O2% = 1.40%) and Ru-NC (n = 3.88, H2O2% = 5.81%). In the Tafel plots (Fig. 6c), RuFe-NC can be seen to display a Tafel slope of 80.7 mV dec−1, which is close to that of Pt/C (79.0 mV dec−1), indicating highly efficient electron-transfer kinetics where the first electron reduction of oxygen was likely the rate-determining step.34,40,48 Notably, the Tafel slope was greater for RuFe-NCmix at 102 mV dec−1, and markedly higher for Ru-NC (189 mV dec−1) and Fe-NC (122 mV dec−1), indicating a diminishing ability to cleave the O–O bonds during the ORR process.49
The effective electrochemical surface areas of the samples were then evaluated by the electrode double-layer capacitance (Cdl). Within the potential range of +0.9 to +1.1 V (i.e., a non-faradaic region), CVs were acquired at scan rates varied from 10 to 60 mV s−1 (Fig. S21, ESI†), from which the corresponding Cdl was derived. One can see from Fig. 6d that RuFe-NC possessed the largest Cdl of 25.1 mF cm−2, over two times those of other samples (ca. 11.6 mF cm−2). This is consistent with the porous structure as manifested in the above BET and TEM measurements, which is beneficial to increase the accessibility to the catalytically active sites.
The robustness of the active sites of RuFe-NC was then tested by repeated potential cycling between +0.6 and +1.0 V. After 5000 cycles, there were only minimal changes of the voltammograms (inset to Fig. 6e) and a slight shift of the diffusion-limited current density (less than 5%) in the corresponding LSV curve, manifesting excellent catalyst durability. After 10000 cycles, the activity of RuFe-NC decayed somewhat with a cathodic shift of E1/2 by 50 mV (Fig. S22, ESI†), but remained better than those of a number of relevant catalysts reported recently in the literature (Table S8, ESI†). Furthermore, the RuFe-NC catalysts exhibited remarkable tolerance against poisoning species like KSCN or EDTA, where negligible changes were discerned upon the addition of such poisoning species into the electrolyte (Fig. 6f). A similar behavior was observed with RuFe-NCmix (Fig. S23, ESI†). Such an anti-poisoning property against SCN− might be due to the unique structure of the Ru-doped Fe2O3 nanoparticles where the O-rich surface rendered the binding of SCN− ligands difficult. Meanwhile, as a hexadentate ligand, EDTA mostly chelates mononuclear metal centers, and the minimal impacts on the ORR performance suggests that Fe single atoms (e.g., FeNx) are unlikely the dominant active sites.37,50 These results further confirm that the Ru-doped Fe2O3 nanoparticles, rather than the FeNx moieties, were responsible for the ORR activity, within the present experimental context.
Mechanistically, the remarkable ORR activity most likely arose from the Fe to Ru charge transfer in Ru-doped Fe2O3, owing to their different electronegativity. In fact, electron depletion of Fe within Fe2O3 has been demonstrated as an effective strategy to manipulate the spin states of the Fe centers, generate partially occupied eg orbital and facilitate the adsorption of O2.26,51–53 In the present study, the atomic dispersion of Ru within RuFe-NC facilitated such charge transfer and hence enhanced the ORR activity, in contrast to the RuFe-NCmix sample where apparent segregation of Ru and Fe occurred.
In fact, when RuFe-NC was subject to acid leaching with 0.5 M H2SO4 at 80 °C for 4 h, the ORR activity was markedly diminished, with Eonset = +0.93 V and E1/2 = +0.74 V (Fig. 7), a negative shift of 130 mV as compared to the as-prepared sample. These results demonstrate, again, the dominant contributions of metal oxide nanoparticles in the RuFe-NC composite to the ORR activity, with a minor contribution from the FeNx moieties (as manifested by the leached sample which contained mainly atomically disperse FeNx moieties but still outperformed the metal-free NC).
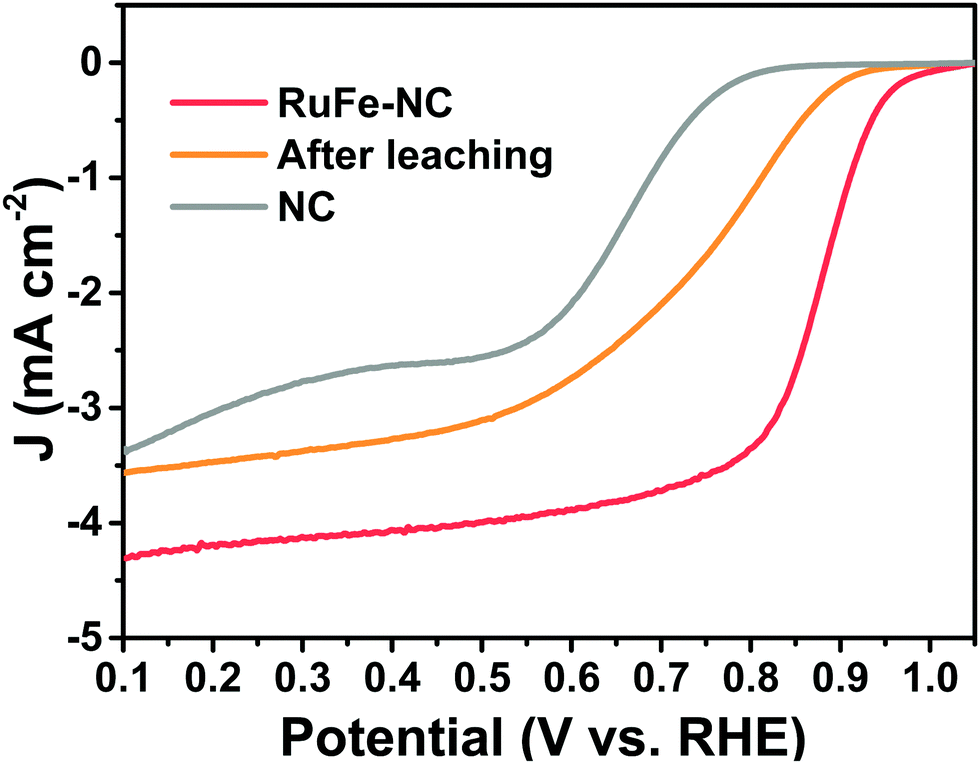 | ||
| Fig. 7 LSV curves of RuFe-NC in 0.1 M KOH before and after acid leaching treatment, in comparison to that of NC. | ||
Conclusions
In this study, a trinuclear Ru(LFe)2 complex was used as the metal precursor to prepare carbon nanocomposites with Ru-doped Fe2O3 nanoparticles, as confirmed in a range of microscopic and spectroscopic measurements. The obtained RuFe-NC nanocomposites exhibited an excellent ORR activity, with an Eonset of +0.99 V and an E1/2 of +0.87 V, which is comparable to that of the state-of-the-art Pt/C catalyst. Notably, the ORR activity was also markedly better than those of the monometal counterparts and samples prepared with a simple physical mixture of the relevant metal salts (where partial segregation of the metal species occurred). This was accounted for by the Fe to Ru charge transfer in the RuFe-NC composites that was advantageous for the adsorption of key reaction intermediates and enhanced electrochemical surface area that facilitated the accessibility of the catalytic active sites. Results from this study suggest that rational design of the metal precursors may be exploited as an effective strategy in manipulating the morphological and electronic structure of carbon for high-performance electrocatalysis.Author contributions
Qiming Liu: writing – original draft, sample preparation, data acquisition and analysis. Hongbo Zhou: preparation of Ru(LFe)2 complex. Forrest Nichols and Frank Bridges: acquisition and analysis of X-ray absorption data. Han-Lin Kuo and Jennifer Q. Lu: acquisition of TEM images and elemental mapping analysis. Rene Mercado, Bingzhang Lu, Weiya Zhu, and Yashu Liu: assistance in data acquisition and results discussion. Shaowei Chen: conception of project, results analysis, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported, in part, by the National Science Foundation (CHE-1900235 and CHE-2003685). TEM, XPS and BET studies were carried out as part of a user project at the National Center for Electron Microscopy and Molecular Foundry of Lawrence Berkeley National Laboratory which is supported by the Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract No. DE-AC02-05CH11231. The XAS experiments were performed at the Stanford Synchrotron Radiation Lightsource (SSRL), which is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The authors also thank Jeremy Barnett for the assistance in sample preparation and data acquisition in XRD measurements using the X-ray Facility at the University of California Santa Cruz funded by the National Science Foundation (MRI-1126845).References
- K. I. Ozoemena and S. W. Chen, Nanomaterials for Fuel Cell Catalysis. in Nanostructure Science and Technology, Springer International Publishing: Cham, 1st edn, 2016 Search PubMed.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- Z. Y. Lu, B. F. Wang, Y. F. Hu, W. Liu, Y. F. Zhao, R. O. Yang, Z. P. Li, J. Luo, B. Chi, Z. Jiang, M. S. Li, S. C. Mu, S. J. Liao, J. J. Zhang and X. L. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 RSC.
- X. J. Zhou, J. L. Qiao, L. Yang and J. J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 CrossRef.
- C. Du, X. H. Gao and W. Chen, Chin. J. Catal., 2016, 37, 1049–1061 CrossRef CAS.
- J. A. Trindell, Z. Y. Duan, G. Henkelman and R. M. Crooks, Chem. Rev., 2020, 120, 814–850 CrossRef CAS PubMed.
- H. S. Wang, Y. Yang, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 4608–4616 CrossRef CAS.
- V. Stamenkovic, T. J. Schmidt, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2002, 106, 11970–11979 CrossRef CAS.
- Y. C. Xing, Y. Cai, M. B. Vukmirovic, W. P. Zhou, H. Karan, J. X. Wang and R. R. Adzic, J. Phys. Chem. Lett., 2010, 1, 3238–3242 CrossRef CAS.
- A. U. Nilekar, Y. Xu, J. L. Zhang, M. B. Vukmirovic, K. Sasaki, R. R. Adzic and M. Mavrikakis, Top. Catal., 2007, 46, 276–284 CrossRef CAS.
- Y. Xiong, Y. Yang, H. Joress, E. Padgett, U. Gupta, V. Yarlagadda, D. N. Agyeman-Budu, X. Huang, T. E. Moylan, R. Zeng, A. Kongkanand, F. A. Escobedo, J. D. Brock, F. J. DiSalvo, D. A. Muller and H. D. Abruna, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1974–1983 CrossRef CAS PubMed.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- E. L. Hu, X. Y. Yu, F. Chen, Y. D. Wu, Y. Hu and X. W. Lou, Adv. Energy Mater., 2018, 8, 1702476 CrossRef.
- X. Han, Z. P. Zheng, J. Y. Chen, Y. K. Xue, H. Q. Li, J. Zheng, Z. X. Xie, Q. Kuang and L. S. Zheng, Nanoscale, 2019, 11, 12610–12618 RSC.
- W. J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L. J. Zhang, J. Q. Wang, J. S. Hu, Z. D. Wei and L. J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- H. S. Park, S. B. Han, D. H. Kwak, J. H. Han and K. W. Park, J. Catal., 2019, 370, 130–137 CrossRef CAS.
- A. Bonnefont, A. S. Ryabova, T. Schott, G. Kerangueven, S. Y. Istomin, E. V. Antipov and E. R. Savinova, Curr. Opin. Electrochem., 2019, 14, 23–31 CrossRef CAS.
- Y. J. Wang, H. B. Fan, A. Ignaszak, L. Zhang, S. Q. Shao, D. P. Wilkinson and J. J. Zhang, Chem. Eng. J., 2018, 348, 416–437 CrossRef CAS.
- Y. Wang, J. Li and Z. D. Wei, J. Mater. Chem. A, 2018, 6, 8194–8209 RSC.
- R. J. Toh, Z. Sofer and M. Pumera, Chem. Phys. Chem., 2015, 16, 3527–3531 CrossRef CAS PubMed.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- Y. J. Ma, H. Wang, J. Key, V. Linkov, S. Ji, X. F. Mao, Q. Z. Wang and R. F. Wang, Int. J. Hydrogen Energy, 2014, 39, 14777–14782 CrossRef CAS.
- Z. Y. Fan, J. Li, W. Yang, Q. Fu, K. Sun, Y. C. Song, Z. D. Wei, Q. Liao and X. Zhu, Chem. Eng. J., 2020, 385, 123393 CrossRef CAS.
- R. J. Gao, J. Wang, Z. F. Huang, R. R. Zhang, W. Wang, L. Pan, J. F. Zhang, W. K. Zhu, X. W. Zhang, C. X. Shi, J. Lim and J. J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- M. R. Wei, S. Huang, Y. Wang, Y. H. Liu, Y. F. He, C. Wang and L. Yang, J. Alloys Compd., 2020, 827, 154207 CrossRef CAS.
- J. Yu, Q. J. He, G. M. Yang, W. Zhou, Z. P. Shao and M. Ni, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- Z. G. Zhu, R. C. Deka, A. Chutia, R. Sahnoun, H. Tsuboi, M. Koyama, N. Hatakeyama, A. Endou, H. Takaba, C. A. Del Carpio, M. Kubo and A. Miyamoto, J. Phys. Chem. Solids, 2009, 70, 1248–1255 CrossRef CAS.
- C. L. Dong, X. L. Zhang, J. Xu, R. Si, J. Sheng, J. Luo, S. N. Zhang, W. J. Dong, G. B. Li, W. C. Wang and F. Q. Huang, Small, 2020, 16, 1905328 CrossRef CAS PubMed.
- M. B. Rossi, K. A. Abboud, P. Albores and L. M. Baraldo, Eur. J. Inorg. Chem., 2010, 5613–5616 CrossRef CAS.
- J. Wang, Z. Q. Huang, W. Liu, C. R. Chang, H. L. Tang, Z. J. Li, W. X. Chen, C. J. Jia, T. Yao, S. Q. Wei, Y. Wu and Y. D. Lie, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- J. Yang, Z. Y. Qiu, C. M. Zhao, W. C. Wei, W. X. Chen, Z. J. Li, Y. T. Qu, J. C. Dong, J. Luo, Z. Y. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS PubMed.
- Q. M. Liu, Y. Peng, Q. X. Li, T. He, D. Morris, F. Nichols, R. Mercado, P. Zhang and S. W. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 17641–17650 CrossRef CAS PubMed.
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee, M. J. Kim, B. S. Mun, S. G. Kwon, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 2035–2045 CrossRef CAS PubMed.
- B. D. Adams and A. C. Chen, Mater. Today, 2011, 14, 282–289 CrossRef CAS.
- B. Z. Lu, L. Guo, F. Wu, Y. Peng, J. E. Lu, T. J. Smart, N. Wang, Y. Z. Finfrock, D. Morris, P. Zhang, N. Li, P. Gao, Y. Ping and S. W. Chen, Nat. Commun., 2019, 10, 631 CrossRef CAS PubMed.
- F. Liu, S. Y. Song, D. F. Xue and H. J. Zhang, Adv. Mater., 2012, 24, 1089–1094 CrossRef CAS PubMed.
- X. Tian, X. Zhao, Y. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. Hensen and X. W. D. Lou, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- B. Z. Lu, T. J. Smart, D. D. Qin, J. E. Lu, N. Wang, L. M. Chen, Y. Peng, Y. Ping and S. W. Chen, Chem. Mater., 2017, 29, 5617–5628 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- D. Flak, Q. L. Chen, B. S. Mun, Z. Liu, M. Rekas and A. Braun, Appl. Surf. Sci., 2018, 455, 1019–1028 CrossRef CAS.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- L. X. Chen, T. Liu, M. C. Thurnauer, R. Csencsits and T. Rajh, J. Phys. Chem. B, 2002, 106, 8539–8546 CrossRef CAS.
- B. K. Teo, EXAFS: basic principles and data analysis, Springer Science & Business Media, 2012 Search PubMed.
- C. H. Zhang, J. W. Sha, H. L. Fei, M. J. Liu, S. Yazdi, J. B. Zhang, Q. F. Zhong, X. L. Zou, N. Q. Zhao, H. S. Yu, Z. Jiang, E. Ringe, B. I. Yakobson, J. C. Dong, D. L. Chen and J. M. Tour, ACS Nano, 2017, 11, 6930–6941 CrossRef CAS PubMed.
- M. A. Hubert, A. M. Patel, A. Gallo, Y. Z. Liu, E. Valle, M. Ben-Naim, J. Sanchez, D. Sokaras, R. Sinclair, J. K. Norskov, L. A. King, M. Bajdich and T. F. Jaramillo, ACS Catal., 2020, 10, 12182–12196 CrossRef CAS.
- B. Lu, Q. Liu, F. Nichols, R. Mercado, D. Morris, N. Li, P. Zhang, P. Gao, Y. Ping and S. W. Chen, Research, 2020, 2020, 9167829 CAS.
- J. Chlistunoff, J. Phys. Chem. C, 2011, 115, 6496–6507 CrossRef CAS.
- J. Wang, X. M. Ge, Z. L. Liu, L. Thia, Y. Yan, W. Xiao and X. Wang, J. Am. Chem. Soc., 2017, 139, 1878–1884 CrossRef CAS PubMed.
- S. Y. Chen, Y. Yan, P. P. Hao, M. H. Li, J. Y. Liang, J. Guo, Y. Zhang, S. W. Chen, W. P. Ding and X. F. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 12686–12695 CrossRef CAS PubMed.
- R. J. Gao, L. Pan, Z. W. Li, C. X. Shi, Y. D. Yao, X. W. Zhang and J. J. Zou, Adv. Funct. Mater., 2020, 30, 1910539 CrossRef CAS.
- G. Q. Shen, R. R. Zhang, L. Pan, F. Hou, Y. J. Zhao, Z. Y. Shen, W. B. Mi, C. X. Shi, Q. F. Wang, X. W. Zhang and J. J. Zou, Angew. Chem., Int. Ed., 2020, 59, 2313–2317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data. See DOI: https://doi.org/10.1039/d2ma00054g |
| This journal is © The Royal Society of Chemistry 2022 |