
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChromatographic detection of low-molecular-mass metal complexes in the cytosol of Saccharomyces cerevisiae†
Trang Q.
Nguyen
a,
Joshua E.
Kim
a,
Hayley N.
Brawley
a and
Paul A.
Lindahl
 *ab
*ab
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843-3255, USA. E-mail: Lindahl@chem.tamu.edu; Fax: +979-845-4719; Tel: +979-845-0956
bDepartment of Biochemistry and Biophysics, Texas A&M University, College Station, TX 77843, USA
First published on 17th April 2020
Abstract
Fluorescence-based chelators are commonly used to probe labile low-molecular-mass (LMM) metal pools in the cytosol of eukaryotic cells, but such chelators destroy the complexes of interest during detection. The objective of this study was to use chromatography to directly detect such complexes. Towards this end, 47 batches of cytosol were isolated from fermenting S. cerevisiae yeast cells and passed through a 10 kDa cut-off membrane. The metal contents of the cytosol and resulting flow-through solution (FTS) were determined. FTSs were applied to a size-exclusion LC column located in an anaerobic refrigerated glove box. The LC system was coupled to an online inductively-coupled-plasma mass spectrometer (ICP-MS) for detection of individual metals. Iron-detected chromatograms of cytosolic FTSs from WT cells exhibited 2–4 major species with apparent masses between 500–1300 Da. Increasing the iron concentration in the growth medium 40-fold increased the overall intensity of these peaks. Approximately 3 LMM cytosolic copper complexes with apparent masses between 300–1300 Da were also detected; their LC intensities were weak, but these increased with increasing concentrations of copper in the growth medium. Observed higher-mass copper-detected peaks were tentatively assigned to copper-bound metallothioneins Cup1 and Crs5. FTSs from strains in which Cup1 or the Cox17 copper chaperone were deleted altered the distribution of LMM copper complexes. LMM zinc- and manganese-detected species were also present in cytosol, albeit at low concentrations. Supplementing the growth medium with zinc increased the intensity of the zinc peak assigned to Crs5 but the intensities of LMM zinc complexes were unaffected. Phosphorus-detected chromatograms were dominated by peaks at apparent masses 400–800 Da, with minor peaks at 1000–1500 Da in some batches. Sulfur chromatograms contained a low-intensity peak that comigrated with a glutathione standard; quantification suggested a GSH concentration in the cytosol of ca. 13 mM. A second LMM sulfur peak that migrated at an apparent mass of 100 Da was also evident.
Significance to metallomicsCells contain low-molecular-mass transition metal complexes that serve critical but poorly defined roles in metal trafficking and regulation. These “labile metal pools” are commonly monitored using fluorescence-based chelator-probes, but there are problems associated with such methods. Here, SEC chromatography with on-line ICP-MS detection was used to detect low-mass metal complexes in yeast cytosol isolated from wild-type cells and various genetic strains. Numerous iron, copper, zinc and manganese complexes with masses between 300–1500 Da were detected. Their concentrations were quantified in cytosol obtained from cells grown under metal-deficient and metal-replete media conditions. Copper and zinc bound metallothioneins were observed. |
Introduction
The concept of a labile iron pool (LIP) in cells was introduced by Jacobs in 1977 and expanded by Williams a few years later.1,2 Based on the available evidence, these researchers proposed that cells contain nonproteinaceous, kinetically labile, low-molecular-mass (LMM) FeII (and other metal) coordination complexes involved in metal ion trafficking, storage, or regulation. They predicted that such iron complexes, with rapidly exchanging ligands, were toxic and could generate reactive oxygen species (ROS) such as hydroxyl radicals via Fenton chemistry. They and others argued that such complexes must be maintained at low concentrations to minimize cellular damage.3These researchers emphasized the difficulty of distinguishing physiologically relevant complexes from artifacts generated during cell lysis and sample preparation. This difficulty stems from the inherent lability (i.e. rapid ligand exchange rates) of classical Werner coordination complexes. Isolating and characterizing dynamically changing species has been, and continues to be, a major challenge. Indeed, the LIP has been compared to the Loch Ness monster, “only to disappear from view before its presence or indeed its nature can be confirmed”.4 Thirty-five years later, “the molecular nature of the labile iron pool (remains) one of the least well understood aspects of iron cell biology”.5
In the mid 1990's, Cabantchik and coworkers developed a powerful method to investigate these mysterious complexes; they exposed intact cells to the membrane-permeable chelator calcein, a derivative of fluorescein whose fluorescence quenched upon iron binding.6 Cells were subsequently incubated with a stronger chelator which caused dequenching as iron dissociated from the initial probe and bound the chelator. This has become the most popular method to quantify and characterize the LIP. Cabantchik reported a LIP concentration in the cytosol of K562 human cells of 0.2–0.5 μM.7 Petrat and coworkers initially reported a cytosolic LIP concentration of 10 ± 3 μM in isolated rat hepatocytes.8 Using Phen Green SK as a probe, they reported 1–3 μM for the cytosolic LIP in cultured rat hepatocytes.9 Epsztejn et al. reported ca. 1–2 μM for the LIP in a similar study.10
Hider and coworkers developed a different approach to study the LIP, namely using thermodynamic binding strengths along with solubility properties and known concentrations of various potential ligands in the cytosol to predict the complexes that constitute the LIP. FeII(glutathionate)1(H2O)5 currently leads the list.11 The oxidation state of iron in the cytosol is almost certainly FeII.12 Thus, Lewis bases in the cytosol that coordinate FeIII but not FeII at physiological pH are not good candidates; this excludes amino acids, ATP/ADP, inositol phosphates, and 2,5-dihydroxybenzoic acid. Citrate coordinates FeII ions but not strong enough to prevent autoxidation to FeIII. In contrast, glutathione (GSH) is present in millimolar concentrations, it binds FeII strongly, and its antioxidant properties help maintain the FeII state.13
Copper, like iron, is essential for cellular function and such ions can also damage the cell. However, the mechanisms of copper trafficking differ from those of iron by exclusively involving proteinaceous copper chaperones. O’Halloran and Culotta argued that cells do not contain “free” copper (where “free” could mean aqueous, chelatable, labile, nonproteinaceous, or LMM).14 Their conclusion that copper is exclusively passed from one protein chaperone to the next in the cell is the current consensus of the field.15
Copper is imported via Ctr1 on the plasma membrane, affording an uncharacterized CuI species in the cytosol.16 Some of this cytosolic copper binds Atx1, a chaperone which delivers copper to Ccc2, a P-type ATPase transporter on the trans-Golgi membrane. Other cytosolic copper binds Ccs1, a chaperone that installs copper into superoxide dismutase. Still other cytosolic copper ions bind and are stored in metallothioneins Cup1 and Crs5.
How cytosolic copper is delivered to mitochondria for installation into the respiratory complex cytochrome c oxidase remains a decades-old puzzle, as no chaperone has been identified. Cox17 is a chaperone in the mitochondrial IMS that delivers copper to two downstream chaperones (Sco1 and Cox11) that ultimately metallate cytochrome c oxidase.17 Cox17 localizes to both the cytosol and mitochondria, initially prompting suggestions that it was the sought-after chaperone that targets mitochondria.18 However, Winge and coworkers excluded this suggestion because anchoring Cox17 to the mitochondria did not prevent cytochrome c oxidase from metallating.19
Despite this, there is some evidence for a transient “labile copper pool” in cells. Fahrni and coworkers used chelators to reveal a small pool of tightly bound copper coordinated to thiols.20 The concentration of this pool increases transiently after cells are exposed to copper salts, especially in mitochondria and Golgi.21 Also interesting is that the phenotypes of cells lacking various copper chaperones (e.g. Cox17, Sco2 and Coa6) can be rescued by including higher-than-normal concentrations of simple copper salts to the growth medium.22–24 This raises the possibility that LMM labile copper can functionally replace copper chaperones and deliver copper into mitochondria for the assembly of cytochrome c oxidase. How they might do this is unknown.
Zinc is found in nearly 600 eukaryotic proteins25 yet no zinc protein chaperones have been identified.26 Thus, it seems unlikely that each Zn-containing protein in the cell is metallated by a dedicated chaperone. The concentration of the LMM nonproteinaceous “labile zinc pool” in the cytosol of mammalian cells is exceedingly low; estimates are in the hundreds of pM.27,28 This translates into less than 1 labile Zn atom per cell. Such low concentrations function to prevent mis-metallating non-zinc metalloproteins in cells.
The transcriptional regulator that controls expression of the major Zn importers in the cell is sensitive to the labile Zn pool. Some cytosolic Zn from this pool is transported to the ER and Golgi for use in the secretory pathway. Other cytosolic Zn is likely bound to metallothioneins.29 In this case, aqueous or LMM Zn might be present at pM concentrations but in equilibrium with a buffering species whose concentration is much higher. In Bacillus subtilis, the buffer that serves this role is bacillithiol, present at ∼1 mM concentration.30 In zinc-shock experiments (adding 1–10 μM Zn salts to Zn-starved cells), chelator-sensitive Zn within the cell increases quickly and then rapidly declines.31 This suggests an expandable pool of mobile Zn ions in the cell, similar to the situation with copper.
Manganese ions enter cells through membrane-bound transporters where they metallate numerous proteins.32 A significant proportion of cellular Mn ions exist as labile LMM complexes. However, the total Mn concentration in the cell, including both protein-bound and the labile manganese pool, is only in the tens of μM.33 Moreover, MnII ions have inherently low binding affinities toward most ligands, and so it has been difficult to develop fluorescence-based probes that are Mn-selective.31 Spectroscopic investigations of intact cells have been more useful in investigating the labile Mn pool.34 Cells compartmentalize metals to control metallation; this allows Mn to metallate proteins that would otherwise be mis-metallated by other more tight-binding metals.13 In mammalian cells, the low level of cytosolic Mn (10−7 M) is achieved by membrane-bound ATPases which pump MnII into the Golgi apparatus and mitochondria. When cells are exposed to high concentrations of Mn, excess Mn is found in the Golgi.31 Whether protein chaperones deliver Mn to cytosolic proteins is unknown.
A major advantage of metal-sensitive fluorescence-based chelators is their ability to penetrate intact cells without disrupting them. This avoids concerns, voiced by Jacobs and Williams and echoed by others, that disrupting the cell might alter labile metal pools and create artifacts.35 The disadvantage of these chelators is that they destroy the complexes of interest as they detect them, releasing the endogenous ligands into the cellular milieu. They also provide unreliable estimates of the size of the metal pools, as the nature of the chelator probe and experimental reaction conditions influence such estimates. Moreover, chelator-probes are not specific for a particular metal much less for a particular metal complex;36 hence the vague notion of a pool has become part of the vernacular in this field.
Here we use chromatography to detect LMM labile metal complexes in the cytosol of budding yeast, S. cerevisiae. We have attempted to exclude artifacts by isolating the cytosol under refrigerated anaerobic conditions. We found that the intensity of detected species varied as expected when the growth medium was supplemented with different metals. We also employed various genetic strains to evaluate whether the detected species varied as expected from the genetic modification. This study demonstrates the presence of LMM iron, copper, zinc and manganese complexes in the cytosol of S. cerevisiae, as well as the presence of LMM phosphorus and sulfur compounds. It also highlights the feasibility of using chromatography to study such pools in the future, as a complement to using fluorescent chelators.
Experimental procedures
Cell growth
The yeast strains used are listed in Table S1 (ESI†). All strains were grown in 2 L of minimal medium (MM) except for cup1Δ cells which were grown in 1 L of complete synthetic media (CSM). The compositions of MM and CSM have been described.37 Cells were harvested at early-exponential phase OD600 = 0.6 ± 0.2 for MM and OD600 = 3.0 ± 0.1 for CSM. Additional conditions for individual batches are listed in Table 1.| Strain | FeIII citrate | CuSO4 | ZnCl2 | MnCl2 | Media | Num. of batches |
|---|---|---|---|---|---|---|
| W303 | 1 | 10 | 0 | 0 | MM | 8 |
| W303 | 40 | 10 | 0 | 0 | MM | 4 (plus 2) |
| W303 | 100 | 10 | 0 | 0 | MM | 4 |
| W303 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
10 | 0 | 0 | MM | 2 |
| W303 | 40 | 0 | 0 | 0 | MM | 3 |
| W303 | 40 | 50 | 0 | 0 | MM | 2 (plus 2) |
| W303 | 40 | 10 | 100 | 0 | MM | 2 |
| W303 | 40 | 10 | 100 | 40 | MM | 2 |
| fet3Δ | 100 | 10 | 0 | 0 | MM | 4 |
| ccc1Δ | 40 | 10 | 0 | 0 | MM | 2 |
| mrs3Δ mrs4Δ | 40 | 10 | 0 | 0 | MM | 2 |
| vma2Δ | 40 | 10 | 0 | 0 | MM | 2 |
| cox17Δ | 40 | 10 | 0 | 0 | MM | 2 |
| cup1Δ | 40 | 10 | 0 | 0 | CSM | 2 |
| CUP1R | 40 | 10 | 0 | 0 | MM | 2 |
Isolation of cytosol
Harvested cells (typically 4–5 grams of wet pellet) were washed 2 times with 1 mM EDTA, pH ∼ 8 followed by 2 additional washes with DI water. Five mL of each wash was used per gram of wet pellet. Cell suspensions were centrifuged at 4000 × g for 5 min. All buffers and solutions were chilled on ice. For 45 batches, washed cells were suspended in 5 mL of Buffer A (10 mM of dithiothreitol in 100 mM Tris, pH = 9.4) per g wet cell mass. After 10 min incubation at 30 °C, cells were centrifuged and brought into a refrigerated nitrogen-atmosphere glovebox (Mbraun Labmaster 130, ∼7 °C, ∼5 ppm of O2) where cytosol was isolated following the protocol of Herrera et al.,38 albeit with some modifications. Buffers B (0.6 M sorbitol, 10 mM KPi, pH = 7.4) and C (10 mM Tris, 10 mM MES, 0.1 mM MgCl2, 12% Ficoll, pH ∼ 6.8) were degassed on a Schlenk line. Cells were resuspended in 5 mL of Buffer B per g of wet cells, and 10 mM PMSF was added. Fresh zymolyase (Amsbio 120493-1; with a specific activity of 100 KU g−1), at a mass of 0.5 mg per g of wet cells, was dissolved in 1 mL of Buffer B, and the resulting solution was mixed into the cell suspension. After mixing, a 1 mL aliquot of the resulting cell suspension was removed to monitor spheroplasts formation. The aliquot and the cell suspension were removed from the glovebox; the suspension was in a sealed centrifuge bottle and the aliquot was in a microcentrifuge tube. Both were incubated at 30 °C. Periodically, 10 μL of the aliquot was mixed with 990 μL of water, and OD600 was measured. This process was repeated until the OD600 decreased to 50% of its initial value (usually after 15 min). Then the cell suspension was centrifuged at 2200 × g for 4 min and returned to the glovebox. The supernatant was discarded and the spheroplast pellet (ca. 5–6 mL) was resuspended in a six-fold volume excess of Buffer C. Half of the resulting suspension (typically 15–17 mL) was loaded into a 40 mL Kimble® Dounce tissue grinder and homogenized with a glass pestle A (clearance 0.0030–0.0060 inches) for 10 strokes. The same was done to the other half. The two homogenates were combined in a centrifuge bottle, which was then sealed, removed from the glovebox, and centrifuged at 1000 × g for 15 min. The bottle was returned to the box, and the supernatant was transferred to a clean centrifuge bottle. The bottle was sealed, removed from the box, and centrifuged at 15000 × g for 30 min. The bottle was returned to the box and the supernatant was transferred to three 16 × 76 mm polycarbonate bottles (Beckman Coulter, 355603) that fit a 70.1 Ti Fixed-Angle titanium rotor (Beckman Coulter). After assembling the outer canisters, the sealed tubes were removed from the box and centrifuged at 112000 × g for 60 min. Bottles were returned to the box and the supernatants were distributed into two clean polycarbonate tubes. The tubes were sealed, removed from the box, and centrifuged at 185000 × g for 150 min. They were returned to the box and the supernatant fraction located between the top lipid layer and the pellet was collected and defined as dilute isolated cytosol. The cytosol occupies 68% of the volume of a spheroplast,39 and the spheroplasts represent ca. 70% of the wet cell pellet.40 Since the dilute isolated pellet was resuspended in 6-fold volume of buffer, undiluted cytosol in cells would be approximately 7 ÷ (0.68 × 0.7) ≈ 14.7 times more concentrated. For 1 batch of WT cells, Tris, MES, Ficoll, and MgCl2 were excluded from buffers, and 20 mM ammonium acetate pH 6.5 was used.
Western blots
Protein concentrations in isolated cytosol were quantified using the 96-well plate Pierce™ BCA Protein Assay Kit (Thermo Scientific™). Whole-cell protein extracts and cytosolic fractions were run on a NuPAGE™ 10% Bis-Tris protein gel (Invitrogen™) at a constant voltage of 40 V for 40 min then switched to 110 V for 110 min. A semidry Trans-Blot cell (Bio-Rad) was used to transfer separated proteins to activated PVDF membranes using a constant voltage of 23 V for 60 min. Membranes were blocked with a solution of 5% milk dissolved in Tris-buffered saline with 0.1% Tween (TBST-milk) for 1 h at RT. Then the membranes were incubated with different primary antibodies overnight at 4 °C. All primary antibodies were diluted in TBST-milk solution as followed: 1:2000 dilution of anti-PGK antibody as cytosol marker (Life Technologies, H0460), 1:2000 dilution of anti-Kar2 antibody as endoplasmic reticulum marker (Santa Cruz Biotechnology, sc-33630), 1:2000 dilution of anti-CPY antibody as vacuole marker (Life Technologies, A-6428), and 1:1000 dilution of anti-VDAC antibody as mitochondria marker (Thermo Fisher, 16G9E6BC4). Membranes were washed and incubated with 1:5000 dilution of either goat anti-rabbit IgG HRP-conjugated secondary antibody (Santa Cruz Biotechnology, clone sc-2004) or goat anti-mouse IgG HRP-conjugated secondary antibody (Invitrogen, clone G-21040) for 1 h at RT. The blots were developed with Clarity™ Western ECL Substrate (Bio-Rad) for 2 min at RT and imaged using FujiFilm LAS-4000 mini with a 15 s exposure under chemiluminescence setting.
Preparation of cytosolic FTS and LC-ICP-MS chromatography
The following procedures were performed in a refrigerated anaerobic glove box. Four mL of dilute isolated cytosolic fractions in Buffer C were passed through an Ultracel regenerated cellulose 10 kDa NMWL membrane (EMD Millipore) installed in an Amicon® EMD Millipore Stirred Cell (Model 8003, 3 mL) and pressurized with 70 psi Argon. In follow-up studies, cytosol FTS was passed through an Amicon® Ultra 2 mL Centrifugal filter centrifuged at 3858 × g for 2 hr. Approximately 75% of the isolated cytosol fractions passed through the 10 kDa membrane in ca. 2.5 h. The resulting flow-through solution (FTS), with a volume of 150 μL, was loaded onto either a single or double SuperdexTM Peptide 10/300 GL (GE Life Sciences) size-exclusion (SEC) column connected to an Agilent 1260 Bioinert quaternary pump (G5611A). The double column consisted of two single columns connected in series. A mobile phase of 20 mM ammonium acetate pH = 6.5 was used with a flow rate of 0.33–0.35 mL min−1 for the double columns and a flow rate of 0.6 mL min−1 for the single column. Other LC-ICP-MS parameters, SEC calibration procedures have been described.41Elemental analysis
Elemental analysis was conducted using ICP-MS (Agilent 7700X) as described37,41 albeit with minor modifications. Three 100 μL aliquots of cytosol and cytosolic FTSs from 4 independent batches were transferred into 15 mL polypropylene screw-top vials. Three hundred μLs of trace-metal-grade 70% w/v nitric acid (Fisher Scientific) were added to each vial. Vials were capped and sealed with electrical tape, then incubated at 70 °C for ca. 15 h. Each replicate was cooled to room temperature, diluted with 5.60 mL high-purity trace-metal-free double distilled-deionized water and mixed before ICP-MS analysis.Results
Forty-seven independent batches of cytosol were isolated from yeast cells as described in Experimental procedures (Table 1). Most batches were passed through a 10 kDa cut-off membrane, and the resulting flow-through solutions (FTSs) were passed down a size-exclusion chromatography column located in a refrigerated anaerobic glove box. The eluate from the column flowed into an ICP-MS for real-time metal detection.The purity of six of batches of dilute isolated cytosol was investigated by Western blot (Fig. S1, ESI†). A representative of these batches, from WT cells grown with 40 μM supplemented Fe-citrate and 10 μM CuSO4, is shown in Fig. 1. The blots exhibited an intense band for the cytosolic marker protein PGK1. Minor contaminations of endoplasmic reticulum (Kar2) and vacuoles (CPY) were evident, but there was no contamination of mitochondria (VDAC). Some intensity of the Anti-CPY band was likely due to a precursor form of CPY (pro-CPY) which is synthesized in the cytosol prior to maturation in the vacuolar lumen.42 Anti-CPY also detects pro-CPY, and so our isolated cytosol preparations were probably less contaminated with vacuoles than is apparent from the blot.
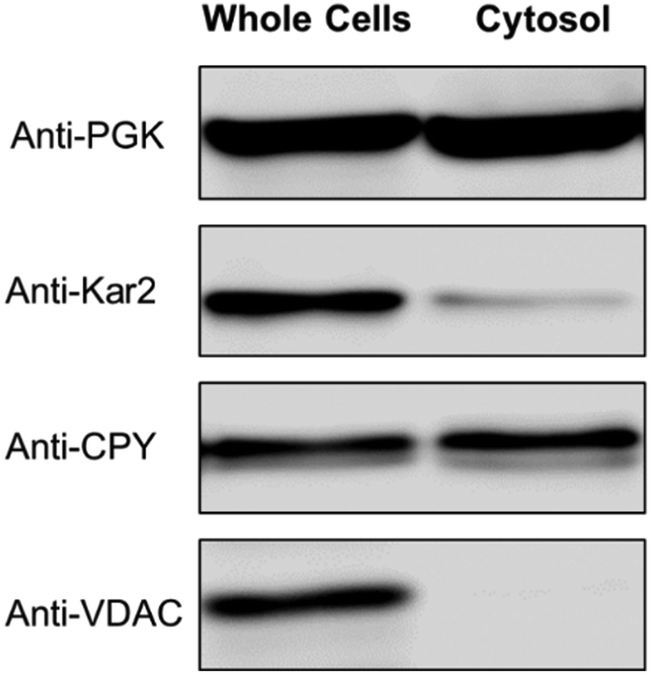 | ||
| Fig. 1 Western blot of isolated cytosol. Each lane contained 22 μg protein. PGK, Kar2, CPY, and VDAC are markers for the cytosol, ER, vacuoles, and mitochondria, respectively. | ||
We determined concentrations of select elements in 16 batches of cytosol isolated from WT cells grown in fermenting minimal media that contained 3 different concentrations of FeIII citrate. Cytosol isolated from cells grown with 1 μM FeIII citrate contained (after multiplying by a dilution factor of 14.7) 98 ± 24 μM Fe, 196 ± 98 μM Cu, 514 ± 29 μM Zn, 5 ± 2 μM Mn, and 122 ± 48 mM P (n = 8). Cytosol isolated from WT cells supplemented with 40 μM FeIII citrate contained higher concentrations of Fe (265 ± 44 μM) and Cu (265 ± 147 μM) but unchanged concentrations of Zn and Mn. The overall protein concentration of the cytosol was ∼100 mg mL−1. The concentration of iron in cytosol from cells supplemented with 100 μM FeIII citrate declined modestly (to 176 ± 74 μM) as did that of Zn (to 29 ± 15 μM); in contrast, the Cu concentration nearly doubled (to 485 ± 250 μM). The number of data points in this series of iron concentrations is too few to establish trends, but the data suggests higher iron concentrations in the cytosol of cells grown on iron-replete media vs. those grown on iron-deficient media.
We wanted to determine the fraction of cytosolic iron due to LMM complex vs. high-molecular-mass iron-containing proteins. The iron concentration was 48 ± 24 μM in FTS from WT cells grown on 1 μM FeIII citrate, 191 ± 74 μM in FTS from WT cells grown on 40 μM FeIII citrate, and 54 ± 4 μM in FTS from WT cells grown on 100 μM FeIII citrate (after multiplying by 14.7). The data points are again too few to draw definitive conclusions, but they suggest that the concentration of the LMM pool is ca. 50 μM under low-iron growth conditions, and between 50 and 200 μM under high-iron conditions.
Most batches of cytosolic FTS were analyzed by LC-ICP-MS. The FTS from fermenting WT cells grown in MM supplemented with 1 or 40 μM iron exhibited 3–4 partially resolved iron-containing peaks, with masses between 700–1300 Da (Fig. 2 upper panel). (The word “apparent” is implied for all chromatography-based masses quoted in the article.)
 | ||
| Fig. 2 Iron-detected chromatograms of FTS from dilute isolated cytosol. Top panel: The average of 2, 4, 4, 8, and 1 independent traces are shown, respectively. The top three samples were prepared from WT cells grown with 10000, 100, 40, and 1 μM FeIII citrate added to the growth medium. Trace of buffer C alone is also shown. Bottom panel: The average of 4, 2, and 2 traces are shown for fet3Δ, mrs3Δmrs4Δ, and ccc1Δ cells. These cells were grown in medium supplemented with 100, 40, and 40 μM FeIII citrate (see Table 1). The FTS from the fet3Δ cells were incubated with acid phosphatase (PPX) as described.37 | ||
The intensity of the peaks from the FTS of WT cells grown with 40 μM iron was approximately twice that from the same cells grown with 1 μM iron. Cytosolic FTS from W303 cells grown in media supplemented with 100 μM iron exhibited an intense peak with a mass of 700 Da (along with less-intense peaks at 800–1300 Da). Fig. 2, upper panel, shows average traces; individual traces for 40 and 100 μM Fe FTSs are shown in Fig. S2 (ESI†). Traces obtained by injecting Buffer C alone exhibited some baseline structure but less intense than observed for the iron-detected features in sample traces.
Similar traces were obtained for cytosolic FTSs from various genetic strains of yeast, including fet3Δ, mrs3Δmrs4Δ, and ccc1Δ (Fig. 2, lower panel). Mrs3/4 are high affinity iron importers located on the mitochondrial inner membrane. The traces from these three strains also exhibited peaks between 600–1500 Da, but with different distributions of species. We could interpret some but not all traces. The iron peaks in traces from these strains were clearly related to those of WT traces, but they were perturbed. FTS from mrs3Δmrs4Δ cells exhibited an intense peak at 700 Da.
Deleting Mrs3/4 should cause the iron species imported by these proteins to accumulate in the cytosol.43 The accumulation of the 700 Da species raises the possibility that this species is imported into mitochondria via Mrs3/4. In contrast, the 700 Da species did not dominate chromatograms of ccc1Δ FTSs. Ccc1 is the major importer of cytosolic iron into vacuoles.44 Deletion of CCC1 causes a 20% decrease in total intracellular iron45 but this is not enough to explain the low intensity iron peaks in traces of FTS from Ccc1Δ cytosol. Fet3 is part of the high-affinity iron importer on the plasma membrane, and the cytosolic FTS from fet3Δ cells exhibited numerous rather intense LC iron peaks in the LMM region. We suspect that the iron species represented by these peaks entered the cell via Fet4, the low affinity iron transporter.46
Adding the phosphatase PPX to these solutions shifted the iron LC peaks, implying that the original peaks might be iron polyphosphate complexes, as is found in vacuoles.37 However, polyphosphate chains are built as they are inserted into vacuoles and are generally not abundant in the cytosol47 discounting this possibility. Nevertheless, the same result was observed in 4 independent preparations.
Cytosolic FTSs isolated from WT cells grown with 10 μM CuSO4 included in the medium exhibited copper traces dominated by an intense peak at ca. 5200 Da (Fig. 3, upper panel). In most batches, a low-intensity copper peak in the region between 1300–1000 Da was evident when the ordinate scale was expanded. The peak at ∼5200 Da was absent in traces from cup1Δ cells (Fig. 3, lower panel), suggesting that the peak arose from Cup1 metallothionein (molecular mass 6650 Da). If so, there would be a minor mis-calibration of the column. Interestingly, the peak at 1300 Da increased intensity in FTS from cup1Δ cells. Deleting CUP1 increases expression of the other metallothionein Crs5,48 which is consistent with our hypothesis that the intense broad peak at ∼8500 Da in the cup1Δ trace originates from copper-bound Crs5 (molecular mass 7192 Da). Traces of cytosolic FTS isolated from WT cells grown without copper supplementation were devoid of peaks. Traces of cytosolic FTS from WT cells grown on 50 μM of CuSO4 (Fig. 3) exhibited the 5200 Da peak as well as three rather intense peaks between 300–1300 Da.
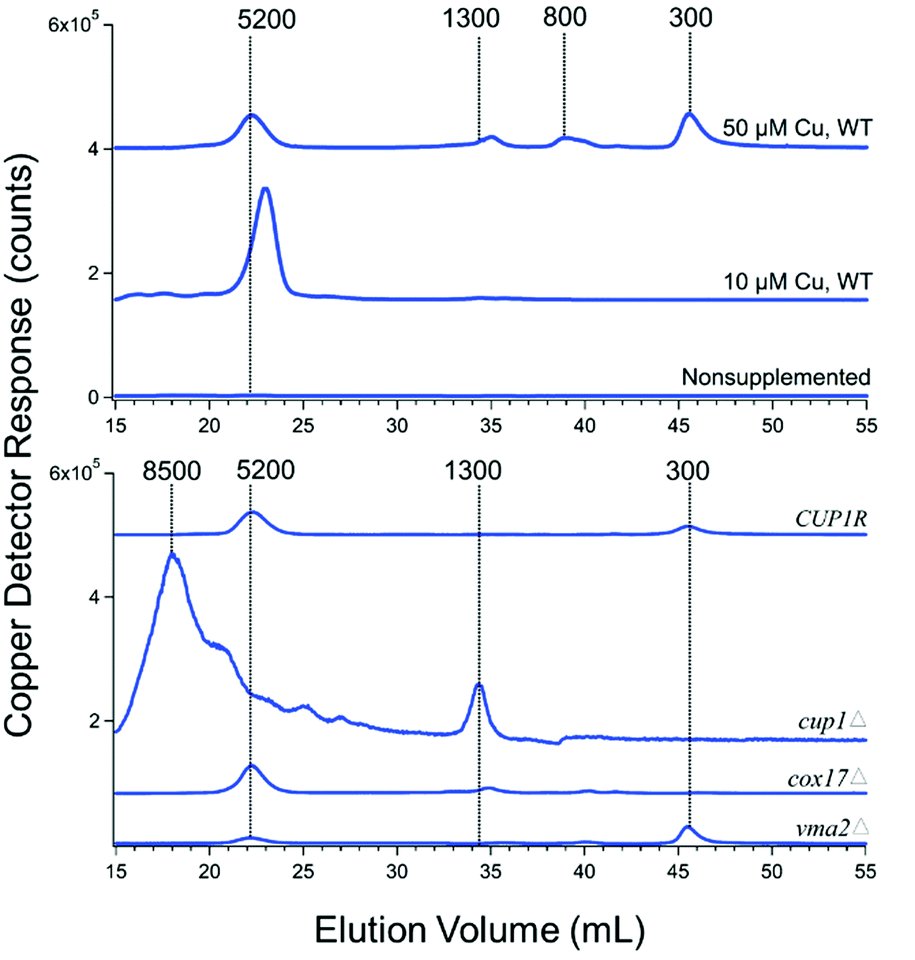 | ||
| Fig. 3 Copper-detected chromatograms from FTS of isolated cytosol. Each trace is the average from two independent batches (Table 1). | ||
The copper peaks in the LMM region (between 300–1300 Da) likely arises from nonproteinaceous complexes. We have tried to evaluate whether these peaks are artifacts or physiologically relevant. The intensities of these peaks increased strongly in traces of cytosolic FTS from genetic strains in which copper metabolism was perturbed, including vma2Δ, cup1Δ, and cox17Δ. Their intensities also increased in traces of cytosolic FTSs from WT cells grown in high concentrations (50 μM) of copper (Fig. 3). VMA2 encodes a subunit of vacuolar H+/ATPase49 such that its deletion leads to alkalization of vacuoles and disruption of copper trafficking to post-Golgi compartments.50,51 In some batches, deletion of either VMA2 or COX17 resulted in the accumulation of LMM copper peaks at 300 and 1300 Da, suggesting that one or more of these LMM nonproteinaceous copper species might traffic copper.
Cytosolic FTS exhibited 1–2 zinc peaks with masses between 1100–1300 Da (Fig. 4). This was the case for both WT cells and the various genetic strains mentioned above grown without zinc supplementation. The intensities of these peaks were unaffected by growing cells in medium supplemented with 100 μM of zinc. However, under these conditions, the intensity of the peak at 8500 Da increased. This peak is likely due to Crs5, which binds stronger to zinc than to copper.50 Crs5 is also overexpressed in cup1Δ cells, increasing the intensity of the 8500 Da species signal in Zn and also Cu cytosolic FTSs chromatograms (indicated by the blue line in Fig. 4).
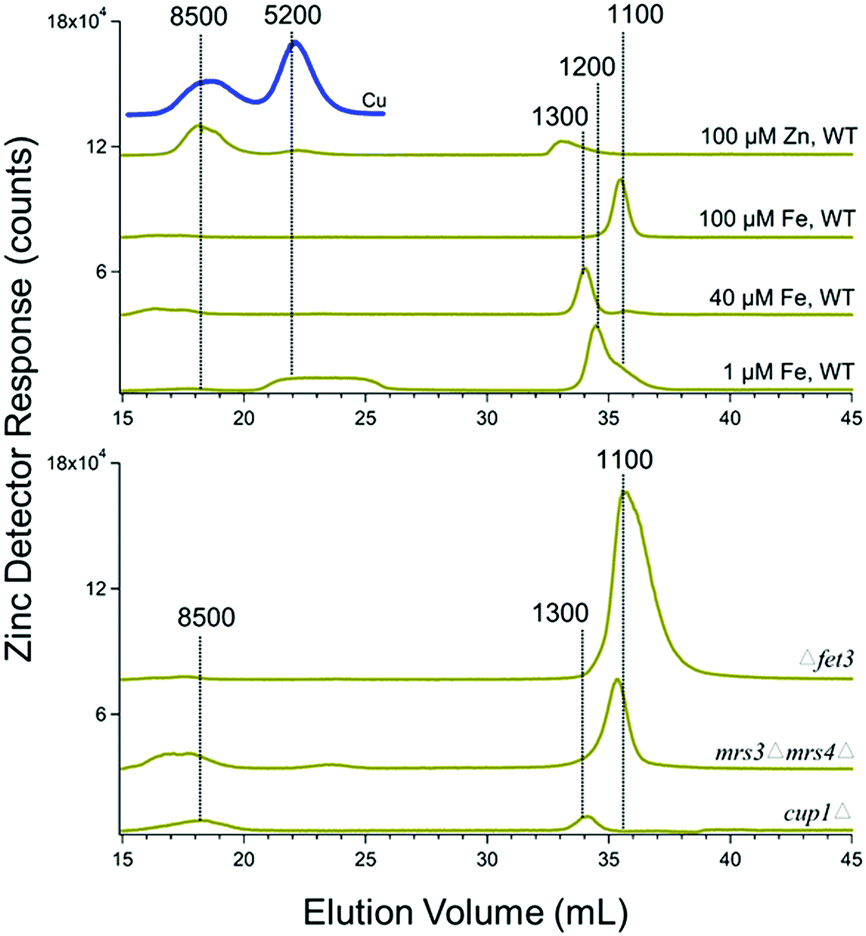 | ||
| Fig. 4 Zinc-detected chromatograms of cytosolic FTS. Average traces are shown, as indicated in Table 1. The blue line is the Cu chromatogram of cytosolic FTS isolated from cells grown with 100 μM ZnCl2 added to the medium. | ||
Cytosolic FTSs exhibited multiple Mn peaks, with masses ranging between 100–1200 Da (Fig. 6, upper panel). Mn peak positions exhibited more batch-to-batch variability than did the peaks for other metals. According to the Irving–Williams series, the binding of Mn to most biological ligands is generally not as strong as the binding of other metals. Curiously, FTSs of fet3Δ, mrs3Δmrs4Δ, and cup1Δ FTSs were dominated by a single intense Mn species at 1000 Da (Fig. 5, lower panel). A Mn peak at 200 Da was also observed in cup1Δ manganese trace which is in the range expected for hexa-aqua manganese.
 | ||
| Fig. 5 Manganese-detected chromatograms from FTS of isolated cytosol. Growth conditions and replicates for cytosol isolation of different yeast strains are given in Table 1. | ||
Phosphorus traces of FTS were dominated by a peak between 700–900 Da (Fig. 6). There were other minor phosphorus peaks with masses between 1000–1500 Da. Curiously, the position of the major peak varied from 700–900 Da, depending on the iron concentration in the medium; in contrast, the minor peak positions were invariant and served as an internal calibrant. Each trace of Fig. 6 is the average of 4–8 individual traces, and the major peaks are relatively sharp. Certain minor phosphorus peaks comigrated with particular iron, copper, zinc and manganese peaks in the 700–1300 Da region; whether this was coincidental is uncertain.
 | ||
| Fig. 6 Phosphorus-detected chromatograms from FTS of isolated cytosol. Growth conditions and replicates for cytosol isolation of different yeast strains are described in Table 1. | ||
Follow-up studies
Prompted by reviewers’ comments, we conducted additional experiments using a column that was treated to minimize the adsorption and desorption of metals (unpublished). We also replaced Tris and MES buffers with ammonium acetate (for buffer C) in hopes of minimizing metal:buffer interactions and eliminating S peaks from the MES buffer. Doing so resulted in quieter baselines (buffer injections) and improved reproducibility. Elution volumes were half of those plotted in other figures because a single column was used rather than the double column used in the initial studies. In recent studies we examined FTS of cytosol isolated from four batches. The cytosol of one batch was first passed through a 10 kDa membrane using an Amicon stirred cell, and the resulting FTS was dominated by an iron peak at the void volume, followed by two LMM peaks at masses of 900 and 500 Da, and a broad feature at a very low mass (Fig. 7A). This batch was isolated replacing the MES and Tris in buffer C with ammonium acetate pH 6.5. The remaining FTS solution from this batch was then passed through a 10 kDa centrifugal filter, affording the second trace in the series (Fig. 7B). The void peak was gone, while the two LMM Fe peaks (now at 800 and 500 Da) remained (as did the broad feature at very low mass). Traces C and D of Fig. 7 were of two independent batches isolated using buffer C. In these traces, the major LMM Fe peak was at 500 Da but with an asymmetry suggesting that a minor peak at ca. 800 Da contributes. The trace of the buffer was devoid of signals. | ||
| Fig. 7 Iron-detected chromatograms of cytosolic FTSs. (A) From WT cells grown on 40 μM Fe and 10 μM Cu; isolated using 20 mM ammonium acetate and filtered using an Amicon stirred cell. (B) A portion of the FTS in (A) after passage through a centrifugal filter. (C) and (D) From WT cells as in (A) but cytosol was isolated using buffer (C). The black line is a simulation of the major Fe species at 500 Da. (E) Buffer trace (20 mM ammonium acetate). FTSs were passed through a single treated column. | ||
We also incubated cytosolic FTS with 100 μM of 2,2-bipyridine, a well-known FeII chelator, for 2 h. The broad Fe peak at very low mass (ca. 28 mL in Fig. 7B) disappeared and was replaced by a peak due to Fe(BPY)3 (Fig. S3, ESI†). Whether iron was displaced from all LMM Fe species in the FTS could not be determined, as the Fe(BPY)3 peak comigrated with some of the peaks. Overall this experiment suggests that the majority of LMM iron in the sample was labile and could be coordinated by BPY.
Copper traces from the two additional batches of cytosolic FTS isolated from WT cells grown with 10 μM Cu (Fig. 8C and D) were again dominated by a peak that probably originated from the Cup1 metallothionein (mis-calibrated to 4000 Da on the single column). Inspection of the LMM region to the right of that peak revealed low intensity peaks with masses between 400–1000 Da. The intensity and composition of all Cu peaks increased dramatically in two batches grown with 50 μM Cu (Fig. 8A and B). Simulations (see insets) indicate approximate masses of 1000, 600, and 400 Da for the low-mass copper species in the cytosol. The same species were observed in FTS from cytosol isolated with ammonium acetate rather than Tris/MES (Fig. 8E). The buffer trace (Fig. 8F) was devoid of Cu peaks including any peaks in the region where the three LMM Cu peaks appeared.
 | ||
| Fig. 8 Copper-detected chromatograms of cytosolic FTSs. (A) and (B) From WT cells grown with 40 μM Fe and 50 μM Cu; isolated using buffer (C). Green, purple, and red lines are simulations of peaks at 1000, 600, and 400 Da, respectively. The black line is a composite simulation. (C) and (D) From WT cells grown with 40 μM Fe and 10 μM Cu; isolated using buffer (C). (E) From WT cells grown with 40 μM Fe and 10 μM Cu; isolated using 20 mM ammonium acetate. (F) Buffer only (20 mM ammonium acetate). (A) Single treated column was used. | ||
Zinc traces for the additional batches (Fig. 9E–H) were dominated by a peak at 700 Da, with a minor peak at ca. 500 Da. Mn traces from the additional batches exhibited a single broad peak at a mass of 200 Da (Fig. 9A–D). The broadness of the peak in some batches suggests that more than one species contributes. The improved reproducibility of these traces favor a single LMM Mn species in cytosol–perhaps a complex closely related to aqueous Mn.
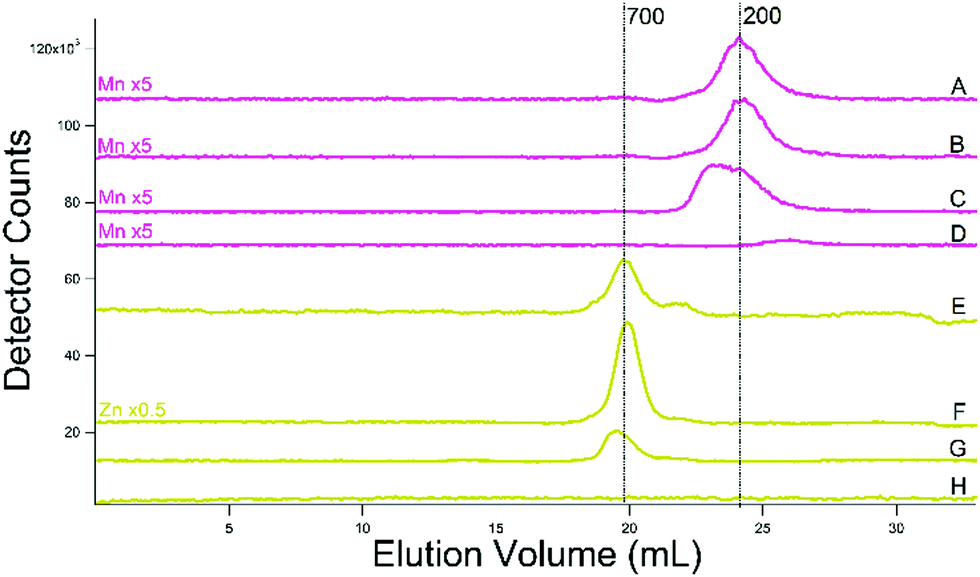 | ||
| Fig. 9 Manganese- (pink, (A)–(D)) and zinc- (yellow-green, (E)–(H)) detected chromatograms of cytosolic FTSs. (A) and (B) From WT cells grown on 40 μM Fe and 10 μM Cu; isolated using buffer (C) and filtered using an Amicon stirred cell. (C) Same but isolated in 20 mM ammonium acetate. (D) Buffer (ammonium acetate). (E) and (F) Same as (A) and (B). (G) Same as (C). (H) Same as (D). (A) Single treated column was used. | ||
The phosphorus-detected traces for all of the additional batches were indistinguishable from the one shown in Fig. 10A; the dominant peak was at ca. 400 Da regardless of buffer or conditions used. No minor phosphorus peaks were detected. For the additional batches prepared using MES (molecular mass of 195 Da) buffer, sulfur traces exhibited an intense peak at 700 Da (Fig. 10B and C). For the batch isolated using ammonium acetate rather than MES, the dominant peak was absent, allowing a minor peak at 500 Da to be observed (Fig. 10D). Glutathione (molecular mass of 307 Da) comigrated with this peak (Fig. 10E) and we assign the FTS peak as such. We estimate from the relative areas that the glutathione concentration in the cytosol is ∼13 mM. (The ICP-MS is less sensitive to sulfur such that traces tend to have lower signal/noise than metals at the same concentration.) The concentration of GSH in the cytosol of yeast has been reported to be nearly the same.52 A second sulfur peak, migrating at a mass of ca. 100 Da, is present (Fig. 10D). The same peak is evident in the sulfur trace from the batch containing MES (Fig. 10C). The GSH peak is also observable as a small shoulder to the right of the MES peak. We are currently investigating the origin of the second sulfur peak.
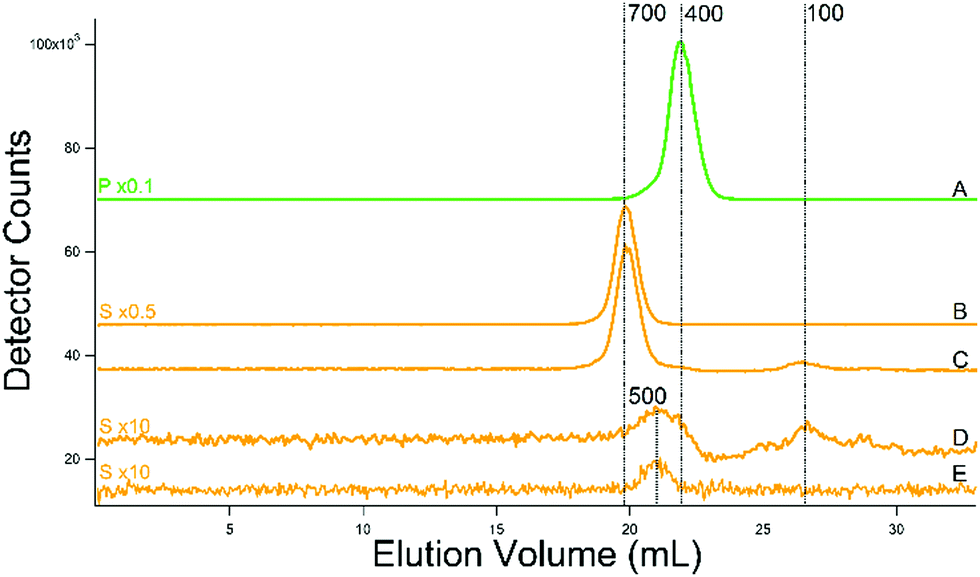 | ||
| Fig. 10 Phosphorus (green, (A)) and sulfur (yellow, (B)–(E)) -detected chromatograms of cytosol FTSs. (A) From WT cells grown with 40 μM Fe and 10 μM Cu; isolated using buffer (C). (B) 10 mM MES buffer. (C) Same as in (A). (D) WT cells grown as in (A) but cytosol was isolated using 20 mM ammonium acetate. (E) Reduced glutathione (250 μM). A single treated column was used. | ||
Discussion
To the best of our knowledge, this is the first study to report the chromatography-based detection of LMM iron, copper, zinc, and manganese (and sulfur and phosphorus) species present in the cytosol of a eukaryotic cell. We focused on LMM metal complexes by isolating the portion of the cytosol (the flow-through solution – FTS) that contains species with masses <10 kDa.We isolated many batches of cytosol in an effort to distinguish reproducible species (presumed to be generated in each batch) from artifacts (presumed to be generated sporadically). We also evaluated how the detected species were affected by supplementing the growth medium with various metal salts, and how the detected species were perturbed when various genetic strains (generally with iron-transporters or copper chaperones/storage proteins deleted) were used. This was done to determine whether the detected peaks would behave as expected if the associated metal species were physiologically relevant. In some but not all cases, we were able to interpret the changes based on known properties of the genetic mutation.
One related challenge is that the chromatography column employed in this study (SuperdexTM Peptide 10/300 GL from GE Life Sciences) adsorbs and desorbs labile metal species. We initially minimized the effects of this by daily rigorous cleaning with a chelator cocktail solution.36 The ICP-MS is highly sensitive so even minor absorption/desorption events are observed. Between the time that this paper was submitted and reviews were returned, we discovered a treatment for the column that diminishes the adsorption/desorption effect. We were able to include a few traces obtained with the treated column in the revised resubmission, and they largely confirm and extend our earlier studies.
We are more optimistic than Jacobs and many others in the field that physiologically relevant labile LMM metal complexes can eventually be isolated and characterized, and that chromatography-based methods are, or soon will be, a useful complement to chelator probes. The lability of Werner type coordination complexes with O, S, and N donors can be controlled by the denticity of the ligands, choice of donor atoms, etc. We conjecture that evolutionary pressures optimized the lability of LMM trafficking and regulatory complexes. We reason that the lability of labile metal pools in cells must be fast enough for the metal to transfer efficiently to target proteins, but slow enough to remain coordinated while being trafficked. The 3D shape of a trafficking complex likely dictates its binding properties to membrane-bound metal-ion transporters such that if the complexes did not “hold together” long enough for binding and recognition to these transporters, metal ion traffic between various cellular compartments would be chaotic, which it is not.
In this study, we measured the concentration of iron and other metals in the cytosol of yeast and flow-through solutions that contain LMM iron species. Roughly half of the cytosolic iron is present as LMM species. The overall iron concentration in the cytosol FTS was higher than we had anticipated (between 50–190 μM), given our LC results and previously published results (for mammalian cells) which suggest values an order of magnitude lower than this. The lower concentration in mammalian cells may arise from a greater reliance in these cells on the iron trafficking chaperone proteins PCBP1/2;53 yeast cells may rely more heavily on the labile iron pool for trafficking. However, this does not resolve the issue entirely.
Iron-replete yeast cells contain 440 ± 30 μM iron,54 and ca. 6% of Mössbauer spectral intensity of whole cells is due to nonheme high spin FeII, similar to the state expected for the LIP in the cytosol. In 4 measurements, the high-spin FeII concentration in yeast cells was 24 ± 2 μM. This includes contributions from nonheme high-spin FeII species in other regions of the cell. If all of this arose from the cytosolic LIP, then converting from cellular concentration to cytosol concentration (which represent ca. 68% of the cell volume) affords a maximum cytosolic LIP concentration, estimated from Mössbauer spectra, of 35 μM. This is lower than the iron FTS concentrations directly measured here (50–190 μM), and an order-of-magnitude higher than the iron concentrations evident from our LC peaks. We are currently working to resolve this internal discrepancy.
Treatment of the cytosolic FTSs with 2,2′-bipyridine chelated much of the iron in the sample, demonstrating its lability. Kosman and coworkers exposed growing yeast cells to the same chelator, and this arrested growth (temporarily).55 They proposed that the chelator coordinated the LIP in the cytosol, and that the LIP was necessary for cell growth. In light of their results, the LMM iron complexes that we have detected here may be the LIP that is essential for cell physiology.
The dominant copper-detected peak in LC traces of yeast cytosol FTSs was assigned to metallothionein Cup1. The peak was absent in cytosol traces from Cup1Δ cells and its intensity varied as expected in cytosol isolated from copper-starved vs. copper-overloaded cells. The peak was absent in FTSs prepared from cells grown in the absence of copper supplementation, it was intense in FTSs from cells grown with 10 μM Cu supplementation, and was very intense in cytosol FTSs from cells grown with 50 μM Cu supplementation. This is the behavior expected for the Cup1 metallothionein which serves to store excess copper.
Cytosol FTS from cells grown on medium supplemented with zinc exhibited a Zn and Cu peak at higher molecular mass (calibrated to 8500 Da on the double column). Both peaks probably arise from the second metallothionein Crs5, which is thought to be more critical for Zn homeostasis than for Cu homeostasis. Cup1 plays the dominant role in Cu homeostasis.
One of the most intriguing results of this study is that cytosol contains 3 copper species whose masses are too low to be copper-bound proteins. These species, whose peak intensities varied with the concentration of copper in the growth medium, have masses of approximately 400, 600 and 1000 Da. Their intensities were strongest in cytosol isolated from cells grown with 50 μM Cu supplementation; in this case, they represented 16% of the Cu in the trace (the remaining 84% was Cup1). In cytosol from cells grown with 10 μM Cu in the medium, only ∼1 percent of the copper is associated with these LMM peaks. The concentration of Cu in such cytosolic FTS was 90 ± 50 μM, suggesting that the concentration of these LMM Cu species is <1 μM (the vast majority would be Cup1). Related LMM copper peaks were observed in LC traces of cytosolic FTS isolated from vma2Δ, cup1Δ, and cox17Δ cells. Copper trafficking is dysregulated in all of these strains, and so the accumulation of copper species between masses of 300–1300 Da suggests that their concentration increases due to copper dysregulation.
The broader question, which we cannot currently answer, is whether the detected LMM Cu species are toxic to the cell or whether they are essential for copper trafficking or regulation. The growth of yeast is not affected by supplementing medium by up to 100 μM CuCl2,24 which leaves open the latter exciting possibility. We considered that the LMM cytosolic copper might dynamically bind Cup1. However, traces of CUP1R cytosolic FTSs exhibited peaks from both Cup1 and a LMM peak at 300 Da. If the LMM Cu species bound metallothioneins, we would have expected that overexpressed Cup1 would have bound any available LMM cytosolic copper.
The intensity of the dominant LMM Zn species in the cytosolic FTSs of WT cells did not increase by supplementing Zn in the growth medium. However, the intensity of the peak assigned to Crs5 did increase. Zinc homeostasis might be tightly regulated by Crs5 expression to prevent higher concentrations of the LMM Zn complex thereby minimizing the toxic effects of Zn mis-metallation.
In FTSs traces of fet3Δ cells, cytosolic iron and phosphorus peaks with masses of 800, 900, 1000, and 1300 Da comigrated. The binding of iron was disrupted after treatment with acid phosphatase, raising the possibility that polyphosphate is an endogenous ligand to LMM cytosolic iron.56 Further studies are required to consider this possibility further.
We observed a GSH peak in traces from a batch prepared in the absence of MES buffer. We did not observe a sulfur peak at higher masses, as might be expected for metal–GSH complexes. However, the sensitivity of the ICP-MS is low for sulfur and so such species might be undetectable if their concentration was in the low μM range. The second sulfur peak is intriguing and we are investigating its origin. From its intensity, it appears to be present in the cytosol at mM concentrations.
In summary, we have detected low-molecular-mass iron, copper, zinc, manganese, phosphorus, and sulfur molecular species with apparent masses between 1500–200 Da in cytosol isolated from yeast. These metal complexes are present in μM concentrations (S and P compounds are present in mM concentrations). We hypothesize that some, most, or all of these metal complexes serve essential cellular roles, e.g. in metal ion trafficking or regulatory homeostasis. Admittedly, we cannot currently exclude the possibility that some of these species are artifacts of isolation, but we took significant steps to avoid forming these species.
Despite future challenges, we are optimistic that chromatographic characterization of labile metal pools will ultimately complement efforts to characterize such pools using fluorescence-based chelator-probes. Together, these two approaches may finally, 40 years after the discovery of these mysterious little labile metal complexes, yield their chemical structures and cellular functions.
Authors’ contribution
TQN developed the large-scale isolation procedure for cytosol and performed experiments on 43 batches. JEK performed the recent experiments, with help from HNB who maintained the LC-ICP-MS and improved the LC experimental methods. PAL offered advice and wrote the initial draft of the paper. All authors edited the paper and agreed to its submission.Abbreviations
| FTS | Flow-through solution |
| LC | Liquid chromatography |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| LIP | Labile iron pool |
| LMM | Low-molecular-mass |
| MM | Minimal medium |
| WT | Wild type |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (GM127021), the National Science Foundation (MCB-1817389), the Robert A. Welch Foundation (A1170), and a T3 grant from Texas A&M University. We thank Danish Khan (Stanford U.) for preparing the fet3Δ strain, Andrew Dancis (U. of Pennsylvania) for preparing the vma2Δ strain, Vishal Gohil (Texas A&M U.) for providing the cox17Δ strain, Dennis Thiele (Duke U.) for providing the cup1Δ and CUP1R strains, Diane Ward (U. of Utah) for providing the ccc1Δ strain and Ccc1p antibody, Nathaniel Dziuba (Texas A&M) for operating and maintaining the LC-ICP-MS, and Carmen Wiggin (Texas A&M) for preparing two cytosol batches. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, NSF, or Welch Foundation.Notes and references
- A. Jacobs, Low-molecular weight intracellular iron transport compounds, Blood, 1977, 50, 433–439 CrossRef CAS PubMed.
- R. J. P. Williams, Free manganese(II) and iron(II) cations can act as intracellular cell controls, FEBS Lett., 1982, 140, 3–10 CrossRef CAS PubMed.
- F. Petrat, H. de Groot, R. Sustmann and U. Rauen, The chelatable iron pool in living cells: a methodically defined quantity, Biol. Chem., 2002, 383, 489–502 CAS.
- R. R. Crichton, Iron uptake and utilization by mammalian cells II. Intracellular iron utilization, Trends Biochem. Sci., 1984, 9, 283–286 CrossRef CAS.
- M. U. Muckenthaler, S. Rivella, M. W. Hentze and B. Galy, A Red Carpet for Iron Metabolism, Cell, 2017, 168, 344–361 CrossRef CAS PubMed.
- W. Breuer, S. Epsztejn and Z. I. Cabantchik, Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II), J. Biol. Chem., 1995, 270, 24209–24215 CrossRef CAS PubMed.
- M. Shvartsman and Z. Ioav Cabantchik, Intracellular iron trafficking: role of cytosolic ligands, Biometals, 2012, 25, 711–723 CrossRef CAS PubMed.
- F. Petrat, U. Rauen and H. de Groot, Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK, Hepatology, 1999, 29, 1171–1179 CrossRef CAS PubMed.
- F. Petrat, H. de Groot and U. Rauen, Determination of the Chelatable Iron Pool of Single Intact Cells by Laser Scanning Microscopy, Arch. Biochem. Biophys., 2000, 376, 74–81 CrossRef CAS PubMed.
- S. Epsztejn, O. Kakhlon, H. Glickstein, W. Breuer and Z. I. Cabantchik, Fluorescence Analysis of the Labile Iron Pool of Mammalian Cells, Anal. Biochem., 1997, 248, 31–40 CrossRef CAS PubMed.
- R. C. Hider and X. L. Kong, Glutathione: a key component of the cytoplasmic labile iron pool, Biometals, 2011, 24, 1179–1187 CrossRef CAS PubMed.
- A. Egyed’ and P. Saltman, Iron is maintained as Fe(II) under aerobic conditions in erythroid cells, Biol. Trace Elem. Res., 1984, 6, 357–364 CrossRef PubMed.
- R. C. Hider and X. Kong, Iron speciation in the cytosol: an overview, Dalton Trans., 2013, 42, 3220–3229 RSC.
- T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O’Halloran, Undetectable Intracellular Free Copper: The Requirement of a Copper Chaperone for Superoxide Dismutase, Science, 1999, 284, 805–808 CrossRef CAS PubMed.
- A. M. Keller, J. J. Benítez, D. Klarin, L. Zhong, M. Goldfogel, F. Yang, T.-Y. Chen and P. Chen, Dynamic Multibody Protein Interactions Suggest Versatile Pathways for Copper Trafficking, J. Am. Chem. Soc., 2012, 134, 8934–8943 CrossRef CAS PubMed.
- N. J. Robinson and D. R. Winge, Copper Metallochaperones, Annu. Rev. Biochem., 2010, 79, 537–562 CrossRef CAS PubMed.
- D. Horn and A. Barrientos, Mitochondrial copper metabolism and delivery to cytochrome c oxidase, IUBMB Life, 2008, 60, 421–429 CrossRef CAS PubMed.
- M. Glerum, D. Shtanko and A. Tzagoloff, Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase, J. Biol. Chem., 1996, 271, 14504–14509 CrossRef PubMed.
- A. B. Maxfield, D. N. Heaton and D. R. Winge, Cox17 is functional when tethered to the mitochondrial inner membrane, J. Biol. Chem., 2004, 279, 5072–5080 CrossRef CAS PubMed.
- M. T. Morgan, D. Bourassa, S. Harankhedkar, A. M. McCallum, S. A. Zlatic, J. S. Calvo, G. Meloni, V. Faundez and C. J. Fahrni, Ratiometric two-photon microscopy reveals attomolar copper buffering in normal and Menkes mutant cells, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12167–12172 CrossRef CAS PubMed.
- L. Yang, R. McRae, M. M. Henary, R. Patel, B. Lai, S. Vogt and C. J. Fahrni, Imaging of the intracellular topography of copper with a fluorescent sensor and by synchrotron x-ray fluorescence microscopy, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11179–11184 CrossRef CAS PubMed.
- D. Moira Glerum, A. Shtanko and A. Tzagoloff, Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase, J. Biol. Chem., 1996, 271, 14504–14509 CrossRef PubMed.
- D. M. Glerum, A. Shtanko and A. Tzagoloff, SCO1 and SCO2 Act as High Copy Suppressors of a Mitochondrial Copper Recruitment Defect in Saccharomyces cerevisiae, J. Biol. Chem., 1996, 271, 20531–20535 CrossRef CAS PubMed.
- A. Ghosh, P. P. Trivedi, S. A. Timbalia, A. T. Griffin, J. J. Rahn, S. S. L. Chan and V. M. Gohil, Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency, Hum. Mol. Genet., 2014, 23, 3596–3606 CrossRef CAS PubMed.
- Y. Wang, E. Weisenhorn, C. W. MacDiarmid, C. Andreini, M. Bucci, J. Taggart, L. Banci, J. Russell, J. J. Coon and D. J. Eide, The cellular economy of the Saccharomyces cerevisiae zinc proteome, Metallomics, 2018, 10, 1755–1776 RSC.
- A. Krężel and W. Maret, The biological inorganic chemistry of zinc ions, Arch. Biochem. Biophys., 2016, 611, 3–19 CrossRef PubMed.
- Y. Qin, J. G. Miranda, C. I. Stoddard, K. M. Dean, D. F. Galati and A. E. Palmer, Direct Comparison of a Genetically Encoded Sensor and Small Molecule Indicator: Implications for Quantification of Cytosolic Zn2+, ACS Chem. Biol., 2013, 8, 2366–2371 CrossRef CAS PubMed.
- M. C. Carpenter, M. N. Lo and A. E. Palmer, Techniques for measuring cellular zinc, Arch. Biochem. Biophys., 2016, 611, 20–29 CrossRef CAS PubMed.
- R. A. Colvin, W. R. Holmes, C. P. Fontaine and W. Maret, Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis, Metallomics, 2010, 2, 306 RSC.
- Z. Ma, P. Chandrangsu, T. C. Helmann, A. Romsang, A. Gaballa and J. D. Helmann, Bacillithiol is a major buffer of the labile zinc pool in Bacillus subtilis, Mol. Microbiol., 2014, 94, 756–770 CrossRef CAS PubMed.
- S. Choi, Y.-M. Hu, M. E. Corkins, A. E. Palmer and A. J. Bird, Zinc transporters belonging to the Cation Diffusion Facilitator (CDF) family have complementary roles in transporting zinc out of the cytosol, PLoS Genet., 2018, 14, e1007262 CrossRef PubMed.
- S. Das, K. Khatua, A. Rakshit, A. Carmona, A. Sarkar, S. Bakthavatsalam, R. Ortega and A. Datta, Emerging chemical tools and techniques for tracking biological manganese, Dalton Trans., 2019, 48, 7047–7061 RSC.
- G. P. Holmes-Hampton, N. D. Jhurry, S. P. McCormick and P. A. Lindahl, Iron content of Saccharomyces cerevisiae cells grown under iron deficient and iron-overload conditions, Biochemistry, 2013, 52, 105–114 CrossRef CAS PubMed.
- R. L. McNaughton, A. R. Reddi, M. H. S. Clement, A. Sharma, K. Barnese, L. Rosenfeld, E. B. Gralla, J. S. Valentine, V. C. Culotta and B. M. Hoffman, Probing in vivo Mn2+ speciation and oxidative stress resistance in yeast cells with electron-nuclear double resonance spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15335–15339 CrossRef CAS PubMed.
- C. C. Philpott and M.-S. Ryu, Special delivery: distributing iron in the cytosol of mammalian cells, Front. Pharmacol., 2014, 5, 173 Search PubMed.
- S. P. McCormick, M. J. Moore and P. A. Lindahl, Detection of Labile Low-Molecular-Mass Transition Metal Complexes in Mitochondria, Biochemistry, 2015, 54, 3442–3453 CrossRef CAS PubMed.
- T. Nguyen, N. Dziuba and P. A. Lindahl, Isolated Saccharomyces cerevisiae vacuoles contain low-molecular-mass transition-metal polyphosphate complexes, Metallomics, 2019, 11, 1298–1309 RSC.
- R. Herrera, M. C. Álvarez, S. Gelis and J. Ramos, Subcellular potassium and sodium distribution in Saccharomyces cerevisiae wild-type and vacuolar mutants, Biochem. J., 2013, 454, 525–532 CrossRef CAS PubMed.
- M. Yamaguchi, Y. Namiki, H. Okada, Y. Mori, H. Furukawa, J. Wang, M. Okhusu and S. Kawamoto, Structure of Saccharomyces cerevisiae determined by freeze-substitution and serial ultrathin-sectioning electron microscopy, J. Electron Microsc., 2011, 60, 321–335 CrossRef PubMed.
- P. A. Lindahl, J. G. Morales, R. Miao and G. P. Holmes-Hampton, Isolation of Saccharomyces Cerevisiae Mitochondria for Mössbauer, EPR, and Electronic Absorption Spectroscopic Analyses, in Methods in Enzymology, ed. W. S. Allison and I. E. Scheffler, Burlington, Academic Press, 2009, vol. 456, pp. 267–285 Search PubMed.
- N. Dziuba, J. Hardy and P. A. Lindahl, Low-molecular-mass iron in healthy blood plasma is not predominately ferric citrate, Metallomics, 2018, 10, 802–817 RSC.
- N. J. Bryant and T. H. Stevens, Vacuole biogenesis in Saccharomyces cerevisiae: protein transport pathways to the yeast vacuole, Microbiol. Mol. Biol. Rev., 1998, 62, 230–247 CrossRef CAS PubMed.
- E. M. Froschauer, R. J. Schweyen and G. Wiesenberger, The yeast mitochondrial carrier proteins Mrs3p/Mrs4p mediate iron transport across the inner mitochondrial membrane, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1044–1050 CrossRef CAS PubMed.
- L. Li, O. S. Chen, D. M. V. Ward and J. Kaplan, CCC1 Is a Transporter That Mediates Vacuolar Iron Storage in Yeast, J. Biol. Chem., 2001, 276, 29515–29519 CrossRef CAS PubMed.
- A. L. Cockrell, G. P. Holmes-Hampton, S. P. McCormick, M. Chakrabarti and P. A. Lindahl, Mössbauer and EPR Study of Iron in Vacuoles from Fermenting Saccharomyces cerevisiae, Biochemistry, 2011, 50, 10275–10283 CrossRef CAS PubMed.
- L. Li and J. Kaplan, Defects in the yeast high affinity iron transport system result in increased metal sensitivity because of the increased expression of transporters with a broad transition metal specificity, J. Biol. Chem., 1998, 273, 22181–22187 CrossRef CAS PubMed.
- R. Gerasimaite and A. Mayer, Enzymes of yeast polyphosphate metabolism: structure, enzymology and biological roles, Biochem. Soc. Trans., 2016, 44, 234–239 CrossRef CAS PubMed.
- A. Pagani, L. Villarreal, M. Capdevila and S. Atrian, The Saccharomyces cerevisiae Crs5 Metallothionein metal-binding abilities and its role in the response to zinc overload, Mol. Microbiol., 2007, 63, 256–269 CrossRef CAS PubMed.
- M. Forgac, Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology, Nat. Rev. Mol. Cell Biol., 2007, 8, 917–929 CrossRef CAS PubMed.
- D. S. Yuan, A. Dancis and R. D. Klausner, Restriction of Copper Export in Saccharomyces cerevisiae to a Late Golgi or Post-Golgi Compartment in the Secretory Pathway, J. Biol. Chem., 1997, 272, 25787–25793 CrossRef CAS PubMed.
- T. Marsh and J. Debnath, Ironing out VPS34 inhibition, Nat. Cell Biol., 2015, 17, 1–3 CrossRef CAS PubMed.
- M. Deponte, The incomplete glutathione puzzle: Just guessing at numbers and figures?, Antioxid. Redox Signaling, 2017, 27, 1130–1161 CrossRef CAS PubMed.
- A. G. Frey, A. Nandal, J. H. Park, P. M. Smith, T. Yabe, M. S. Ryu, M. C. Ghosh, J. Lee, T. A. Rouault, M. H. Park and C. C. Philpott, Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8031–8036 CrossRef CAS PubMed.
- G. P. Holmes-Hampton, N. D. Jhurry, S. P. McCormick and P. A. Lindahl, Iron content of Saccharomyces cerevisiae cells grown under iron-deficient and iron-overload conditions, Biochemistry, 2013, 52, 105–114 CrossRef CAS PubMed.
- R. Hassett, A. M. Romeo and D. J. Kosman, Regulation of high affinity iron uptake in the yeast Saccharomyces cerevisiae, J. Biol. Chem., 1998, 273, 7628–7636 CrossRef CAS PubMed.
- O. S. Chen and J. Kaplan, CCC1 suppresses mitochondrial damage in the yeast model of Friedreich's ataxia by limiting mitochondrial iron accumulation, J. Biol. Chem., 2000, 275, 7626–7632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: yeast strains used; Appendix A: generation of the fet3Δ deletion strain; Fig. S1: Western blot of 6 isolated cytosol batches; Fig. S2: individual iron chromatograms. Fig. S3: results of BPY experiment. See DOI: 10.1039/c9mt00312f |
| This journal is © The Royal Society of Chemistry 2020 |