
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Piecing together the puzzle: nanopore technology in detection and quantification of cancer biomarkers
Trang Vua,
Shanna-Leigh Davidsona,
Julia Borgesia,
Mowla Maksudula,
Tae-Joon Jeonb and
Jiwook Shim *a
*a
aDepartment of Biomedical Engineering, Henry M. Rowan College of Engineering, Rowan University, 201 Mullica Hill Road, Glassboro, New Jersey 08028, USA. E-mail: shimj@rowan.edu; Tel: +1 856 256 5393
bDepartment of Biological Engineering, Inha University, Incheon 22212, Republic of Korea
First published on 4th September 2017
Abstract
Cancer is the result of a multistep process, including various genetic and epigenetic alterations, such as structural variants, transcriptional factors, telomere length, DNA methylation, histone–DNA modification, and aberrant expression of miRNAs. These changes cause gene defects in one of two ways: (1) gain in function which shows enhanced expression or activation of oncogenes, or (2) loss of function which shows repression or inactivation of tumor-suppressor genes. However, most conventional methods for screening and diagnosing cancers require highly trained experts, intensive labor, large counter space (footprint) and extensive capital costs. Consequently, current approaches for cancer detection are still considered highly novel and are not yet practically applicable for clinical usage. Nanopore-based technology has grown rapidly in recent years, which have seen the wide application of biosensing research to a number of life sciences. In this review paper, we present a comprehensive outline of various genetic and epigenetic causal factors of cancer at the molecular level, as well as the use of nanopore technology in the detection and study of those specific factors. With the ability to detect both genetic and epigenetic alterations, nanopore technology would offer a cost-efficient, labor-free and highly practical approach to diagnosing pre-cancerous stages and early-staged tumors in both clinical and laboratory settings.
1. What is nanopore (NP) technology?
Formal definitions of NP-based technologies typically feature devices that contain a nanometer-scale pore embedded in a thin membrane. Originating from the Coulter counter and ion channels, NP-based devices can detect various charged biomolecules that are slightly smaller than the diameter of the pore. In the NP-based analysis, a biological or a solid-state membrane separates the experimental chamber into two compartments, referred to as the cis and trans sides, to which the cathode and anode are attached, respectively. Negatively charged biomolecules, such as DNA, are then introduced into the cis side of the chamber. Under the electrophoretic force exerted by the external voltage, the biomolecule transports through the NP to the trans chamber. As the molecule moves through the NP, it interrupts the current signal, causing ionic current blockages. Physical and chemical properties of the targeted molecule can be analyzed using the amplitude and duration of current blockages through the NP (Fig. 1).1,2 | ||
| Fig. 1 Schematic view of the NP-based experimental setup. The two chambers (cis and trans) are separated by a membrane, which is usually either biological or solid-state. A nanopore, which was embedded/fabricated into the membrane, acts as the single channel connecting the two chambers. DNA is added to the cis side. Under the electrophoretic force exerted by applied voltage, DNA strands translocate through the NP to the trans chamber, creating characteristic current blockages.2 | ||
Both biological and solid-state NPs, which can be obtained or fabricated in numerous ways,3–8 offer a wide range of biomolecule detection. Biological NP is secreted from different bacteria, in which the two most popular types come from α-Hemolysin and MspA porin. These biological NPs are then usually inserted into different biological substrates, such as a phospholipid bilayer, liposomes, or polymer films. Biological membranes are structurally well-defined and easily reproducible. Biological NP is mostly used for the detection of single-stranded DNA (ssDNA), microRNA (miRNA), and disease diagnostics.2 Most solid-state NPs are fabricated in membranes made of silicon oxide (SiO), silicon nitride (SiNx), hafnium oxide (HfO2), graphene, aluminum oxide (Al2O3) and hybrid materials.9–11 With controllable pore size and membrane thickness, solid-state NPs have been beneficial for use in RNA sequencing, single-stranded and double-stranded DNA sequencing, DNA–protein complex detection, and other biomolecule detection.
2. Key concepts of cancer development
The development of a malignant cancerous tumor frequently results from a multistep process, rather than just a single genetic change.12 This multistep process originates from various genetic and epigenetic modifications. Genetic modifications include structural variants, aberrant transcription factors activity, and telomere oxidative damage. The epigenetic modifications include DNA methylation, histone modifications, and aberrant expression of miRNAs. Both genetic and epigenetic alterations exert their pathological effects by causing defects in genes in one of two ways: (1) an enhanced expression or activation of oncogenes – gain in function- and/or (2) repression or inactivation of tumor-suppressor genes – loss of function.13 Several methods have been used in research for detecting various cancer-causing factors and different techniques are applied depending on the particular type of cancer.3. Toward personalized genomic medicines for cancer studies
Using the concept of massively parallel sequencing, the development of high-throughput next-generation sequencing (NGS) devices, particularly Illumina, Complete Genomics, and Roche Applied Science 454 among many others, have revolutionized personalized genomic medicines. In fact, for the first time, several clinical samples of patients were obtained and analyzed with NGS, providing an insight to the high complexity of diseases such as cancer.14–21 While providing high-throughput and high accuracy reads, NGS still requires multiple processing steps and file formats for their outputs (e.g. FASTQ, BAM, SAM and VCF).22,23 The high volumes of data generated by NGS make managing and storing results one of the major challenges for clinical laboratories.24 Furthermore, NGS technologies employ several enrichment, amplification, and labeling steps, such as polymerase chain reaction (PCR) and bisulfite conversions, causing the performance to be time and cost intensive, as well as increasing the possibility of false positive results.25 Due to the need of a label-free, high throughput system, there has been a growing interest toward using third- and fourth-generation sequencing, specifically NP technologies, in cancer prevention and detection in the past decades.Since its development and publication in 1996, the NP has become an emergent and powerful technology for a direct and inexpensive method for DNA sequencing, biosensing, and detecting biological or chemical modifications on single molecules, as well as the kinetics of DNA and protein folding. NP technology offers many advantages that NGS devices are incapable of. For example, NP has demonstrated the ability to detect CpGs methylation (one of the earliest epigenetic biomarkers in cancer hallmarks) without the need of PCR amplification and bisulfite conversion.26,27 Thus, NP technology strives to be a potential genomic tool that is label-free, has a high throughput, a small sample volume requirement, flexible runtime, and minimal footprint.2 However, despite the past twenty years of significant progress in single molecular sequencing and analysis, NP technologies have not yet been translated into even distantly comparable advances in clinical settings. There are two general, synergistic goals that have been striven for to increase the efficacy of single molecule analysis using NP: to decrease the translocation time of biomolecules through the pore and to increase the base-calling accuracy. An ideal single molecule analysis system would be highly accurate, have a high throughput, and be sensitive to both genetic and epigenetic changes of the cancer genome. Many previous articles have extensively discussed the potential and development of NP technology, especially in DNA sequencing.2,3,28–32 However, to the best of our knowledge, an article focusing solely on the application of NP technology in the early detection of various types of cancer biomarkers and causal factors has yet to be published. We believe this review will contribute to the further understanding of the potentials and challenges of applying NP technology in cancer research. Herein, we provide a brief overview of the six main cancer-causing factors, along with methods conventionally used in detecting cancer at the molecular level. We then focus on reviewing NP technology with a focus on its development as a method for specific molecular detection, as well as its future potential and challenges in the clinical domain. All studies presented here are not intended to form an exhaustive list, but rather, to illustrate the totality of our major achievements and challenges of applying NP technology in early cancer detection.
4. Detection of genetic modifications using nanopores
4.1 Structural variants
Structural variants (SVs) are one of the first recognized causal factors of cancer. A structural variant is a form of somatic DNA mutation, whereby the SV promotes the development and progression of cancer while contributing to all the important hallmarks of the instability in cancer genomes.33 The four main types of SVs are large deletions, amplifications, inversions and translocations of nucleotides within a DNA sequence. They are often responsible for the creation of fusion genes, copy number, and other regulatory changes that lead to activation or overexpression of oncogenes, as well as inactivation of tumor suppressor genes.34–38 In many cases, different SVs occur simultaneously in a specific pathway that amplifies their genetic effects on cell instability. For example, with head and neck cancers, it was found that when the deletion of CDKN2A and amplification of CCND1 happen together, there is a higher risk of recurrence, metastasis, and death rather than when either genetic alteration occurs alone.37,39SVs are important indicators of human cancers.33,41–43 Complex SVs have been found to cause approximately half of nucleotide deletions in pancreatic ductal adenocarcinoma (PDAC).44–46 Furthermore, CDKN2A/p16 and SMAD4/DPaazC4 have been identified as two of the most common deleted tumor suppressor genes. The ability to detect these mutations is critically important to the healthcare industry, allowing the monitoring of cancer patients for early detection of possible relapse.33,44–47 In mammalian cells with highly repetitive genomes, studies of SVs frequently use a resequencing approach, in which the read from the target genome is independently aligned from the reference genome to search for SVs.48 In general, besides specificity and sensitivity, when detecting SVs, a method's quality is further judged by its ability to accurately predict breakpoint locations, the size of variants, and changes in copy count.33,49
Norris et al. demonstrated the value of detecting long SVs using Oxford MinION™, to detect a series of well-characterized SVs, including large deletions, inversions, and translocations that inactivate the CDKN2A/p16 and SMAD4/DPC4 tumor suppressor genes in pancreatic cancer.33 Using Oxford Nanopore barcodes, the Norris et al. produced libraries for all 12 PCR amplicons in one run, yielding reads with PHRED scores of 10.9–11.50. PHRED, invented back in 1998 by Ewing and Green, was originally a base-calling program for automated sequencer traces. In later research, the term “PHRED score” has been used for the determination of quality and accuracy between consensus sequences. The higher the PHRED score, the higher the accuracy. For example, a PHRED score of 10 stands for a 90% base call accuracy, and a PHRED score of 20 is correlated with 99% base call accuracy.50 For this specific study, the readings were averaged at 640 bps long with a PHRED score of 11.50. It was also found that these reads are consistent for the entire bps length. The amplicons mapped with an overall percentage of 99.6% for regions of hg19, while 79% of aligned reads accurately matched to bases. Notably, the representation of amplicons does not change accuracy based on the complexity of the sequence. Additionally, the researchers wanted to test their method with low frequency SVs. In a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilutions, the run produced 4058 2D reads from 270 of 512 channels. The average read length was 650 bps and had a PHRED score of 10.9. Overall, the researchers proved their methods can be conducted in a timely manner. For the two sequences (CDKN2A/p16 and SMAD4/DPC4) in this study, it took 15 minutes and 33 minutes respectively, to generate 450 reads.33 In comparison, 2nd generation sequencers could generate millions of reads simultaneously, but it would take hours to days to complete. The experiment indicated the ability of NPs to serve as a reliable and efficient method of sequencing, allowing rapid detection of tumor-associated structural variants. The two limitations of MinION™, as noted by the researchers, were (1) a relatively high mismatch and index error rate and (2) a limited yield (on the scale of Mb or Gb) (Fig. 2).
:100 dilutions, the run produced 4058 2D reads from 270 of 512 channels. The average read length was 650 bps and had a PHRED score of 10.9. Overall, the researchers proved their methods can be conducted in a timely manner. For the two sequences (CDKN2A/p16 and SMAD4/DPC4) in this study, it took 15 minutes and 33 minutes respectively, to generate 450 reads.33 In comparison, 2nd generation sequencers could generate millions of reads simultaneously, but it would take hours to days to complete. The experiment indicated the ability of NPs to serve as a reliable and efficient method of sequencing, allowing rapid detection of tumor-associated structural variants. The two limitations of MinION™, as noted by the researchers, were (1) a relatively high mismatch and index error rate and (2) a limited yield (on the scale of Mb or Gb) (Fig. 2).
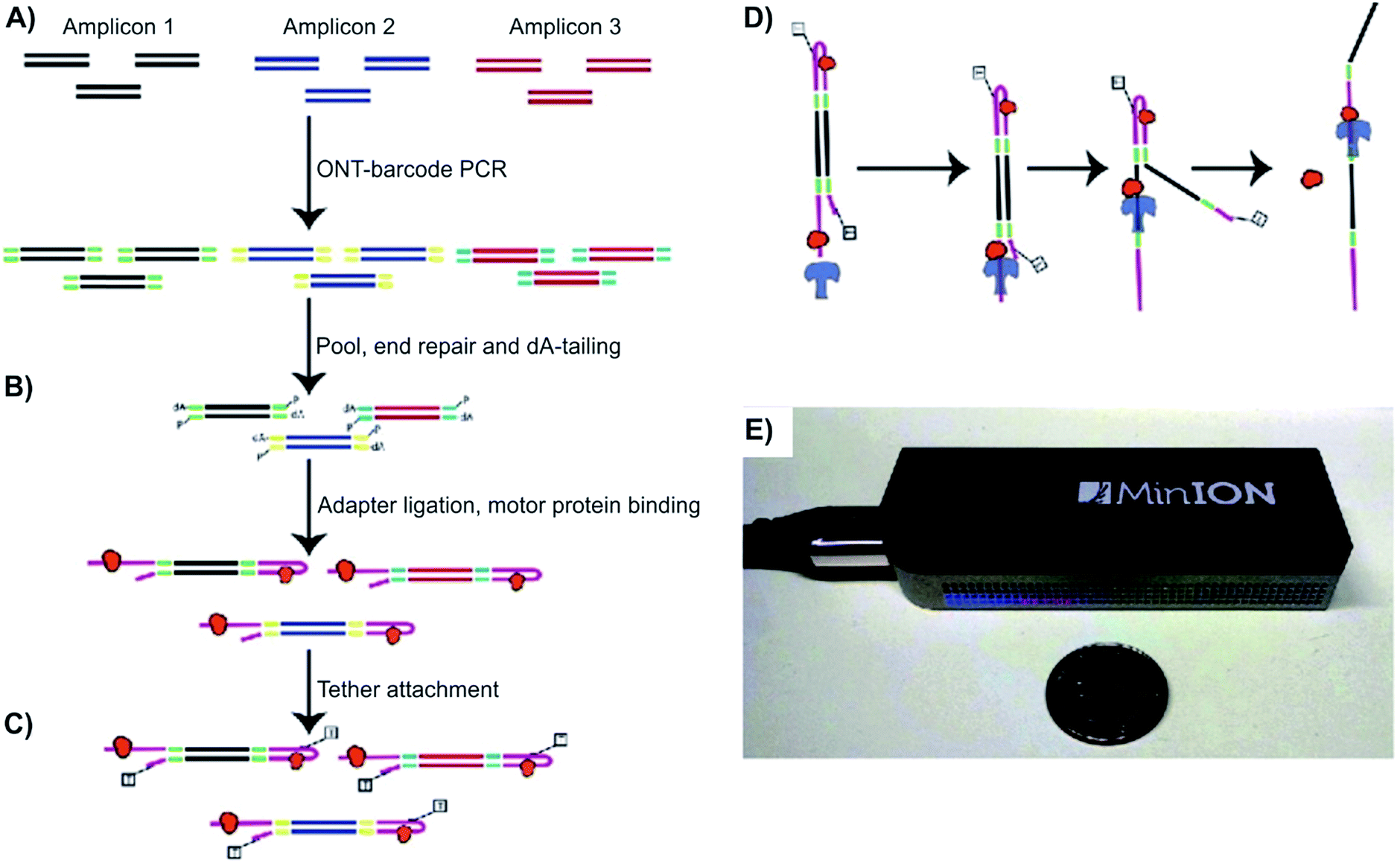 | ||
| Fig. 2 Detecting structural variants with nanopore. (A–D) Schematic of the Oxford MinION Nanopore Library Prep workflow. Oxford Nanopore barcodes were pooled into amplicons by PCR. After NEB End Repair and dA-tailing modules, hairpin and leader adapters were ligated on, each containing a motor protein (orange). Then, tether attachment allowed DNA molecules to attach to the membrane of MinION flowcell. Within the flowcell, molecules, each with attached motor proteins, were pulled through a nanopore, producing 2D consensus read. (E) Size comparison between an Oxford MinION and a quarter coin.33 | ||
Comparing to conventional genome-based methods, such as fluorescence in situ hybridization (FISH), fiber-FISH, array comparative genomic hybridization (aCGH) and paired-end mapping (PEM), which have a read length of approximately 35–400 base pairs (bps),40,49 NP allows a much more flexible read lengths (of a few bps to kbps). However, the average PHRED score of reads generated by MinION is still relatively low compared to other sequencers (e.g. Illumina, 454, Ion Torrent, and PacBio). At the moment, Illumina is the most popular DNA sequencer on the market. Still, depending on the equipment model and sample size, sequencing using Illumina can take from 3–12 days to complete. Additionally, the current market price of Illumina ranges from $50000 (MiniSeq) to over $6M (Illumina HiSeq X Five), costing tremendously more than the NP-based sequencers.
4.2 Transcriptional factors
The second most well-known causal factor of cancer is aberrant activity of transcription factors (TFs), which are often members of multigene families with common structural domains.12 TFs are the main regulators of gene expression and signaling pathways in all biological systems and bind to a specific sequence of DNA to promote or inhibit gene expression. In cells, a major portion of oncogenes and tumor-suppressor genes are encoded by TFs.13,51 Aberrant TF activity can occur due to changes in expression, protein stability, protein–protein interactions, post-translational modifications, and numerous other mechanisms.52 In a healthy cell, upstream transcriptional regulators highly regulate all genes with similar functions. However, changes in TF activity leads to deregulation of genes involved in promoting cancer cell proliferation, survival, and inducing angiogenesis and metastasis of tumors.13,51 For example, nuclear TFs, the signal transducer and activator of transcription, has been linked to various human cancer cell lines and primary tumors, including leukemia and lymphoma, as well as breast, lung, pancreas, and prostate cancers.34–38,53–57Various direct and indirect techniques have been used to characterize TFs, along with other sequence-specific DNA binding proteins, including electrophoresis, electrophoretic mobility shift assay, nuclear magnetic resonance, X-ray crystallography, atomic force microscopy, optical tweezers, and direct fluorescent visualization, among others.58–63 However, most of these methods require some combination of chemical cross-linking between TFs and DNA, modification or tagging of the TF and DNA, and amplification assays. Furthermore, due to the complicated requirements, these methods would lack the ability to resolve fine details of the TF and DNA complex (i.e. partial versus full binding of the TF domains to DNA).63 The specific mechanism of TFs binding to DNA sequences is still under invasive study and is a major area of interest in molecular biology.63,64
Squires et al. used solid-state NPs as biosensors for the characterization of DNA, RNA, and proteins. With the use of an electric field, the researchers could guide the polymers through a NP and identify individual molecules. The current-blockage patterns generated during translocation of charged molecules provides an abundance of information about TF local properties, as well as TF–DNA interactions.63 As previously noted, the regulation of TFs has not been well investigated, hence the use of solid-state NPs could be a novel technique in describing these molecular interactions. As proof of technique, the Squires et al. has shown that their NPs can distinguish between specific and nonspecific binding of TF, by analyzing the ion current of the canonical zinc-finger DNA-binding domain of early growth response 1 (zif268). Characterization of the zif268 was accomplished using the distinct blockage patterns of the current within the nanopore.65 Through analyzing the data, the researchers found that there are three main types of blockages, existing mostly in five distinct patterns rather than randomly. These patterns have a direct correlation to preexisting data. Hence, the NP presents great potential in characterizing DNA complexes because of its ability to detect complex structures and protein conformations, with the possibility of removing TFs as needed. Squires et al. note that their NP sensor can identify small TFs in DNA as well as distinguish between specific and nonspecific binding. This research technique allows information-gathering availability with respect to TF–DNA interactions (Fig. 3).
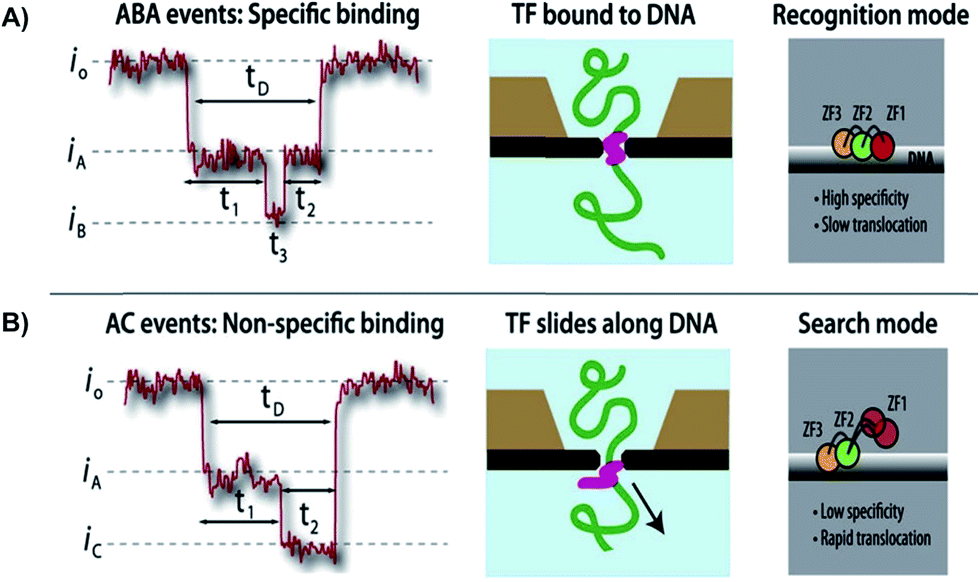 | ||
| Fig. 3 Distinguishing between specific and non-specific binding of TF—DNA with solid-state nanopore. Translocation event traces and proposed mechanisms for (A) specific binding, and (B) non-specific binding of TF to DNA.63 | ||
4.3 Telomeres
On average, telomeres are shortened by 19 bps per year due to aging, oxidation, stress, mitotic activity of tissues, and lifestyle.66–69 When shortened to a critical length, telomeres lose their ability to protect the DNA chromosomes70 and restrict the proliferation of normal somatic cells.69 This leads to chromosomal fusion and degradation.71,72 In contrast, approximately 85% of human cancer cells can achieve an “immortality” status by maintaining and elongating telomeres via the de novo synthesis of telomeric DNA.71 A study was conducted on 47102 individuals from the general population, where these individuals were followed for up to 20 years to find out the relationship between telomere length and cancer. Although short telomere length is not an indication of cancer,69 it was observed that cancer patients with shorter telomere length had increased risk of early death. This result was observed in patients with lung and esophagus cancer, malignant melanoma, and leukemia.69
Even though it has been years since the first research, the kinetics of telomeres in cancer cells remains elusive. At present, measuring the length of telomeres and observing the kinetics of folding are still challenging, as there is no gold-standard technique.73 In order to fully understand the role of telomeres in cancer prediction or therapy, it is essential to understand the kinetics of telomere folding and other conformational changes as a response to different living and environmental conditions.
Work is currently underway to apply NP sensor in tracking the telomeric DNA G-quadruplex folding/unfolding. Several research groups have used biological NP to capture some or all four folded-structures of G-quadruplex, including hybrid (hybrid-1 and hybrid-2), basket, and propeller structures.10,74–76 Findings from these studies reported that even though the four G-quadruplex structures all folded from the same DNA sequence, they produced very different electrical signatures.76 This was attributed to the overall shape and volume of each secondary structure. It was observed that both hybrid-1, -2, and basket forms had a diameter of 2.7 nm and 2.4 nm, respectively. Since the cis opening of the α-hemolysin pore has a diameter of 3.0 nm, these three folds can enter the large vestibule. However, the propeller fold, with a disk-shaped structure and diameter of 4.0 nm, exceeds the diameter of the NP cis opening and was unable to enter the vestibule.77
Another inventive solution to capture and unravel G-quadruplexes is to employ a 25-mer poly-2′deoxyadenosine tail (d25A-tail) on the 5′ end of the telomeric DNA. Applying this method, the Burrows group reported the analysis of various folding motifs of the telomere sequence, with and without the 5′-d25A-tails.76 Among the four loop topologies, only the basket fold was able to translocate through the NP without the addition of the homopolymer tail to the 5′ end. For the G-quadruplex to move through the NP, it needs to unravel to a singular strand which would be able to translocate through the narrow β-barrel, and the remaining G-triplex has to roll within the vestibule. This is likely a favorable process for the basket fold because of its nearly spherical shape.78 Even though the volume of the vestibule is large enough to accommodate all four G-quadruplexes within its cavity, the narrow entrance of the vestibule prevented the propeller fold from entering the NP. However, with the addition of the 5′ tail, the propeller fold was able to circumvent the problem of entering the cavity, and yet still had a very fast translocation signature. This is attributed to the fact that the propeller fold was able to roll outside of the vestibule while an electric force was applied to the dA25-tail as it threaded through the ion channel, without having any molecular interactions or steric hindrance that would have been experienced on the interior of the vestibule.76
In the light of those previous studies, for the first time, the unfolding kinetics of human i-motifs were studied using the α-hemolysin NP. Under acidic conditions, cytosine (C)-rich DNA sequences can adopt i-motif folds, since the hemi-protonation of C-rich strands allow C+·C base pairs to form.79 The Ding et al. conducted experiments on the human i-motif sequence at a constant ionic strength, but various pH (5.0–7.2). Since the dimension of an i-motif (2.0 nm × 2.0 nm) is smaller than cis opening (∼3.0 nm) of the α-hemolysin pore, it can enter the pore without unfolding and be captured in the nanocavity.79 Hence, a d25A tail was attached to the sequence, in order to increase the unfolding rate of i-motif. Upon the attachment of d25A, it was observed that at pH 5.0, the folded structure entered the α-HL pore, yielding characteristic current patterns. However, when the pH was at 6.8 and 7.2 (higher than the transition pH 6.15), the percentage of strands still folded was 4% and 2%, respectively. Furthermore, the force applied in this study was analogous to the forces exerted on genomic DNA by RNA polymerases II (5–20 pN) and DNA helicase (6–16 pN).79 Hence, these studies strive to show the potential of α-hemolysin as part of biosensor development, aiding in our knowledge of the lifetimes of i-motifs of telomere sequences, and their biologically relevant structures, which can be used as drug delivery targets for cancer treatments.80
These findings are steps toward a better understanding of the folding and unfolding mechanisms of the telomere. When pre-detecting different cancer types, conventional methods, such as FISH, Southern blot, and quantitative-PCR, require complicated meta-analyses, chemical-crosslinking and intensive preparation; hence the results are inconsistent.69,81–83 Whereas NP analysis, lacking all those complications, allows a better understanding of the kinetics and mechanisms, aiding in the analysis of how different oxidation, stress and factors affect the length of telomeres, as well as the correlation between cancer development and telomere immortality (Fig. 4).
 | ||
| Fig. 4 Capturing unfolding process of the four G-quadruplex structures with biological nanopore. (A) Schematic of the α-hemolysin NP, with the cis opening of 3.0 nm, constriction of 1.4 nm, and trans opening of 2.0 nm. (B) Folding structures and dimensions of G-quadruplex conformations: hybrid-1, hybrid-2, basket, and propeller. (C) G-Quadruplex fold entered and unfold inside the nanocavity of α-hemolysin NP, causing two distinct levels of blockade. (D) Except the propeller fold, all other G-quadruplex can enter the cis opening of α-hemolysin NP without unfolding, but cannot pass through the pore constriction.65 (E) Models of the three conformations with the additional 5′-dA25 tail unraveling through α-hemolysin pore. Both hybrid and basket folds were able to enter the cis opening of the α-hemolysin pore, thus unraveled inside the pore nanocavity. On the other hand, propeller fold, because of its size, could not enter the NP. This conformation unraveled its structure outside of the pore, using the help of the 5′-dA25 additional tail.65 | ||
5. Detection of epigenetic modifications using nanopores
5.1 DNA methylation
The overall level of 5-methylcystosine contained in the cell sample can be quantified using high-performance liquid chromatography (HPLC), high-performance capillary electrophoresis (HPCE), bisulfite sequencing, methylation-specific PCR, among many others.102–108 However, these methods have certain drawbacks. For example, although HPLC and HPCE can accurately quantify the total amount of methylated CpGs, they have incomplete restriction enzyme cutting, offer limited region of study, require substantial amounts of high molecular weight DNA, and are labor intensive. Similarly, with PCR-based methods, only the methylation status of CpG sites that are complementary to the primers can be interrogated. Thus, the predominant methylation patterns in the sample may not necessarily reflect the actual results (false positive results).
With NP analysis, current methods used in the detection of aberrant CpGs methylation usually employ either a methylation specific labeler, or an electro-optical tagging.26,27,109 The first method, as proposed by Shim et al., employs an engineered methyl-CpG-binding domain protein (i.e. MBD1x or Kaiso Zinc Finger proteins) as a selective labeler to detect and quantify hypermethylated CpG sites in double-stranded DNA (dsDNA).26,109 As the DNA translocated through the NP, the presence of 5-mC·labeler complexes caused a signature current blockage, allowing the detection and coarse quantification of 5-mC sites on a single molecule.109 Indeed, this method set an initial application in screening for the presence of hyper- and hypomethylated DNA. Moreover, Shim et al. pointed out that with the versatile binding affinity of KZF to various methylation patterns, the studied assay can allow various patterns to be screened.26 Since NP analysis requires low volumes of DNA for testing, the technique will be more applicable and practical for clinical use. Without the need of DNA replication and amplification, detecting CpG methylation using NPs requires much less labor in comparison to other conventional methods.
The second method, as mentioned before, uses an electro-optical solid-state NP to detect and quantify hypomethylation in DNA.27 In this approach, enzyme DNA MTases was assisted by small molecular weight synthetic cofactors to catalyze a one-step enzymatic reaction. This enzyme–cofactor complex was directly conjugated onto fluorescent probes and attached to the unmethylated CpG sites. The Meller group was able to detect and differentiate between fully methylated, partially methylated and unmethylated dsDNA, using ultrasensitive electro-optical NP sensing as the tool for single-fluorophore multicolor quantification. Unlike MBPs, DNA MTase only labeled unmethylated CpG sites of the target DNA. This allowed the direct targeting of hypomethylated CpG sites in the genome (i.e. promoter regions of oncogenes). Furthermore, this electro-optical solid-state NP showed a high potential for employing multiple DNA MTases and other epigenetic biomarkers. With the aid of those biomarkers, orthogonal labeling/sensing of 5-mC can be achieved in the future.27 Further research must be done in order to develop a calibrated scale to count the number of unmethylated CpGs in the target sequence.
The presence of bulk 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC) on ss- and dsDNA has been successfully detected and distinguished using both solid-state and biological NPs.121–123 For instance, the Drndic group proposed a method using solid-state NP to discriminate two different structures that translocated through the pore (5-mC and 5-hmC). Upon the addition of 3 kbp dsDNA, a sequence of current blockage was generated, in which the magnitude of each spike was related to the excluded volume of biopolymer that occupies the pore. From the differences in ΔImax values, Wanunu et al. was able to discriminate between 5-mC and 5-hmC. Shorter end-to-end distance of the more polar 5-hmC indicated an increased flexibility in 5-hmC comparing to cytosine and 5-mC. Moreover, it was shown that different proportions of 5-hmC in DNA fragment containing cytosine and 5-mC can be quantified using ionic current signal.121 The second device used in the detection of CpG methylation variants employed both the wild-type phi29 DNA polymerase (phi29 DNAP) and MspA in the same assay.122,123 With this unique approach, the Wescoe et al. reported a direction detection of all five cytosine variants (C, mC, hmC, fC and caC). In this single-molecule tool, a phi29 DNA polymerase drew ssDNA through the pore in single-nucleotide steps and the ion current through the pore was recorded.122 Overall, the single-pass call ranged from approximately 91.6% to 98.3% depending on neighboring nucleotides.122,123 Since the knowledge of the five cytosine variants, especially fC and caC, is still very limited, the possibility of these variants having an impact on genome-wide demethylation or other modifications in cancer cells should not be eliminated.
These studies have shown NP analysis potential as a robust and efficient tool for the study of DNA methylation. The technique can directly detect CpG methylation without the need for DNA amplification or complicated preparation processes. Due to its special characteristics, methylation of CpG is usually erased during replication and amplification. Bisulfite conversion, for example, requires large amplification, leading to false positive results. Hence, NP analysis could be a more practical and reliable method to screen and detect aberrant DNA methylation in cancer patients. However, in order to apply NP technology to clinical trials and testing, a genome-wide mapping of CpG methylation needs to be developed with a higher base-call accuracy (Fig. 5).
 | ||
| Fig. 5 Distinguishing variants of cytosine with biological and solid state nanopores. (A) Chemical structures of cytosine and its variants. First row: mC (left) and fC (right). Second row: cytosine. Third row: hmC (left) and caC (right).122 (B) Schematic of the Phi 29 DNAP–MspA complex. MspA pore constriction is shorter and narrower compared to α-hemolysin (as shown in the top), allowing short subtle structural changes to be distinguished. (C) A typical trace of DNA translocation through the Phi 29 DNAP – MspA complex.121 (D) Detection of DNA methylation with methyl binding proteins (MBP) using solid state nanopore. MBPs bind to methylated CpGs on DNA, allow the detection and differentiation between unmethylated, hypermethylated and locally methylated DNAs. (E) Detection of DNA methylation with optical-tagging using solid-state nanopore.27 | ||
5.2 Histone–DNA modifications
Results from several studies have indicated that aberrant DNA–methylations are also linked to the presence of aberrant histone modifications.101,124–127 Histones are the gene activity's dynamic regulators. They go through several post-translational modifications, such as acetylation, methylation, phosphorylation, ubiquitylation and others. Specifically, methylation and acetylation of lysine residues on the nucleosomal core histones play an essential role in gene expression and chromatin structure regulation.128 In normal cells, histones in DNA sequences are hypoacetylated and hypermethylated. The two key regulators of histone modifications are histone deacetylases and histone methyltransferases (MTases), which are associated with methyl-binding proteins (MBPs) and MTases.128–130 However, global alterations of histone modification patterns can interrupt normal gene expression, thus the genome's structure and integrity.131 Structurally, histone can assemble into octamers, where a strand of 146 bps long DNA then wraps around to create a nucleosome. Acetylation or deacetylation of the lysine residue on nucleosomes can promote or suppress DNA replication accordingly. Accumulation of histones and nucleosomes in a cell has also been shown to be the early markers of cell death.132,133 Although it is well known that epigenetic modifications of histones can affect different nucleosome structures, the mechanism(s) of action is still unknown.134–136Several studies have been conducted on the translocation or unravelling of a nucleosome and its subunit structure through NP.136,138,139 Generally, it was found that DNA–histone complexes lead to higher applied voltage required and overall longer time periods to translocate through the NP, most likely due to either: (1) the bulky disk shape nucleosome experienced a higher drag force comparing to a bare dsDNA, (2) the positively charged histone core lowered the total net charge density of nucleosomes, causing the translocation speed in electrophoresis to reduce, and (3) the unwinding process of histone–DNA complex.136,140
As mentioned earlier, epigenetic modifications have been known to affect the structural integrity and stability of nucleosomes. Given this fact, it was hypothesized that methylation of CpGs on dsDNA would affect the way nucleosomes fold and/or unravel. To test this hypothesis, the Langecker et al. investigated the influence of DNA methylation on the stability of unlabeled mononucleosomes.139 Similar to the results reported in other studies, under the electrophoretic force, the nucleosomal DNA tail entered the pore and gradually unraveled under increasing voltage, which was much higher in comparison with free DNA capture.141 This experiment was repeated on nucleosomes with and without methylated DNA sequences, yielding that methylation of CpGs did not affect the nucleosome assembly, stability, or unraveling trajectories. This finding suggested that histone modifications (i.e. acetylation and phosphorylation) play a much more dominant role in nucleosomal maintenance than DNA methylation. The confirmation of methylation-independent nucleosome stability indicated other possible mechanisms by which DNA methylation alters gene expression, for example, modulating the binding of transcription activators/repressor.139
The NP-based studies outlined herein lay the groundwork for understanding and predicting the influence of different histone core modifications on the nucleosome structure,139 in which our knowledge is still quite limited. Unlike conventional methods (i.e., single-gene chromatin immunoprecipitation (ChIP), ChIP with a DNA array (ChIP-on-chip),142,143 HPLC, HPCE, and many others), NP devices is more versatile, because they do not rely heavily on the quality of the polyclonal antibodies or antibodies that are available.101 Although the study here indicated that DNA methylation does not affect the nucleosome assemble, further studies need to be done in order to confirm the role of DNA methylation in other processes (i.e. regulating transcription activators/repressors binding, or gene expressions), as well as the relationship between acetylation and phosphorylation on nucleosome assembly, and chromatin stability.
5.3 MicroRNA
MiRNAs are small endogenous biomolecules that are in length of 18–22 bps. They play an important role in embryonic differentiation, hematopoiesis, cardiac hypertrophy and numerous cancer-related processes, including proliferation, apoptosis, differentiation, migration and metabolism.144,145 Since a single miRNA can target up to hundreds of mRNAs,137 an aberrant miRNA expression may affect several transcripts and cancer-related signaling pathways. In cancer cells, because of the genetic diversity of tumors and cancer cell lines, an individual miRNA can be up-regulated in one type of cancer and down-regulated in another.137 Overall, miRNAs function depends on their targets within the specific tissue.12 Usually, the up-regulated miRNAs function as oncogenes by down-regulating tumor-suppressor genes, while the down-regulated miRNAs function as tumor-suppressor genes by down-regulating oncogenes.Detection of miRNAs faces several challenges, mainly due to the shortness of miRNAs. Some quantitative methods have been applied to miRNA detection with enhanced sensitivity and/or selectivity, including quantitative reverse transcription real-time polymerase chain reaction (qRT-PCR) assays, microarrays, colorimetry, bioluminescence, enzyme turnover, electrochemistry, molecular beacons, deep sequencing and single-molecule fluorescence.146–149 Unfortunately, these techniques incur DNA amplification errors, unavailable internal controls, and cross-hybridization. Also, the short sequence of miRNAs makes the designing of probes and primers even more challenging.146,148
MiRNAs have been investigated as potential molecular biomarkers, because their expression levels are associated with various diseases.150 For instance, each year, lung cancer causes approximately 1.2 million deaths worldwide.151 Since there is no effective screening procedure available, more than 70% of lung cancer patients were diagnosed with less than a 15% chance of a 5 year survival rate.151 More than 100 types of miRNAs have been identified to deregulate lung cancer progression.150 Noticeably, high levels of miR155 and low levels of let-7a-2 have been associated with a significantly poor prognosis and shorter survival times in lung cancer patients.152,153 Many research groups have used biological and solid-state NPs for the detection of miRNAs in different tissues. For example, the solid-state NP was used for rapid detection of probe-specific miRNAs (miRNA-122a and miRNA-153).154 Specifically, for every 1 fmol of miRNA duplex per mL solution, the capture rate was 1 molecule per second. In this study, the p19 protein from the Carnation Italian ringspot virus was used to enrich miRNA-122a and miRNA-153. Since miRNA concentrations were 1% relative to other cellular RNAs, to detect a specific miRNA using a NP sequence, an enrichment step was required.154 P19 binds 21–23 bps dsRNA in a size-dependent, but sequence-independent manner. Additionally, the highly affinitive and selective viral p19 protein does not bind ssRNA, tRNA or rRNA. This eliminates the possibility of false results from mismatched binding.155 Detection of 250 molecules in 4 minutes was sufficient to determine miRNA concentration with 93% confidence.154
A different approach from using viral proteins for probe-specific miRNAs detection is to employ an engineered-probe with a programmable sequence to differentiate single nucleotide differences in miRNA family members.150 The Wang et al. proposed a system that enabled sensitive, selective, and direct quantifications of cancer-associated miRNAs in the blood. In this study, the group constructed a robust protein nanopore-based sensor that utilized an oligonucleotide probe (P155) to detect aberrant expression of miRNA-155 and let-7a-2 from lung cancer patients.150 The generated signature electrical signals provided a direct and label-free detection of the target miRNA in a fluctuating background, such as plasma RNA extract.150 Probe (P155) has a programmable sequence and can be optimized to achieve high sensitivity and selectivity. Additionally, using chemical modifications, distinct probes can further be engineered with specific barcodes, allowing multiple miRNAs to be simultaneously detected. Furthermore, with the development of miRNA markers, manipulatable miRNA profile detection NP arrays can be constructed for a noninvasive screening and early diagnosis of cancer.150
Comparing to qRT-PCR assays, microarrays, colorimetry, bioluminescence, and other current methods,146–149 NP arrays is a simpler, faster methods to detect miRNAs in cancer patients. This approach lacks all the complications that conventional methods have, such as DNA amplification errors, unavailable internal controls, and cross-hybridization. Early detection is one of the most crucial contributors to a higher survival rate, especially lung cancer patients (Fig. 6).151
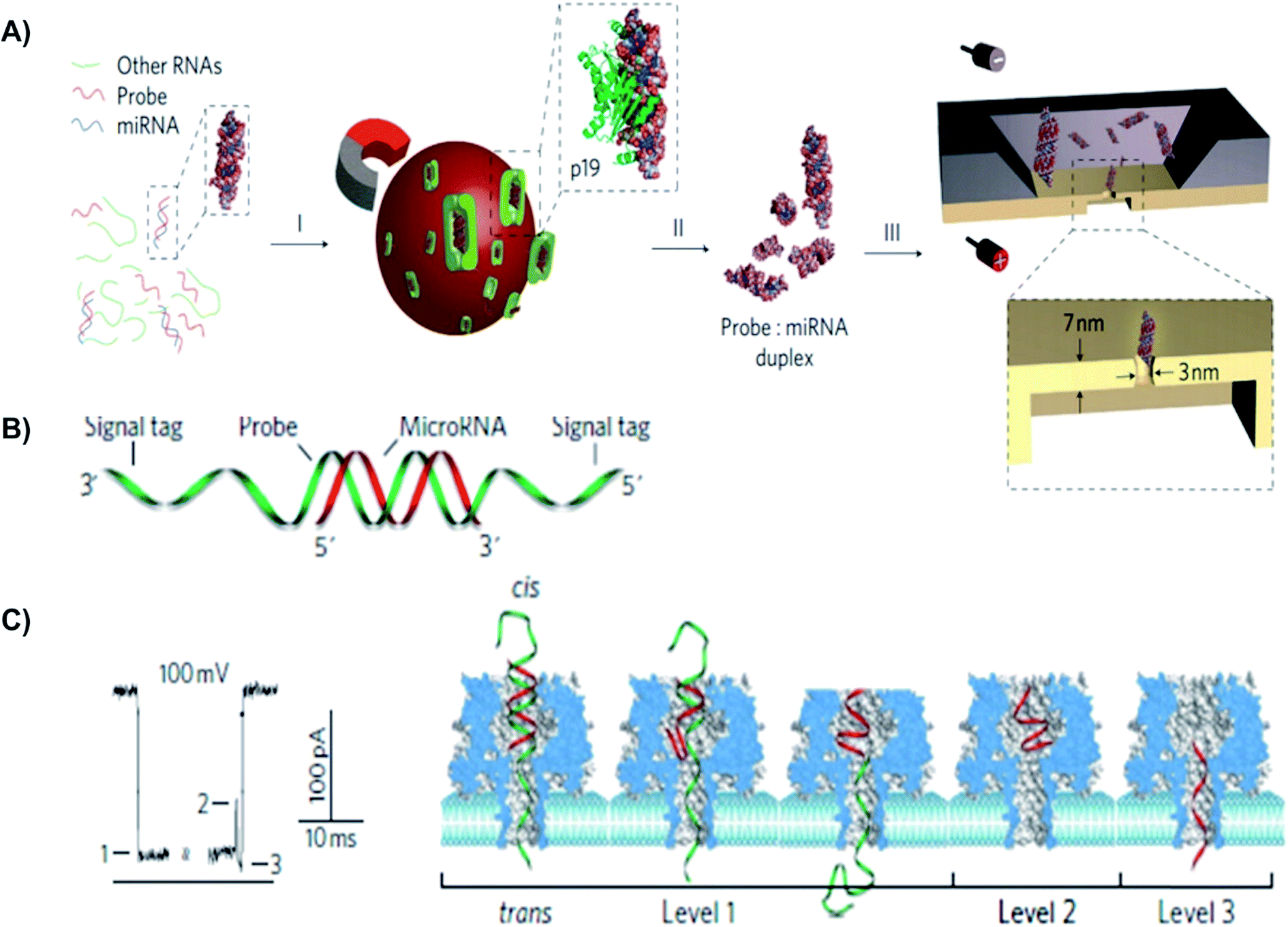 | ||
| Fig. 6 Detection of a miR-155 using using solid-state and biological α-hemolysin nanopores. (A) Schematic of miRNA detection with viral proteins for probe-specific miRNA, using solid-state nanopore. Protein from Carnation Italian ringspot virus was used to enrich miRNA form background fluid. (B) Detection of probe-specific miRNA using alpha-hemolysin biological nanopore. MiRNA-155 (shown in red) was attached to a DNA P155 probe (shown in green). (C) At 8.0 pH and 100 mV, translocation of the miRNA-155·P155 resulted in various current blockage patterns. (C) A typical current blockade with three characteristic blocking levels, representing the mechanism of miRNA-155·P155 complex dissociation and translocation through the pore (as shown in the right-hand side).154 | ||
6. Summary and conclusions
In this paper, we have concisely reviewed the main genetic and epigenetic causal factors of cancer, as well as summarized how NPs have been used in the research of each factor. Thanks to several unique features of this emerging technology, the NP-based analysis offers four main benefits: firstly, NP analysis offers long reads of genomic DNA (>10 kB). Therefore, linkages between modified cytosines may be revealed that are biologically significant and otherwise difficult to discern. For example, it was shown that histone–DNA interaction is not affected by methylation of DNA. Also, for the first time, differences between caC, fC, and hmC from mC were successfully distinguished. Secondly, the genomic DNA is read directly as it transports through the NP. Thus, errors (false-positive results) caused by copying do not occur. Thirdly, with biological NP membranes, the study of biomolecules' folding-unfolding kinetics and mechanisms are possible to accomplish. Furthermore, the DNA fragment can be retained in NP indefinitely, allowing rereads of a captured DNA fragment.123 Lastly, many conventional methods are still impractical for clinical testing, because these methods require highly trained experts, intensive labor, a high capital cost, and a large footprint. With NP technology, there are no such requirements, offering more flexibility and practicality for research labs and clinics (Table 1).| Cancer biomarkers | Type of nanopore | Support needed | Advantages | Limitations | Ref. | |
|---|---|---|---|---|---|---|
| BL | SS | |||||
| a BL = biological nanopore. SS = solid-state nanopore. (*) label-free and do not require additional aid, in order to detect biomolecules. | ||||||
| Structural variants | ✓ | * | Does not require multiple processing steps and file output formats. Low capital cost and short sequencing time | Low sensitivity and accuracy, high mismatch rate. Thus, PHRED score is not high enough for cancer detection yet | 33 | |
| Transcriptional factors | ✓ | * | Label- and tether-free. Does not require chemical-crosslinking or tagging. Hence, allow direct detection and distinguishing between full versus partial, and specific versus nonspecific bindings | Not able to predict the exact binding site TFs on an unknown DNA sequence | 63 | |
| Telomere | ✓ | * | Does not require complicated sample preparation and labeling, thus, allow the kinetics and folding/unfolding mechanism of the four different G-quadruplex structures to be studied | The correlation between telomere folding/unfolding and shortening has not been clarified | 10, 74–76, 78 and 79 | |
| ✓ | Poly-adenosine tail | |||||
| Aberrant methylation of CpGs | ✓ | ✓ | Methyl specific labelers | Does not require PCR or complicated sample amplification. Hence, eliminate false-positive results and allow epigenetic changes to be captured. Real-time detection for all four variants of cytosine (mC, cacC, fC, hmC) | Only provide coarse quantification of methylation on CpGs. Thus, it is still challenging to finely quantify and map the methylation profile of CpGs at the promoter region (essential for precancerous and tumorous detection) | 26, 27, 107 and 119–121 |
| ✓ | Optical-tagging | |||||
| Histone–DNA modification | ✓ | * | Show that methylation of DNA does not affect the ability of nucleosome to ravel and unravel. Moreover, NP allows nucleosomes to unravel with the double-stranded DNA remain unzipped | Has not proved the ability to detect and/or map global changes of post-translational modifications (i.e. methylation, acetylation) on nucleosome | 135–138 | |
| Expression of miRNA | ✓ | ✓ | Engineered probe | Does not require amplification and cross-hyberdization of samples | Multiple miRNAs can be up- or downregulated at the same time in cancerous cells. Hence, several miRNAs have to be tested and clustered together. However, since the sensitivity and accuracy of NP still low, this will potentially lead to false results | 149 and 153 |
| Viral proteins | Multiple miRNAs can potentially be detected using engineered probe. Data were consistent with good statistical distribution | |||||
Although the concepts of NP analysis in early cancer detection are exceptionally promising, several key technological challenges must be addressed before this method can be implemented in clinical uses. First and foremost, the biggest drawback of NP-based methods is high mismatch and error rates. Because the NP membrane thickness, especially biological ones, is relatively large comparing to a nucleotide, NP sensitivity is still low at the single-nucleotide level. Furthermore, even though different DNA conformations and foldings yield distinguishing characteristic current blockades, information about the molecular structure cannot be determined by NP membrane alone. In order to confirm the exact structure that causes a signature blockades in NP, researchers need the aid of other equipment, such as circular dichroism (CD), FRET, FISH, among many others. This limits the use of NP membranes as an independent, stand-alone tool for molecular studies in general, and early cancer detection, specifically. Moreover, since one single biological molecule can quickly adopt multiple, complex conformations under different environments, many research groups choose to use short/simplified sequences in their NP studies. Hence, the complexity of cancer cells has not yet been demonstrated and/or fully investigated with NP membranes.
With this review paper, we hope to give our readers an overview of the essential genetic and epigenetic modifications in cancerous tissue and the progression of cancer cells. With the complexity of the human body and more specifically cancer tissues, many of the mechanisms for cancer proliferation remain unknown. NP-based membranes have shown their ability to detect various biomolecules chemical and structural modifications, as well as genetic and epigenetic modifications. Thus, NP technology could be the one simple solution replacing many costly, labor-intensive conventional cancer screening methods. With the complexity of the field, there is growing opportunity for more significant research to be conducted in the next few decades.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Rowan University Startup Fund.References
- B. M. Venkatesan, B. Dorvel, S. Yemenicioglu, N. Watkins, I. Petrov and R. Bashir, Adv. Mater., 2009, 21, 2771–2776 CrossRef CAS PubMed.
- Y. Feng, Y. Zhang, C. Ying, D. Wang and C. Du, Genomics, Proteomics Bioinf., 2015, 13, 4–16 CrossRef PubMed.
- M. Rhee and M. A. Burns, Trends Biotechnol., 2007, 25, 174–181 CrossRef CAS PubMed.
- T. Deng, M. Li, Y. Wang and Z. Liu, Sci. Bull., 2015, 60, 304–319 CrossRef CAS.
- H. Kwok, K. Briggs and V. Tabard-Cossa, PLoS One, 2014, 9, e92880 Search PubMed.
- B. N. Miles, A. P. Ivanov, K. A. Wilson, F. Doğan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15–28 RSC.
- T. A. Desai, D. J. Hansford, L. Kulinsky, A. H. Nashat, G. Rasi, J. Tu, Y. Wang, M. Zhang and M. Ferrari, Biomed. Microdevices, 1999, 2, 11–40 CrossRef CAS.
- L.-Q. Gu and J. W. Shim, Analyst, 2010, 135, 441–451 RSC.
- S. Banerjee, J. Shim, J. Rivera, X. Jin, D. Estrada, V. Solovyeva, X. You, J. Pak, E. Pop and N. Aluru, ACS Nano, 2012, 7, 834–843 CrossRef PubMed.
- J. Shim and L.-Q. Gu, Methods, 2012, 57, 40–46 CrossRef CAS PubMed.
- S. Banerjee, J. Wilson, J. Shim, M. Shankla, E. A. Corbin, A. Aksimentiev and R. Bashir, Adv. Funct. Mater., 2015, 25, 936–946 CrossRef CAS PubMed.
- C. M. Croce, N. Engl. J. Med., 2008, 358, 502–511 CrossRef CAS PubMed.
- T. A. Libermann and L. F. Zerbini, Curr. Gene Ther., 2006, 6, 17–33 CrossRef CAS PubMed.
- C.-S. Ku and D. H. Roukos, Expert Rev. Med. Devices, 2013, 10, 1–6 CrossRef CAS PubMed.
- B. Tran, J. E. Dancey, S. Kamel-Reid, J. D. McPherson, P. L. Bedard, A. M. Brown, T. Zhang, P. Shaw, N. Onetto and L. Stein, J. Clin. Oncol., 2012, 30, 647–660 CrossRef PubMed.
- M. L. Metzker, Nat. Rev. Genet., 2010, 11, 31–46 CrossRef CAS PubMed.
- S. P. Shah, A. Roth, R. Goya, A. Oloumi, G. Ha, Y. Zhao, G. Turashvili, J. Ding, K. Tse and G. Haffari, Nature, 2012, 486, 395–399 CAS.
- S. Banerji, K. Cibulskis, C. Rangel-Escareno, K. K. Brown, S. L. Carter, A. M. Frederick, M. S. Lawrence, A. Y. Sivachenko, C. Sougnez and L. Zou, Nature, 2012, 486, 405–409 CrossRef CAS PubMed.
- M. J. Ellis, L. Ding, D. Shen, J. Luo, V. J. Suman, J. W. Wallis, B. A. Van Tine, J. Hoog, R. J. Goiffon and T. C. Goldstein, Nature, 2012, 486, 353–360 CAS.
- C. Curtis, S. P. Shah, S.-F. Chin, G. Turashvili, O. M. Rueda, M. J. Dunning, D. Speed, A. G. Lynch, S. Samarajiwa and Y. Yuan, Nature, 2012, 486, 346–352 CAS.
- C. S. Ross-Innes, R. Stark, A. E. Teschendorff, K. A. Holmes, H. R. Ali, M. J. Dunning, G. D. Brown, O. Gojis, I. O. Ellis and A. R. Green, Nature, 2012, 481, 389–393 CAS.
- N. Aziz, Q. Zhao, L. Bry, D. K. Driscoll, B. Funke, J. S. Gibson, W. W. Grody, M. R. Hegde, G. A. Hoeltge and D. G. Leonard, Arch. Pathol. Lab. Med., 2014, 139, 481–493 CrossRef PubMed.
- H. L. Rehm, S. J. Bale, P. Bayrak-Toydemir, J. S. Berg, K. K. Brown, J. L. Deignan, M. J. Friez, B. H. Funke, M. R. Hegde and E. Lyon, Genet. Med., 2013, 15, 733–747 CrossRef PubMed.
- R. Luthra, H. Chen, S. Roy-Chowdhuri and R. R. Singh, Cancers, 2015, 7, 2023–2036 CrossRef PubMed.
- M. R. Schweiger, M. Kerick, B. Timmermann and M. Isau, Cancer Metastasis Rev., 2011, 30, 199–210 CrossRef CAS PubMed.
- J. Shim, Y. Kim, G. I. Humphreys, A. M. Nardulli, F. Kosari, G. Vasmatzis, W. R. Taylor, D. A. Ahlquist, S. Myong and R. Bashir, ACS Nano, 2015, 9, 290–300 CrossRef CAS PubMed.
- T. Gilboa, C. Torfstein, M. Juhasz, A. Grunwald, Y. Ebenstein, E. Weinhold and A. Meller, ACS Nano, 2016, 10, 8861–8870 CrossRef CAS PubMed.
- J. J. Nakane, M. Akeson and A. Marziali, J. Phys.: Condens. Matter, 2003, 15, R1365 CrossRef CAS.
- M. Rhee and M. A. Burns, Trends Biotechnol., 2006, 24, 580–586 CrossRef CAS PubMed.
- M. Wanunu, Phys. Life Rev., 2012, 9, 125–158 CrossRef PubMed.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs and X. Huang, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS PubMed.
- R. D. Maitra, J. Kim and W. B. Dunbar, Electrophoresis, 2012, 33, 3418–3428 CrossRef CAS PubMed.
- A. e. a. Norris, Cancer Biol. Ther., 2016, 17, 246–253 CrossRef CAS PubMed.
- L. C. Strong, V. M. Riccardi, R. E. Ferrell and R. S. Sparkes, Science, 1981, 213, 1501–1503 CAS.
- J. B. Konopka, S. M. Watanabe, J. W. Singer, S. J. Collins and O. N. Witte, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 1810–1814 CrossRef CAS.
- Y. Tsujimoto, J. Gorham, J. Cossman, E. Jaffe and C. M. Croce, Science, 1985, 229, 1390–1393 CAS.
- D. G. Albertson, C. Collins, F. McCormick and J. W. Gray, Nat. Genet., 2003, 34, 369–376 CrossRef CAS PubMed.
- M. Mijušković, S. M. Brown, Z. Tang, C. R. Lindsay, E. Efstathiadis, L. Deriano and D. B. Roth, PLoS One, 2012, 7, e48314 Search PubMed.
- A. Namazie, S. Alavi, O. I. Olopade, G. Pauletti, N. Aghamohammadi, M. Aghamohammadi, J. A. Gornbein, T. C. Calcaterra, D. J. Slamon and M. B. Wang, Laryngoscope, 2002, 112, 472–481 CrossRef CAS PubMed.
- C. Alkan, B. P. Coe and E. E. Eichler, Nat. Rev. Genet., 2011, 12, 363–376 CrossRef CAS PubMed.
- P. C. Nowell and D. Hungerford, J. Natl. Cancer Inst., 1960, 25, 85–109 CAS.
- T. P. Dryja, J. Rapaport, J. M. Joyce and R. A. Petersen, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 7391–7394 CrossRef CAS.
- D. J. Slamon, G. Clark, S. G. Wong, W. J. Levin, A. Ullrich and W. L. McGuire, Science, 1987, 82, 177–182 Search PubMed.
- S. Jones, X. Zhang, D. W. Parsons, J. C. Lin, R. J. Leary, P. Angenendt, P. Man-koo, H. Carter, H. Kamiyama and A. Jimeno, et al., Science, 2008, 1801–1806 CrossRef CAS PubMed.
- A. V. Biankin, N. Waddell, K. S. Kassahn, M. C. Gingras, L. B. Muthuswamy, A. L. Johns, D. K. Miller, P. J. Wilson, A. M. Patch and J. Wu, et al., Nature, 2012, 491, 399–405 CrossRef CAS PubMed.
- A. L. Norris, H. Kamiyama, A. Makohon-Moore, A. Pallavajjala, L. A. Morsberger, K. Lee, D. Batista, C. A. Iacobuzio-Donahue, M. T. Lin and A. P. Klein, et al., Genes, Chromosomes Cancer, 2015, 54, 472–481 CrossRef CAS PubMed.
- F. Diehl, K. Schmidt, M. A. Choti, K. Romans, S. Goodman, M. Li, K. Thornton, N. Agrawal, L. Sokoll and S. A. Szabo, et al., Nat. Med., 2008, 14, 985–990 CrossRef CAS PubMed.
- A. Ritz, A. Bashir, S. Sindi, D. Hsu, I. Hajirasouliha and B. J. Raphael, Bioinformatics, 2014, 30, 3458–3466 CrossRef CAS PubMed.
- P. Medvedev, M. Stanciu and M. Brudno, Nat. Methods, 2009, 6, S13–S20 CrossRef CAS PubMed.
- B. Ewing and P. Green, Genome Res., 1998, 8, 186–194 CrossRef CAS PubMed.
- J. E. Darnell, Nat. Rev. Cancer, 2002, 2, 740–749 CrossRef CAS PubMed.
- P. Blume-Jensen and T. Hunter, Nature, 2001, 411, 355–365 CrossRef CAS PubMed.
- R. Catlett-Falcone, T. H. Landowski, M. M. Oshiro, J. Turkson, A. Levitzki, R. Savino, G. Ciliberto, L. Moscinski, J. L. Fernández-Luna and G. Nuñez, Immunity, 1999, 10, 105–115 CrossRef CAS PubMed.
- T. Bowman, R. Garcia, J. Turkson and R. Jove, Oncogene, 2000, 19, 2474 CrossRef CAS PubMed.
- P. J. Coffer, L. Koenderman and R. P. de Groot, Oncogene, 2000, 19, 2511–2522 CrossRef CAS PubMed.
- J. I. Song and J. R. Grandis, Oncogene, 2000, 19, 2489 CrossRef CAS PubMed.
- J. Bromberg, J. Clin. Invest., 2002, 109, 1139–1142 CrossRef CAS PubMed.
- M. M. Garner and A. Revzin, Nucleic Acids Res., 1981, 9, 3047–3060 CrossRef CAS PubMed.
- C. A. Frederick, J. Grable, M. Melia, C. Samudzi, L. Jen-Jacobson, W. Bi-Cheng, P. Greene, H. W. Boyer and J. M. Rosenberg, Nature, 1984, 309, 327–331 CrossRef CAS PubMed.
- N. P. Pavletich and C. O. Pabo, Science, 1991, 252, 809–817 CrossRef CAS PubMed.
- D. S. Johnson, A. Mortazavi, R. M. Myers and B. Wold, Science, 2007, 316, 1497–1502 CrossRef CAS PubMed.
- Y. Takayama, D. Sahu and J. Iwahara, Biochemistry, 2010, 49, 7998–8005 CrossRef CAS PubMed.
- A. e. a. Squires, Sci. Rep., 2015, 5, 11643 CrossRef CAS PubMed.
- F. Spitz and E. E. M. Furlong, Nat. Rev. Genet., 2012, 13, 613 CrossRef CAS PubMed.
- C. Bettegowda, M. Sausen, R. J. Leary, I. Kinde, Y. Wang, N. Agrawal, B. R. Bartlett, H. Wang, B. Luber and R. M. Alani, et al., Sci. Transl. Med., 2014, 6, 224 Search PubMed.
- S. Brouilette, R. K. Singh, J. R. Thompson, A. H. Goodall and N. J. Samani, Arterioscler., Thromb., Vasc. Biol., 2003, 23, 842–846 CrossRef CAS PubMed.
- C. G. Maubaret, K. D. Salpea, A. Jain, J. A. Cooper, A. Hamsten, J. Sanders, H. Montgomery, A. Neil, D. Nair and S. E. Humphries, J. Mol. Med., 2010, 88, 785–794 CrossRef PubMed.
- M. Weischer, S. E. Bojesen, R. M. Cawthon, J. J. Freiberg, A. Tybjærg-Hansen and B. G. Nordestgaard, Arterioscler., Thromb., Vasc. Biol., 2012, 32, 822–829 CrossRef CAS PubMed.
- M. Weischer, B. G. Nordestgaard, R. M. Cawthon, J. J. Freiberg, A. Tybjærg-Hansen and S. E. Bojesen, J. Natl. Cancer Inst., 2013, 459–468 CrossRef CAS PubMed.
- M. P. Hande, E. Samper, P. Lansdorp and M. A. Blasco, J. Cell Biol., 1999, 144, 589–601 CrossRef CAS PubMed.
- W. C. Hahn, S. A. Stewart, M. W. Brooks, S. G. York, E. Eaton, A. Kurachi, R. L. Beijersbergen, J. H. Knoll, M. Meyerson and R. A. Weinberg, Nat. Med., 1999, 5, 1164–1170 CrossRef CAS PubMed.
- C. P. Morales, S. E. Holt, M. Ouellette, K. J. Kaur, Y. Yan, K. S. Wilson, M. A. White, W. E. Wright and J. W. Shay, Nat. Genet., 1999, 21, 115–118 CrossRef CAS PubMed.
- R. M. Cawthon, Nucleic Acids Res., 2002, 30, e47 CrossRef PubMed.
- J. W. Shim and L.-Q. Gu, J. Phys. Chem. B, 2008, 112, 8354 CrossRef CAS PubMed.
- J. W. Shim, Q. Tan and L.-Q. Gu, Nucleic Acids Res., 2009, 37, 972–982 CrossRef CAS PubMed.
- N. An, A. M. Fleming, E. G. Middleton and C. J. Burrows, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14325–14331 CrossRef CAS PubMed.
- J. W. F. Robertson, J. J. Kasianowicz and S. Banerjee, Chem. Rev., 2012, 112, 6227–6249 CrossRef CAS PubMed.
- Y. Jung, S. Cheley, O. Braha and H. Bayley, Biochemistry, 2005, 44, 8918–8929 CrossRef.
- Y. Ding, A. M. Fleming, L. He and C. J. Burrows, J. Am. Chem. Soc., 2015, 137, 9053–9060 CrossRef CAS PubMed.
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459–3469 CrossRef CAS PubMed.
- G. Liang, A. A. Qureshi, Q. Guo, I. De Vivo and J. Han, Cancer Epidemiol., Biomarkers Prev., 2011, 20, 1043–1045 CrossRef CAS PubMed.
- Z. Liu, H. Ma, S. Wei, G. Li, E. M. Sturgis and Q. Wei, Cancer Epidemiol., Biomarkers Prev., 2011, 20, 2642–2645 CrossRef CAS PubMed.
- L. Hou, X. Zhang, A. J. Gawron and J. Liu, Cancer Lett., 2012, 319, 130–135 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- M. C. Haffner, A. Chaux, A. K. Meeker, D. Esopi, J. Gerber, L. G. Pellakuru, A. Toubaji, P. Argani, C. Iacobuzio-Donahue and W. G. Nelson, Oncotarget, 2011, 2, 627–637 Search PubMed.
- L. Holmfeldt and C. G. Mullighan, Cancer Cell, 2011, 20, 1–2 CrossRef CAS PubMed.
- C.-X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C.-H. Chen, W. Zhang and X. Jian, Nat. Biotechnol., 2011, 29, 68–72 CrossRef CAS PubMed.
- H. Yang, Y. Liu, F. Bai, J. Zhang, S. Ma, J. Liu, Z. Xu, H. Zhu, Z. Ling and D. Ye, Oncogene, 2013, 32, 663–669 CrossRef CAS PubMed.
- A. Bacolla, D. N. Cooper and K. M. Vasquez, Genes, 2014, 5, 108–146 CrossRef PubMed.
- M. Esteller, P. G. Corn, S. B. Baylin and J. G. Herman, Cancer Res., 2001, 61, 3225–3229 CAS.
- G. Strathdee and R. Brown, Expert Rev. Mol. Med., 2002, 4, 1–17 CAS.
- P. W. Laird, Nat. Rev. Cancer, 2003, 3, 253–266 CrossRef CAS PubMed.
- D. K. Vanaja, M. Ehrich, D. Van den Boom, J. C. Cheville, R. J. Karnes, D. J. Tindall, C. R. Cantor and C. Y. Young, Cancer Invest., 2009, 27, 549–560 CrossRef CAS PubMed.
- N. Nakamura and K. Takenaga, Clin. Exp. Metastasis, 1998, 16, 471–479 CrossRef CAS PubMed.
- A. M. Adrianadecapoa, S. Della rosa, P. Caiafa, L. Mariani, F. Del nonno, A. Vocaturo, R. A. P. Donnorso, A. Niveleau and C. Grappelli, Oncol. Rep., 2003, 10, 545–549 Search PubMed.
- Y. Akiyama, C. Maesawa, S. Ogasawara, M. Terashima and T. Masuda, Am. J. Pathol., 2003, 163, 1911–1919 CrossRef CAS PubMed.
- C. A. Iacobuzio-Donahue, A. Maitra, M. Olsen, A. W. Lowe, N. T. Van Heek, C. Rosty, K. Walter, N. Sato, A. Parker and R. Ashfaq, Am. J. Pathol., 2003, 162, 1151–1162 CrossRef CAS PubMed.
- Y. Oshimo, H. Nakayama, R. Ito, Y. Kitadai, K. Yoshida, K. Chayama and W. Yasui, Int. J. Oncol., 2003, 23, 1663–1670 CAS.
- N. Sato, A. Maitra, N. Fukushima, N. T. van Heek, H. Matsubayashi, C. A. Iacobuzio-Donahue, C. Rosty and M. Goggins, Cancer Res., 2003, 63, 4158–4166 CAS.
- J. Shim, G. I. Humphreys, B. M. Venkatesan, J. M. Munz, X. Zou, C. Sathe, K. Schulten, F. Kosari, A. M. Nardulli and G. Vasmatzis, Sci. Rep., 2013, 3, 1389 CrossRef PubMed.
- M. Esteller, Nat. Rev. Genet., 2007, 8, 286–298 CrossRef CAS PubMed.
- J. C. Susan, J. Harrison, C. L. Paul and M. Frommer, Nucleic Acids Res., 1994, 22, 2990–2997 CrossRef.
- J. G. Herman, J. R. Graff, S. Myöhänen, B. D. Nelkin and S. B. Baylin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- C. A. Eads, K. D. Danenberg, K. Kawakami, L. B. Saltz, C. Blake, D. Shibata, P. V. Danenberg and P. W. Laird, Nucleic Acids Res., 2000, 28, e32 CrossRef CAS PubMed.
- M. F. Fraga and M. Esteller, BioTechniques, 2002, 33, 632–649 CAS.
- M. Weber, J. J. Davies, D. Wittig, E. J. Oakeley, M. Haase, W. L. Lam and D. Schuebeler, Nat. Genet., 2005, 37, 853–862 CrossRef CAS PubMed.
- I. Keshet, Y. Schlesinger, S. Farkash, E. Rand, M. Hecht, E. Segal, E. Pikarski, R. A. Young, A. Niveleau and H. Cedar, Nat. Genet., 2006, 38, 149–153 CrossRef CAS PubMed.
- B. Khulan, R. F. Thompson, K. Ye, M. J. Fazzari, M. Suzuki, E. Stasiek, M. E. Figueroa, J. L. Glass, Q. Chen and C. Montagna, Genome Res., 2006, 16, 1046–1055 CrossRef CAS PubMed.
- J. Shim, J. A. Rivera and R. Bashir, Nanoscale, 2013, 5, 10887–10893 RSC.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu and L. Aravind, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- S. Ito, A. C. D'Alessio, O. V. Taranova, K. Hong, L. C. Sowers and Y. Zhang, Nature, 2010, 466, 1129–1133 CrossRef CAS PubMed.
- H. Zhang, X. Zhang, E. Clark, M. Mulcahey, S. Huang and Y. G. Shi, Cell Res., 2010, 20, 1390–1393 CrossRef PubMed.
- H. Wu and Y. Zhang, Genes Dev., 2011, 25, 2436–2452 CrossRef CAS PubMed.
- A. Szwagierczak, S. Bultmann, C. S. Schmidt, F. Spada and H. Leonhardt, Nucleic Acids Res., 2010, 38, e181 CrossRef PubMed.
- M. Wossidlo, T. Nakamura, K. Lepikhov, C. J. Marques, V. Zakhartchenko, M. Boiani, J. Arand, T. Nakano, W. Reik and J. Walter, Nat. Commun., 2011, 2, 241 CrossRef PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef CAS PubMed.
- T. S. Shankar and L. Willems, Vasc. Pharmacol., 2014, 60, 57–66 CrossRef PubMed.
- Y. Fu and C. He, Curr. Opin. Chem. Biol., 2012, 16, 516–524 CrossRef CAS PubMed.
- C. G. Lian, Y. Xu, C. Ceol, F. Wu, A. Larson, K. Dresser, W. Xu, L. Tan, Y. Hu and Q. Zhan, Cell, 2012, 150, 1135–1146 CrossRef CAS PubMed.
- M. Yu, G. C. Hon, K. E. Szulwach, C.-X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen and B. Park, Cell, 2012, 149, 1368–1380 CrossRef CAS PubMed.
- M. Wanunu, D. Cohen-Karni, R. R. Johnson, L. Fields, J. Benner, N. Peterman, Y. Zheng, M. L. Klein and M. Drndic, J. Am. Chem. Soc., 2011, 133, 486–492 CrossRef CAS PubMed.
- A. H. Laszlo, I. M. Derrington, H. Brinkerhoff, K. W. Langford, I. C. Nova, J. M. Samson, J. J. Bartlett, M. Pavlenok and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18904–18909 CrossRef CAS PubMed.
- Z. L. Wescoe, J. Schreiber and M. Akeson, J. Am. Chem. Soc., 2014, 136, 16582–16587 CrossRef CAS PubMed.
- C. T. Nguyen, F. A. Gonzales and P. A. Jones, Nucleic Acids Res., 2001, 29, 4598–4606 CrossRef CAS PubMed.
- J. A. Fahrner, S. Eguchi, J. G. Herman and S. B. Baylin, Cancer Res., 2002, 62, 7213–7218 CAS.
- E. Ballestar, M. F. Paz, L. Valle, S. Wei, M. F. Fraga, J. Espada, J. C. Cigudosa, T. H. M. Huang and M. Esteller, EMBO J., 2003, 22, 6335–6345 CrossRef CAS PubMed.
- M. F. Fraga, E. Ballestar, A. Villar-Garea, M. Boix-Chornet, J. Espada, G. Schotta, T. Bonaldi, C. Haydon, S. Ropero and K. Petrie, Nat. Genet., 2005, 37, 391–400 CrossRef CAS PubMed.
- Y. Wang, W. Fischle, W. Cheung, S. Jacobs, S. Khorasanizadeh and C. D. Allis, Novartis Found. Symp., 2004, 259, 3–17 CAS.
- J. Dobosy and E. Selker, Cell. Mol. Life Sci., 2001, 58, 721–727 CrossRef CAS PubMed.
- P. A. Wade, BioEssays, 2001, 23, 1131–1137 CrossRef CAS PubMed.
- D. B. Seligson, S. Horvath, T. Shi, H. Yu, S. Tze, M. Grunstein and S. K. Kurdistani, Nature, 2005, 435, 1262–1266 CrossRef CAS PubMed.
- D. Wu, A. Ingram, J. H. Lahti, B. Mazza, J. Grenet, A. Kapoor, L. Liu, V. J. Kidd and D. Tang, J. Biol. Chem., 2002, 277, 12001–12008 CrossRef CAS PubMed.
- C. Gabler, N. Blank, T. Hieronymus, M. Schiller, J. Berden, J. Kalden and H. Lorenz, Ann. Rheum. Dis., 2004, 63, 1135–1144 CrossRef CAS PubMed.
- K. Luger, A. W. Mäder, R. K. Richmond, D. F. Sargent and T. J. Richmond, Nature, 1997, 389, 251–260 CrossRef CAS PubMed.
- J. L. Workman and R. E. Kingston, Annu. Rev. Biochem., 1998, 67, 545–579 CrossRef CAS PubMed.
- S. C. Andrey Ivankin, J. Am. Chem. Soc., 2016, 15350–15352 Search PubMed.
- M. D. Jansson and A. H. Lund, Mol. Oncol., 2012, 6, 590–610 CrossRef CAS PubMed.
- G. V. Soni and C. Dekker, Nano Lett., 2012, 12, 3180–3186 CrossRef CAS PubMed.
- M. Langecker, A. Ivankin, S. Carson, S. R. Kinney, F. C. Simmel and M. Wanunu, Nano Lett., 2014, 15, 783–790 CrossRef PubMed.
- M. Wanunu, J. Sutin and A. Meller, Nano Lett., 2009, 9, 3498–3502 CrossRef CAS PubMed.
- A. Ivankin, S. Carson, S. R. Kinney and M. Wanunu, J. Am. Chem. Soc., 2013, 135, 15350–15352 CrossRef CAS PubMed.
- H. Suzuki, E. Gabrielson, W. Chen, R. Anbazhagan, M. van Engeland, M. P. Weijenberg, J. G. Herman and S. B. Baylin, Nat. Genet., 2002, 31, 141–149 CrossRef CAS PubMed.
- K. Yamashita, S. Upadhyay, M. Osada, M. O. Hoque, Y. Xiao, M. Mori, F. Sato, S. J. Meltzer and D. Sidransky, Cancer Cell, 2002, 2, 485–495 CrossRef CAS PubMed.
- R. W. Carthew and E. J. Sontheimer, Cell, 2009, 136, 642–655 CrossRef CAS PubMed.
- J. Winter, S. Jung, S. Keller, R. I. Gregory and S. Diederichs, Nat. Cell Biol., 2009, 11, 228–234 CrossRef CAS PubMed.
- C. Chen, D. A. Ridzon, A. J. Broomer, Z. Zhou, D. H. Lee, J. T. Nguyen, M. Barbisin, N. L. Xu, V. R. Mahuvakar and M. R. Andersen, Nucleic Acids Res., 2005, 33, e179 CrossRef PubMed.
- E. A. Hunt, A. M. Goulding and S. K. Deo, Anal. Biochem., 2009, 387, 1 CrossRef CAS PubMed.
- W. Li and K. Ruan, Anal. Bioanal. Chem., 2009, 394, 1117–1124 CrossRef CAS PubMed.
- S. Yendamuri and R. Kratzke, Transl. Res., 2011, 157, 209–215 CrossRef CAS PubMed.
- Y. Wang, D. Zheng, Q. Tan, M. X. Wang and L. Q. Gu, Nat. Nanotechnol., 2011, 6, 668–674 CrossRef CAS PubMed.
- G. A. Silvestri, A. J. Alberg and J. Ravenel, BMJ, 2009, 339, 30–53 CrossRef PubMed.
- N. Yanaihara, N. Caplen, E. Bowman, M. Seike, K. Kumamoto, M. Yi, R. M. Stephens, A. Okamoto, J. Yokota and T. Tanaka, Cancer Cell, 2006, 9, 189–198 CrossRef CAS PubMed.
- S. K. Patnaik, E. Kannisto, S. Knudsen and S. Yendamuri, Cancer Res., 2010, 70, 36–45 CrossRef CAS PubMed.
- M. Wanunu, T. Dadosh, V. Ray, J. Jin, L. McReynolds and M. Drndić, Nat. Nanotechnol., 2010, 5, 807–814 CrossRef CAS PubMed.
- S. G. Jin, S. Kadam and G. P. Pfeifer, Nucleic Acids Res., 2010, 38, e125 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |